

SUMMARY
Asian cultivated rice (Oryza sativa) and African cultivated rice (Oryza glaberrima) originated from the wild rice species Oryza rufipogon and Oryza barthii, respectively. The genomes of both cultivated species have undergone profound changes during domestication. Whole‐genome de novo assemblies of O. barthii, O. glaberrima, O. rufipogon and Oryza nivara, produced using PacBio single‐molecule real‐time (SMRT) and next‐generation sequencing (NGS) technologies, showed that Gypsy‐like retrotransposons are the major contributors to genome size variation in African and Asian rice. Through the detection of genome‐wide structural variations (SVs), we observed that besides 28 shared SV hot spots, another 67 hot spots existed in either the Asian or African rice genomes. Based on gene annotation information of the SVs, we established that organelle‐to‐nucleus DNA transfers resulted in numerous SVs that participated in the nuclear genome divergence of rice species and subspecies. We detected 52 giant nuclear integrants of organelle DNA (NORGs, defined as >10 kb) in six Oryza AA genomes. In addition, we developed an effective method to genotype giant NORGs, based on genome assembly, and first showed the dynamic change in the distribution of giant NORGs in rice natural population. Interestingly, 16 highly differentiated giant NORGs tended to accumulate in natural populations of Asian rice from higher latitude regions, grown at lower temperatures and light intensities. Our study provides new insight into the genome divergence of African and Asian rice, and establishes that organelle‐to‐nucleus DNA transfers, as potentially powerful contributors to environmental adaptation during rice evolution, play a major role in producing SVs in rice genomes.
Keywords: de novo assembly, structural variation, organelle‐to‐nucleus DNA transfer, African and Asian rice
Significance Statement
We report whole‐genome de novo assemblies of four Oryza AA genomes and the identification of genome‐wide structural variations in African and Asian rice. We also established that organelle‐to‐nucleus DNA transfers, as potentially powerful contributors to environmental adaptation during rice evolution, participated in the nuclear genome divergence of rice species and subspecies.
INTRODUCTION
Genomic variations are the basis for genetic diversity, genome evolution and speciation. Structural variants (SVs), including copy‐number variations, deletions, duplications, insertions, inversions and translocations, are important components of genetic variation. Many studies have revealed that SVs not only significantly influence cellular molecular processes (Ben‐David et al., 2014; Wu et al., 2017), but also are associated with human diseases and phenotypic changes in animals and plants (Cook et al., 2012; Weischenfeldt et al., 2013; Zhang et al., 2015; Duan et al., 2017; Liu et al., 2017; Wu et al., 2018; Chakraborty et al., 2019). Despite their importance, large swathes of SVs remain to be discovered in the genomes of most organisms because of the limitations of whole‐genome information and the lack of precise computational methods to identify them. Although tremendous efforts have been made to detect SVs embedded in plant genomes based on the resequencing of short reads (Long et al., 2013; Zhang et al., 2015; Torkamaneh et al., 2018; Fuentes et al., 2019), both high false‐positive rates and high false‐negative rates remain major unresolved problems in SV detection (Huddleston et al., 2017; Nattestad et al., 2018; Fuentes et al., 2019). Recently, advanced long‐read sequencing technologies, including those developed by PacBio and Oxford Nanopore (Audano et al., 2019; De Coster et al., 2019), coupled with updated algorithms and methods (English et al., 2014; Sedlazeck et al., 2018), have paved the way towards the accurate identification of genome‐wide SVs, and especially large SVs, thereby enhancing our understanding of their contribution to genome evolution.
Previous studies have revealed that the movement of transposable elements (TEs) in many animal and plant genomes can lead to large SVs, which have shaped genome structure and have affected gene expression and function. Notably, organelle‐to‐nucleus DNA transfer also plays an important role in producing SVs. In eukaryotes, the chloroplast and mitochondrion were derived by endosymbiosis from a cyanobacterium and an α‐proteobacterium, respectively. Compared with their evolutionary progenitors, contemporary organelle genomes are substantially smaller, through the massive loss of dispensable sequences and the transfer of DNA fragments from their genomes to the nuclear genome (Blanchard and Lynch, 2000; Kleine et al., 2009). Previous reports have shown that organelle‐to‐nucleus DNA transfer is a common and continuous process, giving rise to many nuclear integrants of plastid DNA (NUPTs) and mitochondrial DNA (NUMTs) that have shaped nuclear genome architecture and have profoundly affected genome evolution (Matsuo et al., 2005; Hazkani‐Covo et al., 2010; Liang et al., 2018).
Nuclear integrants of plastid DNA (NUPTs) and/or NUMTs have been identified in almost all eukaryotic genomes sequenced, and both the copy number and content of nuclear integrants of organelle DNA (NORGs) are highly diverse between species, based on the analysis of single representative reference genomes (Hazkani‐Covo et al., 2010; Hazkani‐Covo and Martin, 2017). For example, the number of NUMTs detected ranges from zero in the nuclear genome of Anopheles gambiae to more than 4000 in the nuclear genome of Ornithorhynchus anatinus (Richly and Leister, 2004a; Calabrese et al., 2017). Additionally, NUPTs are detected rarely in Plasmodium falciparum but frequently both in Oryza sativa (rice) and in Arabidopsis thaliana (Richly and Leister, 2004b). Interestingly, DNA fragments derived from mitochondrion‐to‐nucleus transfers show a high degree of presence/absence polymorphism in the human population (Lang et al., 2012; Dayama et al., 2014). In rice, several NUPT and NUMT events occur specifically in one subspecies of Asian cultivated rice but are absent in another (Huang et al., 2005; Guo et al., 2008; Wang and Timmis, 2013). The evolutionary trajectory of NORGs at the population level remains unclear, however.
Rice is an agriculturally important crop, grown widely around the world. Cultivated rice includes two species, O. sativa and Oryza glaberrima. Asian cultivated rice O. sativa has been further classified into two subspecies, japonica and indica, based on their morphometric and genetic differences. Previous studies suggest that O. sativa was domesticated from Oryza rufipogon (perennial wild rice) around 9000 years ago in Asia, and that O. glaberrima was domesticated from Oryza barthii, independently, around 3000 years ago in West Africa (Wang et al., 2014; Choi et al., 2020). Notably, Oryza nivara, as the annual form of O. rufipogon, might be an important contributor for the domestication of indica rice through introgression hybridization (Choi et al., 2017). Therefore, African and Asian rice are ideal model plants for comparing genome variations occurring in the complex domestication process.
To compare the differences in genome‐wide SVs between African and Asian rice, we assembled three high‐quality genomes of the wild relatives of cultivated rice, O. barthii, O. nivara and O. rufipogon, and one African cultivated rice (O. glaberrima) belonging to the AA genome group, and then integrated the reference genomes of two subspecies of Asian cultivated rice (O. sativa L. ssp. japonica var. Nipponbare and indica var. R498). We then accurately identified genome‐wide SVs between wild and cultivated rice and compared the distributions of SV hot spots in African and Asian rice. We further detected genome‐wide NORG events, which revealed a significant difference in the evolution of African and Asian rice. Sixteen giant NORGs (>10 kb) tend to accumulate in natural populations of rice from higher latitude regions, grown at lower temperatures and light intensities, indicating that NORG events may have contributed to environmental adaptation during the evolution of Asian rice.
RESULTS
Four high‐quality de novo assemblies of wild and cultivated rice genomes
To uncover the full spectrum of SVs in African and Asian rice genomes, we collected four wild and cultivated rice samples belonging to the AA genome group for high‐throughput genome sequencing: a perennial wild rice, O. rufipogon accession DXCWR, from China; an annual wild rice, O. nivara accession W2014, from India; an annual wild rice, O. barthii accession W1411, from Sierra Leone; and an African cultivated rice, O. glaberrima accession IRGC104165, from Guinea (Figure S1). For each sample, we generated on average 24.8 Gb of Illumina paired‐end reads and 10.3 Gb of PacBio single‐molecule long reads. The average sequencing depth of the Illumina reads ranged from 55.7‐ to 72.1‐fold genome coverage, and the average sequencing depth of the PacBio reads, with 7.6‐ to 9.6‐kb read lengths, ranged from 22.5‐ to 31.5‐fold genome coverage (Figure S2; Table S1).
The PacBio subreads were processed using the canu (Koren et al., 2017) pipeline to generate contigs. The raw PacBio contigs were incorporated to order and link the contigs using bacterial artificial chromosome (BAC) end sequences (BESs) collected from the Oryza Map Alignment Project (OMAP, http://www.omap.org) and used to generate scaffolds. The N50 size of the scaffolds ranged from 0.39 to 2.46 Mb (Table S2). After the removal of redundant sequences, the final assemblies comprised 349.75 Mb for O. barthii, 352.35 Mb for O. glaberrima, 365.94 Mb for O. nivara and 359.28 Mb for O. rufipogon (Figure 1; Table S2). The contiguity of the four de novo assemblies for the African and Asian rice species was 6–44 times longer than that of previously reported assemblies based on short reads, as evaluated by contig N50 values (Figure S3) (Huang et al., 2012; Wang et al., 2014; Zhang et al., 2014; Stein et al., 2018). Additionally, the genome sizes of the two Asian cultivated rice subspecies, japonica Nipponbare and indica R498, were 374.47 and 390.32 Mb, respectively (Kawahara et al., 2013; Du et al., 2017). These results show that Asian rice species have larger genome sizes than African rice species.
Figure 1.
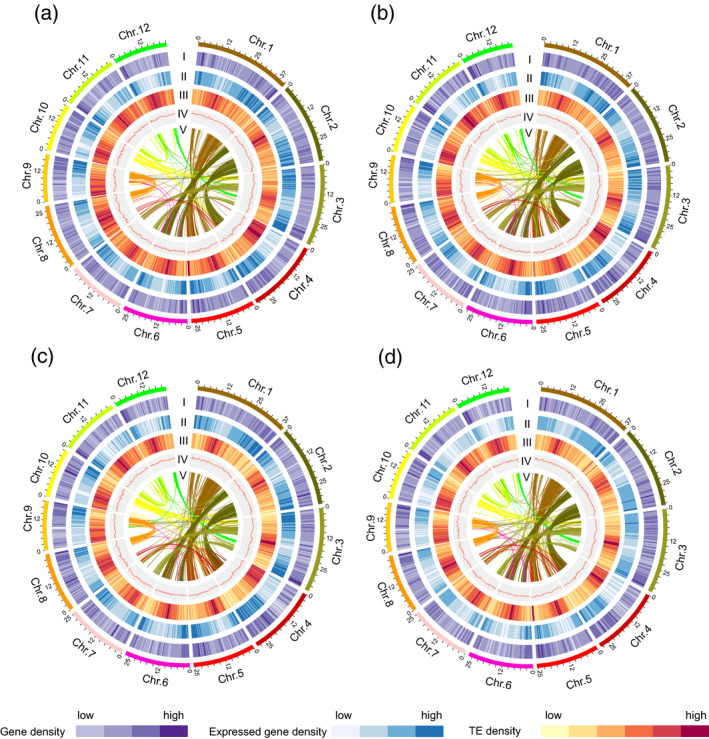
Genomic landscapes of the pseudochromosomes of four de novo assemblies of African and Asian rice.
