

Abstract
Multiple sulfatase deficiency (MSD) is an ultra‐rare neurodegenerative disorder caused by pathogenic variants in SUMF1. This gene encodes formylglycine‐generating enzyme (FGE), a protein required for sulfatase activation. The clinical course of MSD results from additive effect of each sulfatase deficiency, including metachromatic leukodystrophy (MLD), several mucopolysaccharidoses (MPS II, IIIA, IIID, IIIE, IVA, VI), chondrodysplasia punctata, and X‐linked ichthyosis. While it is known that affected individuals demonstrate a complex and severe phenotype, the genotype‐phenotype relationship and detailed clinical course is unknown. We report on 35 cases enrolled in our retrospective natural history study, n = 32 with detailed histories. Neurologic function was longitudinally assessed with retrospective scales. Biochemical and computational modeling of novel SUMF1 variants was performed. Genotypes were classified based on predicted functional change, and each individual was assigned a genotype severity score. The median age at symptom onset was 0.25 years; median age at diagnosis was 2.7 years; and median age at death was 13 years. All individuals demonstrated developmental delay, and only a subset of individuals attained ambulation and verbal communication. All subjects experienced an accumulating systemic symptom burden. Earlier age at symptom onset and severe variant pathogenicity correlated with poor neurologic outcomes. Using retrospective deep phenotyping and detailed variant analysis, we defined the natural history of MSD. We found that attenuated cases can be distinguished from severe cases by age of onset, attainment of ambulation, and genotype. Results from this study can help inform prognosis and facilitate future study design.
Keywords: leukodystrophy, mucopolysaccharidoses, multiple sulfatase deficiency, outcomes
Abbreviations
- ARSA
arylsulfatase A
- CT
computed tomography
- DEXA or DXA
dual‐energy X‐ray absorptiometry
- EEG
electroencephalogram
- EKG
Electrocardiogram
- ECHO
Echocardiogram
- FGE
formylglycine generating enzyme
- G‐tube
gastrostomy tube
- GAG
glycosaminoglycan
- GJ‐tube
gastrojejunostomy tube
- GMFCS
Gross Motor Functional Classification System
- HSM
Hepatosplenomegaly
- LSD
lysosomal storage disorder
- MSD
multiple sulfatase deficiency
- MLD
metachromatic leukodystrophy
- MRI
magnetic resonance imaging
- ND
not done
- SUMF1
Sulfatase Modifying Factor 1
1. INTRODUCTION
Multiple Sulfatase Deficiency (MSD, MIM# 272200) is an ultra‐rare autosomal recessive neurometabolic disorder. Because there are less than 150 individuals reported in the literature, we do not fully understand the natural history of the disorder. MSD results from deficient activity of the enzyme responsible for activating all cellular sulfatases, formylglycine‐generating enzyme (FGE), which is encoded by SUMF1. 1 , 2 Without this key step, newly synthesized sulfatases are not activated in the endoplasmic reticulum. 3 The additive effects of each individual sulfatase deficiency, 4 , 5 which includes seven types of lysosomal storage diseases (LSDs), (ie, metachromatic leukodystrophy (MLD) and six mucopolysaccharidoses (MPS) subtypes), X‐linked ichthyosis, and X‐linked chondrodysplasia punctata 5 , 6 drives MSD pathology. Furthermore, deficiencies of sulfatases without known associated monogenic disorders likely contribute to the complex and variable MSD phenotype.
Proposed MSD subgroups include neonatal, severe late infantile, mild infantile, and juvenile forms. 7 , 8 , 9 In general, classification is stratified by age of onset before or after 2 years to discriminate neonatal/severe infantile cases from mild infantile/juvenile cases. However, these classification schemes were developed based on a limited number of case reports and case series. The relevance of this age‐of‐onset classification system to outcomes is unknown, and a thorough, systematic examination of age of symptom onset in MSD is lacking.
Recently, families of MSD patients have formed advocacy groups with the aim to fund preclinical research on small‐molecule, cell‐based, and gene therapies (https://curemsd.org, http://www.savingdylan.com). As targeted therapeutics are developed, it will be essential to understand the clinical presentation and evolution of MSD. Specifically, we need to identify sub‐populations that share clinical features within this heterogeneous disorder in order to guide clinical trial design. 10 , 11 A centralized, large‐scale prospective natural history studies may not be feasible in this unique and severely affected population. Such natural history efforts are essential to prepare for MSD clinical trials, and have proven beneficial in other rare disorders. 12 , 13 In this study, we analyzed molecular, biochemical and longitudinal clinical data to define the progression of MSD and provide an important foundation for future MSD clinical trials.
2. METHODOLOGY
2.1. Cohort identification
We identified 35 individuals (Table 1, Table S1) with MSD through our natural history study. Subjects were referred from 10 countries including the US, Germany, Ireland, United Kingdom, Israel, Poland, Argentina, Brazil and China. Clinical records were reviewed by investigators at two study sites: The Children's Hospital of Philadelphia (CHOP) and University Medical Centre Göttingen. We conducted this study as part of the Global Leukodystrophy Initiative (GLIA) Clinical Trials Network, through the Myelin Disorders Bioregistry Project (MDBP, clinical trials identifier: NCT03047369), an IRB‐approved research biorepository based at CHOP. Individuals were enrolled directly under MDBP or through institutionally‐approved protocols at each local site.
TABLE 1.
Demographics and diagnostics within the SUMF1 deficient cohort
| Cases: N | 35 |
| Cases with clinical records: N (%) | 32 (91.4%) |
| Sex (F): N (%) | 15 (42.9%) |
| Clinical presentation (from n = 30) | |
| Average age in years (SD) | 0.7 years (±1.0 years) |
| Median age in years (range) | 0.25 years (0‐3.5 years) |
| Survival: Deceased N (from n = 8) | |
| Average age in years (SD) | 14.9 years (±11.6 years) |
| Median age in years (range) | 13 years (1.5–41 years) |
| Age at diagnosis (from n = 28) | |
| Average age in years (SD) | 4.2 years (±3.7 years) |
| Median age in years (range) | 2.7 years (0.7‐14 years) |
Abbreviation: SD, standard deviation.
