

Abstract
The mechanisms of carotenoid accumulation in yellow-fleshed sweetpotato cultivars are unclear. In this study, we compared the transcriptome profiles of a yellow-fleshed cultivar, Beniharuka (BH) and two of its spontaneous white-fleshed mutants (WH2 and WH3) to reveal the genes involved in yellow flesh. As a result of RNA sequencing, a total of 185 differentially expressed genes (DEGs) were commonly detected in WH2 and WH3 compared to BH. Of these genes, 85 DEGs and 100 DEGs were commonly upregulated and downregulated in WH2 and WH3 compared to BH, respectively. g1103.t1, a paralog of zeaxanthin epoxidase (ZEP), was only DEG common to WH2 and WH3 among 38 genes considered to be involved in carotenoid biosynthesis in storage roots. The expression level of g1103.t1 was also considerably lower in five white-fleshed cultivars than in five yellow-fleshed cultivars. Analysis of carotenoid composition in the storage roots showed that the epoxidised carotenoids were drastically reduced in both WH2 and WH3. Therefore, we propose that the ZEP paralog, g1103.t1, may be involved in carotenoid accumulation through the epoxidation of β-carotene and β-cryptoxanthin in sweetpotato.
Subject terms: Plant breeding, Plant molecular biology, Transcriptomics
Introduction
The flesh colour of the storage roots of the sweetpotato Ipomoea batatas (L.) Lam is one of the most important targets in breeding programs. Sweetpotato cultivars show various flesh colours such as white, yellow, orange and purple depending on their carotenoid or anthocyanin components. In Asia, especially in Japan, yellow-fleshed cultivars are popular for table use and processed products1. In addition to the commercial value of their appearance, yellow-fleshed cultivars may have healthcare functions. Some plant carotenoids have antioxidant activity and are thought to contribute to preventing cancer, heart disease and eye diseases2,3, and carotenoids in yellow-fleshed sweetpotato have been shown to have such activity4,5.
Yellow-fleshed sweetpotato mainly contains unique carotenoids in the form of β-cryptoxanthin epoxides and β-carotene epoxides such as β-cryptoxanthin 5,8-epoxide, β-carotene 5,8;5′,8′-diepoxide, etc.4,6. These pigments cause a yellow colour in storage roots. Figure 1 shows a proposed biosynthetic pathway of carotenoids in sweetpotato7–9. A lot of genes regulate the biosynthesis of carotenoids. The flesh colour (white or yellow) of sweetpotato could be determined by the balance of the synthesis and metabolisation of these yellow pigments1,5. Recently, genes related to carotenoid accumulation were identified in sweetpotato and their functions were revealed8. The carotenoid content in sweetpotato calli was found to be increased by suppressing the gene expression of IbLCYE, IbLCYB and IbCHYB individually10–12, and overexpression of IbOr also increased carotenoid content in calli and storage roots13–15. However, the molecular mechanisms involved in the biosynthesis of β-cryptoxanthin epoxides and β-carotene epoxides remain unclear.
Figure 1.

Proposed carotenoid biosynthetic pathway in sweetpotato. DXS 1-deoxy-d-xylulose 5-phosphate synthase, DXR 1-deoxy-d-xylulose 5-phosphate reductoisomerase, MCT 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase, CMK 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol kinase, MDS 2-C-methyl-d-erythritol 2,4-cyclodiphosphate synthase, HDS 4-hydroxy-3-methylbut-2-enyl diphosphate synthase, HDR 4-hydroxy-3-methylbut-2-enyl diphosphate reductase, DMAPP dimethylallyl diphosphate, IPP isopentenyl pyrophosphate isomerase, GGPS geranylgeranyl pyrophosphate synthase, GGPP geranyl geranyl diphosphate, PSY phytoene synthase, Or orange, PDS phytoene desaturase, Z-ISO 15-cis-ζ-carotene isomerase, ZDS ζ-carotene desaturase, CRTISO carotenoid isomerase, LCYB lycopene β-cyclase, LCYE lycopene ε-cyclase, CHYB carotenoid β-hydroxylase, CHYE carotenoid ε-hydroxylase, CCD carotenoid cleavage dioxygenase, ZEP zeaxanthin epoxidase, VDE violaxanthin de-epoxidase, NXS neoxanthin synthase, NCED 9-cis-epoxycarotenoid dioxygenase.
In the carotenoid metabolism of plants, the cyclisation of lycopene is an essential step16. β-Carotene is synthesised through the addition of β-rings to lycopene by lycopene β-cyclase (LCYB) (Fig. 1). Subsequent hydroxylation of β-carotene β-rings leads to the generation of zeaxanthin via β-cryptoxanthin. Zeaxanthin epoxidase (ZEP) then catalyses the epoxidation of β-rings in zeaxanthin and antheraxanthin. On the other hand, β-cryptoxanthin epoxides and β-carotene epoxides in yellow-fleshed sweetpotato are generated by the epoxidation of β-rings in β-cryptoxanthin and β-carotene, respectively. Khan et al. (2016)7 suggest that these epoxidation steps may be catalysed by a novel enzyme highly homologous to ZEP. However, ZEP paralogs encoding such an enzyme have not yet been identified in sweetpotato.
Flesh-coloured mutants of various sweetpotato cultivars have contributed to revealing the genetic and physiological mechanisms underlying the pigmentation of storage roots. Tanaka et al. (2012)17 studied the gene structure and function of IbMYB1 genes using a white-fleshed mutant (AYM96) of a purple-fleshed cultivar, Ayamurasaki. McGregor and LaBonte (2006)18 compared gene expression for β-carotene accumulation between Jewel and its mutant (White Jewel). Recently, several spontaneous white-fleshed mutants of the cultivar Beniharuka (BH), whose flesh colour is pale yellow, were found. BH and its mutant series would be useful in an exploration of the accumulation mechanism of yellow pigments like β-cryptoxanthin epoxides and β-carotene epoxides.
