

Abstract
Fungal secondary metabolites (SMs) are an important source of numerous bioactive compounds largely applied in the pharmaceutical industry, as in the production of antibiotics and anticancer medications. The discovery of novel fungal SMs can potentially benefit human health. Identifying biosynthetic gene clusters (BGCs) involved in the biosynthesis of SMs can be a costly and complex task, especially due to the genomic diversity of fungal BGCs. Previous studies on fungal BGC discovery present limited scope and can restrict the discovery of new BGCs. In this work, we introduce TOUCAN, a supervised learning framework for fungal BGC discovery. Unlike previous methods, TOUCAN is capable of predicting BGCs on amino acid sequences, facilitating its use on newly sequenced and not yet curated data. It relies on three main pillars: rigorous selection of datasets by BGC experts; combination of functional, evolutionary and compositional features coupled with outperforming classifiers; and robust post-processing methods. TOUCAN best-performing model yields 0.982 F-measure on BGC regions in the Aspergillus niger genome. Overall results show that TOUCAN outperforms previous approaches. TOUCAN focuses on fungal BGCs but can be easily adapted to expand its scope to process other species or include new features.
INTRODUCTION
Secondary metabolites (SMs) are specialized bioactive compounds primarily produced by plants, fungi and bacteria. They represent a vital source for drug discovery: from anticancer, antiviral and cholesterol-lowering medications to antibiotics and immunosuppressants (1). Genes involved in the biosynthesis of many SMs in fungi are co-localized in the genome, organized as clusters of genes (2), and known as biosynthetic gene clusters (BGCs). Typically, BGCs are minimally composed of one or more synthase or synthetase genes encoding backbone enzymes, which produce the core structure of the compound, and genes that encode tailoring enzymes, which modify the core compound to generate variants (3). Backbone enzymes determine the class of SM produced by a BGC. BGCs may also contain other genes such as those encoding cluster-specific transcription factors, mitigating toxic properties, transporters, tailoring enzymes and genes with hypothetical functions (4). Identifying new fungal BGCs can potentially lead to the discovery of new compounds that can serve as vital source for drug discovery (5,6). Despite the availability of a large volume of fungal genome sequence data, BGC discovery remains a challenging task (1) due to the diversity of fungal BGCs. Fungal BGCs have been shown to present noticeable differences in synteny and non-conservation of sequences even in related species or different strains of the same species (3), where clustered genes of the same SM can appear in different scaffolds among evolutionarily close species.
Several studies have presented approaches to discover BGCs (1). Most approaches to identify fungal BGCs rely on probabilistic or data-driven methods, requiring as input genomic data (7) combined with gene functional annotations (8) and/or transcription data (9,10). Previous works also analysed fungal gene expression levels (9), motif co-occurrence in promoters around anchor genes (containing backbone enzymes) (8), compared expression levels of virtual gene clusters in conditions favourable to SM production (10) and analysed homologous genes through sequence alignment and filtering syntenic blocks (7). fungiSMASH (11) combines a probabilistic method (profile hidden Markov models from proteins) and curated BGC detection rules, and can use tools such as CASSIS (cluster assignment by islands of sites) (8) and ClusterFinder (12) to predict fungal BGC boundaries. These previous approaches present several limitations: overprediction of BGC length (11,13); dependence on manual curation (9), which is expensive; or a very limited scope, potentially affecting the ability to process different BGC types or organisms (7,13).