The diagnosis of MSD was defined as a clinical history consistent with MSD and one or more of the following 1 : biochemical testing (low enzymatic activity of at least two sulfatases), 2 genetics (presence of two likely pathogenic or pathogenic variants in SUMF1), or 3 disease in a first degree relative with biochemical or genetic confirmation. REDCap was used to create a central database. Data were extracted from each clinical case and entered into REDCap by a study physician. Because data were extracted by retrospective medical record review, not all key variables were available across the full population. As such the number of individuals for whom information was available is noted for each parameter.
2.2. Clinical characterization
We collected information on age and symptoms at presentation (age at onset = AAO) and demographic information, including age at last evaluation. Microcephaly was identified in medical records and defined as more than 2 standard deviations from the mean for head circumference or as noted “microcephalic”.
MLD‐specific functional scales, the Gross Motor Function Classification (GMFC‐MLD) and Expressive Language Function Classification (ELFC‐MLD), 14 as well as the Eating and Drinking Ability Classification System (EDACS) 15 were applied retrospectively using information available in the medical record. The EDACS is a simple, validated scale, with Level I representing safe and efficient oral intake to Level V where individuals require tube feeding. In these scales, higher function is represented by lower numbers. For longitudinal mapping, the function between two scores was assigned the score of the prior encounter. Time to event measures, such as loss of ambulation or seizures were also collected.
2.3. Genetic and biochemical characterization of SUMF1 variants
For each individual, we assigned a SUMF1 “genotype score” (Table S1). Variants of unknown significance were awarded one point each as were missense variants with biochemical or in silico modeling data supporting mild functional disruption. Missense severe and nonsense alleles were each assigned two points each. As such, each individual could be assigned a total genotype score of 2 to 4. Individuals with a score of 2 to 3 were grouped as “moderate genotype” vs “severe” for the individuals with a genotype score of 4.
Full details of biochemical and computational methods are available in the Supplementary Methods. In short, expression constructs harboring each novel SUMF1 variant (reference sequence NG_016225.2) were transfected into HT1080 fibrosarcoma cells and immortalized MSD fibroblasts (MSDi, a gift from Andrea Ballabio, Italy). Sulfatase activities, including steroid sulfatase (STS) and arylsulfatase A (ARSA), were measured for each. The stability of each FGE variant was measured using a cycloheximide chase assay. To evaluate the molecular consequence of each novel variant, in silico analysis of variant FGE structure was computed using the X‐ray coordinates of FGE crystal structure (PDB:1Y1E). Analysis of the polar contacts between residues in the native structure (FGE‐WT) and respective mutated residue generated by in silico mutagenesis were compared using PyMOL.
2.4. Statistical analysis
The total number of cases analyzed for each parameter varied based on the availability of the pertinent medical information. Novel cohorts were grouped by genotype classification and by age at first clinical symptom. In order to compare achievement and loss of milestones, we created Kaplan‐Meier curves for key developmental milestones. Values were presented as means with SD and medians with ranges. Statistical analysis was performed in Microsoft Excel and GraphPad Prism, and figures were prepared in Prism 8.0. Log‐rank (Mantel‐Cox) tests were used to compare acquisition of developmental milestones between subgroups. The Mann‐Whitney test was used to compare differences between two independent groups, using a two tailed P value.
3. RESULTS
3.1. Clinical characterization
Our cohort included 32 individuals of whom eight individuals were deceased at the time of record collection. Death occurred at an average age of 14.9 years (±11.6 years SD; median 13 years, range 1.5‐41 years) (Table 1, Figure 1A, Table S1).
FIGURE 1.
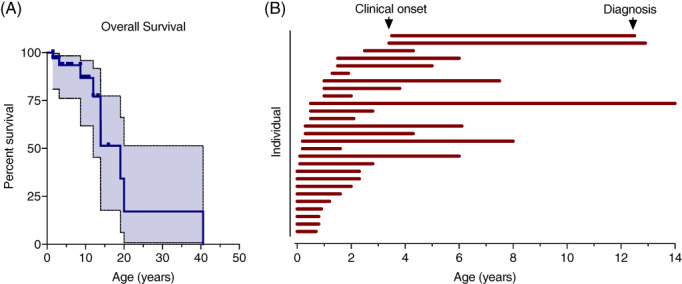
Survival and age at clinical onset and diagnosis in MSD. A, Overall survival within the MSD cohort presented by Kaplan‐Meier curve with 95% confidence interval (shaded). B, Paired age at clinical onset and age at diagnosis were available for 27 individuals, presented as a swim lane plot. Each individual is represented by a horizontal bar beginning at the age at presentation and ending at diagnosis
We obtained information on both the age at diagnosis and age at clinical presentation for 27 individuals. Review of clinical records revealed that 24/27 individuals had clinical features of MSD before 2 years of age (Figure 1B), with a mean age at symptom onset of 0.7 years (Table 1). Next, we characterized the time from symptom onset to diagnosis. In our cohort, there was a mean delay to diagnosis of 3.6 years (SD ± 3.2 years, median 2.3 years, range 0.6‐13.5 years) (Figure 1B).
Patients presented with neurologic symptoms as the most common first clinical feature; 23 of 27 children presented with developmental delay, of whom 79% demonstrated motor delay and 60% had language delay. Somatic features were also common with at least 16 individuals manifesting non‐neurologic symptoms, including ichthyosis and orthopedic complications, at presentation.