(a–d) Characterization of the Oryza rufipogon (a), Oryza nivara (b), Oryza barthii (c) and Oryza glaberrima (d) genomes. Tracks from the outer to the inner circles represent gene density (I), distribution of expressed genes (II), transposable element (TE) density (III), distribution of GC content (IV) and segmental duplication (V), respectively.
Quality assessment of the four genome assemblies indicated that at least 97.4% of the Illumina reads mapped back to the corresponding genome assemblies (Table S1), and approximately 95.2–97.4% of benchmarking universal single‐copy orthologs (BUSCO) (Simao et al., 2015) could be successfully searched in the four genome assemblies, which is similar to the percentage for the japonica Nipponbare reference genome (Table S3). Additionally, core eukaryotic genes mapping approach (CEGMA) (Parra et al., 2007) analysis showed that 96.4% (99.6%), 96.0% (99.2%), 95.6% (98.8%) and 95.2% (98.4%) of the complete (partial) gene sets could be aligned with the assembled O. barthii, O. glaberrima, O. nivara and O. rufipogon genomes, respectively (Table S4). Finally, both collinearity and nucleotide identity scores were high, with nucleotide identity scores ranging from 97.1 to 99.9% when each of the four de novo assemblies were aligned with eight BAC sequences deposited in GenBank (Figure S4). Taken together, these results indicate that the four rice genomes are high‐quality de novo assemblies, thus ensuring the reliability of the subsequent genomic analyses in this study.
Comparison of genomic divergence in African and Asian rice
To accurately predict genes, we generated a total of 245.3 Gb of RNA‐sequencing (RNA‐seq) data from different tissues and developmental stages of the four rice samples, with an average of 5.98 Gb per tissue (Table S5). Through integrating the transcripts assembled from RNA‐seq data with the gene predictions obtained by ab initio prediction and evidence‐based methods using the de novo assembled genomes, 44 938, 44 763, 45 346 and 44 592 protein‐coding genes (excluding transposon‐coding genes) were predicted in O. barthii, O. glaberrima, O. nivara and O. rufipogon, respectively (Table S2). To understand the potential biological functions of the predicted protein‐coding genes, we further annotated the functions of each predicted gene using the bioinformatic databases and tools currently available. More than 75.9% of the predicted protein‐coding genes captured functional descriptions for each genome, including protein domains, motifs, and homologs (Table S6).
Transposable elements (TEs) are an important component of plant genomes. The O. barthii, O. glaberrima, O. nivara and O. rufipogon assemblies contained 35.76, 35.89, 39.16, and 39.28% repetitive sequences, respectively (Table S7), showing that the proportion of repetitive sequences was significantly higher in rice assemblies produced using long reads than that detected in previous assemblies using short reads (27.65–29.83%) (Zhang et al., 2014), which highlights the advantage of using long‐read technology for assembling repetitive regions. Notably, the O. nivara and O. rufipogon genomes harbored more repetitive sequences than the O. barthii and O. glaberrima genomes.
An investigation of TE class revealed that Gypsy‐like retrotransposons were the most abundant class and occurred more frequently in O. nivara (15.44%) and O. rufipogon (15.43%) than in O. barthii (12.17%) and O. glaberrima (12.10%), indicating that these retrotransposons may be an important contributor to genome size variation between African and Asian rice (Table S7). Estimating the insertion time of Gypsy elements in six Oryza AA genomes, including the four de novo assemblies and two Asian cultivated rice subspecies (japonica Nipponbare and indica R498) reference genomes, showed that the amplification bursts of Gypsy elements occurred earlier in Asian rice than in African rice (Figure S5), further suggesting that the recent bursts of Gypsy elements might contribute to the larger Asian rice genome size.
Widespread SVs within Oryza AA genomes
To precisely identify SVs (≥50 bp) in the six Oryza AA genomes, we used three complementary methods based on PacBio long reads and de novo assemblies (English et al., 2014; Nattestad and Schatz, 2016; Marcais et al., 2018; Sedlazeck et al., 2018). Concurrently, we developed a custom validation pipeline that improved the accuracy of SV identification and extracted the insertion sequences from the corresponding assembled genomes. In total, we detected 17 085/21 246, 16 730/20 604, 15 457/16 331, 9050/8396 and 15 046/14 099 insertions/deletions (indels) in O. barthii, O. glaberrima, O. nivara, O. rufipogon and O. sativa ssp. indica, respectively, against the japonica Nipponbare reference genome, spanning 42.4–85.5 Mb of genomic regions (Table S8). At least 71% of the identified SVs were shorter than 2 kb, and on average each genome had 2719/1432 indels longer than 5 kb (Figure 2a,b; Table S8).
Figure 2.
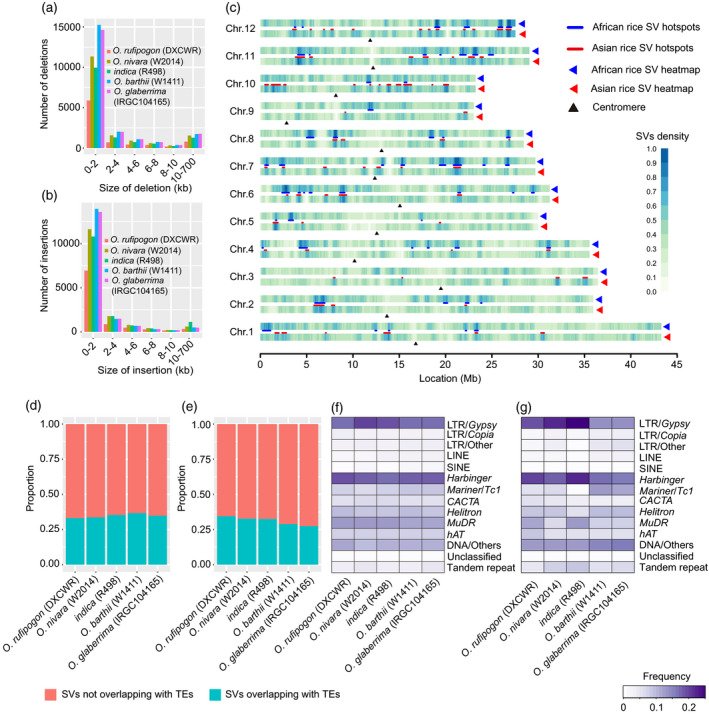
Characteristics of structural variations (SVs) and distribution of SV hot spots in African and Asian rice genomes.
(a) Frequency distribution of deletion size.
(b) Frequency distribution of insertion size.
(c) Whole‐genome distribution of merged SVs and SV hot spots. The colors from light to dark blue indicate an increase of normalized SV density in each window of 500 kb with a step size of 50 kb. Blue and red lines represent locations of SV hot spots in African and Asian rice genomes, respectively.
(d, e) Proportions of deletions (d) and insertions (e) overlapping with transposable elements (TEs) annotated in each genome.
(f, g) Frequencies of deletions (f) and insertions (g) overlapping with various classes of TEs in the five Oryza AA genomes.
Among the SVs identified, we uncovered several well‐characterized SVs associated with important phenotypic variations. For example, we detected the 383‐bp deletion in the semi‐dwarf1 (sd1) gene known as the ‘Green Revolution’ gene in the indica R498 genome (Monna et al., 2002) (Figure S6a). We also identified a 1212‐bp deletion approximately 5 kb upstream of the known quantitative trait locus (QTL) GRAIN WIDTH AND WEIGHT ON CHROMOSOME 5 (GW5)/GRAIN SIZE ON CHROMOSOME 5 (GSE5) in the japonica Nipponbare genome (Duan et al., 2017; Liu et al., 2017) (Figure S6b). Notably, we identified two insertions in O. rufipogon and one insertion in O. nivara linked to this 1212‐bp causal variation (Figure S6b). Additionally, we identified novel SVs in several known genes, such as PROSTRATE GROWTH 1 (PROG1) (Jin et al., 2008; Tan et al., 2008), which influences plant architecture, and RICE FLOWERING LOCUS T1 (RFT1), which regulates flowering time (Komiya et al., 2008).
Two adjacent long terminal repeat (LTR)/Copia elements were specifically inserted in the coding region of PROG1 in the O. barthii genome, resulting in the loss of function of PROG1, whereas a large segment deletion harboring the PROG1 gene existed in the O. glaberrima genome, consistent with a previous report (Figure S6c) (Wu et al., 2018). As compared with the japonica Nipponbare reference genome, multiple TEs were inserted into the RFT1 genic regions in the O. barthii and O. glaberrima genomes, producing a truncated RFT1 mRNA, and thereby resulting in a loss of function (Figure S6d). These results indicate that the SVs played important roles in the changes in gene function that occurred during rice domestication and improvement.
Varied structural variation hot spots between African and Asian rice
To investigate the divergence of SVs in African and Asian rice, we merged SVs and obtained 24 943/25 631 and 35 015/27 908 indels, respectively, in African and Asian rice with at least 70% reciprocal overlap. Notably, approximately 33.3% of the deletions that we identified in Asian rice were identified in the present study and had not been previously reported (Fuentes et al., 2019), suggesting that the advanced long‐read sequencing technology that we used is an effective means of detecting SVs.
The SVs that we identified were unevenly distributed along 12 chromosomes and clustered into certain specific chromosomal regions (Figure 2c). Based on our evaluation of SV density, we identified 55 and 68 putative SV hot spots in the African and Asian rice genomes, respectively. Comparison of the locations of these SV hot spots showed that a total of 28 hot spots occurred at similar chromosome positions in both African and Asian genomes, whereas the remaining 67 hot spots were embedded in the African or Asian rice genomes alone (Figure S7a). Additionally, 34 (50%) of the SV hot spots identified in Asian rice were also detected in the 3K Rice Genomes Project by high‐depth short‐read sequencing (Fuentes et al., 2019), indicating that these genomic regions harboring common SV hot spots might be prone to the formation of SVs (Figure S7b).
The SVs that we identified were enriched for different classes of repetitive DNA. We examined the intersections of SVs through TE annotation and found that more than 27% of indel events overlapped TEs across species, with at least 80% reciprocal overlap (Figure 2d,e), suggesting that TE mobility played a crucial role in SV formation. Of these overlapping TEs, the LTR/Gypsy and Harbinger superfamilies were the most frequent among class‐I and class‐II TEs, respectively (Figure 2f,g). Unlike LTR/Gypsy among class‐I TEs, the Harbinger superfamily is not the most abundant class‐II TE family in rice genomes (Table S7), suggesting that this superfamily had higher transposition activity than other classes of DNA transposons in rice. Both LTR/Gypsy and Harbinger TEs showed lower frequencies of insertion in African rice than in Asian rice, implying that they might have lower transposition activity in African rice genomes (Figure 2g).
Abundance and characteristics of NUPTs and NUMTs in African and Asian rice
Based on the locations of SV breakpoints, we established that among 43 011 unique deletions identified in the present study, approximately 39.9% were located either within or spanning a total of 20 659 genes, resulting in changes in gene structure in 16 876 genes. Gene ontology (GO) analysis showed that these genes were involved in a variety of biological processes, including DNA metabolic processes, macromolecule metabolic processes and cell death (Table S9). In addition, we also surveyed genes located within insertions in each genome and found that similar biological processes were over‐represented (Table S9). A set of terms related to chloroplast or mitochondrion, including NADH dehydrogenase activity and photosystem II, was shared between African and Asian rice (Table S9). Alignment analysis showed that the sequences of several SVs were completely or partially homologous to the chloroplast or mitochondrial genomes (Figure S8), indicating that NUPTs and NUMTs caused varied SVs during the course of rice genome evolution.
