

Abstract
The opioid crisis has underscored the urgent need to identify safe and effective therapeutic strategies to overcome opioid-induced liabilities. We recently reported that LY2828360, a slowly signaling G protein-biased cannabinoid CB2 receptor agonist, suppresses neuropathic nociception and attenuates the development of tolerance to the opioid analgesic morphine in paclitaxel-treated mice. Whether beneficial effects of LY2828360 are dependent upon the presence of a pathological pain state are unknown and its impact on unwanted opioid-induced side-effects have never been investigated. Here, we asked whether LY2828360 would produce synergistic anti-allodynic effects with morphine in a paclitaxel model of chemotherapy-induced neuropathic pain and characterized its impact on opioid-induced reward and other unwanted side-effects associated with chronic opioid administration. Isobolographic analysis revealed that combinations of LY2828360 and morphine produced synergistic anti-allodynic effects in suppressing paclitaxel-induced mechanical allodynia. In wildtype (WT) mice, LY2828360 blocked morphine-induced reward in a conditioned place preference assay without producing reward or aversion when administered alone. The LY2828360-induced attenuation of morphine-induced reward was absent in CB2 knockout (CB2KO) mice. In the absence of a neuropathic pain state, LY2828360 partially attenuated naloxone-precipitated opioid withdrawal in morphine-dependent WT mice, and this withdrawal was itself markedly exacerbated in CB2KO mice. Moreover, LY2828360 did not reliably alter morphine-induced slowing of colonic transit or attenuate tolerance to morphine antinociceptive efficacy in the hot plate test of acute nociception. Our results suggest that cannabinoid CB2 receptor activation enhances the therapeutic properties of opioids while attenuating unwanted side-effects such as reward and dependence that occur with sustained opioid treatment.
Keywords: CB2, morphine, reward, withdrawal, physical dependence
Graphical Abstract
1. Introduction
The opioid crisis remains a major public health issue (Wilson et al., 2020). Opioid abuse can culminate in addiction, physical dependence and, in extreme cases, an overdose death (Compton et al., 2015; Paulozzi et al., 2006). There is, therefore, an urgent need to identify analgesic strategies that circumvent opioid abuse liability and unwanted side-effects (Trang et al., 2007). One such approach aims to harness crosstalk between the endocannabinoid and endogenous opioid systems (Perron et al., 2015; Pertwee, 2001).
The endocannabinoid system consists of lipid signaling molecules that bind to two G protein-coupled cannabinoid receptors 1 and 2 (CB1 and CB2). The clinical use of CB1 agonists is however limited by adverse side-effects (Jones, 2008; Kirilly et al., 2012). Cannabinoid CB2 receptors represent an alternative therapeutic target that avoids these unwanted side-effects (see review in (Guindon and Hohmann, 2008)). Cannabinoid CB2 receptors are localized to cells within the immune system (Galiegue et al., 1995; Schatz et al., 1997) and are often upregulated in response to injury and inflammation (Maresz et al., 2005; Wotherspoon et al., 2005). In preclinical studies, cannabinoid CB2 receptor activation attenuates neuropathic pain (Gutierrez et al., 2011; Lin et al., 2018), inhibits inflammation (Gutierrez et al., 2007; Nackley et al., 2003) and reduces thermal or mechanical hypersensitivities and edema in persistent pain states (Hohmann et al., 2004; Kinsey et al., 2011). Further, selective CB2 agonists do not elicit adverse CB1-mediated side-effects (Deng et al., 2015; Nackley et al., 2003). Cannabinoid CB2 receptor activation also facilitates morphine antinociception under normal and inflammatory conditions (Desroches et al., 2014; Merighi et al., 2012). Moreover, cannabinoid CB2 receptors modulate the rewarding and reinforcing effects of other drugs of abuse such as ethanol and cocaine (Ortega-Alvaro et al., 2015; Xi et al., 2011). Thus, CB2 agonists may promote critical aspects of opioid antinociceptive efficacy while potentially reducing abuse liability.
LY2828360, a G protein-biased CB2 agonist exhibiting similar affinity for human and rat cannabinoid CB2 receptors, produced a delayed inhibition of cAMP accumulation, and activated ERK1/2 signaling without internalizing cannabinoid CB2 receptors or recruiting β-arrestin (Lin et al., 2018). Strikingly, LY2828360 suppressed chemotherapy-induced neuropathic nociception, blocked the development of anti-allodynic tolerance to morphine, and attenuated opioid withdrawal in morphine-dependent mice (Lin et al., 2018). LY2828360 also showed good CNS penetration, oral activity and efficacy in a preclinical model of joint pain (Hollinshead et al., 2013). Moreover, LY2828360 lacked both toxicity and efficacy in a clinical trial for suppressing osteoarthritic knee pain (Pereira et al., 2013) (www.clinicaltrials.gov identifier: NCT01319929). However, the impact of LY2828360 on the detrimental effects of opioids are poorly understood and whether such effects require the presence of a neuropathic pain state is unknown.
We used isobolographic analysis to evaluate whether LY2828360 would act synergistically with morphine to suppress chemotherapy-induced allodynia. We also employed a conditioned place preference (CPP) assay to determine whether LY2828360 would block morphine-induced reward in a CB2-dependent manner. Finally, we examined the impact of LY2828360 on morphine-induced changes in colonic motility, hotplate antinociception and naloxone-precipitated opioid withdrawal.
2. Materials and methods
2.1. Animals
One-hundred and twenty-six adult male WT mice on a C57BL/6J background were bred at Indiana University or purchased from The Jackson Laboratory (Bar Harbor, ME) and used in these studies. Thirty-four adult CB2KO mice (B6.129P2-CNR2 (tm1Dgen/J)), bred at Indiana University for greater than 20 generations onto a C57BL/6J background, were used in these studies. All mice weighed 25-33 g and were ~12-14 weeks old when used in this study. Mice were single housed several days before initiating any pharmacological manipulations. All mice were maintained on a 12 h light/dark cycle (lights on from 7 am to 7 pm) in a temperature and humidity-controlled facility and allowed ad libitum access to food and water throughout the experimental period. The neuropathic pain model, colonic motility/hot plate antinociception, and precipitated withdrawal experiments used n = 6-8 mice per group. The CPP experiments used n = 10 mice per group. All experiments were approved by the Indiana University Bloomington Animal Care and Use Committee (Protocol 19-037) and followed the International Association for the Study of Pain (IASP) Guidelines for the Use of Animals in Research (Zimmermann, 1983).
2.2. Drugs and Chemicals
Paclitaxel (Tecoland Corporation, Edison, NJ) was dissolved in a vehicle consisting of 95% ethanol, emulphor (Alkamuls EL-620; Solvay, Princeton, NJ), and 0.9% saline (Baxter Healthcare Corporation, Deerfield, IL) in a 1:1:18 ratio, respectively, and injected via the intraperitoneal (i.p.) route in a volume of 6.67 mL/kg. LY2828360 (8-(2-chlorophenyl)-2-methyl-6-(4-methylpiperazin-1-yl)-9-(tetrahydro-2H-pyran-4-yl)-9Hpurine) was purchased from Sai Biotech (Mumbai, India). Morphine sulfate was obtained either from the NIDA Drug Supply Program (National Institute on Drug Abuse, Bethesda, MD) or purchased from Sigma Aldrich (St. Louis, MO). Morphine (75 mg) and placebo pellets were obtained from the NIDA Drug Supply Program (National Institute on Drug Abuse, Bethesda, MD). In all studies used to assess paclitaxel-induced mechanical allodynia and CPP, LY2828360 or morphine was dissolved in a vehicle comprised of 20% dimethyl sulfoxide (DMSO 100 % concentration, Sigma Aldrich, St. Louis, MO) with the remaining 80% comprised of ethanol: emulphor: saline in a 1:1:8 ratio, respectively, to maintain consistency with prior studies (Slivicki et al., 2020; Slivicki et al., 2018) used to generate ED50 for suppressing mechanical allodynia and morphine-induced CPP. LY2828360 or morphine was dissolved in a vehicle comprised of 20% dimethyl sulfoxide with the remaining 80% comprised of ethanol: emulphor: saline in a 1:1:18 ratio, respectively for all other studies (Console-Bram et al., 2017). Naloxone (Sigma Aldrich, St. Louis, MO) was dissolved in saline. Control groups always received the same vehicle used to dissolve the drug and only a single vehicle was used in a given experiment. Drugs were administered via intraperitoneal (i.p.) injection to mice in a final volume of 5 ml/kg. Combination doses of morphine + LY2828360 were administered such that overall injection volumes did not exceed 5 mL/kg.
2.3. Paclitaxel-induced allodynia
Paclitaxel (4 mg/kg i.p.) was administered once daily every other day for a dosing regimen consisting of a total of 4 injections (on day 0, 2, 4 and 6) as described in our previous work (Deng et al., 2015; Lin et al., 2018; Slivicki et al., 2017). Animals were tested for responsivity to mechanical stimulation as a measure of paclitaxel-induced allodynia before paclitaxel treatment on days 0, 4, 7, and 15 (data not shown). Assessments of mechanical stimulation were always performed prior to paclitaxel injections whenever injections and behavioral testing were performed on the same day. All other pharmacological manipulations took place beginning on day 16 post-paclitaxel treatment, when behavioral hypersensitivities are stable as reported previously (Deng et al., 2015; Slivicki et al., 2017). Our lab and others (Curry et al., 2018; Donvito et al., 2016; Polomano et al., 2001; Siau et al., 2006; Ward et al., 2011; Zheng et al., 2011) have used this model extensively to characterize effects of distinct approaches to manipulate the endocannabinoid signaling system on the development and maintenance of neuropathic nociception.
2.3.1. Assessment of Mechanical Allodynia
Paw withdrawal thresholds in response to mechanical stimulation were measured in grams (g) using an electronic von Frey anesthesiometer (IITC model Alemo 2390–5, Woodland Hills, CA) as described in our previous work (Li et al., 2019; Slivicki et al., 2020; Slivicki et al., 2018). Mice were allowed to habituate under individual, inverted plastic cages on a mesh table for at least 30 min prior to testing. Following the cessation of exploratory behaviors, a force was applied to the midplantar region of the hind paw with a semiflexible tip connected to the anesthesiometer. Mechanical stimulation was removed when the mouse withdrew its paw from contact with the mechanical stimulator, and the value of the applied force was recorded in grams. Mechanical paw withdrawal thresholds were obtained in duplicate for each paw and are reported as the mean of duplicate determinations from each animal, averaged across animals, for each group.
2.3.2. Dose-response and drug combination studies of LY2828360 and morphine
Dose-response curves assessing suppression of mechanical allodynia induced by LY2828360 and morphine treatments were generated in paclitaxel-treated mice during the maintenance phase of neuropathic nociception. ED50 values for the suppression of paclitaxel-induced mechanical allodynia were generated using a within-subjects design for any given drug condition. The morphine dose response curve for the suppression of paclitaxel-induced mechanical allodynia was reported previously by our group (Slivicki et al., 2020; Slivicki et al., 2018). Dose response curves for LY2828360 alone and morphine in combination with LY2828360 have not been previously reported; data from all experiments was collected by the same experimenter (RAS) in parallel with that reported previously for morphine alone (Slivicki et al., 2020; Slivicki et al., 2018). The experimenter (RAS) was blinded to the experimental conditions in all studies. Escalating doses were administered, using a within-subjects design, to groups receiving either morphine (1, 3, 5, 10, 20, and 30 mg/kg i.p.) or LY2828360 (0.1, 0.3, 1, 3 and 10 mg/kg i.p.) administered alone. In a separate group of paclitaxel-treated mice, combination doses of morphine + LY2828360, based on the 1: 1 ratio of the ED50 for each compound were administered in an escalating fashion every 2-3 days for every treatment. The 1:1 combination doses were used to generate the combination ED50, derived from individual dose-response curves (Table 1). ED50 values were derived to compare anti-allodynic efficacy of each treatment in response to cutaneous stimulation evoked by mechanical stimulation of the hind paw. Mechanical paw withdrawal thresholds (g) were assessed in all groups using identical methods 30 min after each injection (i.p.) of drug or vehicle. Values were converted to % maximal effect (%MPE) using the formula: % MPE = (Experimental Value – Post-paclitaxel baseline)/(Pre-paclitaxel baseline – Post-paclitaxel baseline) x 100.
Table 1.