In our cohort, neurologic disability was universal. All patients demonstrated an abnormal neurologic exam at some point in the disease course, including 27 of 28 with abnormal tone and 27 of 29 with language abnormalities (Figure 2A). Microcephaly was noted in 12 of 21 cases (57%). Within this cohort, no individuals were noted to have macrocephaly, although three were noted to have hydrocephalus.
FIGURE 2.
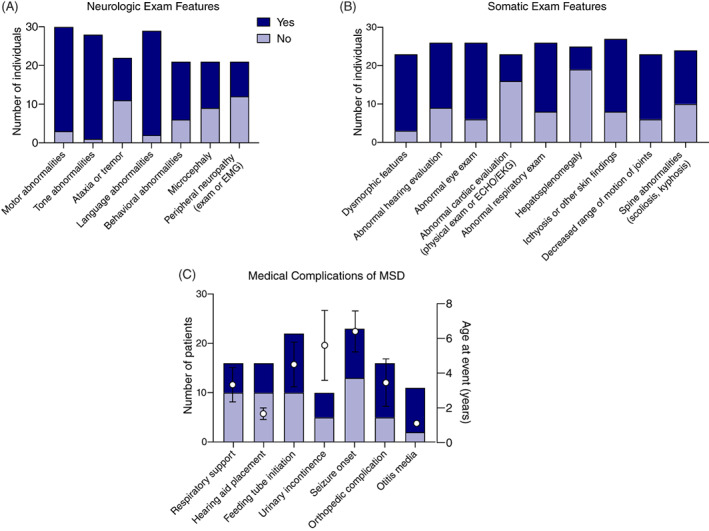
Key time to event measures and clinical features in MSD. A, Key disease‐specific clinical outcomes, and B, C, exam features were collected in the medical records as available. Events or features were classified as either yes (event/feature noted at any point in the medical record; dark blue) or no (event/feature noted not to have occurred; light blue). The total number of records with available information is noted on the left y‐axis. For medical complications (time to event measures), the mean age (± SE of the mean) at which the first event occurred is shown (right axis, black circles)
Next, we characterized the extra‐neurologic symptoms in our cohort (Figure 2B). Within this cohort, the skin examination at presentation was described in only 14 cases, of which 5 had pathologic findings (35.7%). Within all available medical records, skin findings consistent with MSD were noted in 19 of 27 cases (70%). Hepatosplenomegaly was noted in 6 of 25 individuals (24%). Seven of 23 individuals were noted to have an abnormal cardiac evaluation (Figure 2). These findings ranged from incidental findings of a patent foramen ovale to left ventricular hypertrophy to one subject with Tetralogy of Fallot. We collected information regarding key respiratory time to event measures related to the pneumonias and the use of airway support (Figure 2C). We found 6 of 16 individuals required airway support during their lifetime (37.5%). Two individuals presented in the neonatal period with pulmonary hypertension. To better characterize progressive swallowing dysfunction, we collected information on swallowing function and rated the level of function using the EDACS (Figure 4B). Children affected by MSD demonstrated progressive swallowing difficulty, which correlated with early onset disease. At the last clinical encounter, 55% (n = 12/22) of the MSD population was reliant on non‐oral feeds. Worsening EDACS scores correlated with not having attained independent ambulation.
To quantify the rate of neurologic regression in MSD, we retrospectively classified gross motor and language skills in all available clinical records using the GMFC‐MLD and ELFC‐MLD, 14 , 16 scales previously validated in MLD cohorts (Figures 3 and 4). We selected these scales because MLD and MSD share a deficiency of ARSA and some clinical features. A score of 0 represents normal neurologic function for both the GMFC‐MLD and ELFC‐MLD, while the maximum score of 6 or 4, respectively, represents complete lack of mobility or communication.
FIGURE 3.
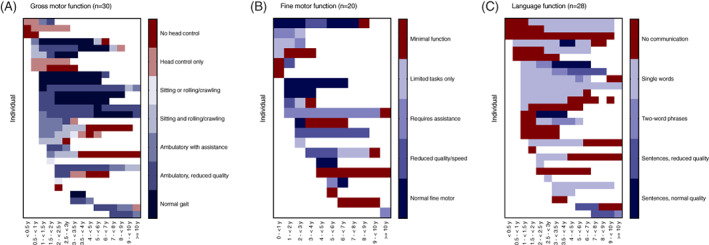
Developmental heat maps in MSD. For each available medical encounter, gross motor, fine motor, and language skills were retrospectively classified using the GMFC‐MLD, MACS, and ELFC, respectively
FIGURE 4.