We further identified genome‐wide NUPTs and NUMTs in the six Oryza AA genomes (Figure S9) and found that the total combined lengths of NUPTs and NUMTs ranged from 0.47 to 0.94 Mb and from 0.54 to 1.27 Mb (Figure 3a), with average individual lengths of 545 and 729 bp, respectively. These NORGs constituted 0.33–0.50% of total nuclear genomes in the six Oryza AA genomes. Notably, both the quantity and the degree of fragmentation of NUPTs in japonica Nipponbare genome showed obvious differences from those in the other five Oryza AA genomes (Figure 3a,b). In addition, the total length of NUMTs in O. barthii (approximately 1.3 Mb) was longer than that in other rice species (Figure 3a), and both African rice species also possessed more NUMTs (>200 bp) in pericentromeric regions than Asian rice species (two‐tailed Student’s test, P < 0.003) (Figure 3c).
Figure 3.
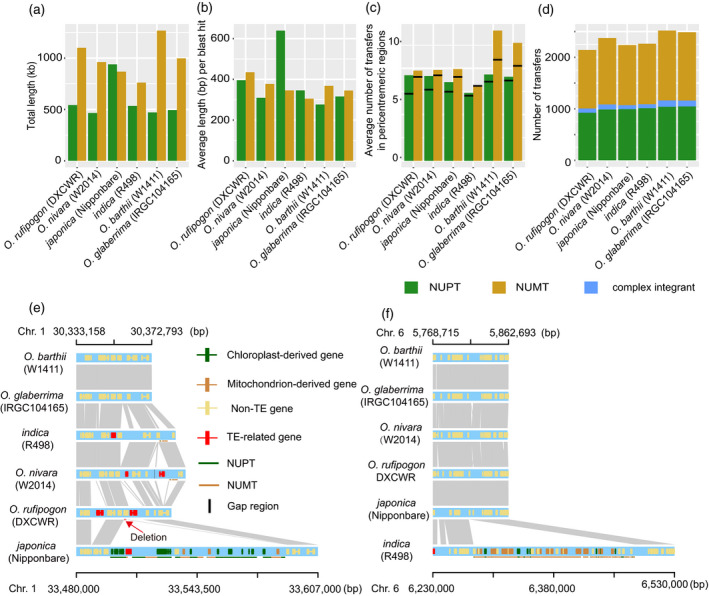
Characteristics of DNA transfers from organellar to nuclear genomes in African and Asian rice.
(a) Total lengths of nuclear integrants of plastid DNA (NUPTs) and mitochondrial DNA (NUMTs) in the six Oryza AA genomes.
(b) Fragmentation status of NUPTs and NUMTs in the six Oryza AA genomes.
(c) Average numbers of NUPTs and NUMTs (>200 bp in length) in the pericentromeric regions of each genome. Black lines within the colored bars represent the expected numbers of NUPTs and NUMTs.
(d) Number of NUPTs, NUMTs and complex integrants in the six Oryza AA genomes.
(e) A unique complex integrant in the japonica Nipponbare genome. The grey regions represent sequences sharing sequence collinearity.
(f) A unique complex integrant in the indica rice R498 genome.
In general, NUPTs and NUMTs are located at different genomic regions; however, approximately 4% of all NORGs contained both NUPTs and NUMTs (Figure 3d). For example, four small NUMTs were inserted into an approximately 93‐kb NUPT on chromosome 1 in the japonica Nipponbare genome (Figure 3e), and an approximately 172‐kb NUMT on chromosome 6 in the indica R498 genome was broken up by several small NUPTs (Figure 3f). These observations suggest that NUMTs can insert into NUPTs and vice versa, thereby generating mixed NORGs.
To investigate whether there were preferred insertion sites for NORGs, we performed motif enrichment analyses of the 100‐bp flanking sequences surrounding NORG insertion sites. Of the motifs over‐represented in these flanking regions, only the AT/TA repeat motif was repeatedly enriched at the flanking regions of both NUPTs and NUMTs (Figures S10 and S11), implying that pieces of organellar DNA might prefer to integrate into AT/TA‐enriched regions. For example, as compared with the japonica Nipponbare genome, a giant NUPT (nupt3) was found inserted into an (AT)43 repeat sequence in the indica R498 genome, coupled with an increase of 278 AT repeats, and the flanking sequences of a NUMT (NivM_153) insertion in the O. nivara W2014 genome were also an AT/TA enrichment region (Figure 4a,b).
Figure 4.
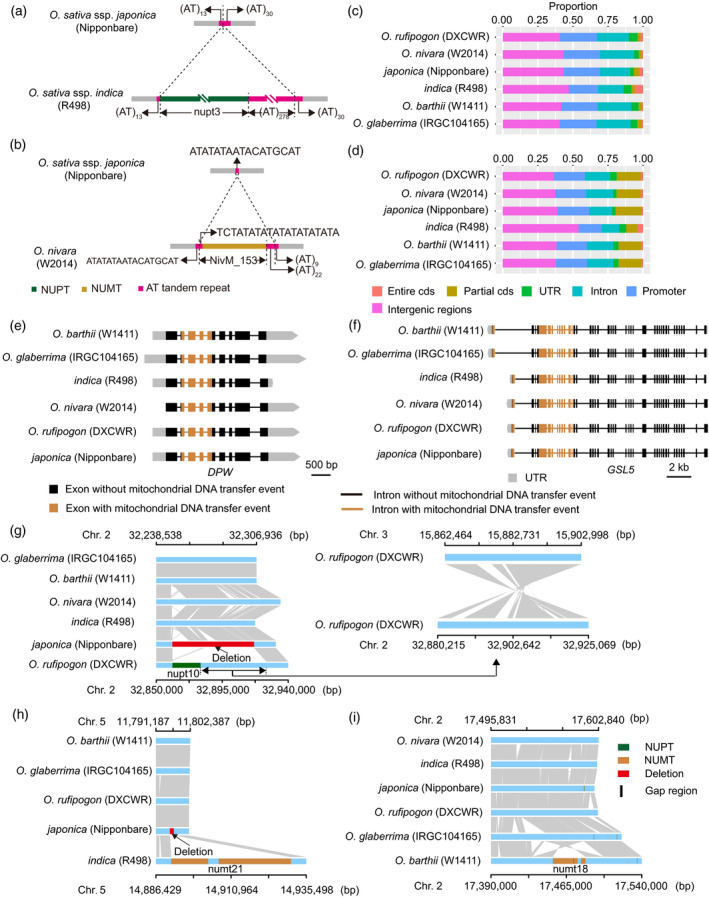
Insertion preferences and consequences of organelle‐to‐nucleus DNA transfers in African and Asian rice.
(a, b) A chloroplast DNA (a) and a mitochondrial DNA (b) fragment are integrated into the AT/TA enrichment regions of indica and into the AT/TA enrichment regions of Oryza nivara, respectively.
(c, d) Insertion preferences of nuclear integrants of plastid DNA (NUPTs) (c) and nuclear integrants of mitochondrial DNA (NUMTs) (d) in the six Oryza AA genomes. cds, coding sequence; UTR, untranslated region.
(e) A mitochondrial DNA fragment is integrated into the coding region of the gene DEFECTIVE POLLEN WALL (DPW) in all six Oryza AA genomes.
(f) Two mitochondrial DNA fragments are integrated into the gene GLUCAN SYNTHASE‐LIKE 5 (GSL5).
(g) A giant nuclear integration of chloroplast DNA (nupt10) caused a large deletion and a translocation in the Oryza rufipogon DXCWR genome. The grey regions represent sequences sharing sequence collinearity.
(h) A giant nuclear integration of mitochondrial DNA (numt21) was coupled with a deletion in the indica R498 genome.
(i) A giant nuclear integration of mitochondrial DNA (numt18) caused a chromosomal segment inversion in the Oryza barthii W1411 genome.
Further investigation of the locations of NORGs showed that approximately 42.7% of NUPTs and 40.5% of NUMTs occur in intergenic regions, 25.2% of NUPTs and 21.1% of NUMTs in promoter regions, and 32.1% of NUPTs and 38.3% of NUMTs in genic regions. In genic regions, approximately 4.9% of NUPTs and 17.4% of NUMTs were found in exons, indicating that, as compared with NUPTs, NUMTs preferentially integrate into genomic DNA in locations that allow them to reconstruct existing exons (Figure 4c,d). For example, we identified a mitochondrial DNA (mtDNA) fragment that is integrated into the coding region of the gene DEFECTIVE POLLEN WALL (DPW), which controls rice anther development (Shi et al., 2011), thereby creating four exons encoding an NAD‐binding 4 domain (Figure 4e).
Likewise, GLUCAN SYNTHASE‐LIKE 5 (GSL5), regulating rice male fertility, contains two mtDNA fragments in its coding regions, respectively (Figure 4f) (Shi et al., 2015). Like GSL5, 10 of the 11 glucan synthase‐like genes in the rice genome contained NUMTs (Table S10). The wheat orthologs of DPW and GSL5 also carry these mitochondrially derived DNA segments, suggesting that these NUMTs are ancient events that happened prior to the divergence between rice and Triticum aestivum (wheat) (Figure S12). Taken together, these findings indicate that organelle‐to‐nucleus DNA transfers have contributed to the generation of de novo genes during genome evolution.
Giant NUPTs and NUMTs contributed to the divergence of the rice nuclear genome
To investigate whether NUPTs and NUMTs were involved in the divergence of rice nuclear genomes, we focused on the giant NUPTs and NUMTs of >10 kb in length along the anchored 12 chromosomes. We identified a total of 25 giant NUPTs and 27 giant NUMTs in the six Oryza AA genomes, which we refer to as nupt1–nupt25 and numt1–numt27 (Figure S9). We designed primers across the nuclear–organellar genomic junctions and confirmed a subset of the giant NORG events by PCR (Figure S13). Among the 52 giant NORGs, nine giant NUPTs and eight giant NUMTs were located in pericentromeric regions (Figure S9), consistent with previous reports that the pericentromeric region might have a stronger tolerance for the integration of a large organelle DNA fragment (Matsuo et al., 2005). Among the six Oryza AA genomes surveyed, four giant NUPTs and 11 giant NUMTs occurred in two or more genomes, and the remaining giant NORGs existed in only one genome (Figure 5a,b). For instance, the giant NUPT nupt6 was found in all Asian rice genomes, and three giant NUMTs, numt10, numt15 and numt16, were shared by both African and Asian rice genomes. Additionally, the surveyed genomes of O. barthii, O. glaberrima and O. nivara each harbored one or two giant NUPTs, whereas the genomes of O. rufipogon, indica and japonica carried seven, nine and 11 giant NUPTs, respectively (Figure 5a). The distribution of giant NUMTs was a little less heterogeneous, with 14 found in the genome of O. barthii and with between five and nine found each in the other five Oryza AA genomes (Figure 5b). These results implied that giant NORG events exhibited considerable divergence both between and among African and Asian rice genomes.
Figure 5.