Doses of LY2828360 and morphine used in combination to determine combination ED50 used in isobologram.
| Doses used to calculate ED50 of Morphine + LY2828360 combination treatment | |
|---|---|
| Morphine (mg/kg i.p.) | LY2828360 (mg/kg i.p.) |
| 0.42 | 0.04 |
| 0.84 | 0.09 |
| 1.67 | 0.19 |
| 3.34 | 0.38 |
| 6.68 | 0.77 |
2.4. Impact of LY2828360 on morphine-induced CPP
We evaluated whether LY2828360 would alter the rewarding effects of morphine using the conditioned place preference approach described in our previously published work (Slivicki et al., 2020). In brief, the CPP testing apparatus consisted of a three-chamber device with two side chambers having distinct visual cues (vertical or horizontal black and white stripes) and a center neutral (grey) chamber. The apparatus was used to assess CPP or conditioned place aversion in an unbiased fashion (Fig. 2A). The time the animal spent in each chamber was recorded over a 30 min test interval. On days 1 and 2, mice were allowed to freely explore and habituate to the entire apparatus. On day 3, an initial baseline preference assessment was conducted to confirm that mice did not show any bias for any specific chamber prior to drug pairings. Animals spending more than 1440 s (i.e. 80% of time) or less than 360 s (i.e. 20% of time) in either distinct chamber were excluded from the experiment and never received any pharmacological manipulations. Next, mice were restricted to a single, randomly selected side chamber after receiving each of four repeated pairings of the assigned drug condition on alternate days (i.e. on day 4, 6, 8 and 10) with vehicle administered in the opposite chamber on other days (i.e. on day 5, 7, 9 and 11). The drug conditions evaluated were morphine alone (8 mg/kg i.p.; data shown in (Slivicki et al., 2020)), LY2828360 alone (3 mg/kg i.p.), or morphine (8mg/kg i.p.) + LY2828360 (3 mg/kg i.p.). A separate group of mice received vehicle in both chambers (data shown in (Slivicki et al., 2020)). Mice in the morphine + LY2828360 group were pre-treated (20 mins prior) with LY2828360 (3 mg/kg i.p.) 20 min before morphine (8 mg/kg i.p.). The total injection volume and vehicle used was the same across all groups. Treatment assignments were randomized to ensure equal baseline chamber times and unbiased pairings to the left and right chambers (counterbalanced). On day 12, all mice were evaluated in the drug free state for the time spent in either chamber to assess the impact of LY2828360 on morphine-induced CPP. A drug chamber preference score (Time in drug chamber post-conditioning minus time in drug chamber pre-conditioning) was also calculated to compare the effect of the different pharmacological treatments between groups of mice.
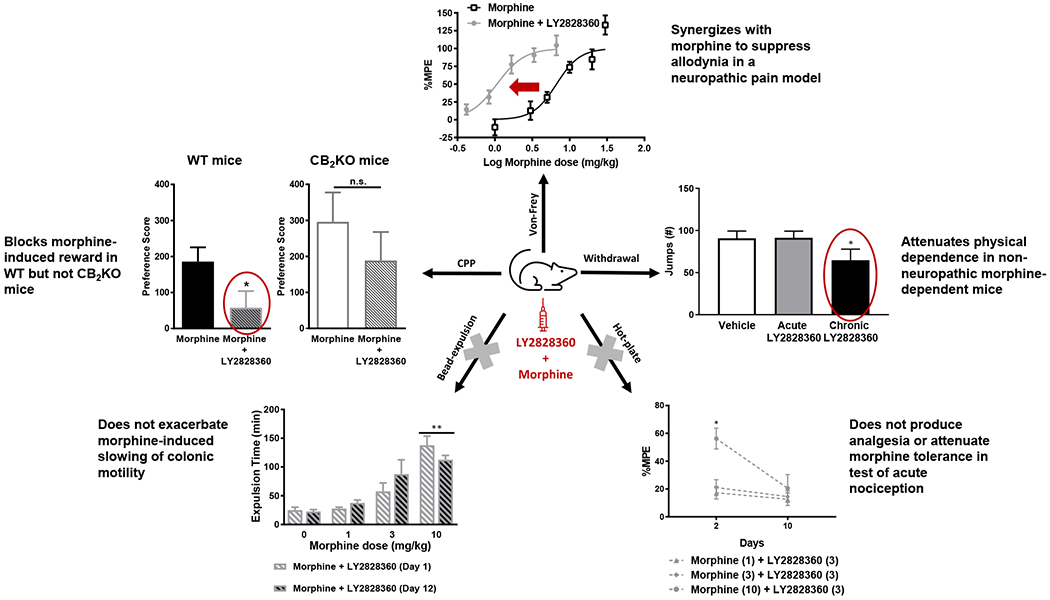
Fig. 2. Pre-treatment with LY2828360 blocks morphine-induced CPP in WT mice.
(A) The schematic shows the timeline of the CPP study. The black vertical arrows show the days when mice received an i.p. drug injection prior to placement in the drug-paired chamber whereas the grey vertical arrows show the days when mice receive an i.p. vehicle injection prior to placement in the vehicle-paired chamber. (B) Morphine (8 mg/kg i.p.) increased the time spent in the drug-paired chamber relative to the vehicle-paired chamber on the test day indicative of CPP and consistent with the development of opioid-induced reward (previously published in (Slivicki et al., 2020)). (C) Vehicle-vehicle pairings did not result in preference or aversion for any chamber (previously published in (Slivicki et al., 2020)). (D) LY2828360 (3 mg/kg i.p.) treatment in the absence of morphine also did not result in the development of preference or aversion for any chamber. (E) Pre-treatment with LY2828360 (3 mg/kg i.p.) 20 minutes prior to morphine (8 mg/kg i.p.) blocked morphine-induced CPP. No difference in chamber preference times were observed pre-conditioning (baseline) in any cohort of mice. Data are expressed as mean ± S.E.M. (n = 10 per group) *P < 0.05 vs. vehicle-paired chamber, two-way repeated measures ANOVA followed by Bonferroni’s post hoc test.
2.5. Impact of LY2828360 on gastrointestinal function following acute and chronic morphine dosing
Gastrointestinal function was assessed using the glass bead expulsion test (Raffa et al., 1987) to assess colonic motility and by measurements of fecal boli accumulation as described in our previously published work (Slivicki et al., 2018). The glass bead expulsion test was used to examine the impact of LY2828360 on morphine-induced slowing of colonic motility following acute and chronic morphine treatment and was performed on days 1 and 12 of repeated injections (Fig. 5A). Separate groups of mice received vehicle, LY2828360 (3 mg/kg i.p.), morphine (1, 3 or 10 mg/kg i.p.), or a combination of morphine (1, 3 or 10 mg/kg i.p.) + LY2828360 (3 mg/kg i.p.) over 12 consecutive days. Mice were fasted for 24 h prior to testing. On the test day, animals were allowed to habituate for 30 min prior to testing. Twenty min following drug administration, a glass bead of approximately 2 mm diameter (Fine Science Tools, Foster City, CA) was inserted 2 cm in the rectal colon using a semi-flexible rubber filament. The time to expel the bead (min) was recorded. The weight of fecal boli produced (mg) was also recorded during a 6 h period beginning at the time of the glass bead insertion.
Fig. 5. LY2828360 does not alter impact of morphine on colonic motility following acute or chronic morphine dosing.
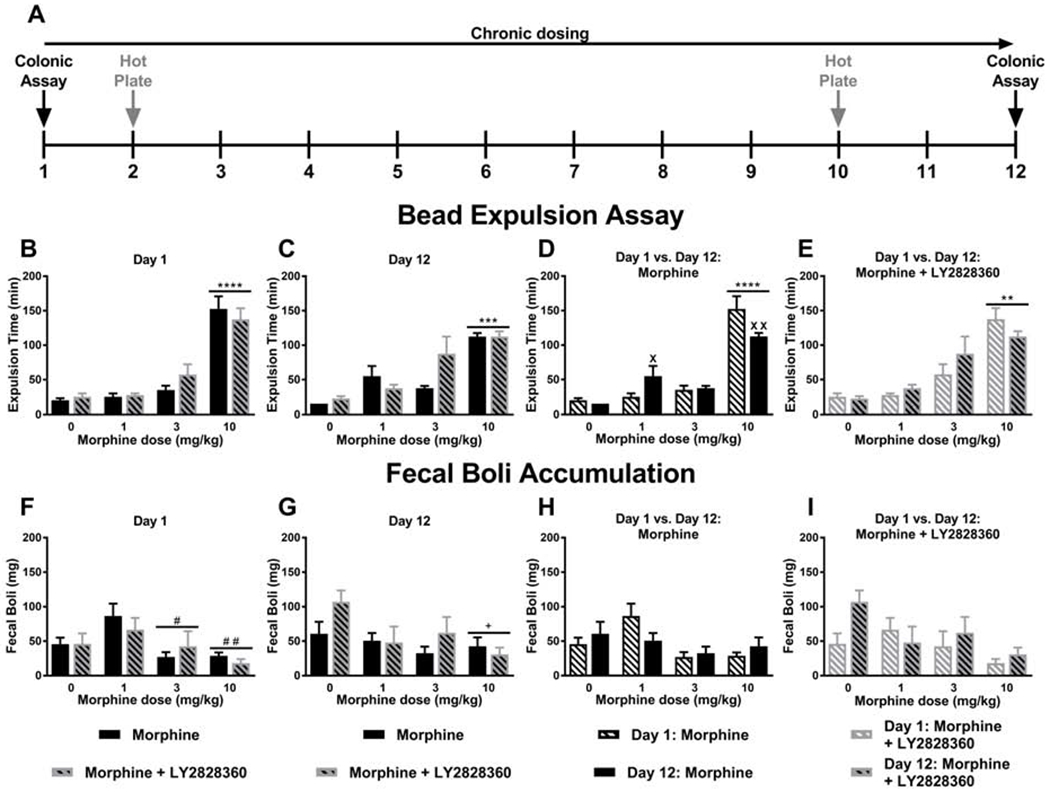
(A) The schematic shows the timing of experimental procedures. Mice received once daily injections (i.p.) of morphine for 12 days. The black vertical arrows show the days when the colonic motility assay was performed, and the grey vertical arrows show the days when the hot plate assay (Fig. 6) was performed. (B) Acute high dose morphine (10 mg/kg i.p. x 1 day) produced longer bead expulsion times compared to all other doses, consistent with opioid-induced slowing of colonic motility and was not affected by LY2828360 treatment. (C) Chronic high dose morphine (10 mg/kg i.p. x 12 days) increased glass bead expulsion times, consistent with absence of tolerance, and was not affected by LY2828360 treatment. (D) High dose morphine (10 mg/kg i.p.) slowed glass bead expulsion time irrespective of injection day. Low dose morphine (1 mg/kg i.p. x 12 days) produced longer bead expulsion times compared to acute treatment on day 1. A modest, but reliable attenuation of bead expulsion time was observed following chronic high dose morphine (10 mg/kg i.p. x 12 days) compared to acute treatment on day 1. (E) High dose morphine (10 mg/kg i.p.) prolonged glass bead expulsion time irrespective of injection day and the presence or absence of LY2828360 treatment. (F) Acute high (10 mg/kg i.p. x 1 day) and middle dose (3 mg/kg i.p. x 1 day) morphine reduced fecal boli accumulation compared to the low dose (1 mg/kg i.p. x 1 day), consistent with the development of morphine-induced constipation and was not affected by LY2828360 treatment. (G) Chronic high dose morphine (10 mg/kg i.p. x 12 days) reduced fecal boli accumulation compared to vehicle , consistent with the development of morphine-induced constipation and was not affected by LY2828360 treatment. (H) Fecal boli accumulation on day 1 and day 12 of morphine alone treatment showed a main effect of the dose but no reliable post hoc differences between groups for the differing morphine doses. (I) Fecal boli accumulation on day 1 and day 12 of morphine + LY2828360 treatment did not differ based upon dose of morphine or injection day. Data are expressed as mean ± S.E.M. (n = 6 per group) **** P < 0.0001 vs. all other groups, *** P < 0.001 vs. all other groups, # # P < 0.01 vs. low dose morphine group, # P < 0.05 vs. low dose morphine group, + P < 0.05 vs. vehicle group, two-way ANOVA followed by Bonferroni's post hoc test. ****P < 0.05 vs. all other groups,**P < 0.01 vs. all other groups, XX P < 0.01 vs. acute treatment, X P < 0.05 vs. acute treatment, two-way repeated measures ANOVA followed by Bonferroni’s post hoc test.
2.6. Impact of LY2828360 on hotplate antinociception following acute and chronic morphine dosing
The hot plate test was used to assess the impact of LY2828360 in a test of acute nociception in the same mice used to assess colonic motility (Fig. 5A). Separate groups of mice received once daily injections of vehicle, LY2828360 (3 mg/kg i.p.), morphine (1, 3 or 10 mg/kg i.p.), or a combination of morphine (1, 3 or 10 mg/kg i.p.) + LY2828360 (3 mg/kg i.p.) over 12 consecutive days. A single drug or vehicle condition was tested in each group of mice. Hotplate antinociception was measured on day 2 of repeated dosing to assess the acute effects of LY2828360 and on day 10 of repeated dosing to assess impact of LY2828360 on morphine tolerance in a test of acute thermal nociception. Colonic transit and hotplate antinociception were always measured on different days to minimize unnecessary stress that could otherwise potentially impact assessments (Fig. 5A). Mice were placed on a 56° C hotplate until jumping, paw shaking, or paw licking behaviors were observed or until the maximum cut-off latency time of 30 s was reached. Baseline (pre-injection response) was assessed approximately 7 h prior to drug injections, and post-injection antinociceptive effects were measured 30 min after drug administration. Antinociception was expressed as a percentage of the maximum possible nociceptive effect (% MPE) calculated according to the following equation: % MPE = [(post morphine latency – baseline latency) / (cut-off latency – baseline latency)] *100 (Liu et al., 2011). Lower antinociceptive effects of day 10 compared to day 2 of repeated dosing was defined as indicative of the development of tolerance.