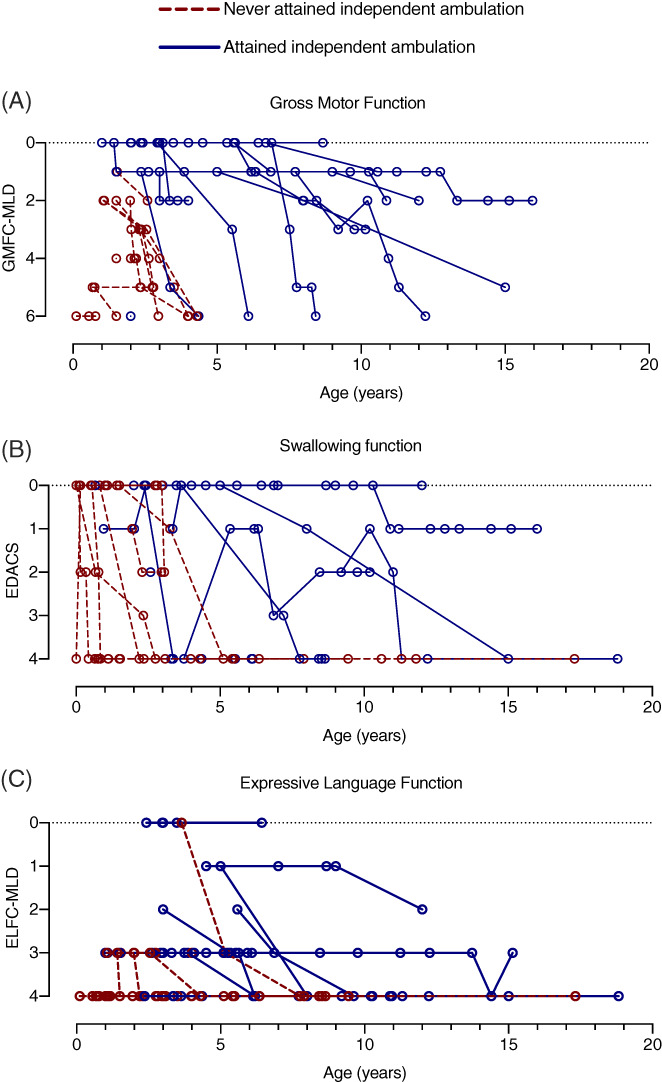
Gross motor, swallowing and expressive language function in MSD. A, Gross motor function was scored at each available medical encounter using the GMFC‐MLD in 29 individuals. Score corresponds to function as follows: 0 = walking without support; 1 = walking without support reduced quality of performance; 2 = walking with support; 3 = sitting without support AND crawling/rolling; 4 = either sitting without support OR crawling/rolling; 5 = no sitting without support AND no crawling/rolling; 6 = no head or trunk control. B, Swallowing function was scored at each available medical encounter using the EDACS in 26 individuals. Score corresponds to function as follows: 0 = eats and drinks safely and efficiently; 1 = some limitations to efficiency; 2 = some limitations to safety; 3 = significant limitations to safety; 4 = unable to eat or drink safely. C, Expressive language was scored at each available medical encounter using the ELFC‐MLD in 29 individuals. Score corresponds to function as follows: 0 = speaks in complete sentences of normal quality; 1 = speaks in complete sentences of reduced quality; 2 = speaks in maximum of 2‐word phrases; 3 = speaks in single words; 4 = complete loss of expressive language. Individuals who never attained independent ambulation are noted with red, dotted lines, while those who were able to independently ambulate are noted in solid blue lines
In terms of gross motor skills, within our cohort, 18/31 children attained unassisted ambulation, but later neurologic regression was noted in 83%. Fourteen of 30 subjects lost at least two points by the GMFC‐MLD scale, representing a major change in neurologic function. Six individuals had severe developmental delay at baseline and their function was below the threshold of measurement by these retrospective tools. As such, regression was not measurable in these individuals. Based on GMFC‐MLD score trajectories, subjects that attained independent ambulation had a later onset of disease and more protracted course of gross motor regression as compared to those who never walked independently (Figure 4A), supporting the existence of subcohorts of MSD patients that have distinct clinical courses.
Overall, language development was more impaired compared to motor function, with only 8 of 29 children ever attaining multi‐word speech (Figures 3 and 4). Speech development in MSD can be further complicated by hearing loss, which was noted in 17 of 26 individuals (65%). Six of 16 individuals were noted have hearing aids (Figure 2C).
3.2. Genetic and biochemical characterization of SUMF1 variants
To understand how genotype correlates with outcomes in MSD, we analyzed each SUMF1 variant in our cohort. Confirmatory biochemical testing and/or SUMF1 molecular testing results were available for n = 31 individuals, with specific molecular results available from 24 individuals (Table S1). Of the 48 alleles present in the cohort, 5 were severe stop‐gain variants. Sixteen unique missense variants were identified (Table 2). While the functional effects of many of these missense variants had been previously reported, 9 , 17 , 18 we identified 11 variants that had limited functional or severity information available.
TABLE 2.
Functional analysis of missense SUMF1 variants in cohort
| SUMF1 Variant | ARSA activity (% control) | STS activity (% control) | Estimated FGE half‐life (h) | Missense variant classification |
|---|---|---|---|---|
| c.797C > T (p.Pro266Leu) | 72.7 | 42.6 | 3 | Mild |
| c.1034G > A (p.Arg345His) | 51.2 | 31.5 | 4 | Mild |
| c.817G > A (p.Asp273Asn) | 52.1 | 12.7 | 5 | Mild |
| c.836C > T (p.Ala279Val) b | 26.8 | 54.1 | Decreased | Mild |
| c.529G > C (p.Ala177Pro) b | Reduced | Reduced | Normal | Mild |
| c.1091G > A (p.Arg364His) | 1.0 | 1.0 | 3 | Severe |
| c.536G > C (p.Trp179Ser) | 3.4 | 51.4 | 2 | Severe |
| c.890A > C (p.Asn297Thr) | 1.0 | 0.9 | 3 | Severe |
| c.305G > 5 (p.Gly102Val) | 14.2 | 15.9 | 4 | Severe |
| c.776A > T (p.Asn259Ile) | 1.2 | 5.5 | 2.5 | Severe |
| c.464C > T (p.Ser155Phe) | 0.9 | 7.5 | 3 | Severe |
| c.739G > C (p.Gly247Arg) a | 2.3‐11 | 0.3‐2.7 | Decreased | Severe |
| c.463 T > C (p.Ser155Pro) a | 3.4‐12 | 14‐32 | Decreased | Severe |
| c.1043C > T (p.Ala348Val) c | 1.0 | 1.0 | 2 | Severe |
| c.640G > A (p.Ala214Thr) | Unknown | Unknown | Unknown | Likely mild (in silico minimal strain on secondary structure) |
| c.539G > T (p.Trp180Leu) | Unknown | Unknown | Unknown | Likely severe (in silico loss of substrate binding) |
Abbreviations: ARSA, arylsulfatase A; FGE, formylglycine generating enzyme; STS, steroid sulfatase.