Approaches derived from supervised learning have shown to perform well when predicting bacterial BGCs (14,15). To our knowledge, such methods have not been applied to identifying fungal BGCs. For instance, RiPPMiner (14) based on support vector machine (SVM) and random forest achieves 0.91 F-measure (F-m) in binary classification of ribosomally synthesized and post-translationally modified peptides. A recent approach, called DeepBGC, was designed to exploit Pfam (16) domain embeddings to represent bacterial BGCs (12) to feed a bidirectional long short-term memory (BiLSTM) neural network (15). DeepBGC relies also on post-processing methods such as merging consecutive BGC genes or filtering regions without known BGC protein domains. DeepBGC achieved a 0.923 area under the curve when predicting BGC positions in a set of 65 experimentally validated BGCs from six bacterial genomes, outperforming previous studies (15). When handling fungal BGC data, DeepBGC in its original version yielded performance no higher than 0.2 F-m (17), and when trained on fungal data underperformed previous methods such as fungiSMASH (11), as we show in the ‘Results’ section. This could indicate that BGC discovery methods developed for bacteria may not be suitable for fungi due to the high diversity of fungal BGCs that are found to vary even among closely related species (3). Hence, it is important to develop BGC discovery approaches dedicated to fungi, taking into account the specific characteristics of fungal BGCs, such as high diversity, BGC components, and BGC and genome lengths that are usually longer than bacteria. Here, we propose TOUCAN, a supervised learning framework to tackle BGC discovery in fungi that is based on a combination of heterogeneous biological feature types: k-mers, protein domains and Gene Ontology (terms) to represent protein motifs and functions relevant to fungal BGCs.
MATERIALS AND METHODS
TOUCAN classification models were built based on a set of six open-access fungal BGC datasets of varying distributions, a total of six classifiers and two post-processing methods. In this section, we present the methodology adopted to develop TOUCAN models. TOUCAN predictions are validated based on a set of curated fungal BGCs.
Datasets
TOUCAN classification models were developed with comprehensive and exhaustive fungal BGC datasets presented in (17) that are publicly available to support benchmarking of BGC discovery methods. The six fungal BGC training datasets are composed of different distributions of positive instances obtained from the MIBiG (Minimum Information about a Biosynthetic Gene cluster) (2) repository and synthetic negative instances generated from OrthoDB (18) orthologues. Fungal orthologous genes were previously applied in BGC discovery (7). Orthologues can be a relevant source of negative instances since they represent conserved genes across species, while BGCs are known to show large genomic diversity even in closely related species (3).
To build negative instances, the amino acid sequences of OrthoDB fungal orthologous genes were concatenated using a fixed window size of 7000 amino acids, which corresponds to the average amino acid length of all positive instances from the fungal subset in MIBiG. This process generated a pool of training samples of 693 195 synthetic negative clusters [see (17) for details]. Studying datasets of various distributions could shed light on the impact of class imbalance in fungal BGC discovery, which by nature presents a highly imbalanced scenario where only a small fraction of fungal genomes actually corresponded to BGCs (17). To account for genomic diversity in fungi, positive instances in the six datasets represent >10 different BGC types and >100 fungal species, while negative instances were generated from a pool of orthologous genes representing ≈300 fungal species. To build and validate our models, we performed a random fixed split in each training dataset for which 80% of instances are dedicated to training and 20% for validation. Supplementary Table S1 shows the positive versus negative distribution, and the training and validation splits in the six training fungal BGC datasets. A random fixed split allows us to evaluate the performances of the same training and validation sets under different parameters.
In the test phase, we evaluated our classification models with six test datasets, generated similarly to (17), from a manually curated genome sequence of Aspergillus niger NRRL3, available at https://gb.fungalgenomics.ca/portal. Aspergillus niger is an organism of interest for BGC discovery due to its relevance to industrial processes, and its ubiquitous distribution (6). In this work, 85 manually curated BGCs (19) in A. niger will be considered as gold standard. Test candidate BGCs are generated by sequentially extracted genomic regions of A. niger with a sliding window of 5000, 7000 or 10 000 amino acids, with a 30% or 50% overlap. The overlap of genomic regions allows us to cover BGC fragmented by the sliding windows. Multiple test datasets allow to analyse the impact of window lengths and overlaps when handling input data of test organisms, helping to determine recommended parameters to obtain BGC predictions in new genome sequences. By generating test candidates based on a fixed sliding window length, new sequence data can be processed without requiring curation, genome annotation or gene models as input, unlike that of other BGC discovery tools. In the ‘Results’ section, we report the performance obtained by the models using different window lengths and overlaps.