Sweetpotato has a complex genome with hexaploidy (2n = 6x = 90) and high heterozygosity, and little is known about its genome or genes. Recently, the draft genome sequences of sweetpotato19 and I. trifida, a diploid wild relative of sweetpotato20,21 were released and became available for researchers and breeders. These databases make genomic and transcriptomic approaches easier in studies on sweetpotato. Transcriptome sequencing using a next-generation sequencer is effective to analyse the gene expression profile involved in flesh colour. RNA sequencing (RNA-seq) by de novo assembly has revealed the expression of genes related to anthocyanin accumulation in purple-fleshed sweetpotato cultivars22,23 and β-carotene accumulation in orange-fleshed cultivars24,25. Gemenet et al. (2020)26 also revealed that the phytoene synthase gene (PSY) played an important role in the carotenoid accumulation in orange-fleshed storage roots by combining transcriptome analysis and QTL analysis. However, it remains unknown what genes are essential for the accumulation of carotenoids in yellow-fleshed sweetpotato cultivars. In the present study, we used a yellow-fleshed cultivar (BH) and its two white-fleshed mutants (WH2 and WH3) and compared the gene expression profiles in their storage roots using Illumina paired-end sequencing technology to identify the genes involved in yellow flesh.
Results
Characteristics of BH, WH2 and WH3
The storage roots of BH, WH2 and WH3 used in this experiment are shown in Fig. 2. The a* (red/green) and b* (yellow/blue) values indicated a significant difference in the flesh colours of BH, WH2 and WH3 (Supplementary Table S1) in accordance with their visible appearance. The a* value of BH was lower than those of WH2 and WH3, and its b* value was higher. Measured traits other than flesh colour did not differ among these lines (Supplementary Table S1).
Figure 2.
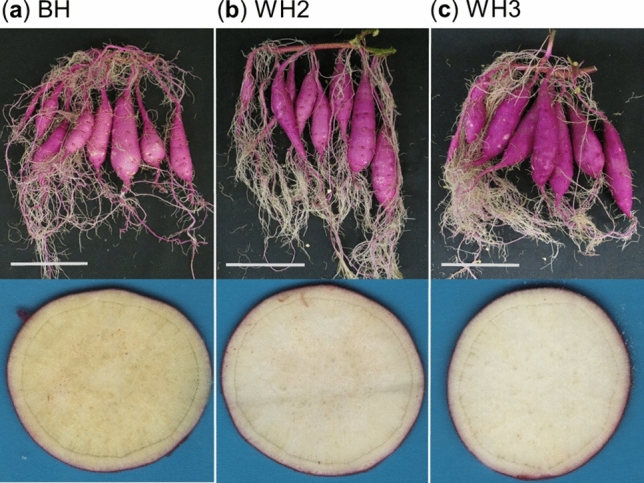
Phenotypic characteristic appearance and cross sections of the storage roots of (a) the Beniharuka cultivar (BH), and its two white-fleshed mutants (b) WH2 and (c) WH3. Bars = 10 cm.
The total carotenoid contents in WH2 and WH3 estimated from the total peak area detected by high performance liquid chromatography (HPLC) chromatograms were more than 85% lower than that in BH (Fig. 3, Supplementary Table S2). Also, the peaks corresponding to the major carotenoids in BH, β-carotene-5,8;5′,8′-diepoxide and β-cryptoxanthin 5,8-epoxide (Fig. 3a, Supplementary Table S2), were not detected in WH2 or WH3 (Figs. 3b,c, Supplementary Table S2). These results suggest that the white flesh of WH2 and WH3 is caused by this large decrease in carotenoids.
Figure 3.

Chromatograms of carotenoids extracted from (a) BH, (b) WH2 and (c) WH3. Peaks were identified by comparing with Ishiguro et al. (2010)4: unknown (1), unknown (2), unknown (3), ipomoeaxanthin C1 (4), β-cryptoxanthin 5,8-epoxide (5), unknown (6), β-carotene 5,8;5′,8′-diepoxide (cis-isomer) (7), β-carotene 5,8;5′,8′-diepoxide (diastereomer) (8, 9), β-carotene 5,8-epoxide (10), β-carotene (11).
Transcriptome profiling in BH, WH2 and WH3 storage roots
RNA-seq data of BH, WH2 and WH3 storage roots with three biological replicates were generated using Illumina NovaSeq 6000. On average, 44 million reads were obtained after removing low-quality reads and Illumina adaptor sequences. Of these reads, approximately 90% were mapped to the reference genome using HISAT2.
To identify differentially expressed genes (DEGs) in WH2 and WH3, gene expression was compared between BH and WH2, and between BH and WH3 (Supplementary Fig. S1) using featureCounts and edgeR. Totals of 249 and 906 DEGs in WH2 and WH3, respectively, were detected after filtering with |log2 fold changes (logFC)|≥ 2, average log2-counts-per-million (logCPM) ≥ 1 and false discovery rate (FDR) ≤ 0.01. Of these, 111 DEGs in WH2 and 468 in WH3 were upregulated compared to BH (Fig. 4a, Supplementary Table S3 and S4). Among the upregulated DEGs, 85 were common to WH2 and WH3 (Supplementary Table S5). In contrast, 138 DEGs in WH2 and 438 in WH3 were downregulated compared to BH (Fig. 4b, Supplementary Table S6 and S7). Among the downregulated DEGs, 100 were common to WH2 and WH3 (Supplementary Table S8).
Figure 4.

Venn diagrams showing the number of differentially expressed genes (DEGs) identified by RNA-seq analysis. (a) Number of upregulated genes in WH2 and WH3 compared to BH. (b) Number of downregulated genes in WH2 and WH3 compared to BH.