Schlotawa et al. Eur J Hum Gen 2011.
Schlotawa et al. Hum Mutat 2008.
Staretz‐Chacham et al. Mol Genet Genom Med 2020.
To confirm pathogenicity and predict molecular severity, in vitro steroid sulfatase (STS) and arylsulfatase A (ARSA) levels were measured for nine of the under‐characterized SUMF1 variants (Figure 5A, Table 2, Figure S1). All variants suppressed sulfatase activity to varying degrees. It is known that many pathogenic SUMF1 variants result in decreased protein stability. 9 Consistent with these previous reports, each of the novel SUMF1 variants reduced the half‐life of the FGE enzyme (Figure 5B, Figure S2). In silico structural modeling was completed for 11 of the novel missense variants and demonstrated that variants resulted in a loss of key interactions between residues and structural destabilization (Figure S3). This analysis further supports the pathogenicity of each SUMF1 variant.
FIGURE 5.
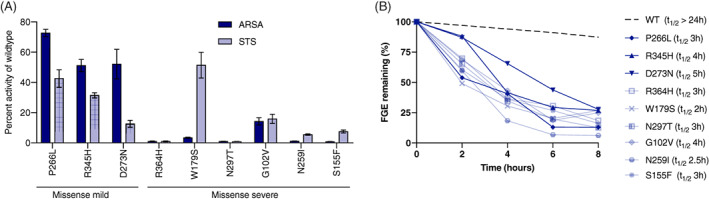
In vitro residual sulfatase activity and FGE stability of novel SUMF1 variants. A, Steroid sulfatase (STS) and arylsulfatase A (ARSA) activities were measured in cells harboring each of the 9 novel SUMF1 variants. Bar diagram depicting the comparison of the specific activity of STS and ARSA relative to activity of cells expressing FGE‐WT (Also see Figure S1). The values represent mean ± SEM of three independent experiments. B, The stability of the novel FGE variants was compared to wildtype and the half‐life (t1/2) was calculated (see legend). Plot depicts the percentage of FGE remaining after cycloheximide chase for the indicated time points. The amount of FGE was determined by quantification of signals corresponding of FGE in western blots (see Figure S2). After normalization of the anti‐HA antibody signals (corresponding to FGE) in cell lysates to anti‐Hsc70 signals, the total amount of FGE (μg/mg of total protein in lysate) in the cells and media were combined and expressed as the percentage of that at the start of the chase (0 hours)
We used previously published variant data and our biochemical and in silico data to annotate the severity of each SUMF1 variant (Table 2, Table S1). Mild variants had substantial residual ARSA activity, while severe variants had ARSA activities of <20% of control. We calculated a genotype score for each subject based on the severity of each allele. Six subjects had a severe genotype (score = 4, two severe alleles); while, 18 had an attenuated genotype, that is, they harbored at least one mild missense allele. Of the attenuated cases, 9 had a score of 3 (one severe and one mild allele), and 9 had a score of 2 (two mild alleles).
3.3. Clinical biochemical testing and outcomes
To investigate if results of clinical sulfatase testing in blood correlates with outcomes, we reviewed available biochemical results from our cohort. Residual sulfatase activity varied between individual enzymes and between individuals (Figure 6A). Most individuals demonstrated arylsulfatase A activity that was less than 10% of control activities; other enzymes were more variable. Unlike our in silico and in vitro data activity data which correlated with clinical severity, clinical testing of residual sulfatase activity in blood did not correspond to genotype severity score (Figure 6A) or clinical outcome as measured by age of onset of regression (Figure 6B).
FIGURE 6.
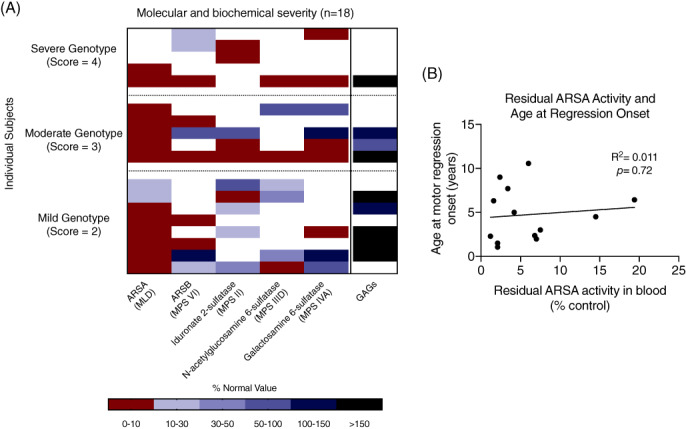
Relationship between clinical biochemical testing and genotypic severity in MSD. A, For each individual with at least two documented sulfatase activity results, genotype severity score, residual enzyme activity, and glycosaminoglycan (GAG) results are shown. B, There is no significant correlation between residual ARSA activity in blood and age at onset of motor regression (ie, last age at best GMFC‐MLD score before regression)
Many individuals with MSD may also demonstrate elevated excretion of urinary sulfatides and glycosaminoglycans (GAGs). Consistent with prior reports, we found that neither normal GAG excretion nor does the presence of high residual activity levels of some sulfatases exclude a diagnosis of MSD. 19
3.4. Predicting outcomes from early clinical and molecular features
As described above, motor regression scoring suggests two subpopulations exist that can be differentiated by attainment of independent ambulation (Figure 4). We sought to identify additional, earlier features that correlate with these distinct sub‐cohorts. Identification of such features could help guide prognostication in newly‐diagnosed patients and inform subject selection for clinical trials.