2.7. Impact of LY2828360 on naloxone-precipitated opioid withdrawal symptoms in morphine-dependent mice
Separate groups of otherwise normal (i.e. paclitaxel naïve) mice were rendered tolerant to morphine by either surgically implanting a morphine pellet subcutaneously or by employing an escalating i.p. morphine dosing regimen. A single dose of naloxone was administered to all mice 30 min after the final morphine/vehicle dose to assess the effects of LY2828360 on naloxone-precipitated opioid withdrawal symptoms. Immediately following the naloxone injection, mice were placed in individual plexiglass observation cylinders. Naloxone-precipitated withdrawal jumping was video recorded over 30 mins and scored by a blinded scorer using the Boris open source software (Friard and Gamba, 2016). Changes in body weight (g) and in body temperature (°C) were calculated as difference scores (i.e. between post assessment measurements for each dependent measure ~30 min following naloxone challenge and the corresponding dependent measure assessed prior to pharmacological manipulations). Positive values represent greater body weight loss or increased hypothermia in response to the pharmacological manipulation. No other withdrawal behaviors were quantified.
2.7.1. Opioid-dependence induced by subcutaneous implantation of a morphine pellet
Mice were anesthetized with isoflurane and surgically implanted with a morphine (75 mg) pellet subcutaneously just below the nape of the neck. The chronic LY2828360 injection group received once daily i.p. injections of LY2828360 (3 mg/kg i.p.) beginning 30 min prior to the implantation surgery and then for three consecutive days (Fig. 7A). Subsequently, three days (71.5 h) following pellet implantation, all groups (chronic LY2828360, acute LY2828360 and vehicle) were injected with either LY2828360 (3 mg/kg i.p.) or an equivalent volume of vehicle and subsequently received a saline challenge. Then, 30 min later (i.e. at 72 h post-surgery) mice in all groups were challenged with the opioid antagonist naloxone (2 mg/kg i.p.) to precipitate a μ-opioid receptor-dependent withdrawal syndrome as described previously (Lichtman et al., 2001; Ramesh et al., 2011). The number of jumps was video recorded over 30 min following both the saline challenge and the naloxone challenge and subsequently quantified by a blinded scorer. Body weight change (g) and body temperature change (°C) were assessed 30 min following the naloxone challenge and 30 min after pharmacological manipulations as described above.
Fig. 7. Repeated LY2828360 dosing reduces the somatic expression of morphine dependence in morphine-pelleted WT mice.
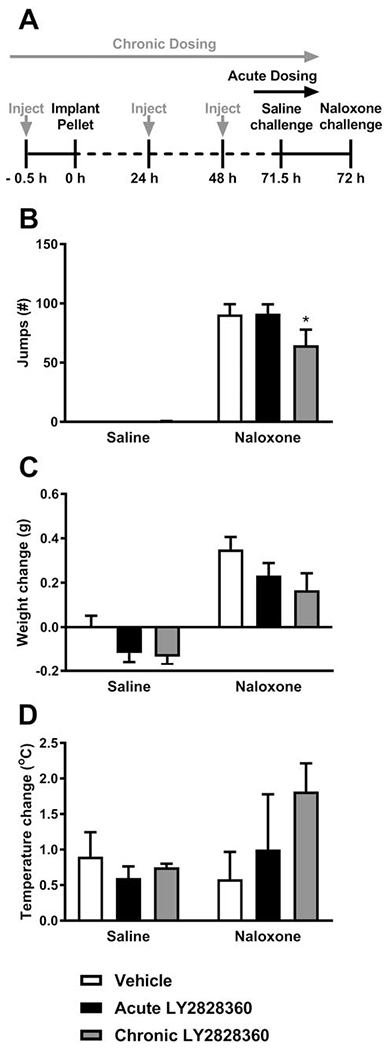
(A) The schematic shows the experimental timeline. The grey vertical arrows show the timing of the chronic LY2828360 injections. The black vertical arrows show the timing of the acute LY2828360/vehicle injection. After 72 h, naloxone was injected to precipitate opioid withdrawal. (B) In morphine pelleted mice, naloxone (2 mg/kg i.p.) produced characteristic jumping behavior compared to the saline challenge. Naloxone-precipitated jumps were lower in the chronic LY2828360-compared to the acute LY2828360- and vehicle-treated mice. (C) Naloxone challenge produced weight loss irrespective of drug treatment. (D) Body temperature change did not differ between the groups as a function of challenge condition, indicating no significant loss of body temperature following the naloxone challenge and showed no significant effect of LY2828360 treatment. Data are expressed as mean ± S.E.M. (n = 6 per group) *P < 0.05 vs. acute LY2828360- and vehicle-treated mice, two-way repeated measures ANOVA followed by Bonferroni’s post hoc test.
In a separate study, WT and CB2KO mice were similarly implanted with identical 75 mg morphine pellets (Fig. 8A). Similarly, three days (71.5 h) following pellet implantation, all groups received a challenge with saline, administered i.p. Thirty min later (i.e. at 72 h post-surgery), mice in all groups were challenged with the opioid antagonist naloxone (2 mg/kg i.p.) to precipitate a μ-opioid receptor-dependent withdrawal syndrome. The number of jumps was video recorded over 30 min following both the saline challenge and the naloxone challenge and subsequently quantified by a blinded scorer.
Fig. 8. Morphine-pelleted CB2KO mice show greater levels of somatic expression of morphine dependence compared to WT mice.
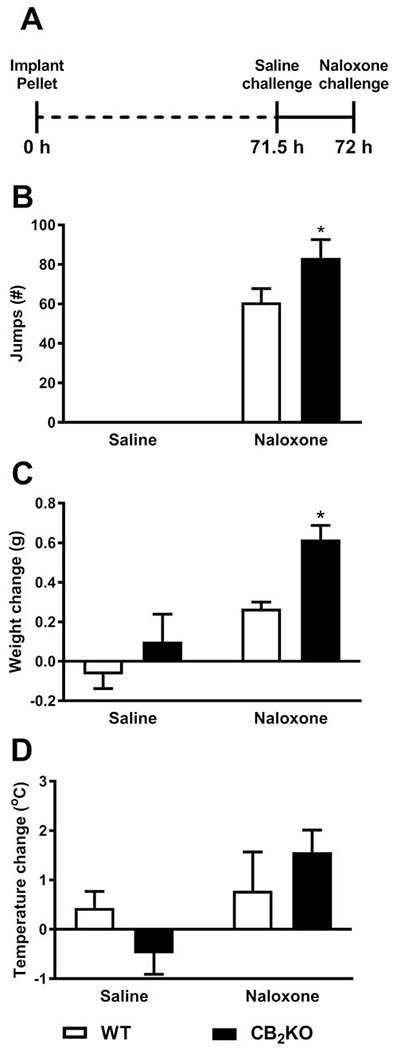
(A) The schematic shows experimental timeline. After 72 h, naloxone was injected to precipitate opioid withdrawal. (B) Naloxone, but not saline, challenge precipitated jumping in WT and CB2KO mice, consistent with the development of physical dependence. While there was no main effect of genotype, post hoc tests showed that CB2KO mice had a significantly higher number of jumps when compared to WT mice. (C) CB2KO mice exhibited significantly more weight loss compared to WT mice in response to a naloxone challenge. (D) No differences in body temperature change was observed between the groups as function of time or genotype. Data are expressed as mean ± S.E.M. (n = 6 per group) *P < 0.05 vs. WT mice, two-way repeated measures ANOVA followed by Bonferroni’s post hoc test.
2.7.2. Opioid-dependence induced by an escalating i.p. morphine dosing schedule
Morphine dependence was induced in a separate group of otherwise normal (i.e. paclitaxel-naïve) mice by repeated injections for three consecutive days using an escalating dose schedule (Liu et al., 2011). Mice received morphine injections twice daily (9 am and 5 pm) for 2 consecutive days (day 1: 7.5 and 15 mg/kg i.p.; day 2: 30 and 30 mg/kg i.p.). The morphine + LY2828360 group received LY2828360 (3mg/kg i.p.) concurrently with each morphine dose (Fig. 9A). Separate groups received vehicle or LY2828360 alone. On the testing day (48 h following first injection), a final dose of morphine (30 mg/kg i.p.) in the presence or absence of LY2828360 was administered. Then, 30 min (48.5 h) after the final pharmacological treatment, all mice were challenged with naloxone (10 mg/kg i.p.) to precipitate a μ-opioid receptor-dependent withdrawal syndrome as described above. The number of jumps was video recorded over 30 min following both the saline challenge and the naloxone challenge and subsequently quantified by a blinded scorer. Change in body weight (g) and change in body temperature (°C) was evaluated before and 30 min following the naloxone challenge as described above. In a separate study, WT and CB2KO mice were similarly rendered morphine dependent using the escalating dosing schedule described above. Separate groups received of WT and CB2KO mice received saline injections as controls. Similarly, on the testing day a final dose of morphine or saline was administered followed 30 min later by a naloxone challenge to precipitate a μ-opioid receptor-dependent withdrawal syndrome. The number of jumps, body weight change and temperature change were evaluated before and 30 min following the naloxone challenge as described above.
Fig. 9. LY2828360 treatment did not impact the somatic expression of morphine dependence induced by an escalating i.p. morphine dosing.
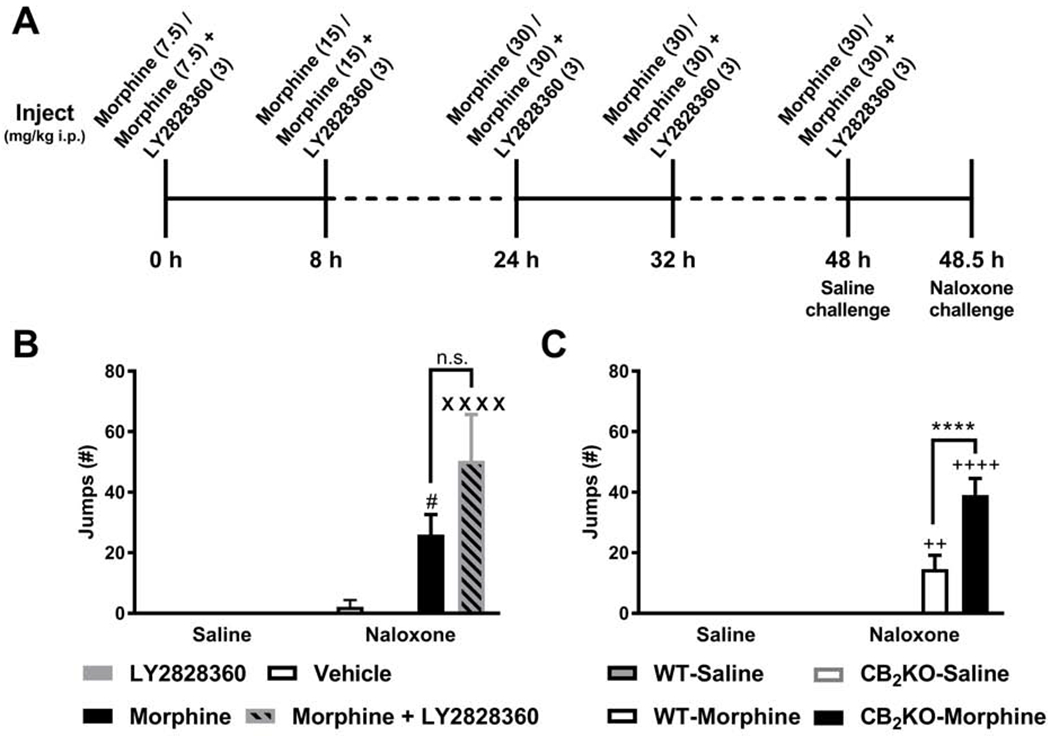
(A) The schematic shows the timing and doses of the escalating i.p. morphine dosing schedule. After 48.5 h, naloxone was injected to precipitate opioid withdrawal. (B) Naloxone (10 mg/kg i.p.) produced characteristic jumping behavior consistent with the development of dependence. Withdrawal jumps did not differ between the morphine + LY2828360 and the morphine alone group. (C) Naloxone-precipitated withdrawal jumps were higher in CB2KO mice compared to WT mice receiving identical morphine (i.p.) treatments. Data are expressed as mean ± S.E.M. (n = 6 per group in Fig. 9B and n=8 per group in Fig. 9C) ****P < 0.0001 vs. WT mice, # P < 0.05 vs. LY2828360-treated mice, XXXX P < 0.0001 vs. LY2828360- and vehicle-treated mice, ++ P < 0.01, ++++ P < 0.0001 vs. saline-treated WT and CB2KO mice, two-way repeated measures ANOVA followed by Bonferroni’s post hoc test, n.s., non-significant.
2.8. Statistical Analysis
Nonlinear regression analyses were used to generate ED50 values with 95% confidence limits. For isobolographic analyses, the combination ED50, based on the 1:1 ratio of individual ED50 values of either morphine or LY2828360 alone, in suppressing responses to mechanical stimulation were generated. Doses of both compounds were administered in combination in an ascending fashion. The observed combination ED50 values (derived from the group receiving morphine + LY2828360 in a 1:1 ratio based upon the individual ED50s for morphine and LY2828360 alone) was calculated and plotted against the theoretical ED50 values. Theoretical ED50 values (Tallarida, 2006) were derived as the expected sum of the two compounds when administered based on their independent ED50 values as described previously by our group (Slivicki et al., 2020; Slivicki et al., 2018; Slivicki et al., 2017). Two-way repeated measures (2 x 2) ANOVA followed by Bonferroni post hoc tests were used to compare chamber preference times in CPP studies and withdrawal behaviors following the naloxone challenge. Two-way ANOVA followed by Bonferroni post hoc tests were used to compare bead expulsion times in colonic motility studies and % MPE in hot plate tests. A priori planned comparisons following an ANOVA were also performed using Bonferroni post hoc tests, as appropriate for within-group comparisons. Unpaired two-tailed t-tests were performed to compare the drug chamber preference scores in CPP studies in the case of two group comparisons. All data was analyzed using GraphPad Prism version 7.05 (GraphPad Software Inc., La Jolla, CA, USA). P < 0.05 was considered statistically significant.