Features
To represent the fungal BGC dataset instances as feature vectors, we relied on heterogeneous biological features extracted from the protein sequences of dataset instances: k-mers, Pfam protein domains and GO terms. Several feature types are combined to better represent the diverse genomic profiles in fungal BGCs and help build relevant discriminative models. Feature vectors are composed of number of occurrences of features per training instance. K-mers (a contiguous number of K amino acids appearing sequentially) are common features in genomic classification tasks (20). We have extracted k-mers with varying lengths of 3 ≤ K ≤ 9. K-mers appearing less than three times were discarded to reduce feature dimensionality, because presence of rare features could introduce bias (21). K-mer lengths were evaluated separately using validation sets to identify the K value yielding the best performance. Further details on validation of K values are provided in the ‘Results’ section.
Pfam protein domains were previously applied in BGC discovery in both fungi (13) and bacteria (12,15). Protein domains are relevant features for BGC classification and can indicate the presence of backbone enzymes, a key component of BGCs (3,19). We performed an analysis of protein domain distribution among positive instances in our datasets to understand their relevance as features. In our analysis, Pfam protein domains extracted from positive instances were manually labelled by us as high (corresponding to a domain usually only present in BGCs) and medium (a domain usually present in, but not limited to, BGCs). The complete lists of medium and high annotated Pfam domains are presented in Supplementary Tables S2 and S3. Then, we analysed all positive instance datasets for the presence or absence of such domains, shown in Supplementary Figure S1. This analysis highlights two important aspects: first, the protein domain diversity in fungal BGCs; and second, the presence of high domains shared by most BGCs suggesting that they share a structural pattern, most likely related to the presence of a backbone enzyme. The structural pattern yielded by the distribution of manually annotated protein domains in positive instances suggests that this feature type might carry an important discriminating power. Pfam domain features were extracted from our training datasets using the Pfam database.
GO term annotations were also modelled as features and obtained from our training instances using Swiss-Prot (22). To identify corresponding GO terms, we performed a BLAST analysis of amino acid sequences from our dataset instances against the Swiss-Prot database composed of 560 292 reviewed entries (as of June 2019). BLAST parameters considered were evalue (expected value) ≤ 1e−4 and qcovs (query coverage per subject) ≥ 50. A qcovs ≥ 50 could indicate relevant sequence similarity (23), since the alignment length would correspond to at least 50% of 7000 amino acids for each match. We considered GO terms from all classes. GO term matches found were filtered for duplicates, and only unique GO terms were kept to represent dataset instances.
The number of unique features per type extracted from each training dataset and used to build our classification models is shown in Supplementary Table S4. At this point, extracted features were all kept (except for k-mers that occur less than three times in a dataset), without relying on feature selection methods. The feature order is not necessarily conserved during classification, and it is by all purposes processed in a bag-of-words manner. Consider that all extracted features can be relevant at this point since the experiments performed in our work are still a learning space of suitable parameters to tackle BGC discovery. Feature selection could therefore limit the exploration of potentially relevant attributes or combinations of features, but it might be valuable as the next step.
Classification methods
TOUCAN classification models were built with a total of six classifiers. We performed experiments with different classification algorithms to assess the performance of heterogeneous features and post-processing methods, and then identified the best configuration to tackle the BGC discovery task. Three classifiers were SVM classifiers: C support vector (svc), linear support vector (lsvc) and nu support vector (nusvc) classifiers. SVM classifiers were previously applied in BGC discovery (14). Default parameters were used for svc and lsvc during experiments, while for the nusvc classifier the nu parameter was adjusted in connection with the percentage of positive instances pos in a given dataset:
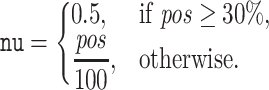 |
(1) |
The other three classifiers were a multilayer perceptron (mlp), logistic regression (logit) and random forest (randomf). While logit classifier can provide a baseline model for the task, neural networks (15) and randomf (14) were also previously applied in BGC discovery. Also for mlp, logit and randomf, default parameters were kept but could, however, be optimized to suit specific experiments if needed. These six classifiers were evaluated independently during our experiments.