We focused on two parameters available across our population: age at first MSD‐related clinical symptom and genotype severity. The previously‐reported classification schema would not be informative in our MSD cohort as 89% of our cohort demonstrated symptoms before 2 years of age (Figure 1). As such, we segregated our cohort by presentation before and after 1 month of age. We also segregated the population by genotype severity, either severe (two severe alleles) and attenuated (at least one mild missense allele) (Figure 7A). There was significant overlap between the populations using these two classification schemes. All subjects with onset after 1 month of age had attenuated SUMF1 genotypes. While all individuals with severe genotypes demonstrated symptoms before 1 month of age.
FIGURE 7.
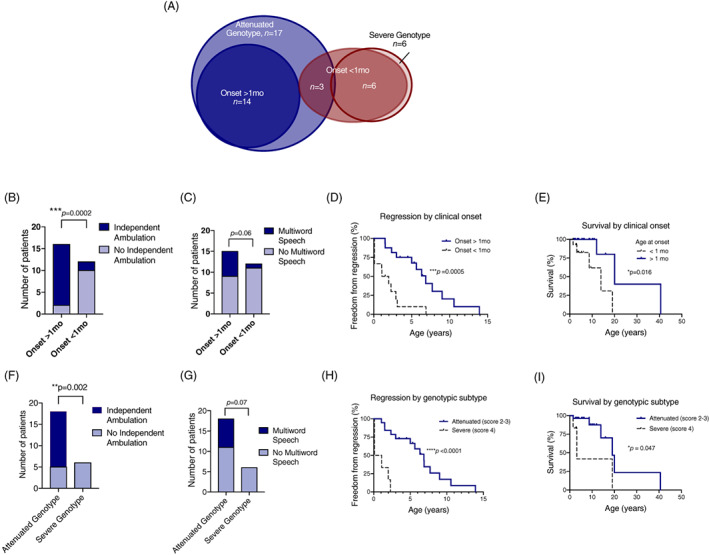
Early features that correlate with outcomes in MSD. A, Venn diagram showing the overlap between subjects presenting in the neonatal period (<1 mo, dark red, n = 9), subjects with a severe genotype (light red, n = 6), subjects presenting after the neonatal period (>1 mo, dark blue, n = 14), and subjects with attenuated genotypes (light blue, n = 17). The cohort was divided by age of onset before or after 1 month, and the number of subjects that attained, B, independent ambulation, and C, multiword speech are shown (chi‐square test, P values as labeled). D, Freedom from regression divided by age of onset is shown (log‐rank Mantel‐Cox test, P = .0005). E, Survival divided by age of onset is shown (log‐rank Mantel‐Cox test, P = .016). The cohort was also divided by genotype severity (attenuated = score of 2‐3, severe = score of 4), and the number of subjects that attained (F) independent ambulation, and G, multiword speech are shown (chi‐square test, P values as labeled). Freedom from regression divided by genotype severity is shown (log‐rank Mantel‐Cox test, P < .0001). Neurologic regression (H) and survival (I) are presented by Kaplan‐Meier curves, with subcohorts defined by genotype severity (log‐rank Mantel‐Cox test, P < .0001 and P = .047, respectively)
Onset of symptoms before 1 month of life correlated with a number of clinical outcomes (Figure 7B‐E) including failure to develop independent ambulation, age of regression onset (median 1.5 vs 6.8 years, log‐rank Mantel‐Cox test, P = 0.0005), and survival (median 13.9 vs 20.0 years, log‐rank Mantel‐Cox test, P = 0.0005). There was also a trend toward improved language development in individuals who present after a month of life.
Like age of onset before 1 month, presence of a severe genotype also correlated with poorer clinical outcomes (Figure 7F‐I). Specifically, individuals with severe variants were less likely to develop independent ambulation or multiword speech, had earlier onset of motor regression (defined as loss of one point on the GMFC‐MLD scale) (median 0.6 vs 6.9 years, log‐rank Mantel‐Cox test, P < 0.0001), and reduced survival (median 3.1 vs 19.1 years, log‐rank Mantel‐Cox test, P = 0.047).
4. DISCUSSION
Multiple sulfatase deficiency is an ultra‐rare neurodegenerative disorder. 5 , 8 , 20 The phenotype of MSD is more than a direct summation of individual sulfatase deficiencies as the simultaneous loss of several sulfatases may result in novel pathophysiologic effects. 5 Additionally, several sulfatases have uncharacterized phenotypes, adding to the clinical uncertainty surrounding this rare disorder. 5 The ultra‐rare nature of MSD has limited our understanding of clinical features and progression.
There are several limitations of the retrospective approach employed here including missing or conflicting information from available medical records. Some key outcomes, such as hearing loss and subtle findings on physical examination were not noted consistently in the medical records. These omissions may represent a lack of targeted investigation or a lack of presence. As such, for outcome measures and severity classification, we selected features that were consistently described and easily measured. While not all information was available for all individuals, the depth of information collected allowed for the identification of key variables associated with neurodevelopmental outcomes and overall survival. Another limitation of this work is the small number of patients, as such much of the data is descriptive. However, given the ultra‐rare nature of MSD, this is the largest cohort of MSD patients reported to date. As our cohort includes individuals from around the world, we anticipate that this cohort accurately reflects the global disease burden of MSD. Regional differences in standard of care are likely reflected in the variable use of interventions, such as gastrostomy tube placement and respiratory support.
4.1. Clinical characterization
We found that MSD is a severe, progressive neurologic disease with a high burden of somatic disease. Functional scales, including the GMFC‐MLD, developed and validated for MLD could be easily applied retrospectively to MSD patients and successfully captured regression in this population. As is common in rare diseases, many individuals experience a delay in diagnosis. We found that many of the children demonstrated unique clinical features that accompanied the early developmental delay, including ichthyosis, hepatosplenomegaly, and hearing loss. Identification of this characteristic constellation of signs and symptoms can facilitate earlier diagnosis of this rare disorder and prevent application of an inappropriate therapy. This is especially crucial as new treatments are being developed for single sulfatase disorders such and MLD and MPSII. We are optimistic that awareness will increase and diagnostic delays will decrease because of the important activities of MSD advocacy foundations and general rising awareness of orphan diseases.