3. Results
3.1. LY2828360 produces a leftward shift in the dose-response of morphine to suppress paclitaxel-induced mechanical allodynia
Prior to pharmacological manipulations, paclitaxel lowered mechanical paw withdrawal thresholds relative to baseline (pre-injection) thresholds in all groups (F1, 10= 221.1, P < 0.0001; Fig. 1A and F1, 10= 148, P < 0.0001; Fig. 1C). The interaction between the treatment and time was not significant (F1, 10= 1.725, P = 0.2183; Fig. 1A and F1, 10= 0.152, P = 0.7048; Fig. 1C). Paw withdrawal thresholds did not differ between groups at baseline (P = 0.0842; Fig. 1A and P = 0.5661; Fig. 1C) or following the establishment of paclitaxel-induced neuropathy (P > 0.05; Fig. 1A, C). Thus, groups did not differ prior to initiation of dose response studies. LY2828360 increased mechanical paw withdrawal thresholds in paclitaxel-treated mice (Fig. 1A) and suppressed paclitaxel-induced mechanical allodynia with an ED50 of 0.7764 (0.4489 - 1.343) mg/kg i.p. (Fig. 1B). Similarly, as reported previously (Slivicki et al., 2020; Slivicki et al., 2018), morphine suppressed paclitaxel-induced mechanical allodynia with an ED50 of 6.682 (4.905 – 9.103) mg/kg i.p (Fig. 1A–D). Co-administration of LY2828360 with morphine increased mechanical paw withdrawal thresholds in paclitaxel-treated mice (Fig. 1C) and produced a leftward shift of the ED50 of morphine, reducing the ED50 for suppressing paclitaxel-induced hypersensitivity to mechanical stimulation from 6.682 (4.905 – 9.103) mg/kg i.p. to 1.069 (0.4489 – 1.343) mg/kg i.p. (Fig. 1D). Raw scores (i.e. paw withdrawal thresholds in g) and transformed scores (i.e. converted to % MPE) are shown in Fig. 1A, C and Fig. 1B, D, respectively.
Fig. 1. The CB2 agonist LY2828360 synergizes with the opioid analgesic morphine in suppressing paclitaxel-induced mechanical allodynia.
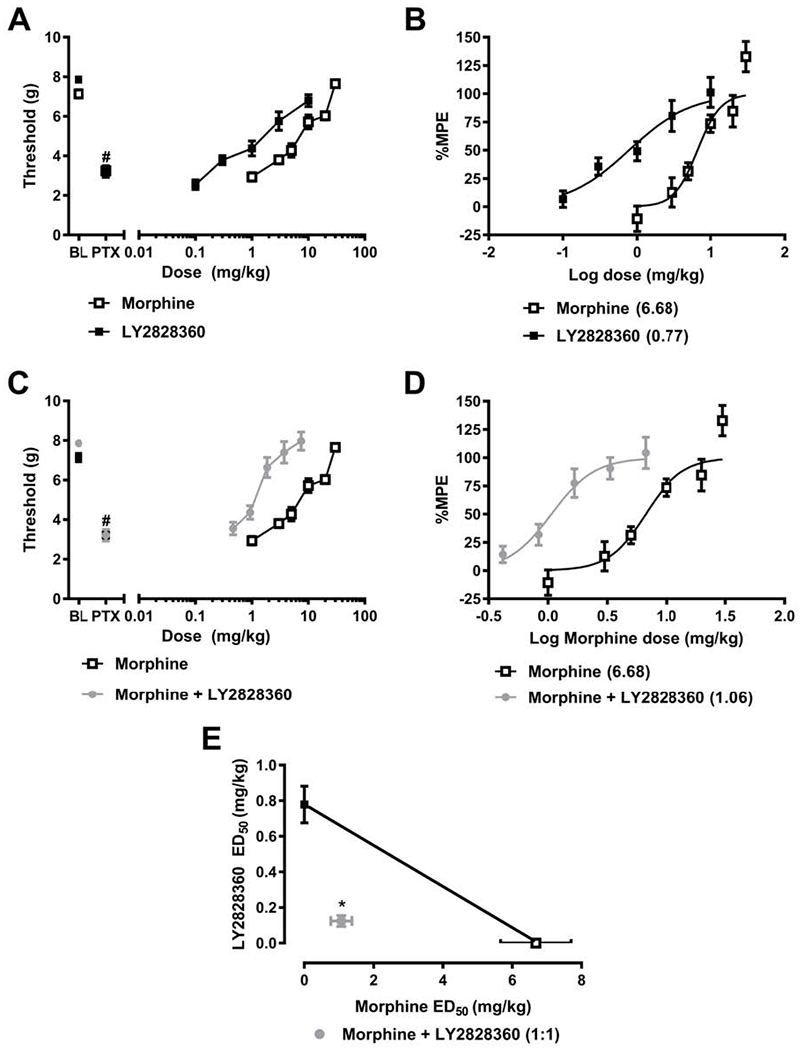
(A,B) Paclitaxel reduces mechanical paw withdrawal thresholds prior to pharmacological manipulations, consistent with development of allodynia. Morphine and LY2828360 (A) increase paw withdrawal thresholds (g) in paclitaxel-treated mice and (B) suppress paclitaxel-induced mechanical allodynia with ED50s of 6.682 mg/kg i.p. and 0.7764 mg/kg i.p., respectively. Co-administration of LY2828360 with morphine (C) increases paw withdrawal thresholds in paclitaxel-treated mice and (D) produces a leftward shift in the dose-response curve of morphine to reduce paclitaxel-induced mechanical allodynia. (D) LY2828360 combination treatment reduced the ED50 of morphine from 6.682 (4.904 – 9.105) mg/kg i.p. to 1.069 (0.4489 – 1.343) mg/kg i.p. (E) Isobolographic analysis reveal a synergistic interaction of the combination of LY2828360 with morphine when administered in doses based upon in a 1:1 ratio of the ED50s (mg/kg i.p.) of each agent alone. Data are expressed as mean ± S.E.M. (n = 6 per group) #P < 0.0001 vs. baseline (pre-paclitaxel responding), *P < 0.05, two-tailed t-test vs. theoretical additive values.
3.1.1. LY2828360 synergizes with morphine to reduce paclitaxel-induced mechanical allodynia
LY2828360 and morphine synergistically suppressed paclitaxel-induced mechanical allodynia (Fig. 1E). The observed ED50 of LY2828360 with morphine (1.193 (0.8996 - 1.583) mg/kg i.p.) in suppressing mechanical allodynia was lower (P < 0.05, two-tailed t-test) than the theoretical additive value (ED50: 3.728 (3.390 –4.066) mg/kg i.p.) of the 1:1 ED50 combination (Fig. 1E).
3.2. LY2828360 blocks morphine CPP and does not cause place preference or aversion when administered alone in WT mice
Morphine (8 mg/kg i.p.) produces a robust CPP relative to the vehicle-paired chamber (Fig. 2B) (Slivicki et al., 2020). The interaction between the conditioning phase and drug treatment was significant (F1, 18= 17.03, P = 0.0006). No main effect of drug treatment (F1,18= 2.944, P = 0.1034) or conditioning phase (F1,18= 1.773, P = 0.1996) was observed, as expected. Post hoc comparisons revealed that WT mice exhibited increased time in the morphine-paired chamber vs. the vehicle-paired chamber on the CPP test day (P = 0.0089) but no such difference was observed on the baseline day prior to conditioning, consistent with the development morphine-induced CPP. In a separate group of mice, time spent in each chamber did not differ when mice received vehicle in both chambers (i.e. vehicle-vehicle pairings; Fig. 2C) (Slivicki et al., 2020). The interaction between the conditioning phase and drug treatment was not significant (F1,18= 0.4332, P = 0.5188) and no main effects of vehicle treatment (F1,18= 0.2187, P = 0.6457) or conditioning phase (F1,18= 2.403, P = 0.1385) were observed. Similarly, LY2828360 (3 mg/kg i.p.) treatment alone did not alter time spent in the drug-paired chamber vs. the vehicle-paired chamber on the CPP test day (Fig. 2D). The interaction between the conditioning phase and drug treatment was not significant (F1,18= 0.0543, P = 0.8184) and no main effect of drug treatment (F1,18= 0.006251, P = 0.9379) or conditioning phase (F1,18= 1.654, P = 0.2147) were observed. In a separate group of mice, pre-treatment with LY2828360 (3 mg/kg i.p.) blocked CPP to morphine (8mg/kg i.p.) (Fig. 2E). The interaction between the conditioning phase and drug treatment was not significant for the morphine + LY2828360 combination treatment (F1,18= 0.3683, P = 0.5515) and no main effect of drug treatment (F1,18= 0.256, P = 0.6190) or conditioning phase (F1,18= 1.089, P = 0.3105) were observed. Planned comparisons revealed neither an increase nor a decrease in the time spent in the drug-paired chamber vs. the vehicle-paired chamber on the CPP test day in groups receiving either LY2828360 alone (P > 0.05; Fig. 2D) or morphine + LY2828360 (P > 0.05; Fig. 2E). Thus, LY2828360 effectively blocked the development of morphine-induced CPP without producing conditioned place aversion.
3.2.1. CB2KO mice show morphine-induced CPP and this CPP is not blocked by LY2828360
CB2KO mice showed robust morphine-induced CPP (8 mg/kg i.p.) (Fig. 3A). The interaction between the conditioning phase and drug treatment was significant (F1,18= 27.18, P < 0.0001) and there was a significant main effect of drug treatment (F1,18= 5.147, P = 0.0358) but no overall effect of the conditioning phase (F1,18= 0.02107, P = 0.8862). Post hoc comparisons revealed that CB2KO mice exhibited increased time (P = 0.0002) in the morphine-paired chamber vs. the vehicle-paired chamber on the CPP test day, but no such difference was observed on the baseline day prior to conditioning, consistent with the development of morphine-induced CPP. In a separate group of CB2KO mice, pre-treatment with LY2828360 (3 mg/kg i.p.) prior to morphine (8 mg/kg i.p.) failed to block morphine-induced CPP (Fig. 3B). While the interaction between the conditioning phase and drug treatment was significant (F1,18= 9.169, P = 0.0072), no significant main effects of either drug treatment (F1,18=2.436, P = 0.1360) or conditioning phase (F1,18= 0.4942, P = 0.4910) were observed. Post hoc comparisons revealed that, in CB2KO mice, morphine + LY2828360-pairing increased the time spent in the drug-paired chamber vs. the vehicle-paired chamber (P = 0.0137) on the CPP test day, but no such difference was observed on the baseline day prior to conditioning, consistent with the development of morphine-induced CPP.
Fig. 3. LY2828360 blockade of morphine-induced CPP is absent in CB2KO mice.
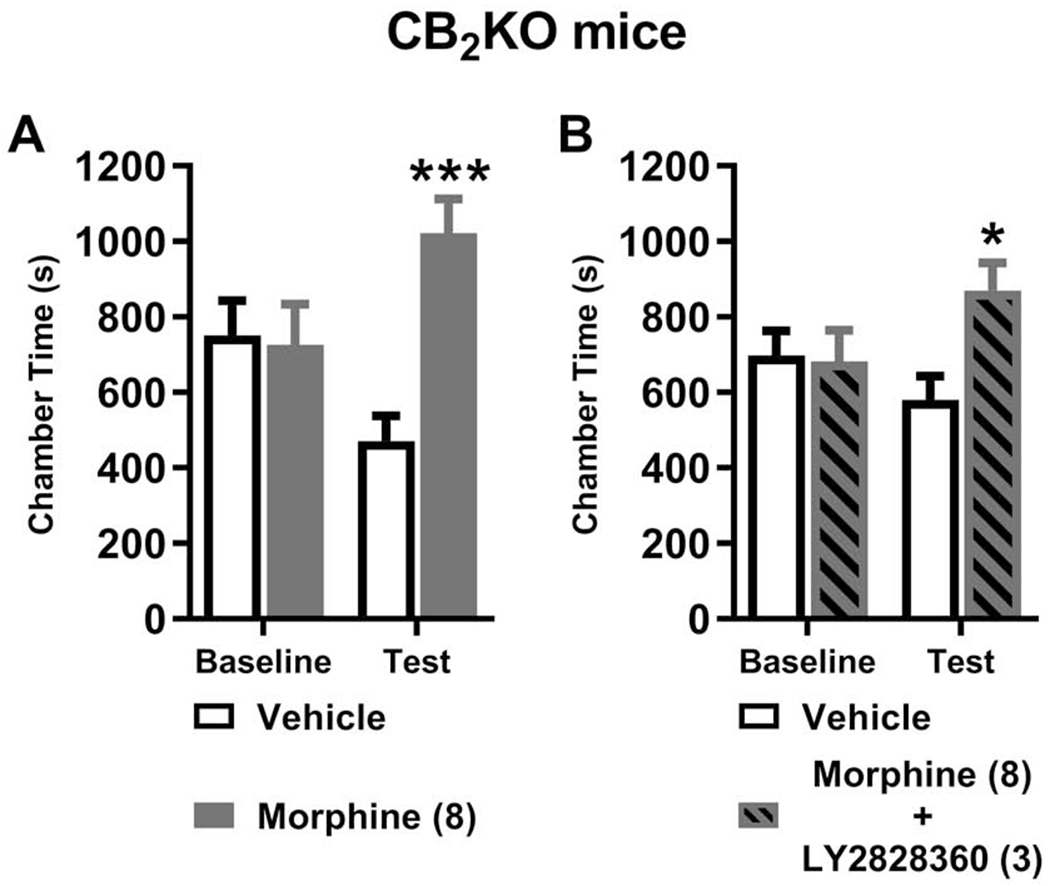
(A) Morphine (8 mg/kg i.p.) increased the time spent in the drug-paired chamber relative to the vehicle-paired chamber in CB2KO mice, indicative of opioid-induced CPP. (B) In a separate set of CB2KO mice, pre-treatment with LY828360 (3 mg/kg i.p.) prior to morphine (8 mg/kg i.p.) did not block the development of morphine-induced CPP. No difference in chamber preference times were observed pre-conditioning (baseline) in any cohort of mice. Data are expressed as mean ± S.E.M. (n = 10 per group). ***P < 0.001 vs. vehicle-paired chamber, *P < 0.05 vs. vehicle-paired chamber, two-way repeated measures ANOVA followed by Bonferroni’s post hoc test.