Post-processing methods
Predictions of candidate BGCs outputted by TOUCAN are post-processed to improve output precision. Post-processing methods adopted in our work were greedy approaches, such as in PRISM (24) that identifies bond-forming domains and expands cluster boundaries on either ends of such domains. Unlike PRISM, TOUCAN does not require curation as input, and relies on classification models to identify potential BGC regions in which post-processing methods can be applied, facilitating its use on newly sequenced or not yet annotated genomes. The post-processing methods succ and merge are shown in the SuccessiveMerge algorithm, and aim to address potential cluster boundary limitations (over- or underestimation) common in previous approaches (11,13).
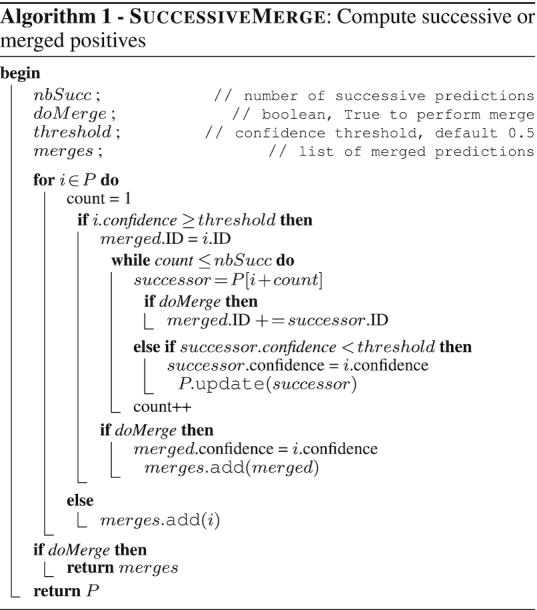
BGC region length can vary greatly among fungal MIBiG BGCs: for an x number of amino acids, x can vary such as 195 ≤ x ≤ 62 079, with a standard deviation σ(x) ≈ 6013.73 and mean 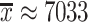 . In this work, a fixed amino acid length to generate test candidate instances from an organism genome is applied. Both succ and merge post-processing help to overcome the shortcoming in cases where cluster regions have limited boundaries. The succ post-processing gives to an nbSucc of successive predictions the same confidence prediction score of a positive prediction (confidence ≥ threshold). The merge post-processing merges an nbSucc of successive predictions of a positive prediction (confidence ≥ threshold) into a single positive prediction. For both succ and merge, we considered 0 ≤ nbSucc ≤ 3, set as an arbitrary parameter for the first evaluation of post-processing methods. Both post-processing methods were applied only if nbSucc successive predictions were also not positive.
. In this work, a fixed amino acid length to generate test candidate instances from an organism genome is applied. Both succ and merge post-processing help to overcome the shortcoming in cases where cluster regions have limited boundaries. The succ post-processing gives to an nbSucc of successive predictions the same confidence prediction score of a positive prediction (confidence ≥ threshold). The merge post-processing merges an nbSucc of successive predictions of a positive prediction (confidence ≥ threshold) into a single positive prediction. For both succ and merge, we considered 0 ≤ nbSucc ≤ 3, set as an arbitrary parameter for the first evaluation of post-processing methods. Both post-processing methods were applied only if nbSucc successive predictions were also not positive.