To estimate the function of DEGs, transcript sequences obtained from the Ipomoea Genome Hub website (https://ipomoea-genome.org/) were annotated by sequence-similarity searches against the UniProtKB database using Hayai-Annotation Plants. Out of the total 64,295 predicted genes, 38,788 (60.3%) genes were matched to sequences in UniProtKB with a sequence identity > 40%, query coverage > 40% and e-value < 10–6. In addition, 19,272 (49.7%), 32,095 (82.7%) and 30,017 (77.4%) genes of the annotated 38,788 genes were assigned to gene ontology (GO) terms in the biological process, cellular component and molecular function, respectively. Of the DEGs of WH2 and WH3, 211 and 785 genes were assigned to GO terms, respectively, and further classified into 44 GO term categories using WEGO2 software (Supplementary Fig. S2). Although the numbers of DEGs were different between WH2 and WH3, the percentage of DEGs classified to each category were similar between them. In the cellular component category, most WH2 and WH3 DEGs were classified to cell, organelle, membrane and membrane part. In the molecular function category, DEGs in WH2 and WH3 were largely in catalytic activity and binding. In the biological process category, cellular process and metabolic process were representative classes of WH2 and WH3 DEGs. This similarity between DEGs in WH2 and WH3 suggests that WH2 and WH3 share the molecular mechanism of absence of carotenoids.
DEGs in carotenoid biosynthesis
In order to reveal the genes responsible for the white-flesh mutation, we focused on previously known genes for carotenoid biosynthesis. Based on the functional annotation of UniProtKB and GO terms, 69 genes were identified as being involved in the carotenoid biosynthesis pathway shown in Fig. 1. Although g20008.t1 was not annotated by Hayai-Annotation Plants, its sequence showed high similarity to the lycopene ϵ-cyclase gene (IbLCYE, GenBank: HQ828093.1) of sweetpotato. Of these 70 genes including g20008.t1, 38 showed an average logCPM larger than 1 and are considered to constitute the expressed gene set of carotenoid biosynthesis in sweetpotato storage roots. In this gene set, only g1103.t1 (ZEP) was counted as a common DEG for both WH2 and WH3, showing logFC values of -5.33 and -5.76, respectively. Although g16874.t1 (PSY) was significantly downregulated in both WH2 and WH3 (Fig. 5), it was not counted as DEG, because its |logFC| values were less than 2. A few DEGs for either WH2 or WH3 were also observed: g21348 (CCD8) was counted as a DEG for only WH2, while g6783.t1 (DXS), g21335.t1 (NCED) and g7563.t1 (DXS) were DEGs for only WH3. Gene expression of g1103.t1 was extremely suppressed in both WH2 and WH3 in all DEGs (Supplementary Table S8). Thus, g1103.t1 was assumed to be one of the most promising candidate genes for yellow flesh, and we therefore conducted further characterization of this gene.
Figure 5.

Heatmap of log2 fold change (logFC) values for the 38 genes related to carotenoid biosynthesis. The functional annotation of each gene is described in Fig. 1. *, ** and *** indicate DEGs in WH2, WH3 and both WH2 and WH3, respectively.
Phylogenetic analysis of ZEP
In the set of genes expressed in carotenoid biosynthesis, g1103.t1, g1106.t1, g41700.t1 and g55667.t1 were annotated as ZEP (Fig. 5). Of these, g1103.t1 and g1106.t1 are closely related to known functional ZEP genes in other plants. Although each ZEP gene was found in other plant species, sweetpotato had two ZEP homologues. Sequence similarity searches on public genome databases of wild diploid sweetpotato (I. trifida)20 and morning glory (I. nil)27 showed that these two species also each have two ZEP genes (I. trifida: Itr_sc000013.1_g00031.1 and Itr_sc000013.1_g00027.1; I. nil: INIL11g18864.t1 and INIL11g18861.t1); Fig. 6a). In the sweetpotato genome, g1103.t1 and g1106.t1 were located at a distance of 16.4 kb in the same linkage group (Fig. 6b), and INIL11g18864.t1 and INIL11g18861.t1 were located at a distance of 20.9 kb in the same chromosome 11 of the morning glory genome. These genome structures support the idea that g1103.t1 and g1106.t1 are paralogous genes.
Figure 6.

Characterisation of zeaxanthin epoxidase (ZEP) homologs. (a) Phylogenetic analysis of the deduced amino acid sequences of g1103.t1 and g1106.t1 with 1,000 bootstrap replications. The deduced amino acid sequences of ZEP homologs in Ipomoea trifida and Ipomoea nil were added to this analysis. ZEPs of eight additional species obtained from the National Centre for Biotechnology Information (NCBI) database are also incorporated into this analysis. The scale bar of 0.1 refers to 10% sequence divergence. (b) The genomic position of ZEP homologs in I. batatas and I. nil. Blue boxes indicate untranslated regions (UTRs) and orange boxes indicate exons in each gene. Arrows show the coding direction of each gene.
Gene expression analysis for ZEP paralogs by quantitative real-time PCR
To confirm the results of RNA-seq, the expression patterns of g1103.t1 (ZEP), g1106.t1 (ZEP) and g16874.t1 (PSY) in BH and its white-fleshed mutants were analysed using quantitative real-time polymerase chain reaction (qRT-PCR) (Supplementary Table S9). The qRT-PCR results showed that the expression levels of g1103.t1 in WH2 and WH3 were much lower than that in BH. The expression levels of g1106.t1 and g16874.t1 in BH and its white-fleshed mutants were almost similar, which was also consistent with RNA-seq results. These results indicate the high reproducibility of the RNA-seq in this study.
Next, differences in the expression levels of g1103.t1, g1106.t1 and g16874.t1 were measured using storage roots at 11 weeks after planting and leaves at 7 weeks after planting from five yellow-fleshed cultivars and five white-fleshed cultivars (Table 1). The expression of g1103.t1 was detected in all yellow-fleshed cultivars and was higher in storage roots at 11 weeks after planting than in leaves at 7 weeks after planting. On the other hand, g1103.t1 expression was much lower than in all five white-fleshed cultivars. In three of the white-fleshed cultivars in particular, East Cape 4, Joy White and Lingnan 1, the expression of g1103.t1 was too low to detect. In contrast, g1106.t1 was similarly expressed in white- and yellow-fleshed cultivars. Expression levels of g1106.t1 were significantly higher in leaves at 7 weeks after planting than in storage roots at 11 weeks after planting. Significant difference in the expression level of g16874.t1 were not also detected between storage roots of yellow-fleshed cultivars and white-fleshed cultivars although g16874.t1 showed higher expression in BH than in WH2 and WH3 in RNA-seq analysis. This suggests that g16874.t1 is not involved in carotenoid accumulation in storage roots.