3.2.2. LY2828360 reduced preference scores for the morphine-paired chamber in WT but not CB2KO mice
Drug chamber preference scores were higher in WT mice that received morphine alone in the drug-paired chamber compared to morphine + LY2828360 (t18= 2.106, P = 0.0495; Fig. 4A). In contrast, drug chamber preference scores did not significantly differ in CB2KO mice that received repeated drug pairings with either morphine alone or morphine + LY2828360 (t18= 0.9459, P = 0.3567; Fig. 4B). Preference scores for the morphine-paired chamber did not differ reliably between WT and CB2KO mice (t18= 1.218, P = 0.2389; Fig. 4C).
Fig. 4. LY2828360 pretreatment reduced preference for morphine-paired chamber in WT but not CB2KO mice.
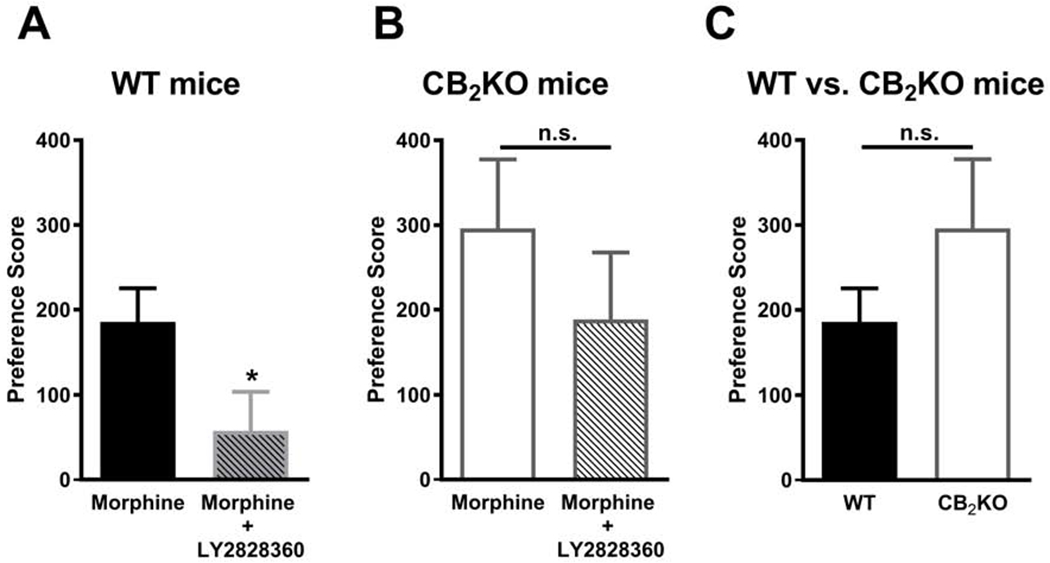
(A) In WT mice, the drug chamber preference score for the morphine + LY2828360 group was lower than the morphine alone groups. (B) In CB2KO mice, drug chamber preference scores did not differ reliably in mice receiving morphine alone or morphine + LY2828360. (C) Preference scores for the morphine-paired chamber did not differ reliably between WT and CB2KO mice. Data are expressed as mean ± S.E.M. (n = 10 per group) *P < 0.05, unpaired two-tailed t-test, n.s., non-significant.
3.3. LY2828360 does not alter colonic motility and fecal boli accumulation following acute and chronic morphine dosing
Acute morphine treatment prolonged the glass bead expulsion time (Fig. 5B) in a dose-dependent manner on day 1 of injections. There was a significant main effect of the morphine dose on the glass bead expulsion time (F3,40= 57.74, P < 0.0001; Fig. 5B), consistent with an acute injection of morphine slowing colonic motility. There was no reliable effect of LY2828360 treatment on bead expulsion time (F1,40= 0.2446, P = 0.6236; Fig. 5B) and the interaction between morphine dose and LY2828360 treatment was not significant (F3,40 = 1.024, P = 0.3924; Fig. 5B). Post hoc analyses revealed that the highest dose of morphine (10 mg/kg i.p.), administered in either the presence or absence of LY2828360, was associated with the longest bead expulsion time compared to either vehicle, the low (1 mg/kg i.p.) or middle (3 mg/kg i.p.) dose of morphine (P < 0.0001 vs. all other doses).
Twelve days of chronic morphine treatment increased glass bead expulsion time (Fig. 5C) in a dose-dependent manner. There was a significant main effect of the morphine dose on the glass bead expulsion (F3,40= 25.52, P < 0.0001; Fig. 5C), consistent with the absence of tolerance to opioid-induced slowing of colonic motility. There was no reliable effect of LY2828360 treatment (F1,40= 1.645, P = 0.2070; Fig. 5C) but the interaction between morphine dose and LY2828360 treatment was significant (F3,40 = 3.376, P = 0.0275; Fig. 5C). Post hoc analyses revealed that the highest dose of morphine (10 mg/kg i.p.), administered in either the presence or absence of LY2828360, was associated with the longest bead expulsion time compared to either vehicle, the low (1 mg/kg i.p.) or middle (3 mg/kg i.p.) dose of morphine (P < 0.0001 vs. all other doses).
Comparisons of bead expulsion times between day 1 and day 12 of repeated dosing with morphine alone (Fig. 5D) revealed a significant main effect of the dose of morphine (F3, 20= 48.57, P < 0.0001; Fig. 5D) but not of injection day (F1, 20= 0.3388, P = 0.5671; Fig. 5D) and the interaction between morphine dose and injection day was significant (F3, 20= 7.204, P = 0.0018; Fig. 5D). Post hoc analyses revealed that the highest dose of morphine (10 mg/kg i.p.), administered either acutely (day 1) or chronically (i.e. on all 12 days of repeated injections), was associated with longer bead expulsion times compared to all other groups (P < 0.0001 vs. all other doses). A modest but reliable increase in the bead expulsion time was also observed in the chronic low dose group (1 mg/kg i.p. x 12 days; p= 0.0448) whereas a modest decrease in bead expulsion time was observed for the chronic high dose group (10 mg/kg i.p. x 12 days; P = 0.0053) compared to the corresponding acute treatment.
Comparisons of bead expulsion times between day 1 and day 12 of the morphine + LY2828360 dosing (Fig. 5E) revealed a significant main effect of the dose of morphine (F3, 20= 22.02, P < 0.0001; Fig. 5E) but not of injection day (F1, 20= 0.1743, P = 0.6807; Fig. 5E) and the interaction between morphine dose and injection day was not significant (F3,20= 2.369, P = 0.1011; Fig. 5E). Post hoc analyses revealed that the highest dose of morphine (10 mg/kg i.p.) + LY2828360, administered on either day 1 or day 12 of repeated injections, was associated with the longest bead expulsion time compared to either vehicle (P < 0.0001), the low (1 mg/kg i.p.; P < 0.0001) or middle (3 mg/kg i.p.; P = 0.0072) dose of morphine + LY2828360.
Acute morphine treatment also decreased fecal boli accumulation (Fig. 5F) in a dose-dependent manner on day 1 of injection. ANOVA revealed a significant main effect of the morphine dose on fecal boli accumulation (F3,40= 5.481, P = 0.0030; Fig. 5F), consistent with an acute injection of morphine producing constipation. There was no reliable effect of LY2828360 treatment on fecal boli accumulation (F1,40 = 0.1337, P = 0.7165; Fig. 5F), and the interaction between morphine dose and LY2828360 treatment was not significant (F3,40 = 0.6083, P = 0.6135; Fig. 5F). Post hoc analyses revealed that fecal boli accumulation was lower in groups receiving either the middle (3 mg/kg i.p.; P = 0.0258) or high (10 mg/kg i.p.; P = 0.0025) dose of morphine either in the presence or absence of LY2828360 compared to the low (1 mg/kg i.p.) dose of morphine.
Twelve days of repeated morphine treatment also reduced fecal boli accumulation (Fig. 5G) in a dose-related manner. There was a significant main effect of the morphine dose on fecal boli accumulation (F3,40= 3.176, P = 0.0343; Fig. 5G), consistent with absence of tolerance to opioid-induced slowing of colonic motility. There was no reliable effect of LY2828360 treatment on fecal boli accumulation (F1,40= 1.795, P = 0.1879; Fig. 5G) and the interaction between morphine dose and LY2828360 treatment was not significant (F3,40= 1.408, P = 0.2547; Fig. 5G). Post hoc analyses revealed that fecal boli accumulation was lower in groups receiving the high (10 mg/kg i.p.; P = 0.0359) dose of morphine in either the presence or absence of LY2828360 compared to the vehicle group.
Comparisons of fecal boli accumulation between day 1 and day 12 repeated dosing of the morphine alone (Fig. 5H) revealed a significant main effect of the dose of morphine (F3, 20= 3.147, P = 0.0478; Fig. 5H) but not of injection day (F1, 20= 0.2359, P = 0.9617; Fig. 5H) and the interaction between morphine dose and injection day approached significance (F3, 20= 3.05 6, P = 0.0520; Fig. 5H). However, post hoc analyses failed to reveal differences among the groups receiving the different doses of morphine.
Comparisons of fecal boli accumulation between day 1 and day 12 of repeated dosing with morphine + LY2828360 (Fig. 5I) did not reveal any main effects of the dose of morphine (F3, 20= 2.285, P = 0.1099; Fig. 5I) although the impact of injection day (F1, 20= 3.264, P = 0.0859; Fig. 5I) and the interaction between the injection day and dose (F3,20= 2.52, P = 0.0871; Fig. 5I) approached significance.
3.4. LY2828360 did not alter antinociceptive effects of morphine in the hotplate test of acute antinociception following either acute or chronic morphine dosing
In the same animals used to assess colonic motility, hotplate antinociception was also evaluated on day 2 and 10 of chronic dosing to ascertain whether morphine tolerance developed in a test of acute thermal nociception (Fig. 5A). On day 2 (Fig. 6A) of injections, no differences in the % MPE were observed between morphine and morphine + LY2828360 treatment for any of the three doses of morphine evaluated. There was no main effect of the LY2828360 treatment (F1,10= 0.0926, P = 0.7625) or interaction (F3,40= 0.2977, P = 0.8268). However, as expected, the dose of morphine impacted the magnitude of acute hotplate antinociception (F3,40= 22.44, P < 0.0001; Fig. 6A). Post hoc analyses revealed that the high dose of morphine (10 mg/kg i.p.) administered in either the presence or absence of LY2828360 produced greater hotplate antinociception compared to all other dose and vehicle groups (P < 0.0090 for all comparisons). The low and middle doses of morphine did not produce acute antinociception relative to vehicle (P > 0.05 vs. vehicle). Antinociceptive effects of morphine did not differ from morphine + LY2828360 at any dose (P > 0.05); thus, LY2828360 did not enhance morphine antinociception in an assay of acute nociception. Following repeated daily dosing of morphine and LY2828360 by day 10 (Fig. 6B), no main effect of morphine dose (F3,40= 1.3 8 5, P = 0.2613) or LY2828360 treatment (F1,40= 0.1797, P = 0.6739) on hotplate antinociception was observed, and the interaction between drug treatment and dose was not significant (F3,40= 1.084, P = 0.3668), consistent with the development of morphine tolerance. Thus, LY2828360 treatment did not alter hotplate antinociception at any morphine dose on any day. Comparisons between % MPE observed on injection day 2 and day 10 across all treatment conditions (Fig. 6C) revealed a significant main effect of day (Fi,30= 21.13, P < 0.0001), treatment (F5, 30= 6.899, P = 0.0002), and interaction (F5, 30= 2.549, P = 0.0489). High dose morphine (10 mg/kg i.p.), administered in either the presence or absence of LY2828360 on day 2, produced greater hot plate antinociception than all other treatments (P < 0.05 vs. all other groups). No differences in hotplate antinociception were detected between groups on day 10 of repeated dosing (P > 0.05 vs. all other groups). Thus, tolerance developed to high dose morphine treatment in the hotplate test irrespective of LY2828360 treatment.