Evaluation metrics
TOUCAN classification models were assessed in terms of precision (P), recall (R), F-m and a clusterScore metric. To compute P, R and F-m, we considered as true positives (TPs) BGC candidates predicted as positive that have at least one gene that matches a gold standard BGC. The clusterScore represents the coverage of expected gold standard BGC genes within a candidate BGC, where 0 ≤ clusterScore ≤ 1, and was computed for each BGC candidate predicted positive. To compute the clusterScore for a BGC candidate C and its gold standard BGC match M, we first counted the number of geneMatches in C, meaning the number of M genes in C. We then computed a similarity value sim between all pairs of genes in the disjunctive union C▵M, and add to the clusterScore the best sim obtained for the unmatched M − C genes. Computing the sim value allows us to account for the possible presence of gold standard orthologues among unmatched genes in a BGC candidate predicted positive. The sim value represents a percent identity pident obtained through a local alignment with BLAST between two genes, using cut-offs of minimum pident ≥ 20 and minimum query coverage qcovs ≥ 10. The clusterScore for a BGC candidate C was normalized by the number of genes in its gold standard BGC match M. The Similarity algorithm shows the computation of sim scores, while the ClusterScore algorithm shows the computation of clusterScore. We analysed the clusterScore of TOUCAN predicted positives compared to state-of-the-art methods in the ‘Results’ section.
State-of-the-art performance comparison
The performance of TOUCAN models was compared to results obtained by two state-of-the-art tools: fungiSMASH (11) and DeepBGC (15) (version 0.1.18 and models as of February 2020 available at https://github.com/Merck/deepbgc). The experiments with fungiSMASM were performed with its three strictness levels: relaxed, strict and loose, and with default parameters for its extra feature options (as of January 2020): ‘KnownClusterBlast’, ‘ActiveSiteFinder’ and ‘SubClusterBlast’. DeepBGC focuses on bacterial data and is based on a BiLSTM neural network and Pfam domain embeddings. A total of three DeepBGC classification models are applied in this work: one with original DeepBGC training dataset and hyperparameters, as in (15); one built with DeepBGC original hyperparameters and our best-performing training dataset; and one built with our best-performing training dataset and fungal optimized hyperparameters (thanks to the authors) (see Supplementary Table S7 for original and fungal optimized hyperparameters).


RESULTS
We present here results obtained with TOUCAN, a supervised learning framework to discover fungal BGCs. To identify the best configuration to tackle BGC discovery in fungi, we designed, trained and assessed several classification models combining heterogeneous biological features, datasets of various distributions, classifiers and post-processing methods, as described in the ‘Materials and Methods’ section. Validation results are drawn on held-out training instances corresponding to 20% of each training dataset. The performance of TOUCAN was assessed on test datasets of a gold standard of 85 manually annotated A. niger BGCs (19). The focus here is BGC discovery; hence, the model is optimized to correctly identify positive instances, rather than the negative ones. Thus, results were reported for the positive class.
Feature importance and performance on validation datasets
To identify the most suitable K for k-mer features within 3 ≤ K ≤ 9, we performed a set of experiments on all six datasets and six classifiers, as presented in the ‘Datasets’ and ‘Classification methods’ sections. Performance of k-mer models on our validation sets is shown in Supplementary Figure S2. In general, better performance was achieved with K = 6, which was thus the K value considered for our following experiments. We also performed an analysis of feature importance across training datasets, obtained with a randomf classifier, with default parameters. Table 1 shows the top 15 ranked features across training datasets.
Table 1.