Table 1.
Gene expression of g1103.t1 and g1106.t1 in storage roots (SRs) at 11 weeks after planting and leaves at 7 weeks after planting of 10 sweetpotato cultivars.
| Cultivara | Flesh colour | − ΔΔCt (g1103.t1)c | − ΔΔCt (g1106.t1)c | − ΔΔCt (g16874.t1)c | |||
|---|---|---|---|---|---|---|---|
| SR | Leaf | SR | Leaf | SR | Leaf | ||
| Beniharuka | Pale yellow | 0 | − 3.04 | 0 | 3.73 | 0 | 7.98 |
| Benimasari | Pale yellow | 0.36 | − 2.46 | 0.54 | 2.95 | − 0.25 | 8.13 |
| Nongdahong | Pale yellow | 0.30 | − 1.30 | 0.51 | 5.74 | 0.34 | 9.22 |
| Tamaotome | Yellow | 0.51 | − 1.61 | 0.97 | 4.76 | 1.43 | 7.96 |
| Cavite | Yellow | − 1.21 | − 2.45 | 1.35 | 4.96 | 2.28 | 8.68 |
| Konamizuki | White | − 9.34 | − 7.45 | 0.16 | 3.25 | − 0.02 | 8.14 |
| East Cape 4 | White | < − 10 | n.m | 0.60 | 3.74 | 2.32 | 8.17 |
| Joy White | White | < − 10 | n.m | 0.80 | 4.60 | − 0.30 | 8.36 |
| Lingnan 1 | White | < − 10 | n.m | − 0.21 | 3.79 | 0.60 | 7.50 |
| Koganemasari | White | − 8.80 | − 4.96 | 0.32 | 4.30 | 0.35 | 8.15 |
| Meanb | Pale yellow & yellow | − 0.01 a | − 2.17 b | 0.67 b | 4.43 a | 0.76 b | 8.39 a |
| White | – | – | 0.33 b | 3.94 a | 0.59 b | 8.06 a | |
aThe flesh colours of all cultivars are shown in Supplementary Fig. S3.
bMean values followed by the same letter in g1103.t1, g1106.t1 and g16874.t1 are not significantly different by Tukey’s honestly significant difference (HSD) test (P < 0.01).
cRelative expression values (− ΔΔCt) were calculated by the following formula: −((ΔCt in the target) − (ΔCt in SR of Beniharuka)). The exact relative expression values of samples with “n.m.” were not measurable because the Ct values of g1103.t1 were more than 30 cycles and amplification of primer dimers was observed.
Discussion
In this study, transcriptome changes between a yellow-fleshed sweetpotato cultivar Beniharuka (BH) and its white-fleshed mutants (WH2 and WH3) were compared to reveal the key genes involved in the yellow pigmentation of the sweetpotato storage roots. We found that the gene expression of g1103.t1, which is considered to be a ZEP paralog, was considerably downregulated in WH2 and WH3 compared to BH. The results in this study provide new knowledge about the molecular mechanism of carotenoid accumulation in sweetpotato storage roots and may serve as a basis for the efficient breeding of yellow-fleshed cultivars.
ZEP, the enzyme in the biosynthetic pathway of the carotenoid metabolism, converts zeaxanthin to antheraxanthin and then converts antheraxanthin into violaxanthin by β-ring epoxidation16,28 (Fig. 1). Therefore, loss of ZEP function leads to zeaxanthin accumulation in the leaves of A. thaliana29 and N. plumbaginifolia30. Additionally, zeaxanthin was accumulated in potato tubers by suppressing ZEP expression, and the tubers showed a slightly deeper yellow colour31. Unique carotenoids in sweetpotato such as β-carotene 5,8;5′,8′-diepoxide and β-cryptoxanthin 5,8-epoxide are also synthesised by β-ring epoxidation of β-carotene and β-cryptoxanthin7,9. It was predicted that this epoxidation was regulated by a novel enzyme encoded by the ZEP paralog7. However, no such ZEP paralog has yet been identified in sweetpotato. In WH2 and WH3, zeaxanthin did not accumulate, despite the fact that the expression of g1103.t1, predicted as ZEP, was downregulated compared to BH (Fig. 3, Supplementary Table S9). This suggests that g1103.t1 is not associated with zeaxanthin epoxidation. Additionally, β-cryptoxanthin epoxides and β-carotene epoxides did not accumulate in WH2 and WH3, supporting the hypothesis that g1103.t1 is a ZEP paralog catalysing the epoxidation of β-carotene and β-cryptoxanthin.
The ZEP enzyme was encoded by a single gene in A. thaliana29 and N. plumbaginifolia30. However, phylogenetic analysis in this study indicated that sweetpotato had two genes, g1103.t1 and g1106.t1, with high homology to ZEP in other plants (Fig. 6a). Additionally, two close relatives of sweetpotato, I. trifida and I. nil, also had two genes each that showed high homology to ZEP and were considered to be paralogous. The two ZEP homologs in sweetpotato and I. nil are located close to each other in LG1 and Chr11, respectively, with the same strand (Fig. 6b). These genome structures imply that sweetpotato and I. nil obtained the ZEP paralog by duplication of the locus including the ZEP gene through evolution. Some plants can also synthesis unique carotenoids by duplication and neofunctionalisation of the genes involved in carotenoid synthesis32. Capsanthin and capsorubin are synthesised by capsanthin-capsorubin synthase (CCS) in only a few species (e.g., Capsicum annuum and Lilium lancifolium)33. Based on sequence similarity, it has been suggested that the CCS gene may have evolved from the lycopene cyclase (LCY) gene because of gene duplication33,34. Likewise, ZEP (g1103.t1-type ZEP) of sweetpotato would obtain a new function such as epoxidation of β-carotene or β-cryptoxanthin via gene duplication.