Fig. 6. LY2828360 does not alter acute antinociceptive efficacy produced by acute or chronic morphine dosing in the hot-plate test.
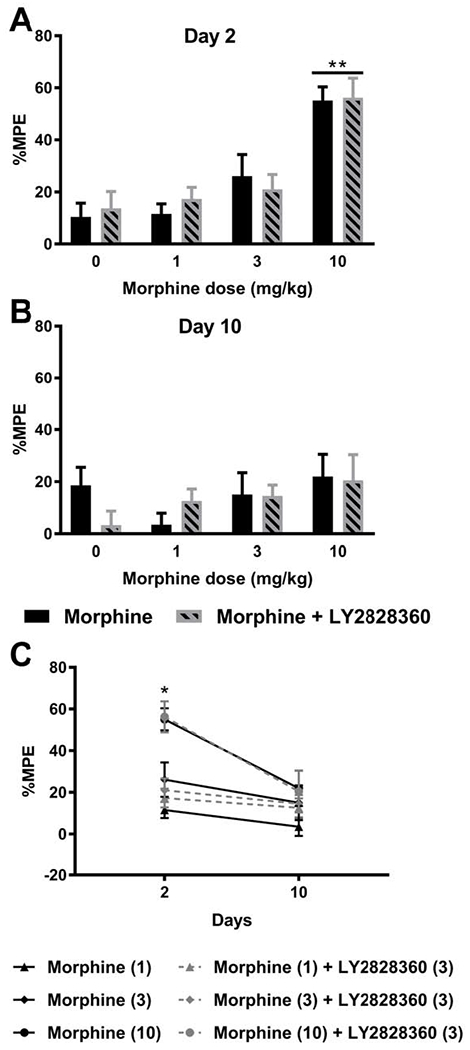
(A) Morphine produced a dose-related acute antinociceptive effect in the hotplate test on day 2 in a manner that was not altered by LY2828360 treatment. (B) Following repeated dosing with morphine for 10 days, hotplate antinociception was no longer observed, consistent with the development of morphine tolerance. (C) Comparisons of all doses and treatments on day 2 and day 10 of repeated injections revealed a significant effect of injection day, drug treatment, and the interaction. Morphine (10 mg/kg i.p.) in the presence or absence of LY2828360 produced greater % maximal possible antinociceptive effect (% MPE) compared to day 10 treatment or any other group. Data are expressed as mean ± S.E.M. (n = 6 per group). *P < 0.05, **P < 0.01, two-way ANOVA followed by Bonferroni’s post hoc test.
3.5. Chronic LY2828360 treatment in morphine-pelleted mice attenuates naloxone-precipitated withdrawal
As expected, in morphine-pelleted WT mice, naloxone challenge produced characteristic jumping behavior (Fig. 7A, B) compared to saline challenge (F1, 15= 192.6, P < 0.0001). Chronic LY2828360 (3 mg/kg i.p. x 4 days), acute LY2828360 (3 mg/kg i.p. x 1 day) and vehicle treatment did not impact naloxone-precipitated jumping (F2,15= 2.225, P = 0.1425) and the interaction between drug treatment and challenge condition was not significant (F2,15 = 2.258, P = 0.1390). However, planned comparisons nonetheless revealed that the number of naloxone-precipitated jumps was lower in the chronic LY2828360-treated group compared to either the acute LY2828360- (P = 0.0405) or vehicle-treated groups (P = 0.0473). Thus, repeated but not acute, LY2828360 treatment reduced, but did not eliminate, naloxone-precipitated jumps in morphine-pelleted mice. Body weight change (Fig. 7C) from baseline (i.e., post-saline challenge) differed as a function of the challenge condition (F1, 15= 49.18, P < 0.0001). There was a significant main effect of LY2828360 treatment between the groups (F2, 15= 5.553, P = 0.0157). However, post hoc comparisons failed to distinguish effects of different experimental treatments following either saline or naloxone challenge. Body temperature change (Fig. 7D) from baseline (i.e., post-saline challenge) did not differ between the groups as a function of challenge condition (F1, 15= 0.9009, P = 0.3576) or LY2828360 treatment (F2, 15= 1.629, P = 0.2291) and the interaction was not significant (F2, 15 = 0.9781, P = 0.3988).
3.5.1. Morphine-pelleted CB2KO mice show higher levels of naloxone-precipitated withdrawal behaviors compared to WT mice
CB2KO mice implanted with morphine pellets exhibited characteristic jumping behavior in response to a naloxone challenge, consistent with the development of physical dependence (Fig. 8A, B). Naloxone challenge significantly increased withdrawal jumps in morphine-pelleted mice (F1,10 = 152.9, P < 0.0001) relative to the saline challenge (Fig. 8B). Both the main effects of genotype (F1,10= 3.724, P = 0.0825) and the interaction between genotype and challenge condition (F1,10 = 3.724, P = 0.0825) approached significance. Post hoc analyses revealed that CB2KO mice had a significantly higher number of jumps when compared to WT mice following the naloxone challenge (P = 0.0259). Body weight change (Fig. 8C) relative to baseline (i.e., post-saline challenge) differed as a function of the challenge condition (F1,10= 32.76, P = 0.0002) and genotype (F1,10= 6.854, P = 0.0257), although the interaction between challenge condition and genotype was not significant (F1,10 = 1.524, P = 0.2452). Post hoc comparisons revealed that CB2KO mice exhibited greater weight loss compared to WT mice following naloxone challenge (P = 0.0205; Fig. 8C). Body temperature change (Fig. 8D) from baseline (i.e., post-saline challenge) approached significance as a function of challenge condition (F1,10= 4.687, P = 0.0556) irrespective of genotype (F1,10= 0.01796, P = 0.8961) and the interaction between challenge condition and genotype was not significant (F1,10 = 2.352, P = 0.1562).
3.5.2. LY2828360 treatment does not impact naloxone-precipitated withdrawal following escalating morphine i.p. dosing
In WT mice subjected to an escalating i.p. morphine dosing regimen, naloxone challenge produced characteristic jumping behavior compared to the saline challenge (F1, 20= 21.7, P = 0.0002; Fig. 9B). There was a significant main effect of treatment on the number of jumps (F3, 20= 7.857, P = 0.0012) and the interaction between drug treatment and challenge condition was significant (F3, 20= 7.857, P = 0.0012). Post hoc analyses revealed that naloxone challenge elicited more jumps in the morphine alone group compared to the LY2828360 alone group (P = 0.0441). Similarly, the morphine + LY2828360 group had significantly higher jumps compared to either the vehicle- (P < 0.0001) or the LY2828360-treated group (P < 0.0001). No differences were observed between the groups following the saline challenge. In the absence of morphine, groups that received vehicle or LY2828360 alone showed almost no jumping behavior in response to either saline or naloxone challenge.
Withdrawal jumping was compared in WT and CB2KO mice following either saline or naloxone challenge in the same escalating i.p. morphine dosing paradigm (Fig. 9C). Both challenge condition (F1, 28= 57.52, P < 0.0001), and treatment (F3, 28= 27.14, P < 0.0001) impacted withdrawal jumping and the interaction between treatment and challenge condition (F3, 28= 27.14, P < 0.0001) was significant. Post hoc comparisons revealed that naloxone precipitated more withdrawal jumps in morphine-treated CB2KO mice compared to morphine-treated WT mice (P < 0.0001) (Fig. 9C). Morphine-treated CB2KO mice also exhibited more jumps compared to saline-treated WT mice (P < 0.0001) and saline-treated CB2KO mice (P < 0.0001) following the naloxone challenge. Morphine-treated WT mice also had significantly higher jumps compared to saline-treated WT mice (P < 0.0001) and saline-treated CB2KO mice (P < 0.0001) following naloxone challenge. No differences were observed between the groups following saline challenge. In the absence of morphine, groups that received saline alone showed no jumping behavior in response to either saline or naloxone challenge.
4. Discussion
Opioid-based therapies are a mainstay of treating chronic pain conditions and are a vital part of the World Health Organization analgesic stepladder (Ballantyne et al., 2016). Unfortunately, these therapies also produce tolerance, abuse liability, physical dependence, respiratory depression, constipation and, in extreme cases, a fatal overdose (Compton et al., 2015; Fields, 2011; Jamison and Mao, 2015). Our results suggest that the CB2 agonist LY2828360 produces synergistic anti-allodynic effects, reduces reward, and physical dependence with morphine while not altering the effects of morphine on acute nociception or colonic motility.
We used isobolographic analysis to show, for the first time, that the CB2 agonist LY2828360 acts synergistically with morphine to suppress neuropathic nociception induced by the chemotherapeutic agent paclitaxel. The CB2 agonists AM1241 and GW405833 suppress capsaicin-evoked release of calcitonin gene-related peptide in spinal cord, suggesting a neuronal mechanism for their anti-allodynic actions (Beltramo et al., 2006) (but see (Li et al., 2017)). Conversely, JWH-015, administered intrathecally, reduced paw incision-induced microglial and astrocytic activation in the spinal cord (Romero-Sandoval and Eisenach, 2007), which may suppress release of inflammatory mediators that sensitize nociceptors (Mazzari et al., 1996). Thus, non-neuronal substrates may also contribute to the anti-allodynic actions of CB2 agonists. Pathological pain states and injury are associated with an upregulation of cannabinoid CB2 receptor protein and mRNA in the spinal cord microglia and dorsal root ganglion cell cultures whereas expression levels remain near the threshold for detection in naive animals (Wotherspoon et al., 2005; Zhang et al., 2003). We previously showed that CB2-selective agonists preferentially suppress activity in spinal nociceptive neurons under pathological conditions that cause sensitization (Nackley et al., 2004). The CB2 agonist JWH-133 suppressed mechanically evoked responses in neuropathic rats (Sagar et al., 2005) but these antinociceptive effects were absent in neuropathic CB2KO mice (Yamamoto et al., 2008). These findings parallel our observations that LY2828360 reduced allodynia in mice exhibiting neuropathic nociception but did not alter acute nociception in control (paclitaxel-naïve) mice.
Abuse liability limits the therapeutic utility of an analgesic. In our study, rewarding effects of morphine were blocked by a dose of LY2828360 that did not itself produce place preference or aversion. Moreover, LY2828360-mediated blockade of morphine CPP was absent in CB2KO mice, consistent with mediation by cannabinoid CB2 receptors. We employed naive mice in our CPP paradigm so that the impact of LY2828360 on the positive reinforcing effects (i.e. reward) of morphine could be assessed without the possible confound of negative reinforcing effects (i.e. removal of an aversive pain state). Our data is in line with previous studies demonstrating that CB2 agonists can suppress the rewarding effects of several drugs of abuse without producing any inherent reward or aversion themselves. For example, JWH-133 inhibited intravenous cocaine self-administration, cocaine CPP and elevated levels of dopamine in the nucleus accumbens of WT but not CB2KO mice (Delis et al., 2017; Xi et al., 2011; Zhang et al., 2017). β-caryophyllene, another CB2 agonist, dose-dependently decreased alcohol consumption and preference (Al Mansouri et al., 2014). Moreover, JWH-015 reduced morphine-induced CPP and dopamine release in the nucleus accumbens, although mediation by cannabinoid CB2 receptors was not assessed (Grenald et al., 2017). Importantly, these studies suggest that CB2 agonists show no rewarding or aversive effects when administered by themselves.
CB2KO mice also exhibited robust morphine-induced CPP in our studies and CB2KO mice tended to exhibit higher morphine preference scores as compared to WT mice, although this difference did not reach statistical significance. These findings parallel previous studies which show that CB2KO mice present with higher levels of acquisition, preference, and consumption of ethanol compared with WT mice (Ortega-Alvaro et al., 2015). CB2KO mice also show similar levels of cocaine self-administration as WT mice (Xi et al., 2011; Zhang et al., 2014). Interestingly, CB2xP mice, which globally overexpress CB2, show decreased levels of cocaine self-administration (Aracil-Fernandez et al., 2012). CB2KO mice however, show lower levels of nicotine-induced reward compared to WT mice (Ignatowska-Jankowska et al., 2013; Navarrete et al., 2013).
Approximately 40-50% of patients treated with opioids develop constipation (Choi and Billings, 2002; Pappagallo, 2001). Whereas tolerance develops to antinociceptive effects of opioids, tolerance is reported to not develop to opioid-induced slowing of colonic transit (Bell et al., 2009). Our results show that LY2828360 did not alter the ability of repeated morphine treatment to inhibit colonic transit and fecal boli accumulation. Cannabinoids modulate gastrointestinal transit in rodents through activation of cannabinoid CB1 receptors in vivo, although cannabinoid CB2 receptors may be involved in pathophysiological conditions such as opioid-induced gastrointestinal inhibition and inflammatory bowel disease (Aviello et al., 2008; Wright et al., 2008). The selective CB2 agonist HU-308 inhibited fecal boli production in normal mice and this effect was attenuated by the CB2 antagonist SR144528 (Hanus et al., 1999). JWH-015 has also been shown to attenuate morphine-induced gastrointestinal slowing in rats (Grenald et al., 2017). In our study, acute LY2828360 treatment was associated with increased fecal boli accumulation compared to vehicle treatment but LY2828360 did not alter effects of morphine on colonic transit or fecal boli accumulation at any dose following acute or repeated morphine treatment. More work is necessary to determine whether the relative lack of effect of LY2828360 on morphine-induced slowing of colonic motility reflects its G protein-biased signaling profile.