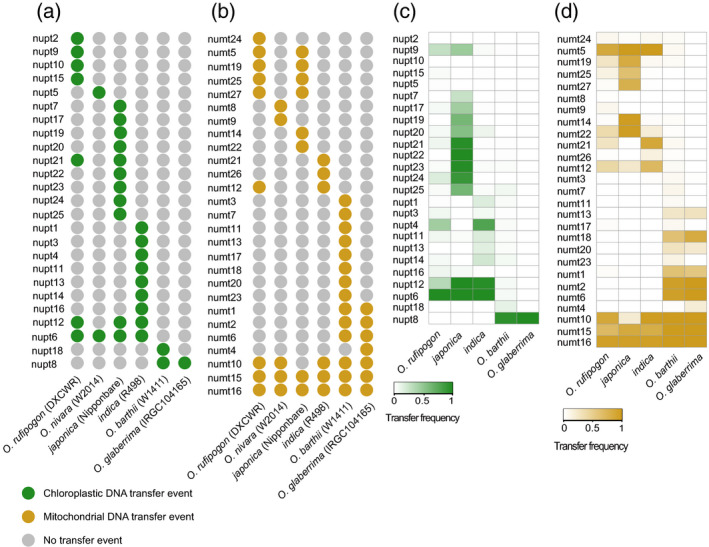
Distribution of giant nuclear integrants of organelle DNA (NORGs) along the 12 anchored chromosomes in the six Oryza AA genomes and rice natural population
(a, b) Giant nuclear integrants of plastid DNA (NUPTs) (a) and nuclear integrants of mitochondrial DNA (NUMTs) (b) identified in the six Oryza AA genomes.
(c, d) Frequency distributions of giant NUPTs (c) and NUMTs (d) in African and Asian rice natural populations.
To analyze the changes in genome structure that occurred in conjunction with these giant NORG events, we compared the sequences surrounding the insertion sites of the 52 giant NORGs in the six Oryza AA genomes and found that these events often resulted in SVs, including deletions, inversions and translocations (Figures 3e and 4g–i; Figure S14). For example, nupt10 on chromosome 2 of the O. rufipogon DXCWR genome appears to have led to a 55.6‐kb deletion and the interchromosomal translocation of a 44.8‐kb segment from chromosome 3 in its descendant japonica Nipponbare (Figure 4g), and numt21 on chromosome 5 resulted in a 1244‐bp deletion in indica R498 (Figure 4h). Similarly, numt18 on chromosome 2 of the O. barthii W1411 genome gave rise to an inversion of the chromosomal segment that harbored the insertion site in O. glaberrima IRGC104165 (Figure 4i). Hence, the organelle‐to‐nucleus DNA transfers also contributed to genome structural variations and promoted the differentiation of rice subspecies and species genomes.
Giant NUPTs and NUMTs were involved in species differentiation and environment adaptation in rice
To investigate the distribution of the 52 giant NORGs in rice natural populations, we reanalyzed the whole‐genome resequencing data for African and Asian rice from previous studies (Huang et al., 2012; Wang et al., 2014; Meyer et al., 2016), including 94 accessions of O. barthii, 113 O. glaberrima cultivars, 446 accessions of O. rufipogon, 519 indica cultivars and 484 japonica cultivars, and genotyped them for the presence and absence of each giant NORG by identifying the reads covering or spanning the insertion or integration sites of the NORGs (Figure S15). The frequency of giant NORGs differed dramatically between African and Asian rice populations (Figure 5c,d). Of 25 giant NUPTs, two (nupt12 and nupt6) had a high frequency in both O. rufipogon and O. sativa, and one (nupt8) showed a high frequency in both O. barthii and O. glaberrima.
Additionally, one giant NUPT (nupt4) showed a high frequency in O. rufipogon and in indica, and four, nupt21, nupt22, nupt23 and nupt24, specifically displayed a high frequency in japonica. No giant NUPTs were identified with high frequency in both African and Asian rice, however, suggesting that these integration events of chloroplast DNA (cpDNA) into the nuclear genome happened after the divergence of the African and Asian Oryza AA genomes. Similarly, of 27 giant NUMTs, only one (numt5) had a high frequency in both O. rufipogon and O. sativa, and four (numt18, numt1, numt2 and numt6) showed a high frequency in both O. barthii and O. glaberrima. Notably, three giant NUMTs (numt10, numt15 and numt16) displayed high frequencies in both African and Asian rice, indicating that they occurred prior to the divergence between African and Asian rice.
To trace the evolutionary trajectory of these three NUMTs, we further analyzed the collinear chromosomal regions in the genomes of wheat and Zea mays (maize). The results showed that there were no giant NUMTs in the syntenic region of wheat and maize (Figure S16), implying that the transfers of these three large segments from the mitochondrial to the nuclear genome happened after the divergence of rice, wheat and maize. Among the 52 giant NORGs detected in the present study, only two NUPTs (nupt2 and nupt10) identified in the O. rufipogon DXCWR genome were not detected in any of these African and Asian rice samples, indicating that the two integration events specifically occurred in O. rufipogon DXCWR. Taken together, these results indicate that organelle‐to‐nucleus DNA transfers were a continuous process during rice evolution and were important contributors to the divergence of rice nuclear genomes.
The frequency of giant NORGs in Asian rice differed noticeably among O. rufipogon, indica and japonica, whereas it was similar between the two African rice species, O. barthii and O. glaberrima (Figure 5c,d). Based on the genotypes (presence/absence) of the 52 giant NORGs in African and Asian rice natural populations, we further calculated the fixation index (F st) of each giant NORG between O. rufipogon and O. sativa, between indica and japonica, between O. barthii and O. glaberrima, and between African and Asian rice species. The F st values of all 37 giant NORGs detected in African rice were <0.2, indicating that none of these 37 NORGs had any obvious differentiation between O. barthii and O. glaberrima. In contrast, a total of four, 18 and 12 giant NORGs with obvious differentiation (F st > 0.3) were, respectively, detected between O. rufipogon and O. sativa, between indica and japonica, and between African and Asian rice (Table S11). Further nucleotide diversity analysis of the 200‐kb flanking regions surrounding these highly divergent NORGs showed that nucleotide diversity was significantly lower in indica and japonica than in O. rufipogon (Figure S17). Thus, these results suggest that the giant nuclear integrants of organelle DNA may have participated in the divergence of rice species or subspecies and undergone directional selection during rice domestication.
Environmental adaptation, with the expansion of rice cultivation regions, is an important factor driving genome divergence and population differentiation. To test whether giant NORG events were involved in rice environmental adaptation, we investigated the geographic distribution of 19 highly differentiated NORGs in Asian rice and found that rice samples harboring NUPTs (10/11, 90.9%) or NUMTs (6/8, 75.0%) trended to distribute in higher latitude regions than those without the corresponding NUPT and NUMT (Figures 6a–d; Figures S18 and S19). Only one NUMT (numt10), found mainly in indica rice, occurred more frequently in rice samples from lower latitude regions (Figures 5d and 6d). Nonetheless, a majority of the highly differentiated NORGs were associated with two bioclimatic variables (annual mean temperature and annual mean solar radiation), as numerous NORG events were found in rice samples from regions with lower temperatures and light intensities (Figure 6e–h). Collectively, these results indicate that changes in environmental conditions might have promoted the survival of organelle‐to‐nucleus DNA transfers and that nuclear integration of organellar DNA might have helped rice to enhance environmental adaptation to higher latitude regions during Asian rice evolution.
Figure 6.
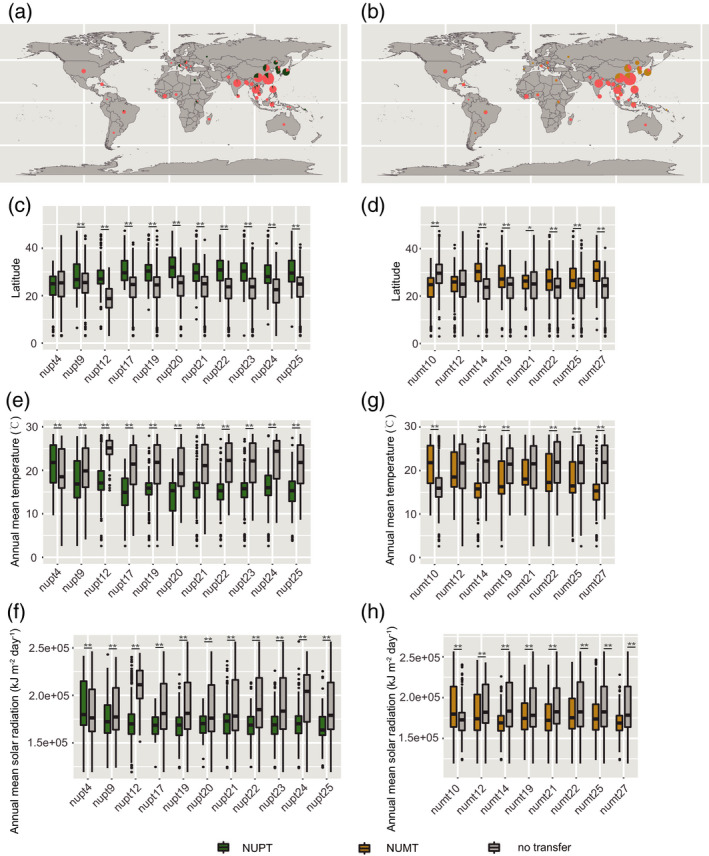
Giant nuclear integrants of plastid DNA (NUPTs) and mitochondrial DNA (NUMTs) were involved in environmental adaptation in Asian rice.
(a, b) Distributions of nupt25 (a) and numt27 (b) in Asian rice natural populations. The colors green, brown and orange in the pie charts represent the presence of NUPT, the presence of NUMT, and the absence of both NUPT and NUMT, respectively. The size of the circles represents the number of rice accessions on a logarithmic scale.
(c, d) The highly differentiated giant NUPTs (c) and NUMTs (d) were found in significantly higher proportions among rice samples from high latitudes than in those from low latitudes.
(e–h) The presence/absence polymorphisms of highly differentiated NUPTs (e and f) and NUMTs (g and h) show strong associations with annual mean temperature (e and g) and annual mean solar radiation (f and h). The center of the box plot and the edges indicate median and 25th or 75th percentiles, respectively, whereas the whiskers indicate the median ± 1.5 × IQR (interquartile range). Single and double asterisks represent significant differences, determined by Mann–Whitney U‐test, at P < 0.05 and P < 0.01, respectively.
DISCUSSION
Although numerous de novo genome assemblies of African and Asian rice have been published in recent years, as high‐throughput sequencing technologies have advanced, these assemblies have been highly fragmented and incomplete because of the limitations of de novo assembly using short‐read platforms (Sakai et al., 2014; Schatz et al., 2014; Wang et al., 2014; Zhang et al., 2014; Stein et al., 2018; Zhao et al., 2018). Thus, it has not been possible to capture the full landscape of structural variations that arose during rice genome evolution. Here, we assembled four high‐quality Oryza AA genomes – from the Asian wild rice O. nivara and O. rufipogon, the African wild rice O. barthii and the African cultivated rice O. glaberrima – using a strategy combining PacBio single‐molecule real‐time (SMRT) and next‐generation sequencing (NGS) technologies. In the assembly process, we invested great effort in filling the gaps in the NGS assembled contigs and raw PacBio reads, and we subsequently ordered and scaffolded the contigs based on long and short reads and BESs collected from OMAP to further improve the genome assemblies. These newly assembled genomes provided a new opportunity for identifying SVs and understanding genome evolution between and within rice subspecies and species through comparative genomics.