Top 15 features ranked by importance for each training dataset, from completely balanced (50% positive, 50% negative) to most imbalanced (5% positive, 95% negative)
| Training dataset distribution | |||||
|---|---|---|---|---|---|
| 50%–50% | 40%–60% | 30%–70% | 20%–80% | 10%–90% | 5%–95% |
| PF00698.21 | PF00698.21 | GO:0008168 | HGTGTQ | PF00109.26 | TACSSS |
| PF00668.20 | HGTGTQ | HGTGTQ | GO:0008152 | GO:0044550 | GTGTQA |
| ADGYCR | GO:0031177 | GQGAQW | PF00550.25 | LYRTGD | GYARGE |
| GO:0016491 | GAGTGG | GYCRAD | IDTACS | VFTGQG | GO:0046148 |
| FDGYRF | VEMHGT | GAGTGG | PF02458.15 | NFSAAG | TGDLAR |
| GO:0016740 | VFTGQG | QQRLLL | DTACSS | VEAHGT | SINSFG |
| MHGTGT | PF00668.20 | TACSSS | VTLSGD | GO:0043041 | DPQQRL |
| DTACSS | GO:0016874 | PF02801.22 | FTGQGA | GHSLGE | LFTSGS |
| GO:1900557 | YKTGDL | GO:0009058 | PF08242.12 | AYEALE | NSFGFG |
| GO:0009058 | GO:0019184 | GO:0046148 | AYGPTE | GO:0016491 | CDTAVA |
| GRFFAA | GO:0043042 | GO:0047462 | GO:0004315 | TQVKIR | FDASFF |
| PF14765.6 | PGRFFA | GEYAAL | GO:0031177 | GO:0046500 | AYGPTE |
| MDPQQR | MHGTGT | GO:0005829 | KLRGFR | DTACSS | YILFTS |
| FTSGST | GO:1900790 | PFAFHS | GO:0016021 | GO:0032259 | AIVLAG |
| GQGAQW | VEIGPH | LHSLEA | PF00067.22 | DTFVRC | AVVGHS |
Highlighted features appeared in multiple datasets.
Features appearing on the top 15 of multiple datasets are highlighted. Protein domain feature names start with PF, GO term feature names start with GO and the other features are 6-mers. We can observe that every protein domain feature appearing among the top ranked of all datasets belonged to either the high or medium manually annotated domains, even though non-high and non-medium domain features are also included in our feature set. Moreover, while GO terms represent 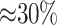 of all top 15 ranked features, they make up for at most 0.7% of total features. This possibly indicates their strong discriminating power in the task. After evaluating feature importance, we trained several classification models combining the feature types for each classifier and training dataset distribution. For each training dataset distribution, a random fixed split, designating 80% of its instances, was selected for training and 20% for validation, as mentioned in the ‘Datasets’ section. The top F-m performances on validation sets per training dataset are shown in Supplementary Table S5. During validation, we noted that models built with three feature types outperformed models using one feature type, such as the ones built when evaluating the most suitable k-mer length.
of all top 15 ranked features, they make up for at most 0.7% of total features. This possibly indicates their strong discriminating power in the task. After evaluating feature importance, we trained several classification models combining the feature types for each classifier and training dataset distribution. For each training dataset distribution, a random fixed split, designating 80% of its instances, was selected for training and 20% for validation, as mentioned in the ‘Datasets’ section. The top F-m performances on validation sets per training dataset are shown in Supplementary Table S5. During validation, we noted that models built with three feature types outperformed models using one feature type, such as the ones built when evaluating the most suitable k-mer length.
Validation performance seems to be overall affected by the instance distribution: more imbalanced datasets show lower F-m compared to more balanced ones. When analysing MIBiG fungal BGCs, only ≈1% of a genome sequence would correspond to cluster regions (17), so utilizing more balanced training data could provide better performance than using real case scenario distributions. We selected the dataset with the best F-m average performance, which was the most balanced (50%–50%), to perform further evaluation with hyperparameter optimization through a grid search, followed by cross-validation (CV) classification for all six classifiers. Best-performing hyperparameters to maximize F-m for each classifier are listed in Supplementary Table S8. A 5-fold CV was performed with optimized hyperparameters on the 50%–50% dataset instances, randomly split between training and validation at each fold. Supplementary Table S6 shows the average performances on the 5-fold CV for each classifier.