4.2. Clinical biochemical testing and outcomes
In addition to capturing clinical data, we functionally characterized several novel SUMF1 missense variants in patients in our cohort by measuring residual sulfatase activity and FGE stability in vitro, followed by in silico modeling of variant FGE structures. We also extracted functional data from published SUMF1 pathogenic variants for all other cases. This allowed a complete genotype analysis and scoring of variant severity. Clinical testing of sulfatase activities in blood and glycosaminoglycan levels in urine did not correlate with genotype severity or outcomes in our cohort. Therefore, we do not recommend that these measures be used to predict individual clinical outcomes or in subgroup classification for clinical trials. Clinical biochemical testing should be used as a diagnostic biomarker, but not as a prognostic biomarker.
4.3. Predicting outcomes from early clinical and molecular features
Importantly, we found that prior classification schemes that separated cohorts into neonatal, severe infantile, mild infantile and juvenile cases did not appropriately predict outcomes in our cohort. 20 , 21 , 22 These approaches traditionally designated neonatal and severe infantile cases as arising before age 2, while mild infantile and juvenile cases presented after age 2. Our approach relied on detailed review of clinical records and thus even in cases with an attenuated clinical course, early signs such as hypotonia and poor feeding were noted before age 2. Our cohort, which included individuals with the full spectrum of neurologic disability, had a mean age of symptom onset of 0.7 years. Despite this clear early onset, there was also a significant delay in diagnosis of 3.6 years on average. As awareness of this rare, but characteristic disorder improves we anticipate earlier diagnosis. Additionally, MSD will likely be detected by newborn screening of lysosomal enzyme disorders, such as MLD. 23 , 24 If a therapy becomes available, early diagnosis and treatment will likely be essential to optimize clinical outcomes.
Through retrospective scoring of neurodevelopment, we identified two major sub‐cohorts: a severe group that never attained independent ambulation, and an attenuated cohort that developed the ability to walk independently followed by regression. To facilitate earlier differentiation of these two subgroups, we searched for additional features that could distinguish severe from attenuated patients. We developed a strategy that included both genotypic and early clinical features, as the functional consequences of novel SUMF1 variants will not always be available. Appearance of signs or symptoms (such as poor feeding or orthopedic anomalies) before 1 month of age and presence two severe SUMF1 alleles each correlated with poor motor and language outcomes, earlier age of regression onset, and decreased survival.
Using deep retrospective phenotyping with biochemical analysis and molecular modeling, we were able to identify two novel clinically coherent subtypes. The key clinical outcomes of independent ambulation and timing of neurologic regression were strongly associated with genotype and early age of onset. We propose that like other lysosomal storage disorders, MSD patients can be classified as “severe” or “attenuated” based on age of onset, variant severity, and early motor development. Additional discriminating factors like involvement of individual organ systems during the progressive course of MSD will refine this classification in future work. This work provides an important foundation for development of a prospective method to define clinically‐coherent groups for future MSD clinical trials.
CONFLICT OF INTEREST
Laura Adang: MLD Foundation and CureMLD scientific advisory board member. Rebecca Ahrens‐Nicklas and Lars Schlotawa: Medical advisors to the United MSD Foundation. Mauricio De Castro: Board of Directors United MSD Foundation. Samuel Groeschel, Christiane Kehrer, Klaus Harzer, Orna Staretz‐Chacham, Thiago Oliveira Silva, Ida Vanessa D. Schwartz, Jutta Gärtner, Carrie Costin, Esperanza Font Montgomery, Thomas Dierks, Karthikeyan Radhakrishnan declare they have no conflicts of interest.
AUTHOR CONTRIBUTIONS
Laura A. Adang, Lars Schlotawa, Rebecca C. Ahrens‐Nicklas, Karthikeyan Radhakrishnan: study design, data collection, analysis, drafting of manuscript. Mauricio De Castro, Carrie Costin, Esperanza Font Montgomery, Samuel Groeschel, Christiane Kehrer, Klaus Harzer, Orna Staretz‐Chacham: data collection, critical revision of manuscript. Thiago Oliveira Silva, Ida Vanessa D. Schwartz, Jutta Gärtner, Thomas Dierks: study design, critical revision of manuscript.
ETHICS STATEMENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study. Proof that informed consent was obtained must be available upon request. The parent IRB protocol is the Myelin Disorders Biorepository Project (ClinicalTrials.gov Identifier: NCT03047369, IRB approval 14‐011236).
Supporting information
Appendix S1: Supporting Information.
ACKNOWLEDGMENTS
We thank the families and patient advocacy groups (MSD Action Foundation and United MSD Foundation) involved in this manuscript. We also thank Dr. Stefanie Beck‐Woedl for assistance with genetic evaluations and Dr. Judith Böhringer for assistance with biochemical evaluations. RCA‐N: Supported by grants from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under Award Number (K08NS105865) and The Children's Hospital of Philadelphia Research Institute. LAA: Supported by grant from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under Award Number K23NS114113. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Adang LA, Schlotawa L, Groeschel S, et al. Natural history of multiple sulfatase deficiency: Retrospective phenotyping and functional variant analysis to characterize an ultra‐rare disease. J Inherit Metab Dis. 2020;43:43:1298–1309. 10.1002/jimd.12298
Karthikeyan Radhakrishnan and Rebecca C. Ahrens‐Nicklas contributed equally to this work.