Previous studies revealed that TE transpositions are an important source of SVs. The proportion of SVs caused by TE transpositions differs dramatically among species, however: 24.2% in human (Sudmant et al., 2015), 9.0% in fruit fly (Zichner et al., 2013), 34.6% in maize (Yang et al., 2017) and 19.2% in Cucumis sativus (cucumber) (Zhang et al., 2015). The 3000 Rice Genomes Project showed that approximately 7.8% of SV events in rice genomes are related to TE mobility (Fuentes et al., 2019); however, we found that TE transposition generated approximately 33.0% of SVs in the rice genomes assessed, suggesting that de novo assembly using long‐read platforms improved our capability for the accurate detection of insertions and deletions resulting from TE elements. The results of our evaluation of the composition of TEs in relation to SVs also showed that the LTR/Gypsy and Harbinger superfamilies, in particular, have played important roles in shaping rice genome divergence.
Organelle‐to‐nucleus DNA transfers (NORGs) of plastid and mitochondrial DNA (NUPTs and NUMTs) also contributed to SV formation, thereby shaping nuclear genome structure during genome evolution (Zhang et al., 2020). In the present study, we identified genome‐wide NORGs in African and Asian rice genomes and found that the AT/TA repeat motif was enriched in the flanking regions surrounding NORG insertion sites, consistent with a similar feature of NUMT insertion in the human genome (Tsuji et al., 2012). The findings supported the possibility that AT‐dinucleotide‐rich sequences promote DNA breakage by potentially forming stable secondary structures and perturbing the progression of DNA replication (Irony‐Tur Sinai et al., 2019), thereby increasing the chances of genomic invasion by exogenous DNA, such as DNA fragments leaking in from the chloroplast or the mitochondrion.
DNA transfers from organelle to nucleus often lead to migrations of organellar genes, especially those encoding ribosomal proteins in plants. The mitochondrial gene Ribosomal Protein S10 was found to have been lost 26 separate times and was frequently integrated into nuclear genomes in a study of 277 diverse flowering plants (Adams et al., 2000). In Populus, the gene Ribosomal Protein L32 was lost from the chloroplast genome and transferred to the nucleus (Ueda et al., 2007). Additionally, previous studies have demonstrated the frequent losses of up to 14 ribosomal protein genes and two succinate dehydrogenase genes in the mitochondrial genomes of 280 angiosperms during evolution, probably as the result of continuing organelle‐to‐nucleus DNA transfers (Adams et al., 2002). The functions of these genes remain unclear, however.
By contrast, in the present study, we identified giant NORGs harboring several entire organellar genes in six Oryza AA genomes and their distribution in African and Asian rice natural population, and found that several exhibited obvious differentiation between Asian cultivated rice subspecies, between Asian wild and cultivated rice, or between African and Asian rice. Additionally, the presence/absence of 16 of the giant NORGs also showed an obvious geographic distribution trend. Taken together, these results indicated that giant NORGs have contributed to rice genome divergence and may also have been involved in environmental adaptation.
These 16 highly differentiated NORGs were more prevalent in rice samples from high‐latitude regions where relatively lower temperature and light intensity result in lower photosynthetic efficiency in rice. Hence, increasing the proportion of chloroplast‐ and mitochondrion‐derived genes in the nucleus via organelle‐to‐nucleus DNA transfer might be an effective strategy to compensate for decreased photosynthetic efficiency. Notably, 10 of the 11 highly differentiated giant NUPTs were linked to previously reported QTLs controlling rice photosynthesis (Table S12) (Hu et al., 2009; Gu et al., 2012; Wang et al., 2015; Adachi et al., 2019; Zhao et al., 2019). Therefore, we speculate that organelle‐to‐nucleus DNA transfers might improve photosynthesis and energy metabolism by increasing the number of chloroplast‐ and mitochondrion‐derived genes in the nucleus, resulting in enhanced adaptability to lower temperature and light intensity and thereby promoting rice cultivation in high‐latitude regions.
In conclusion, we generated four high‐quality newly assembled genomes of African and Asian rice, obtained through the integration of NGS and PacBio SMRT technology, that are potentially important genomic resources for the study of rice genome evolutionary history. Genome‐wide identification of SVs showed that 28 SV hot spots are shared between African and Asian rice genomes, among which 19 reside in rice segmental duplications. Interestingly, some of these SVs were derived from organelle‐to‐nucleus DNA transfers, which are involved in genome divergence within and between species and subspecies. Moreover, several highly differentiated NORGs preferentially occur in rice accessions obtained from high altitudes with lower temperatures and light densities, implying that the organelle‐to‐nucleus DNA transfers participate in both genome divergence and environmental adaptation in rice.
EXPERIMENTAL PROCEDURES
Plant materials and DNA and RNA‐sequencing
Fresh leaves were collected from O. barthii (accession W1411), O. glaberrima (accession IRGC104165), O. nivara (accession W2014) and O. rufipogon (accession DXCWR), grown at the Shangzhuang Experimental Station of China Agricultural University (Beijing, China). High‐quality genomic DNA was isolated from fresh leaf tissues with the modified cetyl trimethylammonium bromide (CTAB) method. PacBio SMRT sequencing was conducted on a PacBio RSII instrument with P5/C3 sequencing chemistry. Illumina sequencing libraries were prepared with 500‐bp insert size and sequenced with the Illumina HiSeq 2500 platform. Illumina short reads were processed to remove adapters and for quality control with the trimmomatic tool (Bolger et al., 2014).
Several tissues at different development stages, including root, leaf sheath, lamina joint, leaf blade, tiller base (50 days after sowing and 100 days after sowing), culm and young panicle (<1, 1–5 and >5 cm), were harvested for RNA extraction (Table S5). Total RNA was extracted using TRizol (ThermoFisher Scientific, https://www.thermofisher.com) and purified using a Qiagen RNeasy kit (Qiagen, https://www.qiagen.com). RNA‐seq libraries with an insert size of about 450 bp were constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, https://international.neb.com) and were then sequenced on an Illumina 2500 platform, yielding a total of 245.3 Gb 150‐bp paired‐end reads (Table S5). The low‐quality RNA‐seq reads were filtered out using the trimmomatic tool (Bolger et al., 2014) to generate clean data.
De novo genome assembly and genome assessment
The raw PacBio long reads were error‐corrected, trimmed and assembled using canu (Koren et al., 2017). Contigs were polished using a module in smrt analysis 2.3.0 of quiver (Chin et al., 2013). Scaffolds were built based on paired‐end reads, BESs from OMAP (http://www.omap.org) and the corrected PacBio reads with sspace‐standard (Boetzer et al., 2011) and sspace‐longread (Boetzer and Pirovano, 2014) packages run with stringent parameters. Gaps in scaffolds were filled by pbjelly (English et al., 2012) (‐minReads = 2) with long corrected reads to improve scaffold completeness. The contigs or scaffolds were anchored, ordered and oriented based on high collinearity between de novo assemblies and the japonica Nipponbare reference sequence with genome puzzle master (gpm) (Zhang et al., 2016a). In order to discard any redundant sequences, adjacent contigs or scaffolds were merged if the end‐to‐end overlap length was >1 kb and the identity was >98%. Furthermore, NGS contigs de novo ‐assembled with sparseassembler (Ye et al., 2012) using corrected short reads were used to fill gaps in scaffolds. Finally, possible errors in the filled gaps and merge regions were corrected again using quiver (Chin et al., 2013). To improve the accuracy of base calling, we aligned the corrected short reads with the corresponding assemblies using bwa‐mem (Li and Durbin, 2009) with default parameters, retained high‐quality alignment reads mapping in proper pairs with samtools (Li et al., 2009) (parameters: ‐Q 30; ‐f 2) and polished the pseudomolecules using pilon (Walker et al., 2014). The completeness of all the assembled genomes was tested with busco (Simao et al., 2015) and cegma (Parra et al., 2007).
Genome annotation
The TE libraries were constructed through a combination of ab initio and homology‐based approaches. We used repeatmodeler (http://www.repeatmasker.org), including two repeat‐detecting programs (recon and repeatscout), to generate TE libraries for each genome. Through integration with the known Repbase library (Bao et al., 2015), final TE libraries were subjected to repeatmasker (Zhi et al., 2006) to mask newly assembled genomes. ltrharvest (Ellinghaus et al., 2008) was used to identify full‐length LTR retrotransposons, which were annotated by ltrdigest (Steinbiss et al., 2009) with the parameters ‐trnas and ‐hmms. To estimate the insertion time of LTR/Gypsy elements, we used mafft (Katoh et al., 2005) to align LTR sequences for each element, and then the alignments were subjected to distmat in the emboss package for calculating the divergence (K). The insertion time was estimated with the formula T = K/(2 × r), where r refers to a synonymous substitution rate of 1.3 × 10−8 per site per year.
Gene predictions were performed by three approaches: (i) ab initio prediction; (ii) protein homology; and (iii) expressed sequence tag (EST), full‐length cDNA (FL‐cDNA) and assembled transcript‐based prediction. augustus (Stanke et al., 2006), snap (Korf, 2004), and glimmerhmm (Majoros et al., 2004) were used to ab initio predict protein‐coding genes with masked genome sequences. The protein sequences from the Oryza species – O. glaberrima CG14, O. sativa ssp. indica 93‐11, O. sativa ssp. indica R498, and O. sativa ssp. japonica Nipponbare – and from A. thaliana (based on the TAIR10 genome release) were aligned to the masked genome with exonerate (Slater and Birney, 2005) to produce gene structures. Additionally, the sequences of ESTs, FL‐cDNAs and assembled transcripts were aligned with the genomes using blat (Kent, 2002) and gmap (Wu and Watanabe, 2005), and the alignments were filtered with a query coverage of <80%. The predicted gene model was processed to generate a consensus model using evm (Haas et al., 2008). Finally, the gene models were updated with pasa (Haas et al., 2003).
Gene function annotation
Functional annotation of the predicted protein‐encoding genes was carried out based on the best alignment through mapping the predicted protein sequences against the Swiss‐Prot and TrEMBL databases (Bairoch and Apweiler, 2000) using blastp (E‐value < 1e–5). Protein motifs, domains, pathway and GO terms for genes were extracted from the results of interproscan (Zdobnov and Apweiler, 2001) and hmmer (Potter et al., 2018), searching against the InterPro (Mitchell et al., 2015) and Pfam (Finn et al., 2014) databases, respectively. Predicted protein sequences were aligned with the National Center for Biotechnology Information (NCBI) non‐redundant (nr) database by blastx (E‐value < 1e–3) and then annotated for GO terms with blast2go 3.2 (Conesa et al., 2005). The final GO annotations for each assembly were generated by integrating the results from interproscan (Zdobnov and Apweiler, 2001) and blast2go (Conesa et al., 2005). Fisher’s exact test was performed to detect significantly enriched GO terms of genes located in SVs against the genome‐wide background in the agrigo 2.0 platform (Tian et al., 2017), and this was followed by multi‐test adjustment (false discovery rate, FDR < 0.05).
The TE‐related genes were annotated based on the alignment results in the MSU Oryza Repeat Database (http://rice.plantbiology.msu.edu/annotation_oryza.shtml) using tblastn with a cut‐off E‐value of < 1e–5 and a coverage of ≥20%. In addition, genes that contained domains associated with TEs were also treated as TE‐related genes using the previous method (Zhang et al., 2016b). The remainders were considered to be non‐TE genes.