The expression of g1103.t1 was extremely low in all white-fleshed cultivars compared to that in yellow-fleshed cultivars (Table 1). The cultivars used in this study were genetically diverse. Nongdahong, Cavite, East Cape 4 and Lingnan 1 are exotic germplasms from China, the Philippines, Papua New Guinea and Taiwan, respectively, and the others are Japanese breeding cultivars. Thus, the relationship between yellow flesh and g1103.t1 expression level is not specific for BH and its white-fleshed mutants. Yellow-fleshed cultivars such as Benimasari and Tamaotome were found to contain sweetpotato carotenoids (i.e,. β-carotene 5,8;5′,8′-diepoxide, β-cryptoxanthin 5,8-epoxide, etc.) as did BH4,6. This suggests that the translated product of g1103.t1 is commonly involved in the synthesis of these carotenoids in yellow-fleshed cultivars.
The other ZEP paralog, g1106.t1, seemed to encode normal ZEP because the relative expression value of g1106.t1 was higher in leaves at 7 weeks after planting than in storage roots at 11 weeks after planting in all cultivars (Table 1). ZEP is the key enzyme for the xanthophyll cycle, which is the mechanism that protects photosynthetic tissues from photodamage35. Therefore, the expression level of the ZEP gene is usually higher in photosynthetic tissues than in non-photosynthetic tissues36–38. In contrast, the expression level of g1103.t1 was found to be higher in storage roots at 11 weeks after planting than in leaves at 7 weeks after planting in yellow-fleshed cultivars (Table 1). The function of the two ZEP paralogs may be separated; one is associated with the epoxidation of β-carotene and β-cryptoxanthin in roots and the other is associated with the epoxidation of zeaxanthin and antheraxanthin in leaves.
The transcription of genes related to carotenoid biosynthesis is known to be regulated by many different genetic factors39. In sweetpotato, it has been reported that IbOr promotes carotenoid accumulation by increasing gene expression in carotenoid biosynthesis13,14. In the present study, the expression level of g20467.t1 annotated as Or did not differ between BH and its white-fleshed mutants (Fig. 5). However, many DEGs other than g1103.t1 were detected in WH2 and WH3 (Fig. 4). Therefore, downregulation of g1103.t1 in WH2 and WH3 might be caused by any of the other DEGs. Furthermore, it has been reported for Citrus species that ZEP expression levels are regulated by differences in cis-elements among ZEP alleles40,41. In potato tubers, ZEP expression and flesh colour is related to the dosage of ZEP alleles42,43. Because sequence diversity in the locus of g1103.t1 among BH, WH2, WH3 and the other cultivars was not examined in this study, it is unclear whether the white flesh is caused by mutation in g1103.t1 itself or in any regulatory genes. In order to reveal the molecular mechanisms of carotenoid accumulation in sweetpotato, further study is necessary to identify the genetic factors that suppress g1103.t1 expression.
In this study, g1103.t1 was identified as a candidate gene involved in yellow colour in sweetpotato storage roots. Although we hypothesise that g1103.t1 regulates flesh colour through the epoxidation of β-carotene and β-cryptoxanthin, the function of the protein encoded by g1103.t1 remains unknown. In further study, the function of the g1103.t1 protein should be revealed by a molecular biological and biochemical approach.
Methods
Plant materials and growth conditions
A sweetpotato cultivar Beniharuka (BH), whose flesh colour is pale yellow, and two spontaneous white-fleshed mutants of BH (WH2 and WH3) were used in this experiment. Two white-fleshed storage roots obtained independently from different BH clones were vegetatively propagated and named WH2 and WH3, respectively. White fleshed mutation of these lines was stable through vegetative propagation of several years.
Stem cuttings of each line were transplanted into 5-L pots filled with vermiculite and grown in a greenhouse for 10 weeks. Following Tanaka et al. (2005)44, water and nutrition were supplied from the bottom of the pots using 1000-fold diluted Hyponex medium (N–P–K = 6.5–6–19; Hyponex Japan Co., Ltd, Osaka, Japan). After measuring the fresh weight of shoots, the number of storage roots, and the weight of total storage roots, International Commission on Illumination (CIE) l*a*b* values on the centre of cross-sections of storage roots were measured using a CM-2600d spectrophotometer (Konica Minolta, Inc., Tokyo, Japan). The storage roots including all internal tissues were then sliced and frozen in liquid nitrogen immediately. Samples were stored at − 80 °C until carotenoids and RNA were extracted. The experiment was carried out in triplicate using three plants (pots) for each line.
In order to study varietal differences in the expression of the candidate genes detected by RNA-seq, the stem cuttings of the five yellow-fleshed cultivars, Beniharuka, Benimasari, Nongdahong, Tamaotome and Cavite, and the five white-fleshed cultivars, Konamizuki, East Cape 4, Joy White, Lingnan 1 and Koganemasari, were cultivated as described above without replication (Supplementary Fig. S3). Unfolded leaves and storage roots were sampled at 7 and 11 weeks after planting, respectively, and frozen in liquid nitrogen immediately. These samples were also stored at -80 °C until RNA extraction.
Evaluation of carotenoid composition
Carotenoid extraction from storage roots was performed following Ishiguro et al. (2010)4. Two grams of freeze-dried powder of storage roots was added to 6 mL acetone and mixed using a vortex mixer. The mixtures were centrifuged at 1500 g for 10 min. After a 2-mL aliquot of the supernatant was removed, 6 mL of acetone was added to the remainder. After re-extraction, 6 mL of the supernatant was taken and combined with the first extract (total 8 mL). The extract was then evaporated by a centrifugal concentrator, and the residue was redissolved in 1 mL tetrahydrofuran containing 0.1% butylated hydroxytoluene (BHT) and filtered through a 0.2-μm membrane filter.
Carotenoid compositions were detected by HPLC following the method described by Ishiguro et al. (2010)4. Relative carotenoid content was estimated from the ratio of the total peak area of each line to that of BH. Carotenoid composition was estimated using authentic reagents (β-carotene) and extracts from the Tamaotome cultivar (analysed by Ishiguro et al. 20104) as standard.