Funding information National Center for Advancing Translational Sciences, Grant/Award Number: KL2TR001879; National Institute of Neurological Disorders and Stroke, Grant/Award Numbers: K08NS105865, K23NS114113
REFERENCES
- 1. Dierks T, Schmidt B, Borissenko LV, et al. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)‐formylglycine generating enzyme. Cell. 2003;113(4):435‐444. [DOI] [PubMed] [Google Scholar]
- 2. Cosma MP, Pepe S, Annunziata I, et al. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 2003;113(4):445‐456. [DOI] [PubMed] [Google Scholar]
- 3. Dierks T, Dickmanns A, Preusser‐Kunze A, et al. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine‐generating enzyme. Cell. 2005;121(4):541‐552. [DOI] [PubMed] [Google Scholar]
- 4. Sardiello M, Annunziata I, Roma G, Ballabio A. Sulfatases and sulfatase modifying factors: an exclusive and promiscuous relationship. Human Mol Genet. 2005;14(21):3203‐3217. [DOI] [PubMed] [Google Scholar]
- 5. Ahrens‐Nicklas R, Schlotawa L, Ballabio A, et al. Complex care of individuals with multiple sulfatase deficiency: clinical cases and consensus statement. Mol Genet Metab. 2018;123(3):337‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khateb S, Kowalewski B, Bedoni N, et al. A homozygous founder missense variant in arylsulfatase G abolishes its enzymatic activity causing atypical usher syndrome in humans. Genet Med: Off J Am College Med Genet. 2018;20(9):1004‐1012. [DOI] [PubMed] [Google Scholar]
- 7. Jaszczuk I, Schlotawa L, Dierks T, et al. Expanding the genetic cause of multiple sulfatase deficiency: a novel SUMF1 variant in a patient displaying a severe late infantile form of the disease. Mol Genet Metab. 2017;121(3):252‐258. [DOI] [PubMed] [Google Scholar]
- 8. Garavelli L, Santoro L, Iori A, et al. Multiple sulfatase deficiency with neonatal manifestation. Italian J Pediat. 2014;40:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schlotawa L, Ennemann EC, Radhakrishnan K, et al. SUMF1 mutations affecting stability and activity of formylglycine generating enzyme predict clinical outcome in multiple sulfatase deficiency. Eur J Hum Genet. 2011;19(3):253‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Helman G, Van Haren K, Bonkowsky JL, et al. Disease specific therapies in leukodystrophies and leukoencephalopathies. Mol Genet Metab. 2015;114(4):527‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Groeschel S, Kuhl JS, Bley AE, et al. Long‐term outcome of allogeneic hematopoietic stem cell transplantation in patients with juvenile metachromatic leukodystrophy compared with nontransplanted control patients. JAMA Neurol. 2016;73(9):1133‐1140. [DOI] [PubMed] [Google Scholar]
- 12. Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile‐onset Pompe disease. J Pediatr. 2006;148(5):671‐676. [DOI] [PubMed] [Google Scholar]
- 13. Pineda M, Jurickova K, Karimzadeh P, et al. Disease characteristics, prognosis and miglustat treatment effects on disease progression in patients with Niemann‐pick disease type C: an international, multicenter, retrospective chart review. Orphanet J Rare Dis. 2019;14(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kehrer C, Groeschel S, Kustermann‐Kuhn B, et al. Language and cognition in children with metachromatic leukodystrophy: onset and natural course in a nationwide cohort. Orphanet J Rare Dis. 2014;9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paulson A, Vargus‐Adams J. Overview of four functional classification systems commonly used in cerebral palsy. Children (Basel, Switzerland). 2017;4(4):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kehrer C, Blumenstock G, Raabe C, Krageloh‐Mann I. Development and reliability of a classification system for gross motor function in children with metachromatic leucodystrophy. Dev Medicine and Child Neurol. 2011;53(2):156‐160. [DOI] [PubMed] [Google Scholar]
- 17. Schlotawa L, Steinfeld R, von Figura K, Dierks T, Gartner J. Molecular analysis of SUMF1 mutations: stability and residual activity of mutant formylglycine‐generating enzyme determine disease severity in multiple sulfatase deficiency. Hum Mutat. 2008;29(1):205. [DOI] [PubMed] [Google Scholar]
- 18. Staretz‐Chacham O, Schlotawa L, Wormser O, et al. A homozygous missense variant of SUMF1 in the Bedouin population extends the clinical spectrum in ultrarare neonatal multiple sulfatase deficiency. Mol Genet Genom Med. 2020;e1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sabourdy F, Mourey L, Le Trionnaire E, et al. Natural disease history and characterisation of SUMF1 molecular defects in ten unrelated patients with multiple sulfatase deficiency. Orphanet J Rare Dis. 2015;10:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Busche A, Hennermann JB, Burger F, et al. Neonatal manifestation of multiple sulfatase deficiency. Eur J Pediatr. 2009;168(8):969‐973. [DOI] [PubMed] [Google Scholar]
- 21. Burk RD, Valle D, Thomas GH, et al. Early manifestations of multiple sulfatase deficiency. J Pediatr. 1984;104(4):574‐578. [DOI] [PubMed] [Google Scholar]
- 22. Nur BG, Mihci E, Pepe S, et al. Neonatal multiple sulfatase deficiency with a novel mutation and review of the literature. Turk J Pediatr. 2014;56(4):418‐422. [PubMed] [Google Scholar]
- 23. Hong X, Kumar AB, Daiker J, et al. Leukocyte and dried blood spot arylsulfatase a assay by tandem mass spectrometry. Anal Chem. 2020;92(9):6341‐6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spacil Z, Babu Kumar A, Liao HC, et al. Sulfatide analysis by mass spectrometry for screening of metachromatic Leukodystrophy in dried blood and urine samples. Clin Chem. 2016;62(1):279‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information.