Genome structure analysis
Representative proteins of each gene from different genomes, including the rice genome (http://rice.plantbiology.msu.edu), wheat genome (https://www.wheatgenome.org) and maize genome (https://www.maizegdb.org), were used to identify collinear regions using mcscanx (Wang et al., 2012) with default parameters. In addition, the segmental duplication in each de novo assembly was identified using mcscanx (Wang et al., 2012). An E‐value of <1e–20 was considered as the alignment threshold for identification of per collinear gene pair.
Identification of structural variants
Structural variants (≥50 bp) were identified through three methods: (i) all subreads were aligned with their reference genomes using blasr (Chaisson and Tesler, 2012), and the alignment results were supplied to pbhoney (English et al., 2014) to detect candidate SVs; (ii) long‐read alignments and potential SV detection were performed with ngmlr and sniffles (Sedlazeck et al., 2018), respectively; (iii) assembly‐based detection was performed by mapping each de novo assembly genome to the japonica Nipponbare reference genome with nucmer (parameters: ‐‐maxmatch ‐l 100 ‐c 500) (Marcais et al., 2018) and the potential structural variants were identified with assemblytics (Nattestad and Schatz, 2016). To further decrease the false‐positive rate, 500 bp each of upstream and downstream sequences surrounding each candidate SV in the japonica Nipponbare genome were extracted and then mapped to another genome using blast. If an alignment was conducted successfully, a one‐to‐one alignment block (in which, when a sequence contained a potential SV in the japonica Nipponbare reference genome, its corresponding sequence in another genome would be analyzed) was produced using nucmer (Marcais et al., 2018), and the boundaries of SVs were determined using show‐diff in mumer package (Marcais et al., 2018). Finally, the SVs identified by the previous three methods were merged if the intervals of SVs have more than 70% reciprocal overlap.
Normalization of SV density is defined by the formula (N − Min)/(Max − Min), where N refers to the number of SVs in one window, Min represents the minimum number of SVs for African rice or Asian rice, and Max indicates the maximum number of SVs for African rice or Asian rice in a sliding window of 500 kb with a step size of 50 kb. The windows with top 5% SV density were defined as SV hot spots.
RNA‐sequencing data analysis
All high‐quality clean RNA‐seq data were mapped to the corresponding genomes using tophat 2.0.12 (Kim et al., 2013). Abundances of transcripts and genes were estimated in fragments per kilobase of exon model per million fragments mapped (FPKM) using cufflinks 2.1.1 (Trapnell et al., 2010). An expressed gene was defined as one with FPKM > 0.5. In addition, transcripts were assembled through the trinity platform (Grabherr et al., 2011) with de novo and genome‐guided assembly modes using RNA‐seq reads after filtering.
Identification and analyses of NUPTs and NUMTs
To identify genome‐wide NUPTs and NUMTs, the complete chloroplast and mitochondrion genomes of japonica Nipponbare were obtained from NCBI GenBank and were mapped to the four de novo assemblies and the japonica Nipponbare nuclear genomes using blast+ with the blastn task and a 4‐bp word size (E‐value < 1e–10). The chloroplast and mitochondrion genomes of indica R498 were mapped to the indica R498 nuclear genomes using the same method. Given the fragmentation of NORGs resulting from recombination and TE transposition, adjacent NUPTs or NUMTs, defined as those <400 bp apart, were merged into a single transfer event. Flanking sequences (100 bp) surrounding the break points of NORGs were extracted from each genome. Motif enrichments in the flanking sequences were performed using meme with the default parameters (Bailey et al., 2009).
Population genetic analysis
Publicly available DNA sequence data sets were collected from previous studies (Huang et al., 2012; Wang et al., 2014; Meyer et al., 2016), including 94 accessions of O. barthii, 113 O. glaberrima cultivars, 446 accessions of O. rufipogon, 484 japonica cultivars and 519 indica cultivars, and aligned with the japonica Nipponbare reference genome using bwa‐mem with default parameters (Li and Durbin, 2009). The alignment results were filtered by mapping quality (>30) and proper pairs using samtools (Li et al., 2009). The presence/absence of 52 giant NORGs in all rice samples were determined by whether pair‐end reads covering or spanning the break points or integration sites of these NORGs were detected or not (Figure S15). Fixation index (F st) values were calculated with vcftools (Danecek et al., 2011), based on genotype information. The RiceHap3 genotype data set (Huang et al., 2012) was downloaded from http://server.ncgr.ac.cn/RiceHap3/ and used to perform nucleotide diversity analysis with a sliding window of 20 kb and a step size of 2 kb using the r package popgenome (Pfeifer et al., 2014).
Geographical and bioclimatic variables analysis
To investigate the geographical distribution patterns of highly differentiated NORGs, the longitude and latitude for each rice sample were retrieved from the previous study (Huang et al., 2012). Two bioclimatic variables, annual mean temperature and solar radiation for each month of the year, were obtained from WorldClim Version 2 (http://www.worldclim.org/version2) at an optimum spatial resolution level of 30 sec (~1 km2). In the present study, solar radiation for each month of the year was transformed into annual mean solar radiation. The bioclimatic variable information was extracted for each accession based on longitude and latitude. The association between environmental conditions and the presence/absence of giant NORG events was determined by Mann–Whitney U‐test.
Primers
The primers used in this study are listed in Table S13.
AUTHOR CONTRIBUTIONS
LT designed the experiments and conceived the project. XM, JF, YW, SZ and XZ prepared the sample material for sequencing. XM performed the data analysis. LT, XM and CS wrote the article. All authors read and approved the final version for publication.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
OPEN RESEARCH BADGE
This article has earned an Open Data badge for making publicly available the digitally‐shareable data necessary to reproduce the reported results.
Supporting information
Figure S1. Geographical distribution of the four Oryza AA species for whole‐genome de novo assembly.
Figure S2. Frequency distribution of subread length for the four rice samples.
Figure S3. Sequence comparisons of de novo assemblies of Oryza rufipogon (a), Oryza nivara (b), Oryza barthii (c) and Oryza glaberrima (d) in present and previous studies (Huang et al., 2012; Wang et al., 2014; Zhang et al., 2014; Stein et al., 2018).
Figure S4. Confirmation of the four de novo assemblies using publicly available bacterial artificial chromosome (BAC) sequences.
Figure S5. The insertion time of LTR/Gypsy elements in the African and Asian rice genomes.
Figure S6. Identification of known and novel structural variations (SVs) in African and Asian rice genomes.
Figure S7. The proportion of common structural variation (SV) hot spots identified in the present and previous studies.
Figure S8. Organelle‐to‐nucleus DNA transfers generating structural variations (SVs).
Figure S9. Genome‐wide identification of nuclear integrants of plastid DNA (NUPTs) and mitochondrial DNA (NUMTs) in African and Asian rice genomes.
Figure S10. Sequence motifs identified in 100‐bp flanking regions surrounding nuclear integrants of plastid DNA (NUPTs) in African and Asian rice genomes.
Figure S11. Sequence motifs identified in 100‐bp flanking regions surrounding nuclear integrants of mitochondrial DNA (NUMTs) in African and Asian rice genomes.
Figure S12. Ancient nuclear integrants of mitochondrial DNA (NUMTs) detected in Triticum aestivum (wheat) genome.
Figure S13. Confirmation of the giant nuclear integrants of plastid DNA (NUPTs) and mitochondrial DNA (NUMTs) identified in this study using PCR analysis.
Figure S14. The nuclear integrant of organelle DNA (NORG) events coupled with the generation of structural variations (SVs).
Figure S15. The method used for the detection of chloroplast or mitochondrial DNA transfer events based on genome assembly in rice natural populations.
Figure S16. Collinearity analysis of the regions harboring giant nuclear integrants of mitochondrial DNA (NUMTs) between Oryza sativa (rice) and Zea mays (maize) (a and c) and between rice and Triticum aestivum (wheat) (b and d).
Figure S17. Comparison of the nucleotide diversity of highly differentiated giant nuclear integrants of plastid DNA (NUPTs) and mitochondrial DNA (NUMTs) (F st values >0.3) among Oryza sativa ssp. indica and japonica and Oryza rufipogon using the public resequencing data (Huang et al., 2012).
Figure S18. Geographical distributions of highly differentiated giant nuclear integrants of plastid DNA (NUPTs) in Asian rice natural populations.
Figure S19. Geographical distributions of highly differentiated giant nuclear integrants of mitochondrial DNA (NUMTs) detected in the Asian rice population.
Table S1. Statistical summary of DNA sequencing reads used in the genome assembly process.
Table S2. Summary of the four Oryza AA genome assemblies and gene annotations.
Table S3. Evaluation of the four de novo Oryza AA genomes as compared with the japonica rice Nipponbare reference genome by busco.
Table S4. Evaluation of four de novo Oryza AA genomes as compared with the japonica rice Nipponbare reference genome by cegma.
Table S5. Summary of RNA‐seq reads used in this study.
Table S6. Summary of gene functional annotations for the four Oryza AA genome assemblies using different databases and bioinformatics tools.
Table S7. Summary of transposable element (TE) annotations of the four Oryza AA genome assemblies.
Table S8. Statistics for structural variations (SVs) of the five other Oryza AA genomes against the japonica rice Nipponbare reference genome.
Table S9. Gene ontology (GO) enrichment analysis for genes harbored in the structural variations (SVs) in African and Asian rice.
Table S10. Ten of 11 glucan synthase‐like family genes contained nuclear integrants of mitochondrial DNA (NUMTs).
Table S11. Fixation index (F st) values of giant nuclear integrants of plastid DNA (NUPTs) and mitochondrial DNA (NUMTs) in pairwise comparisons of rice populations.
Table S12. Highly differentiated nuclear integrants of plastid DNA (NUPTs) co‐localized with quantitative trait loci (QTLs) for photosynthesis identified in previous studies.
Table S13. Primers for validation of the nuclear integrants of organelle DNA (NORGs) investigated in this study.
ACKNOWLEDGEMENTS
We thank the International Rice Research Institute and the National Institute of Genetics (Japan) for providing the rice germplasm. This work was supported by the National Key Research and Development Program of China (grant no. 2016YFD0100400), the National Natural Science Foundation of China (grant nos 91435103 and 31222040) and the Chinese Universities Scientific Fund (grant no. 2020TC162).
DATA AVAILABILITY STATEMENT
Raw PacBio subread and short‐read data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA474415. RNA‐seq data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA478144. All data including the genome assemblies and annotations generated from this study can be found at Zenodo (https://doi.org/10.5281/zenodo.3679049). Scripts related to our study are available in the following GitHub directory https://github.com/xma82/rice_genome_assembly.