LY2828360 did not produce acute antinociception or alter morphine antinociceptive tolerance in the hotplate test. Our studies of anti-allodynic synergy were evaluated in neuropathic mice whereas our assessments of acute hotplate antinociception were performed in otherwise normal (i.e. paclitaxel-naive) mice. Different stimulus modalities were tested in paclitaxel-treated (mechanical) and naive (heat) mice as LY2828360 did not alter mechanical responsiveness in the absence of neuropathic pain in our previous study (Lin et al., 2018). In other studies, AM1241 blocked morphine analgesic tolerance in the hotplate test in tumor bearing rats, but not in assessments of mechanical allodynia (Zhang et al., 2016). JWH-015 potentiated morphine antinociceptive tolerance in the tail-flick and hotplate test in otherwise naive morphine-tolerant rats, although mediation by CB2 was not evaluated (Altun et al., 2015). μ-opioid and cannabinoid CB2 receptor expression and function may be altered following the induction of neuropathic nociception (Bushlin et al., 2010; Zhang et al., 2003), which may in turn change the dynamics and time course of the development of opioid tolerance. Thus, CB2 agonists may diminish tolerance to morphine antinociception depending on their differing signaling profiles, differing pain states, and/or stimulus modalities tested.
Physical dependence is another major opioid-induced side effect, which can lead to a withdrawal syndrome when the user stops taking the drug (Koob et al., 1992). The opioid receptor antagonist naloxone precipitates a range of autonomic and somatic withdrawal signs in morphine-dependent animals (Morgan and Christie, 2011). Jumping (which presumably mimics escape attempts), weight loss and hypothermia are expressed in rodents and are indicative of supraspinal and spinal mediation of naloxone-induced morphine withdrawal (Laschka et al., 1976; Marshall and Buccafusco, 1985). In the present study, chronic LY2828360 treatment reduced, but did not eliminate, naloxone-precipitated withdrawal jumping behavior in morphine-pelleted mice. Due to the higher mortality rates exhibited by C57BL/6J mice following subcutaneous implantation with morphine pellets (Ramesh, 2012), effects of LY2828360 were not tested in CB2KO mice which were also bred onto a C57BL/6J background. Our results are in line with previous work from our laboratory that showed that chronic LY2828360 pre-treatment produced a trend toward reducing naloxone-precipitated withdrawal jumps in paclitaxel-treated WT mice but not in CB2KO mice (Lin et al., 2018). Similarly, the CB2 agonist AM1710 produced a robust suppression of naloxone-precipitated opioid withdrawal in paclitaxel-treated neuropathic mice (Li et al., 2019). However, when administered via an escalating i.p. morphine dosing schedule, LY2828360 did not attenuate naloxone precipitated withdrawal jumps in non-neuropathic mice. More work is necessary to determine how the presence of a pathological pain state impacts effectiveness of LY2828360 in attenuating naloxone-precipitated opioid withdrawal. Differing morphine pharmacokinetics and a pulsatile vs. continuous activation of μ-opioid receptors may also contribute to differences in efficacy of LY2828360 (Lefevre et al., 2020), and have clinical implications. Nonetheless, morphine-dependent (but paclitaxel-naïve) CB2KO mice consistently showed higher levels of naloxone-precipitated jumping compared with WT mice irrespective of the morphine dosing regimen, a finding that is in line with similar observations performed in paclitaxel-treated CB2KO mice (Lin et al., 2018). Stimulation of microglial cannabinoid CB2 receptors by CB2 agonists suppresses microglial activation (Ehrhart et al., 2005; Merighi et al., 2012; Tumati et al., 2012). In rodents, this microglial suppression has been linked to a decrease in opioid-induced withdrawal behaviors without affecting opioid antinociception (Burma et al., 2017). Alternatively, an upregulation of adenylyl cyclase has also been linked to mechanisms of opioid dependence (Bohn et al., 2000). This is noteworthy given that LY2828360 produced a delayed but prominent inhibition of adenylyl cyclase in vitro (Lin et al., 2018). However, more work is necessary to determine whether a CB2-mediated inhibition of adenylyl cyclase by CB2 agonists may counteract morphine-induced adenylyl cyclase upregulation and thereby attenuate morphine withdrawal. Our results highlight the clinical potential of multimodal therapies using CB2 agonists and may accelerate solutions to the current opioid epidemic.
Supplementary Material
Highlights.
LY2828360 is a slowly signaling G protein-biased cannabinoid CB2 receptor agonist
It synergizes with morphine to suppress allodynia in a neuropathic pain model
It blocks morphine-induced reward in wildtype but not CB2 knockout mice
It attenuates naloxone precipitated withdrawal in morphine-dependent mice
It does not alter morphine effects on acute nociception or colonic motility
Acknowledgements:
The authors wish to thank Shahin Saberi and Carmen A. Zavala for their expert assistance in scoring withdrawal jumps in mouse behavior videos under blinded conditions.
Support: This work is supported by the National Institutes of Health National Institute on Drug Abuse (NIDA) [Grants DA047858, DA041229 and DA042584 (A.G.H. and K.M.) and the National Cancer Institute [Grant CA200417 (A.G.H.)], an Indiana Addiction Grand Challenge Grant (to A.G.H. and J.D.C), a T32 NIDA pre-doctoral training grant DA024628 (R.A.S.) and the Harlan Scholars Research Program (V.I. and R.A.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: None of the authors report any conflicts of interest.
References
- Al Mansouri S, Ojha S, Al Maamari E, Al Ameri M, Nurulain SM, Bahi A, 2014. The cannabinoid receptor 2 agonist, beta-caryophyllene, reduced voluntary alcohol intake and attenuated ethanol-induced place preference and sensitivity in mice. Pharmacol Biochem Behav 124, 260–268. [DOI] [PubMed] [Google Scholar]
- Altun A, Yildirim K, Ozdemir E, Bagcivan I, Gursoy S, Durmus N, 2015. Attenuation of morphine antinociceptive tolerance by cannabinoid CB1 and CB2 receptor antagonists. J Physiol Sci 65, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aracil-Fernandez A, Trigo JM, Garcia-Gutierrez MS, Ortega-Alvaro A, Ternianov A, Navarro D, Robledo P, Berbel P, Maldonado R, Manzanares J, 2012. Decreased cocaine motor sensitization and self-administration in mice overexpressing cannabinoid CB(2) receptors. Neuropsychopharmacology 37, 1749–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviello G, Romano B, Izzo AA, 2008. Cannabinoids and gastrointestinal motility: animal and human studies. Eur Rev Med Pharmacol Sci 12 Suppl 1, 81–93. [PubMed] [Google Scholar]
- Ballantyne JC, Kalso E, Stannard C, 2016. WHO analgesic ladder: a good concept gone astray. BMJ 352, i20. [DOI] [PubMed] [Google Scholar]
- Bell TJ, Panchal SJ, Miaskowski C, Bolge SC, Milanova T, Williamson R, 2009. The prevalence, severity, and impact of opioid-induced bowel dysfunction: results of a US and European Patient Survey (PROBE 1). Pain Med 10, 35–42. [DOI] [PubMed] [Google Scholar]
- Beltramo M, Bernardini N, Bertorelli R, Campanella M, Nicolussi E, Fredduzzi S, Reggiani A, 2006. CB2 receptor-mediated antihyperalgesia: possible direct involvement of neural mechanisms. Eur J Neurosci 23, 1530–1538. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG, 2000. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408, 720–723. [DOI] [PubMed] [Google Scholar]
- Burma NE, Bonin RP, Leduc-Pessah H, Baimel C, Cairncross ZF, Mousseau M, Shankara JV, Stemkowski PL, Baimoukhametova D, Bains JS, Antle MC, Zamponi GW, Cahill CM, Borgland SL, De Koninck Y, Trang T, 2017. Blocking microglial pannexin-1 channels alleviates morphine withdrawal in rodents. Nat Med 23, 355–360. [DOI] [PubMed] [Google Scholar]
- Bushlin I, Rozenfeld R, Devi LA, 2010. Cannabinoid-opioid interactions during neuropathic pain and analgesia. Curr Opin Pharmacol 10, 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Billings JA, 2002. Opioid antagonists: a review of their role in palliative care, focusing on use in opioid-related constipation. J Pain Symptom Manage 24, 71–90. [DOI] [PubMed] [Google Scholar]
- Compton WM, Boyle M, Wargo E, 2015. Prescription opioid abuse: Problems and responses. Prev Med 80, 5–9. [DOI] [PubMed] [Google Scholar]
- Console-Bram LM, Zhao P, Abood ME, 2017. Protocols and Good Operating Practices in the Study of Cannabinoid Receptors. Methods Enzymol 593, 23–42. [DOI] [PubMed] [Google Scholar]
- Curry ZA, Wilkerson JL, Bagdas D, Kyte SL, Patel N, Donvito G, Mustafa MA, Poklis JL, Niphakis MJ, Hsu KL, Cravatt BF, Gewirtz DA, Damaj MI, Lichtman AH, 2018. Monoacylglycerol Lipase Inhibitors Reverse Paclitaxel-Induced Nociceptive Behavior and Proinflammatory Markers in a Mouse Model of Chemotherapy-Induced Neuropathy. J Pharmacol Exp Ther 366, 169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delis F, Polissidis A, Poulia N, Justinova Z, Nomikos GG, Goldberg SR, Antoniou K, 2017. Attenuation of Cocaine-Induced Conditioned Place Preference and Motor Activity via Cannabinoid CB2 Receptor Agonism and CB1 Receptor Antagonism in Rats. Int J Neuropsychopharmacol 20, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Guindon J, Cornett BL, Makriyannis A, Mackie K, Hohmann AG, 2015. Chronic cannabinoid receptor 2 activation reverses paclitaxel neuropathy without tolerance or cannabinoid receptor 1-dependent withdrawal. Biol Psychiatry 77, 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desroches J, Bouchard JF, Gendron L, Beaulieu P, 2014. Involvement of cannabinoid receptors in peripheral and spinal morphine analgesia. Neuroscience 261, 23–42. [DOI] [PubMed] [Google Scholar]
- Donvito G, Wilkerson JL, Damaj MI, Lichtman AH, 2016. Palmitoylethanolamide Reverses Paclitaxel-Induced Allodynia in Mice. J Pharmacol Exp Ther 359, 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, Klein T, Fernandez F, Tan J, Shytle RD, 2005. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J Neuroinflammation 2, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, 2011. The doctor's dilemma: opiate analgesics and chronic pain. Neuron 69, 591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friard O, Gamba M, 2016. BORIS: a free, versatile open-source event-logging software for video/audio coding and live observations. Methods in Ecology and Evolution 7, 1325–1330. [Google Scholar]
- Galiegue S, Mary S, Marchand J, Dussossoy D, Carriere D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P, 1995. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem 232, 54–61. [DOI] [PubMed] [Google Scholar]
- Grenald SA, Young MA, Wang Y, Ossipov MH, Ibrahim MM, Largent-Milnes TM, Vanderah TW, 2017. Synergistic attenuation of chronic pain using mu opioid and cannabinoid receptor 2 agonists. Neuropharmacology 116, 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG, 2008. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol 153, 319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez T, Crystal JD, Zvonok AM, Makriyannis A, Hohmann AG, 2011. Self-medication of a cannabinoid CB2 agonist in an animal model of neuropathic pain. Pain 152, 1976–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez T, Farthing JN, Zvonok AM, Makriyannis A, Hohmann AG, 2007. Activation of peripheral cannabinoid CB1 and CB2 receptors suppresses the maintenance of inflammatory nociception: a comparative analysis. Br J Pharmacol 150, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus L, Breuer A, Tchilibon S, Shiloah S, Goldenberg D, Horowitz M, Pertwee RG, Ross RA, Mechoulam R, Fride E, 1999. HU-308: a specific agonist for CB(2), a peripheral cannabinoid receptor. Proc Natl Acad Sci U S A 96, 14228–14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Farthing JN, Zvonok AM, Makriyannis A, 2004. Selective activation of cannabinoid CB2 receptors suppresses hyperalgesia evoked by intradermal capsaicin. J Pharmacol Exp Ther 308, 446–453. [DOI] [PubMed] [Google Scholar]
- Hollinshead SP, Tidwell MW, Palmer J, Guidetti R, Sanderson A, Johnson MP, Chambers MG, Oskins J, Stratford R, Astles PC, 2013. Selective cannabinoid receptor type 2 (CB2) agonists: optimization of a series of purines leading to the identification of a clinical candidate for the treatment of osteoarthritic pain. J Med Chem 56, 5722–5733. [DOI] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Muldoon PP, Lichtman AH, Damaj MI, 2013. The cannabinoid CB2 receptor is necessary for nicotine-conditioned place preference, but not other behavioral effects of nicotine in mice. Psychopharmacology (Berl) 229, 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamison RN, Mao J, 2015. Opioid Analgesics. Mayo Clinic proceedings 90, 957–968. [DOI] [PubMed] [Google Scholar]