RNA extraction, library construction and RNA-seq
Total RNA in storage roots was extracted following the method described by Takahata et al. (2010)45. Total RNA in leaves was extracted using an RNeasy Plant Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol for qRT-PCR. The concentration and purity of extracted RNA were checked by SimpliNano (Biochrom Ltd., Cambridge, UK) and the degree of RNA degradation was checked by electrophoresis on 1.5% agarose gel.
Total RNA extracted from the storage roots of BH, WH2 and WH3 was used for the further step of RNA-seq library construction. RNA integrity confirmation (RNA Integrity Number > 8), library construction and RNA-seq were performed at Macrogen Japan Corp. (Kyoto, Japan). The cDNA library was constructed using a TruSeq Stranded mRNA LT Sample Prep Kit (Illumina, Inc., CA, USA) and sequenced using Illumina NovaSeq 6000.
Analysis of RNA-seq data
Low-quality reads and Illumina adaptor sequences were removed by Trimmomatic (v0.36.6)46. The processed paired-end reads were aligned to the reference genome sequence of sweetpotato (cv Taizhong6) obtained from the Ipomoea Genome Hub (https://ipomoea-genome.org/)19 by HISAT2 (v2.1.0)47. The reads mapped to each gene were counted by featureCounts (v1.6.4)48, and the transcripts per million (TPM) value was calculated49. Differential gene expression between BH and WH2 or WH3 was analysed by edgeR (v3.24.3) and the logFC, average logCPM and FDR were calculated50. Genes with a |LogFC|≥ 2, average logCPM ≥ 1 and FDR ≤ 0.01 were considered DEGs.
Transcript sequences of sweetpotato were also obtained from the Ipomoea Genome Hub and annotated by Hayai-Annotation Plants (v1.0.2)51 with a nucleotide sequence identity > 40%, query coverage > 40% and e-value < 10–6. GO functional classification of DEGs was performed by WEGO (v2.0)52.
Phylogenetic analysis of ZEP
The amino acid sequences deduced from g1103.t1, g1106.t1, g41700.t1 and g55667.t1 were used for phylogenetic analysis. Additionally, the amino acid sequences of ZEP in another eight plants were obtained from the National Centre for Biotechnology Information (NCBI) database. Using the ZEP sequences of Arabidopsis thaliana (GenBank: AAM13144.1) as query, TBLASTN searches were performed on genome databases of Ipomoea trifida (ITR_r1.0: https://sweetpotato-garden.kazusa.or.jp/index.html)20 and Ipomoea nil (Asagao_1.2: https://viewer.shigen.info/asagao/index.php)27, and the deduced amino acid sequences with high similarly to AAM13144.1 were also used. Multiple alignment was conducted by Clustal W53 and phylogenetic analysis was carried out using the neighbor-joining method54. Bootstrap analysis was performed with 1000 replications. The results were drawn using FigTree (v1.4.4) (https://tree.bio.ed.ac.uk/software/figtree/).
qRT-PCR assay for zeaxanthin epoxidase genes
cDNA was synthesised from 0.5 μg of total RNA using a PrimeScript RT Reagent Kit with gDNA Eraser (Takara Bio Inc., Shiga, Japan). Primers for qRT-PCR were designed using specific sequences of g1103.t1 (forward: CGTTCCAACTCGTTTCCATCCTTC; reverse: CGTCTCGGGGCAGTTAATGATTCC), g1106.t1 (forward: CCCTGCATAGTTGGCTCTGT; reverse: CGCAAATCAGTCACGAAGAA) and g16874.t1 (forward: TGAAGCAGAACGAGGAGTAACC; reverse: GCAACAGGCAACATAAGCAAC). Actin (forward: TGTTAGCAACTGGGATGATATGG; reverse: GGATAGCACAGCCTGAATAGC)55 was used as a reference gene to normalise the gene expression of targets. qRT-PCR was conducted in 12.5 μL reaction volume using TB Green Premix Ex Taq II (Takara Bio Inc., Shiga, Japan) and the CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories, Inc., CA, USA). The temperature conditions for real-time qRT-PCR were as follows: 95℃ for 30 s, followed by 40 cycles of 95℃ for 5 s and 64℃ for 30 s (g1103.t1), and 95℃ for 30 s followed by 40 cycles of 95℃ for 5 s and 62℃ for 30 s (g1106.t1, g16874.t1 and Actin). Following the completion of amplification, a melting curve analysis was conducted to validate the uniformity of the amplification product. Data of samples amplified after 30 cycles that seemed to include nonspecific product were excluded from this analysis. The expression of each gene relative to the actin gene was determined by the ΔΔCt method56.
Supplementary information
Acknowledgements
We thank Rieko Gonbori (KARC/NARO) for her technical assistance.
Author contributions
K.S. and M.T. conceived and designed the study. K.S. and R.K. performed the experiments. K.S. analysed the data. Y.K. provided the experimental materials. All authors read and approved the final manuscript.
Data availability
RNA-Seq data are deposited in the Sequence Read Archive (DRA) of DNA Data Bank of Japan (DDBJ) under the accession number of DRA010350.
Competing interests
The authors declare no competing interests.
Footnotes
The original online version of this Article was revised: The original version of this Article contained a repeated error in the Results section, in Table 1, and in the Methods section. In addition, the primer sequence of Actin was incorrect in the Methods section. Full information regarding the corrections made can be found in the correction for this Article.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
9/20/2021
A Correction to this paper has been published: 10.1038/s41598-021-98699-x
Supplementary information
is available for this paper at 10.1038/s41598-020-77293-7.