REFERENCES
- Adachi, S. , Yamamoto, T. , Nakae, T. et al (2019) Genetic architecture of leaf photosynthesis in rice revealed by different types of reciprocal mapping populations. J. Exp. Bot. 70, 5131–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, K.L. , Daley, D.O. , Qiu, Y.L. , Whelan, J. and Palmer, J.D. (2000) Repeated, recent and diverse transfers of a mitochondrial gene to the nucleus in flowering plants. Nature, 408, 354–357. [DOI] [PubMed] [Google Scholar]
- Adams, K.L. , Qiu, Y.L. , Stoutemyer, M. and Palmer, J.D. (2002) Punctuated evolution of mitochondrial gene content: high and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl Acad. Sci. USA, 99, 9905–9912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audano, P.A. , Sulovari, A. , Graves‐Lindsay, T.A. et al (2019) Characterizing the major structural variant alleles of the human genome. Cell, 176, 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, T.L. , Boden, M. , Buske, F.A. , Frith, M. , Grant, C.E. , Clementi, L. , Ren, J.Y. , Li, W.W. and Noble, W.S. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch, A. and Apweiler, R. (2000) The SWISS‐PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, W. , Kojima, K.K. and Kohany, O. (2015) Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐David, U. , Arad, G. , Weissbein, U. et al (2014) Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat. Commun. 5, 4825. [DOI] [PubMed] [Google Scholar]
- Blanchard, J.L. and Lynch, M. (2000) Organellar genes ‐ why do they end up in the nucleus? Trends Genet. 16, 315–320. [DOI] [PubMed] [Google Scholar]
- Boetzer, M. , Henkel, C.V. , Jansen, H.J. , Butler, D. and Pirovano, W. (2011) Scaffolding pre‐assembled contigs using SSPACE. Bioinformatics, 27, 578–579. [DOI] [PubMed] [Google Scholar]
- Boetzer, M. and Pirovano, W. (2014) SSPACE‐LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics, 15, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese, F.M. , Balacco, D.L. , Preste, R. , Diroma, M.A. , Forino, R. , Ventura, M. and Attimonelli, M. (2017) NumtS colonization in mammalian genomes. Sci. Rep. 7, 16357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson, M.J. and Tesler, G. (2012) Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics, 13, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, M. , Emerson, J.J. , Macdonald, S.J. and Long, A.D. (2019) Structural variants exhibit widespread allelic heterogeneity and shape variation in complex traits. Nat. Commun. 10, 4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, C.S. , Alexander, D.H. , Marks, P. et al (2013) Nonhybrid, finished microbial genome assemblies from long‐read SMRT sequencing data. Nat. Methods, 10, 563–569. [DOI] [PubMed] [Google Scholar]
- Choi, J.Y. , Lye, Z.N. , Groen, S.C. , Dai, X. , Rughani, P. , Zaaijer, S. , Harrington, E.D. , Juul, S. and Purugganan, M.D. (2020) Nanopore sequencing‐based genome assembly and evolutionary genomics of circum‐basmati rice. Genome Biol. 21, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, J.Y. , Platts, A.E. , Fuller, D.Q. , Hsing, Y.I. , Wing, R.A. and Purugganan, M.D. (2017) The rice paradox: multiple origins but single domestication in Asian rice. Mol. Biol. Evol. 34, 969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa, A. , Gotz, S. , Garcia‐Gomez, J.M. , Terol, J. , Talon, M. and Robles, M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21, 3674–3676. [DOI] [PubMed] [Google Scholar]
- Cook, D.E. , Lee, T.G. , Guo, X. et al (2012) Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science, 338, 1206–1209. [DOI] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. et al (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayama, G. , Emery, S.B. , Kidd, J.M. and Mills, R.E. (2014) The genomic landscape of polymorphic human nuclear mitochondrial insertions. Nucleic Acids Res. 42, 12640–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coster, W. , De Rijk, P. , De Roeck, A. , De Pooter, T. , D'Hert, S. , Strazisar, M. , Sleegers, K. and Van Broeckhoven, C. (2019) Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res. 29, 1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, H. , Yu, Y. , Ma, Y. et al (2017) Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 8, 15324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, P. , Xu, J. , Zeng, D. et al (2017) Natural variation in the promoter of GSE5 contributes to grain size diversity in rice. Mol. Plant, 10, 685–694. [DOI] [PubMed] [Google Scholar]
- Ellinghaus, D. , Kurtz, S. and Willhoeft, U. (2008) LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics, 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English, A.C. , Richards, S. , Han, Y. et al (2012) Mind the gap: upgrading genomes with Pacific Biosciences RS long‐Read sequencing technology. PLoS One, 7, e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English, A.C. , Salerno, W.J. and Reid, J.G. (2014) PBHoney: identifying genomic variants via long‐read discordance and interrupted mapping. BMC Bioinformatics, 15, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R.D. , Bateman, A. , Clements, J. et al (2014) Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes, R.R. , Chebotarov, D. , Duitama, J. et al (2019) Structural variants in 3000 rice genomes. Genome Res. 29, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr, M.G. , Haas, B.J. , Yassour, M. et al (2011) Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, J. , Yin, X. , Struik, P.C. , Stomph, T.J. and Wang, H. (2012) Using chromosome introgression lines to map quantitative trait loci for photosynthesis parameters in rice (Oryza sativa L.) leaves under drought and well‐watered field conditions. J. Exp. Bot. 63, 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, X. , Ruan, S. , Hu, W. , Ca, D. and Fan, L. (2008) Chloroplast DNA insertions into the nuclear genome of rice: the genes, sites and ages of insertion involved. Funct. Integr. Genomics, 8, 101–108. [DOI] [PubMed] [Google Scholar]
- Haas, B.J. , Delcher, A.L. , Mount, S.M. et al (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J. , Salzberg, S.L. , Zhu, W. , Pertea, M. , Allen, J.E. , Orvis, J. , White, O. , Buell, C.R. and Wortman, J.R. (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazkani‐Covo, E. and Martin, W.F. (2017) Quantifying the number of independent organelle DNA insertions in genome evolution and human health. Genome Biol. Evol. 9, 1190–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazkani‐Covo, E. , Zeller, R.M. and Martin, W. (2010) Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 6, e1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, S.P. , Zhou, Y. , Zhang, L. , Zhu, X.D. , Li, L. , Luo, L.J. , Liu, G.L. and Zhou, Q.M. (2009) Correlation and quantitative trait loci analyses of total chlorophyll content and photosynthetic rate of rice (Oryza sativa) under water stress and well‐watered conditions. J. Integr. Plant Biol. 51, 879–888. [DOI] [PubMed] [Google Scholar]
- Huang, C.Y. , Grunheit, N. , Ahmadinejad, N. , Timmis, J.N. and Martin, W. (2005) Mutational decay and age of chloroplast and mitochondrial genomes transferred recently to angiosperm nuclear chromosomes. Plant Physiol. 138, 1723–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Kurata, N. , Wei, X. et al (2012) A map of rice genome variation reveals the origin of cultivated rice. Nature, 490, 497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston, J. , Chaisson, M.J.P. , Steinberg, K.M. et al (2017) Discovery and genotyping of structural variation from long‐read haploid genome sequence data. Genome Res. 27, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irony‐Tur Sinai, M. , Salamon, A. , Stanleigh, N. , Goldberg, T. , Weiss, A. , Wang, Y.H. and Kerem, B. (2019) AT‐dinucleotide rich sequences drive fragile site formation. Nucleic Acids Res. 47, 9685–9695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, J. , Huang, W. , Gao, J.P. , Yang, J. , Shi, M. , Zhu, M.Z. , Luo, D. and Lin, H.X. (2008) Genetic control of rice plant architecture under domestication. Nat. Genet. 40, 1365–1369. [DOI] [PubMed] [Google Scholar]
- Katoh, K. , Kuma, K. , Toh, H. and Miyata, T. (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara, Y. , de la Bastide, M. , Hamilton, J.P. et al (2013) Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice, 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J. (2002) BLAT ‐ the BLAST‐like alignment tool. Genome Res. 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. and Salzberg, S.L. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleine, T. , Maier, U.G. and Leister, D. (2009) DNA transfer from organelles to the nucleus: the idiosyncratic genetics of endosymbiosis. Annu. Rev. Plant Biol. 60, 115–138. [DOI] [PubMed] [Google Scholar]
- Komiya, R. , Ikegami, A. , Tamaki, S. , Yokoi, S. and Shimamoto, K. (2008) Hd3a and RFT1 are essential for flowering in rice. Development, 135, 767–774. [DOI] [PubMed] [Google Scholar]
- Koren, S. , Walenz, B.P. , Berlin, K. , Miller, J.R. , Bergman, N.H. and Phillippy, A.M. (2017) Canu: scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Res. 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinformatics, 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, M. , Sazzini, M. , Calabrese, F.M. , Simone, D. , Boattini, A. , Romeo, G. , Luiselli, D. , Attimonelli, M. and Gasparre, G. (2012) Polymorphic NumtS trace human population relationships. Hum. Genet. 131, 757–771. [DOI] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , Durbin, R. and Proc, G.P.D. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, B. , Wang, N. , Li, N. , Kimball, R.T. and Braun, E.L. (2018) Comparative genomics reveals a burst of homoplasy‐free numt insertions. Mol. Biol. Evol. 35, 2060–2064. [DOI] [PubMed] [Google Scholar]
- Liu, J. , Chen, J. , Zheng, X. et al (2017) GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nat. Plants, 3, 17043. [DOI] [PubMed] [Google Scholar]
- Long, Q. , Rabanal, F.A. , Meng, D. et al (2013) Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden. Nat. Genet. 45, 884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros, W.H. , Pertea, M. and Salzberg, S.L. (2004) TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene‐finders. Bioinformatics, 20, 2878–2879. [DOI] [PubMed] [Google Scholar]
- Marcais, G. , Delcher, A.L. , Phillippy, A.M. , Coston, R. , Salzberg, S.L. and Zimin, A. (2018) MUMmer4: a fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo, M. , Ito, Y. , Yamauchi, R. and Obokata, J. (2005) The rice nuclear genome continuously integrates, shuffles, and eliminates the chloroplast genome to cause chloroplast‐nuclear DNA flux. Plant Cell, 17, 665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, R.S. , Choi, J.Y. , Sanches, M. et al (2016) Domestication history and geographical adaptation inferred from a SNP map of African rice. Nat. Genet. 48, 1083–1088. [DOI] [PubMed] [Google Scholar]
- Mitchell, A. , Chang, H.Y. , Daugherty, L. et al (2015) The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monna, L. , Kitazawa, N. , Yoshino, R. , Suzuki, J. , Masuda, H. , Maehara, Y. , Tanji, M. , Sato, M. , Nasu, S. and Minobe, Y. (2002) Positional cloning of rice semidwarfing gene, sd‐1: Rice "Green revolution gene" encodes a mutant enzyme involved in gibberellin synthesis. DNA Res. 9, 11–17. [DOI] [PubMed] [Google Scholar]
- Nattestad, M. , Goodwin, S. , Ng, K. et al (2018) Complex rearrangements and oncogene amplifications revealed by long‐read DNA and RNA sequencing of a breast cancer cell line. Genome Res. 28, 1126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattestad, M. and Schatz, M.C. (2016) Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics, 32, 3021–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra, G. , Bradnam, K. and Korf, I. (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Pfeifer, B. , Wittelsburger, U. , Ramos‐Onsins, S.E. and Lercher, M.J. (2014) PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter, S.C. , Luciani, A. , Eddy, S.R. , Park, Y. , Lopez, R. and Finn, R.D. (2018) HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly, E. and Leister, D. (2004a) NUMTs in sequenced eukaryotic genomes. Mol. Biol. Evol. 21, 1081–1084. [DOI] [PubMed] [Google Scholar]
- Richly, E. and Leister, D. (2004b) NUPTs in sequenced eukaryotes and their genomic organization in relation to NUMTs. Mol. Biol. Evol. 21, 1972–1980. [DOI] [PubMed] [Google Scholar]