- Jones D, 2008. End of the line for cannabinoid receptor 1 as an anti-obesity target? Nat Rev Drug Discov 7, 961–962. [DOI] [PubMed] [Google Scholar]
- Kinsey SG, Mahadevan A, Zhao B, Sun H, Naidu PS, Razdan RK, Selley DE, Imad Damaj M, Lichtman AH, 2011. The CB2 cannabinoid receptor-selective agonist O-3223 reduces pain and inflammation without apparent cannabinoid behavioral effects. Neuropharmacology 60, 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirilly E, Gonda X, Bagdy G, 2012. CB1 receptor antagonists: new discoveries leading to new perspectives. Acta Physiol (Oxf) 205, 41–60. [DOI] [PubMed] [Google Scholar]
- Koob GF, Maldonado R, Stinus L, 1992. Neural substrates of opiate withdrawal. Trends Neurosci 15, 186–191. [DOI] [PubMed] [Google Scholar]
- Laschka E, Teschemacher H, Mehraein P, Herz A, 1976. Sites of action of morphine involved in the development of physical dependence in rats. II. Morphine withdrawal precipitated by application of morphine antagonists into restricted parts of the ventricular system and by microinjection into various brain areas. Psychopharmacologia 46, 141–147. [DOI] [PubMed] [Google Scholar]
- Lefevre EM, Pisansky MT, Toddes C, Baruffaldi F, Pravetoni M, Tian L, Kono TJY, Rothwell PE, 2020. Interruption of continuous opioid exposure exacerbates drug-evoked adaptations in the mesolimbic dopamine system. Neuropsychopharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li AL, Carey LM, Mackie K, Hohmann AG, 2017. Cannabinoid CB2 Agonist GW405833 Suppresses Inflammatory and Neuropathic Pain through a CB1 Mechanism that is Independent of CB2 Receptors in Mice. J Pharmacol Exp Ther 362, 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li AL, Lin X, Dhopeshwarkar AS, Thomaz AC, Carey LM, Liu Y, Nikas SP, Makriyannis A, Mackie K, Hohmann AG, 2019. Cannabinoid CB2 Agonist AM1710 Differentially Suppresses Distinct Pathological Pain States and Attenuates Morphine Tolerance and Withdrawal. Mol Pharmacol 95, 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Sheikh SM, Loh HH, Martin BR, 2001. Opioid and cannabinoid modulation of precipitated withdrawal in delta(9)-tetrahydrocannabinol and morphine-dependent mice. J Pharmacol Exp Ther 298, 1007–1014. [PubMed] [Google Scholar]
- Lin X, Dhopeshwarkar AS, Huibregtse M, Mackie K, Hohmann AG, 2018. Slowly Signaling G Protein-Biased CB2 Cannabinoid Receptor Agonist LY2828360 Suppresses Neuropathic Pain with Sustained Efficacy and Attenuates Morphine Tolerance and Dependence. Mol Pharmacol 93, 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Coller JK, Watkins LR, Somogyi AA, Hutchinson MR, 2011. Naloxone-precipitated morphine withdrawal behavior and brain IL-1beta expression: comparison of different mouse strains. Brain Behav Immun 25, 1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN, 2005. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem 95, 437–445. [DOI] [PubMed] [Google Scholar]
- Marshall DC, Buccafusco JJ, 1985. Supraspinal and spinal mediation of naloxone-induced morphine withdrawal in rats. Brain Res 329, 131–142. [DOI] [PubMed] [Google Scholar]
- Mazzari S, Canella R, Petrelli L, Marcolongo G, Leon A, 1996. N-(2-hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur J Pharmacol 300, 227–236. [DOI] [PubMed] [Google Scholar]
- Merighi S, Gessi S, Varani K, Fazzi D, Mirandola P, Borea PA, 2012. Cannabinoid CB(2) receptor attenuates morphine-induced inflammatory responses in activated microglial cells. Br J Pharmacol 166, 2371–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MM, Christie MJ, 2011. Analysis of opioid efficacy, tolerance, addiction and dependence from cell culture to human. Br J Pharmacol 164, 1322–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nackley AG, Makriyannis A, Hohmann AG, 2003. Selective activation of cannabinoid CB2 receptors suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience 119, 747–757. [DOI] [PubMed] [Google Scholar]
- Nackley AG, Zvonok AM, Makriyannis A, Hohmann AG, 2004. Activation of cannabinoid CB2 receptors suppresses C-fiber responses and windup in spinal wide dynamic range neurons in the absence and presence of inflammation. J Neurophysiol 92, 3562–3574. [DOI] [PubMed] [Google Scholar]
- Navarrete F, Rodriguez-Arias M, Martin-Garcia E, Navarro D, Garcia-Gutierrez MS, Aguilar MA, Aracil-Fernandez A, Berbel P, Minarro J, Maldonado R, Manzanares J, 2013. Role of CB2 cannabinoid receptors in the rewarding, reinforcing, and physical effects of nicotine. Neuropsychopharmacology 38, 2515–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Alvaro A, Ternianov A, Aracil-Fernandez A, Navarrete F, Garcia-Gutierrez MS, Manzanares J, 2015. Role of cannabinoid CB2 receptor in the reinforcing actions of ethanol. Addict Biol 20, 43–55. [DOI] [PubMed] [Google Scholar]
- Pappagallo M, 2001. Incidence, prevalence, and management of opioid bowel dysfunction. Am J Surg 182, 11S–18S. [DOI] [PubMed] [Google Scholar]
- Paulozzi LJ, Budnitz DS, Xi Y, 2006. Increasing deaths from opioid analgesics in the United States. Pharmacoepidemiol Drug Saf 15, 618–627. [DOI] [PubMed] [Google Scholar]
- Pereira A, Chappell A, Dethy J, Hoeck H, Arendt-Nielsen L, Verfaille S, Boulanger B, Jullion A, Johnson M, McNearney T, 2013. A proof-of-concept (POC) study including experimental pain models (EPMs) to assess the effects of a CB2 agonist (LY2828360) in the treatment of patients with osteoarthritic (OA) knee pain, Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics, ASCPT. [Google Scholar]
- Perron BE, Bohnert K, Perone AK, Bonn-Miller MO, Ilgen M, 2015. Use of prescription pain medications among medical cannabis patients: comparisons of pain levels, functioning, and patterns of alcohol and other drug use. J Stud Alcohol Drugs 76, 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, 2001. Cannabinoid receptors and pain. Prog Neurobiol 63, 569–611. [DOI] [PubMed] [Google Scholar]
- Polomano RC, Mannes AJ, Clark US, Bennett GJ, 2001. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain 94, 293–304. [DOI] [PubMed] [Google Scholar]
- Raffa RB, Mathiasen JR, Jacoby HI, 1987. Colonic bead expulsion time in normal and mu-opioid receptor deficient (CXBK) mice following central (ICV) administration of mu- and delta-opioid agonists. Life Sci 41, 2229–2234. [DOI] [PubMed] [Google Scholar]
- Ramesh D, 2012. Elevating Endogenous Cannabinoids Reduces Opioid Withdrawal in Mice. [Google Scholar]
- Ramesh D, Ross GR, Schlosburg JE, Owens RA, Abdullah RA, Kinsey SG, Long JZ, Nomura DK, Sim-Selley LJ, Cravatt BF, Akbarali HI, Lichtman AH, 2011. Blockade of endocannabinoid hydrolytic enzymes attenuates precipitated opioid withdrawal symptoms in mice. J Pharmacol Exp Ther 339, 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Sandoval A, Eisenach JC, 2007. Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision. Anesthesiology 106, 787–794. [DOI] [PubMed] [Google Scholar]
- Sagar DR, Kelly S, Millns PJ, O’Shaughnessey CT, Kendall DA, Chapman V, 2005. Inhibitory effects of CB1 and CB2 receptor agonists on responses of DRG neurons and dorsal horn neurons in neuropathic rats. Eur J Neurosci 22, 371–379. [DOI] [PubMed] [Google Scholar]
- Schatz AR, Lee M, Condie RB, Pulaski JT, Kaminski NE, 1997. Cannabinoid receptors CB1 and CB2: a characterization of expression and adenylate cyclase modulation within the immune system. Toxicol Appl Pharmacol 142, 278–287. [DOI] [PubMed] [Google Scholar]
- Siau C, Xiao W, Bennett GJ, 2006. Paclitaxel- and vincristine-evoked painful peripheral neuropathies: loss of epidermal innervation and activation of Langerhans cells. Exp Neurol 201, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slivicki RA, Iyer V, Mali SS, Garai S, Thakur GA, Crystal JD, Hohmann AG, 2020. Positive Allosteric Modulation of CB1 Cannabinoid Receptor Signaling Enhances Morphine Antinociception and Attenuates Morphine Tolerance Without Enhancing Morphine- Induced Dependence or Reward. Frontiers in Molecular Neuroscience 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slivicki RA, Saberi SA, Iyer V, Vemuri VK, Makriyannis A, Hohmann AG, 2018. Brain-Permeant and -Impermeant Inhibitors of Fatty Acid Amide Hydrolase Synergize with the Opioid Analgesic Morphine to Suppress Chemotherapy-Induced Neuropathic Nociception Without Enhancing Effects of Morphine on Gastrointestinal Transit. J Pharmacol Exp Ther 367, 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slivicki RA, Xu Z, Kulkarni PM, Pertwee RG, Mackie K, Thakur GA, Hohmann AG, 2017. Positive Allosteric Modulation of Cannabinoid Receptor Type 1 Suppresses Pathological Pain Without Producing Tolerance or Dependence. Biol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallarida RJ, 2006. An overview of drug combination analysis with isobolograms. J Pharmacol Exp Ther 319, 1–7. [DOI] [PubMed] [Google Scholar]
- Trang T, Sutak M, Jhamandas K, 2007. Involvement of cannabinoid (CB1)-receptors in the development and maintenance of opioid tolerance. Neuroscience 146, 1275–1288. [DOI] [PubMed] [Google Scholar]
- Tumati S, Largent-Milnes TM, Keresztes A, Ren J, Roeske WR, Vanderah TW, Varga EV, 2012. Repeated morphine treatment-mediated hyperalgesia, allodynia and spinal glial activation are blocked by co-administration of a selective cannabinoid receptor type-2 agonist. J Neuroimmunol 244, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SJ, Ramirez MD, Neelakantan H, Walker EA, 2011. Cannabidiol prevents the development of cold and mechanical allodynia in paclitaxel-treated female C57Bl6 mice. Anesth Analg 113, 947–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson N, Kariisa M, Seth P, Smith H.t., Davis NL, 2020. Drug and Opioid-Involved Overdose Deaths - United States, 2017-2018. MMWR Morb Mortal Wkly Rep 69, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotherspoon G, Fox A, McIntyre P, Colley S, Bevan S, Winter J, 2005. Peripheral nerve injury induces cannabinoid receptor 2 protein expression in rat sensory neurons. Neuroscience 135, 235–245. [DOI] [PubMed] [Google Scholar]
- Wright KL, Duncan M, Sharkey KA, 2008. Cannabinoid CB2 receptors in the gastrointestinal tract: a regulatory system in states of inflammation. Br J Pharmacol 153, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi ZX, Peng XQ, Li X, Song R, Zhang HY, Liu QR, Yang HJ, Bi GH, Li J, Gardner EL, 2011. Brain cannabinoid CB(2) receptors modulate cocaine’s actions in mice. Nat Neurosci 14, 1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto W, Mikami T, Iwamura H, 2008. Involvement of central cannabinoid CB2 receptor in reducing mechanical allodynia in a mouse model of neuropathic pain. Eur J Pharmacol 583, 56–61. [DOI] [PubMed] [Google Scholar]
- Zhang HY, Gao M, Liu QR, Bi GH, Li X, Yang HJ, Gardner EL, Wu J, Xi ZX, 2014. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc Natl Acad Sci U S A 111, E5007–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HY, Gao M, Shen H, Bi GH, Yang HJ, Liu QR, Wu J, Gardner EL, Bonci A, Xi ZX, 2017. Expression of functional cannabinoid CB2 receptor in VTA dopamine neurons in rats. Addict Biol 22, 752–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hoffert C, Vu HK, Groblewski T, Ahmad S, O’Donnell D, 2003. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur J Neurosci 17, 2750–2754. [DOI] [PubMed] [Google Scholar]
- Zhang M, Wang K, Ma M, Tian S, Wei N, Wang G, 2016. Low-Dose Cannabinoid Type 2 Receptor Agonist Attenuates Tolerance to Repeated Morphine Administration via Regulating mu-Opioid Receptor Expression in Walker 256 Tumor-Bearing Rats. Anesth Analg 122, 1031–1037. [DOI] [PubMed] [Google Scholar]
- Zheng H, Xiao WH, Bennett GJ, 2011. Functional deficits in peripheral nerve mitochondria in rats with paclitaxel- and oxaliplatin-evoked painful peripheral neuropathy. Exp Neurol 232, 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, 1983. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 16, 109–110. [DOI] [PubMed] [Google Scholar]
- Slivicki RA, Iyer V, Mali SS, Garai S, Thakur GA, Crystal JD, Hohmann AG, 2020. Positive Allosteric Modulation of CB1 Cannabinoid Receptor Signaling Enhances Morphine Antinociception and Attenuates Morphine Tolerance Without Enhancing Morphine- Induced Dependence or Reward. Frontiers in Molecular Neuroscience 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.