References
- 1.Tanaka M, Ishiguro K, Oki T, Okuno S. Functional components in sweetpotato and their genetic improvement. Breed. Sci. 2017;67:52–61. doi: 10.1270/jsbbs.16125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayne ST. Beta-carotene, carotenoids, and disease prevention in humans. FASEB J. 1996;10:690–701. [PubMed] [Google Scholar]
- 3.Krinsky NI, Johnson EJ. Carotenoid actions and their relation to health and disease. Mol. Aspects Med. 2005;26:459–516. doi: 10.1016/j.mam.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Ishiguro, K., Yoshinaga, M., Kai, Y., Maoka, T. & Yoshimoto, M. Composition, content and antioxidative activity of the carotenoids in yellow fleshed sweetpotato (Ipomoea batatas L.). Breed. Sci.60, 324–329 (2010).
- 5.Ishiguro, K. Sweet potato carotenoids. in Sweet Potato (eds. Mu, T. & Singh, J.) 223–241 (Elsevier, 2019).
- 6.Maoka T, Akimoto N, Ishiguro K, Yoshinaga M, Yoshimoto M. Carotenoids with a 5,6-dihydro-5,6-dihydroxy-β-end group, from yellow sweet potato “Benimasari” Ipomoea batatas LAM. Phytochemistry. 2007;68:1740–1745. doi: 10.1016/j.phytochem.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 7.Khan MZ, Takemura M, Maoka T, Otani M, Misawa N. Carotenoid analysis of sweetpotato Ipomoea batatas and functional identification of its lycopene β- and ε-cyclase genes. Z. Nat. C. 2016;71:313–320. doi: 10.1515/znc-2016-0150. [DOI] [PubMed] [Google Scholar]
- 8.Kang L, et al. Metabolic engineering of carotenoids in transgenic sweetpotato. Breed. Sci. 2017;67:27–34. doi: 10.1270/jsbbs.16118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drapal, M. & Fraser, P. D. Determination of carotenoids in sweet potato (Ipomoea batatas L., Lam) tubers: Implications for accurate provitamin A determination in staple sturdy tuber crops. Phytochemistry167, 112102, 10.1016/j.phytochem.2019.112102 (2019). [DOI] [PubMed]
- 10.Kim SH, Ahn YO, Ahn MJ, Lee HS, Kwak SS. Down-regulation of β-carotene hydroxylase increases β-carotene and total carotenoids enhancing salt stress tolerance in transgenic cultured cells of sweetpotato. Phytochemistry. 2012;74:69–78. doi: 10.1016/j.phytochem.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Kim SH, et al. Downregulation of the lycopene ε-cyclase gene increases carotenoid synthesis via the β-branch-specific pathway and enhances salt-stress tolerance in sweetpotato transgenic calli. Physiol. Plant. 2013;143:432–442. doi: 10.1111/j.1399-3054.2012.01688.x. [DOI] [PubMed] [Google Scholar]
- 12.Kim SH, et al. Down-regulation of sweetpotato lycopene β-cyclase gene enhances tolerance to abiotic stress in transgenic calli. Mol. Biol. Rep. 2014;41:8137–8148. doi: 10.1007/s11033-014-3714-4. [DOI] [PubMed] [Google Scholar]
- 13.Kim SH, et al. Cloning and characterization of an Orange gene that increases carotenoid accumulation and salt stress tolerance in transgenic sweetpotato cultures. Plant Physiol. Biochem. 2013;70:445–454. doi: 10.1016/j.plaphy.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Park SC, et al. Enhanced accumulation of carotenoids in sweetpotato plants overexpressing IbOr-Ins gene in purple-fleshed sweetpotato cultivar. Plant Physiol. Biochem. 2016;86:82–90. doi: 10.1016/j.plaphy.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 15.Kim SE, et al. A single amino acid change at position 96 (Arg to His) of the sweetpotato orange protein leads to carotenoid overaccumulation. Plant Cell Rep. 2019;38:1393–1402. doi: 10.1007/s00299-019-02448-4. [DOI] [PubMed] [Google Scholar]
- 16.Nisar N, Li L, Lu S, Khin NC, Pogson BJ. Carotenoid metabolism in plants. Mol. Plant. 2015;8:68–82. doi: 10.1016/j.molp.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka M, Takahata Y, Kurata R, Nakayama H, Yoshinaga M. Structural and functional characterization of IbMYB1 genes in recent Japanese purple-fleshed sweetpotato cultivars. Mol. Breed. 2012;29:565–574. [Google Scholar]
- 18.McGregor CE, LaBonte DR. Differential expression of genes between storage roots of sweetpotato cultivars Jewel and White Jewel. J. Am. Soc. Hortic. Sci. 2006;131:798–805. [Google Scholar]
- 19.Yang J, et al. Haplotype-resolved sweet potato genome traces back its hexaploidization history. Nat. Plants. 2017;3:696–703. doi: 10.1038/s41477-017-0002-z. [DOI] [PubMed] [Google Scholar]
- 20.Hirakawa, H. et al. Survey of genome sequences in a wild sweet potato, Ipomoea trifida (H. B. K.) G. Don. DNA Res.22, 171–179 (2015). [DOI] [PMC free article] [PubMed]
- 21.Wu, S. et al. Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement. Nat. Commun.9, 4580, 10.1038/s41467-018-06983-8 (2018). [DOI] [PMC free article] [PubMed]
- 22.Xie, F. et al. De novo sequencing and a comprehensive analysis of purple sweet potato (Impomoea batatas L.) transcriptome. Planta236, 101–113 (2012). [DOI] [PubMed]
- 23.Ma, P., Bian, X., Jia, Z., Guo, X. & Xie, Y. De novo sequencing and comprehensive analysis of the mutant transcriptome from purple sweet potato (Ipomoea batatas L.). Gene575, 641–649 (2016). [DOI] [PubMed]
- 24.Li, R. et al. De novo transcriptome sequencing of the orange-fleshed sweet potato and analysis of differentially expressed genes related to carotenoid biosynthesis. Int. J. Genomics 843802, 10.1155/2015/843802 (2015). [DOI] [PMC free article] [PubMed]
- 25.Qin Z, et al. Gene identification using RNA-seq in two sweetpotato genotypes and the use of mining to analyze carotenoid biosynthesis. S. Afr. J. Bot. 2017;109:189–195. [Google Scholar]