- Sakai, H. , Kanamori, H. , Arai‐Kichise, Y. et al (2014) Construction of pseudomolecule sequences of the aus rice cultivar Kasalath for comparative genomics of Asian cultivated rice. DNA Res. 21, 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz, M.C. , Maron, L.G. , Stein, J.C. et al (2014) Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica . Genome Biol. 15, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck, F.J. , Rescheneder, P. , Smolka, M. , Fang, H. , Nattestad, M. , von Haeseler, A. and Schatz, M.C. (2018) Accurate detection of complex structural variations using single‐molecule sequencing. Nat. Methods, 15, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, J. , Tan, H. , Yu, X.H. et al (2011) Defective Pollen Wall is required for anther and microspore development in rice and encodes a fatty acyl carrier protein reductase. Plant Cell, 23, 2225–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, X. , Sun, X. , Zhang, Z. , Feng, D. , Zhang, Q. , Han, L. , Wu, J. and Lu, T. (2015) GLUCAN SYNTHASE‐LIKE 5 (GSL5) plays an essential role in male fertility by regulating callose metabolism during microsporogenesis in rice. Plant Cell Physiol. 56, 497–509. [DOI] [PubMed] [Google Scholar]
- Simao, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Slater, G.S. and Birney, E. (2005) Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics, 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Keller, O. , Gunduz, I. , Hayes, A. , Waack, S. and Morgenstern, B. (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, J.C. , Yu, Y. , Copetti, D. et al (2018) Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza . Nat. Genet. 50, 285–296. [DOI] [PubMed] [Google Scholar]
- Steinbiss, S. , Willhoeft, U. , Gremme, G. and Kurtz, S. (2009) Fine‐grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 37, 7002–7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant, P.H. , Rausch, T. , Gardner, E.J. et al (2015) An integrated map of structural variation in 2,504 human genomes. Nature, 526, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, L. , Li, X. , Liu, F. et al (2008) Control of a key transition from prostrate to erect growth in rice domestication. Nat. Genet. 40, 1360–1364. [DOI] [PubMed] [Google Scholar]
- Tian, T. , Liu, Y. , Yan, H. , You, Q. , Yi, X. , Du, Z. , Xu, W. and Su, Z. (2017) agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 45, W122–W129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkamaneh, D. , Laroche, J. , Tardivel, A. , O'Donoughue, L. , Cober, E. , Rajcan, I. and Belzile, F. (2018) Comprehensive description of genomewide nucleotide and structural variation in short‐season soya bean. Plant Biotechnol. J. 16, 749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Williams, B.A. , Pertea, G. , Mortazavi, A. , Kwan, G. , van Baren, M.J. , Salzberg, S.L. , Wold, B.J. and Pachter, L. (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji, J. , Frith, M.C. , Tomii, K. and Horton, P. (2012) Mammalian NUMT insertion is non‐random. Nucleic Acids Res. 40, 9073–9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda, M. , Fujimoto, M. , Arimura, S. , Murata, J. , Tsutsumi, N. and Kadowaki, K. (2007) Loss of the rpl32 gene from the chloroplast genome and subsequent acquisition of a preexisting transit peptide within the nuclear gene in Populus . Gene, 402, 51–56. [DOI] [PubMed] [Google Scholar]
- Walker, B.J. , Abeel, T. , Shea, T. et al (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One, 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. and Timmis, J.N. (2013) Cytoplasmic organelle DNA preferentially inserts into open chromatin. Genome Biol. Evol. 5, 1060–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Yu, Y. , Haberer, G. et al (2014) The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nat. Genet. 46, 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Xie, W. , Xing, H. et al (2015) Genetic architecture of natural variation in rice chlorophyll content revealed by a genome‐wide association study. Mol. Plant, 8, 946–957. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , DeBarry, J.D. et al (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt, J. , Symmons, O. , Spitz, F. and Korbel, J.O. (2013) Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 14, 125–138. [DOI] [PubMed] [Google Scholar]
- Wu, P. , Li, T. , Li, R. , Jia, L. , Zhu, P. , Liu, Y. , Chen, Q. , Tang, D. , Yu, Y. and Li, C. (2017) 3D genome of multiple myeloma reveals spatial genome disorganization associated with copy number variations. Nat. Commun. 8, 1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, T.D. and Watanabe, C.K. (2005) GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics, 21, 1859–1875. [DOI] [PubMed] [Google Scholar]
- Wu, Y. , Zhao, S. , Li, X. et al (2018) Deletions linked to PROG1 gene participate in plant architecture domestication in Asian and African rice. Nat. Commun. 9, 4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, N. , Xu, X. , Wang, R. et al (2017) Contributions of Zea mays subspecies mexicana haplotypes to modern maize. Nat. Commun. 8, 1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, C. , Ma, Z.S. , Cannon, C.H. , Pop, M. and Yu, D.W. (2012) Exploiting sparseness in de novo genome assembly. BMC Bioinformatics, 13(Suppl 6), S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov, E.M. and Apweiler, R. (2001) InterProScan ‐ an integration platform for the signature‐recognition methods in InterPro. Bioinformatics, 17, 847–848. [DOI] [PubMed] [Google Scholar]
- Zhang, G.J. , Dong, R. , Lan, L.N. , Li, S.F. , Gao, W.J. and Niu, H.X. (2020) Nuclear integrants of organellar DNA contribute to genome structure and evolution in plants. Int. J. Mol. Sci. 21, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Kudrna, D. , Mu, T. , Li, W. , Copetti, D. , Yu, Y. , Goicoechea, J.L. , Lei, Y. and Wing, R.A. (2016a) Genome puzzle master (GPM): an integrated pipeline for building and editing pseudomolecules from fragmented sequences. Bioinformatics, 32, 3058–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Chen, L.L. , Xing, F. et al (2016b) Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc. Natl Acad. Sci. USA, 113, E5163–E5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q.J. , Zhu, T. , Xia, E.H. et al (2014) Rapid diversification of five Oryza AA genomes associated with rice adaptation. Proc. Natl Acad. Sci. USA, 111, E4954–E4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Mao, L. , Chen, H. et al (2015) Genome‐wide mapping of structural variations reveals a copy number variant that determines reproductive morphology in cucumber. Plant Cell, 27, 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Q. , Feng, Q. , Lu, H. et al (2018) Pan‐genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 50, 278–284. [DOI] [PubMed] [Google Scholar]
- Zhao, Y. , Qiang, C. , Wang, X. et al (2019) New alleles for chlorophyll content and stay‐green traits revealed by a genome wide association study in rice (Oryza sativa). Sci. Rep. 9, 2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi, D. , Raphael, B.J. , Price, A.L. , Tang, H. and Pevzner, P.A. (2006) Identifying repeat domains in large genomes. Genome Biol. 7, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zichner, T. , Garfield, D.A. , Rausch, T. , Stutz, A.M. , Cannavo, E. , Braun, M. , Furlong, E.E.M. and Korbel, J.O. (2013) Impact of genomic structural variation in Drosophila melanogaster based on population‐scale sequencing. Genome Res. 23, 568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Geographical distribution of the four Oryza AA species for whole‐genome de novo assembly.
Figure S2. Frequency distribution of subread length for the four rice samples.
Figure S3. Sequence comparisons of de novo assemblies of Oryza rufipogon (a), Oryza nivara (b), Oryza barthii (c) and Oryza glaberrima (d) in present and previous studies (Huang et al., 2012; Wang et al., 2014; Zhang et al., 2014; Stein et al., 2018).
Figure S4. Confirmation of the four de novo assemblies using publicly available bacterial artificial chromosome (BAC) sequences.
Figure S5. The insertion time of LTR/Gypsy elements in the African and Asian rice genomes.
Figure S6. Identification of known and novel structural variations (SVs) in African and Asian rice genomes.
Figure S7. The proportion of common structural variation (SV) hot spots identified in the present and previous studies.
Figure S8. Organelle‐to‐nucleus DNA transfers generating structural variations (SVs).
Figure S9. Genome‐wide identification of nuclear integrants of plastid DNA (NUPTs) and mitochondrial DNA (NUMTs) in African and Asian rice genomes.
Figure S10. Sequence motifs identified in 100‐bp flanking regions surrounding nuclear integrants of plastid DNA (NUPTs) in African and Asian rice genomes.
Figure S11. Sequence motifs identified in 100‐bp flanking regions surrounding nuclear integrants of mitochondrial DNA (NUMTs) in African and Asian rice genomes.
Figure S12. Ancient nuclear integrants of mitochondrial DNA (NUMTs) detected in Triticum aestivum (wheat) genome.
Figure S13. Confirmation of the giant nuclear integrants of plastid DNA (NUPTs) and mitochondrial DNA (NUMTs) identified in this study using PCR analysis.
Figure S14. The nuclear integrant of organelle DNA (NORG) events coupled with the generation of structural variations (SVs).
Figure S15. The method used for the detection of chloroplast or mitochondrial DNA transfer events based on genome assembly in rice natural populations.
Figure S16. Collinearity analysis of the regions harboring giant nuclear integrants of mitochondrial DNA (NUMTs) between Oryza sativa (rice) and Zea mays (maize) (a and c) and between rice and Triticum aestivum (wheat) (b and d).
Figure S17. Comparison of the nucleotide diversity of highly differentiated giant nuclear integrants of plastid DNA (NUPTs) and mitochondrial DNA (NUMTs) (F st values >0.3) among Oryza sativa ssp. indica and japonica and Oryza rufipogon using the public resequencing data (Huang et al., 2012).
Figure S18. Geographical distributions of highly differentiated giant nuclear integrants of plastid DNA (NUPTs) in Asian rice natural populations.
Figure S19. Geographical distributions of highly differentiated giant nuclear integrants of mitochondrial DNA (NUMTs) detected in the Asian rice population.
Table S1. Statistical summary of DNA sequencing reads used in the genome assembly process.
Table S2. Summary of the four Oryza AA genome assemblies and gene annotations.
Table S3. Evaluation of the four de novo Oryza AA genomes as compared with the japonica rice Nipponbare reference genome by busco.
Table S4. Evaluation of four de novo Oryza AA genomes as compared with the japonica rice Nipponbare reference genome by cegma.
Table S5. Summary of RNA‐seq reads used in this study.
Table S6. Summary of gene functional annotations for the four Oryza AA genome assemblies using different databases and bioinformatics tools.
Table S7. Summary of transposable element (TE) annotations of the four Oryza AA genome assemblies.
Table S8. Statistics for structural variations (SVs) of the five other Oryza AA genomes against the japonica rice Nipponbare reference genome.
Table S9. Gene ontology (GO) enrichment analysis for genes harbored in the structural variations (SVs) in African and Asian rice.
Table S10. Ten of 11 glucan synthase‐like family genes contained nuclear integrants of mitochondrial DNA (NUMTs).
Table S11. Fixation index (F st) values of giant nuclear integrants of plastid DNA (NUPTs) and mitochondrial DNA (NUMTs) in pairwise comparisons of rice populations.
Table S12. Highly differentiated nuclear integrants of plastid DNA (NUPTs) co‐localized with quantitative trait loci (QTLs) for photosynthesis identified in previous studies.
Table S13. Primers for validation of the nuclear integrants of organelle DNA (NORGs) investigated in this study.
Data Availability Statement
Raw PacBio subread and short‐read data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA474415. RNA‐seq data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA478144. All data including the genome assemblies and annotations generated from this study can be found at Zenodo (https://doi.org/10.5281/zenodo.3679049). Scripts related to our study are available in the following GitHub directory https://github.com/xma82/rice_genome_assembly.