- 26.Gemenet, D. C. et al. Quantitative trait loci and differential gene expression analyses reveal the genetic basis for negatively associated β-carotene and starch content in hexaploid sweetpotato [Ipomoea batatas (L.) Lam.]. Theor. Appl. Genet.133, 23–36 (2020). [DOI] [PMC free article] [PubMed]
- 27.Hoshino, A. et al. Genome sequence and analysis of the Japanese morning glory Ipomoea nil. Nat. Commun.7, 13295, 10.1038/ncomms13295 (2016). [DOI] [PMC free article] [PubMed]
- 28.Ohmiya A, et al. Molecular basis of carotenoid accumulation in horticultural crops. Hort. J. 2019;88:135–149. [Google Scholar]
- 29.Rock CD, Zeevaart JA. The aba mutant of Arabidopsis thaliana is impaired in epoxy-carotenoid biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 1991;88:7496–7499. doi: 10.1073/pnas.88.17.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marin E, et al. Molecular identification of zeaxanthin epoxidase of Nicotiana plumbaginifolia, a gene involved in abscisic acid biosynthesis and corresponding to the ABA locus of Arabidopsis thaliana. EMBO J. 1996;15:2331–2342. [PMC free article] [PubMed] [Google Scholar]
- 31.Römer S, et al. Genetic engineering of a zeaxanthin-rich potato by antisense inactivation and co-suppression of carotenoid epoxidation. Metab. Eng. 2002;4:263–272. doi: 10.1006/mben.2002.0234. [DOI] [PubMed] [Google Scholar]
- 32.Wessinger CA. A genetic route to yellow flowers. New Phytol. 2015;206:1193–1195. doi: 10.1111/nph.13403. [DOI] [PubMed] [Google Scholar]
- 33.Jeknić, Z. et al. Cloning and functional characterization of a gene for capsanthin-capsorubin synthase from tiger lily (Lilium lancifolium Thunb. 'Splendens'). Plant Cell Physiol.53, 1899–1912 (2012). [DOI] [PMC free article] [PubMed]
- 34.Krubasik P, Sandmann G. Molecular evolution of lycopene cyclases involved in the formation of carotenoids with ionone end groups. Biochem. Soc. Trans. 2000;28:806–810. [PubMed] [Google Scholar]
- 35.Latowski D, Kuczyńska P, Strzałka K. Xanthophyll cycle – A mechanism protecting plants against oxidative stress. Redox Biol. 2011;16:78–90. doi: 10.1179/174329211X13020951739938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Audran C, et al. Expression studies of the zeaxanthin epoxidase gene in Nicotiana plumbaginifolia. Plant Physiol. 1998;118:1021–1028. doi: 10.1104/pp.118.3.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang N, et al. Overexpression of zeaxanthin epoxidase gene enhances the sensitivity of tomato PSII photoinhibition to high light and chilling stress. Physiol. Plant. 2008;132:384–396. doi: 10.1111/j.1399-3054.2007.01016.x. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Z, et al. MsZEP, a novel zeaxanthin epoxidase gene from alfalfa (Medicago sativa), confers drought and salt tolerance in transgenic tobacco. Plant Cell Rep. 2016;35:439–453. doi: 10.1007/s00299-015-1895-5. [DOI] [PubMed] [Google Scholar]
- 39.Stanley, L. & Yuan, Y. W. Transcriptional regulation of carotenoid biosynthesis in plants: so many regulators, so little consensus. Front. Plant Sci.10, 1017, 10.3389/fpls.2019.01017 (2019). [DOI] [PMC free article] [PubMed]
- 40.Sugiyama A, et al. Structure and expression levels of alleles of citrus zeaxanthin epoxidase genes. J. Jpn. Soc. Hortic. Sci. 2010;79:263–274. [Google Scholar]
- 41.Sugiyama A, et al. Expression quantitative trait loci analysis of carotenoid metabolism-related genes in citrus. J. Jpn. Soc. Hortic. Sci. 2014;83:32–43. [Google Scholar]
- 42.Wolters AM, et al. Identification of alleles of carotenoid pathway genes important for zeaxanthin accumulation in potato tubers. Plant Mol. Biol. 2010;73:659–671. doi: 10.1007/s11103-010-9647-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCord P, Zhang LH, Brown C. The incidence and effect on total tuber carotenoids of a recessive zeaxanthin epoxidase allele (Zep1) in yellow-fleshed potatoes. Am. J. Potato Res. 2012;89:262–268. [Google Scholar]
- 44.Tanaka M, Takahata Y, Nakatani M. Analysis of genes developmentally regulated during storage root formation of sweet potato. J. Plant Physiol. 2005;162:91–102. doi: 10.1016/j.jplph.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 45.Takahata Y, et al. Inhibition of the expression of the starch synthase II gene leads to lower pasting temperature in sweetpotato starch. Plant Cell Rep. 2010;29:535–543. doi: 10.1007/s00299-010-0842-8. [DOI] [PubMed] [Google Scholar]
- 46.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim D, Langmead B, Salzberg SL. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao Y, Smyth GK, Shi W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 49.Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2014;26:493–500. doi: 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghelfi A, Shirasawa K, Hirakawa H, Isobe S. Hayai-annotation plants: An ultra-fast and comprehensive functional gene annotation system in plants. Bioinformatics. 2019;35:4427–4429. doi: 10.1093/bioinformatics/btz380. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Ye, J. et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res.46, 71–75 (2018). [DOI] [PMC free article] [PubMed]
- 53.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saitou N, Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 55.Tanaka M, Takahata Y, Nakayama H, Nakatani M, Tahara M. Altered carbohydrate metabolism in the storage roots of sweet potato plants overexpressing the SRF1 gene, which encodes a Dof zinc finger transcription factor. Planta. 2009;230:737–746. doi: 10.1007/s00425-009-0979-2. [DOI] [PubMed] [Google Scholar]
- 56.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data are deposited in the Sequence Read Archive (DRA) of DNA Data Bank of Japan (DDBJ) under the accession number of DRA010350.