TOUCAN performance on test datasets
We assessed TOUCAN models on six test datasets with amino acid sliding window lengths of 5000, 7000 and 10 000, with overlaps of 50% and 30%, as described in the ‘Datasets’ section. Candidate BGC predictions on the test data were obtained with TOUCAN classification models built using the six training dataset distributions with fixed training and validation splits, three feature types and six classifiers. We then processed TOUCAN predicted candidate BGCs with post-processing methods succ and merge, considering 0 ≤ nbSucc ≤ 3.
Table 2 shows for the positive class the best F-m obtained for each test dataset among all training dataset distributions. The highest 0.931 F-m was obtained by a model built with a 50%–50% distributed training set, an mlp classifier and a merge3 post-processing. The best F-m was achieved with 10 000-amino acid sliding window test datasets. Regarding classifiers, mlp and logit yielded best performance followed less often by lsvc. As mentioned in the ‘Classification methods’ section, default parameters were used when performing our experiments. Tuning the classifier parameters may affect the performance, but this is not the focus of this study. Overall results showed that a 30% overlap seems to be more advantageous for all sliding window lengths, even though the best F-m was achieved with test candidates generated based on a 50% overlap. The training set distribution seemed to have little influence on test candidates with a sliding window length of 5000 amino acids, showing only a small variation on F-m for both 30% and 50% overlap. Less balanced training distribution seemed to affect performance more for candidates with a sliding window length of 10 000 amino acids, with an F-m varying from 0.618 to 0.931 when using 50% overlap, and from 0.629 to 0.917 when using 30% overlap.
Table 2.
TOUCAN best-performing models per test set sliding windows and overlaps in A. niger
| Sliding | Training | Post- | |||||
|---|---|---|---|---|---|---|---|
| window | Overlap | set | Classifier | process | P | R | F-m |
| 10 000 | 50% | 50%–50% | mlp | merge3 | 1 | 0.871 | 0.931 |
| 10 000 | 50% | 40%–60% | mlp | merge3 | 1 | 0.753 | 0.859 |
| 10 000 | 50% | 30%–70% | mlp | merge2 | 1 | 0.706 | 0.828 |
| 10 000 | 50% | 20%–80% | mlp | merge2 | 1 | 0.706 | 0.828 |
| 10 000 | 50% | 10%–90% | mlp | merge3 | 1 | 0.647 | 0.786 |
| 10 000 | 50% | 5%–95% | mlp | merge3 | 1 | 0.447 | 0.618 |
| 7000 | 50% | 50%–50% | logit | merge3 | 0.929 | 0.765 | 0.839 |
| 7000 | 50% | 40%–60% | logit | merge3 | 1 | 0.741 | 0.851 |
| 7000 | 50% | 30%–70% | mlp | merge3 | 0.969 | 0.729 | 0.832 |
| 7000 | 50% | 20%–80% | mlp | merge3 | 1 | 0.741 | 0.851 |
| 7000 | 50% | 10%–90% | mlp | merge3 | 1 | 0.694 | 0.819 |
| 7000 | 50% | 5%–95% | mlp | merge3 | 1 | 0.647 | 0.786 |
| 5000 | 50% | 50%–50% | logit | merge3 | 0.817 | 0.788 | 0.802 |
| 5000 | 50% | 40%–60% | logit | merge3 | 0.914 | 0.753 | 0.826 |
| 5000 | 50% | 30%–70% | logit | merge3 | 0.953 | 0.718 | 0.819 |
| 5000 | 50% | 20%–80% | logit | merge3 | 1 | 0.718 | 0.836 |
| 5000 | 50% | 10%–90% | mlp | merge3 | 0.913 | 0.741 | 0.818 |
| 5000 | 50% | 5%–95% | mlp | merge3 | 0.923 | 0.706 | 0.800 |
| 10 000 | 30% | 50%–50% | mlp | merge3 | 1 | 0.847 | 0.917 |
| 10 000 | 30% | 40%–60% | mlp | merge3 | 1 | 0.741 | 0.851 |
| 10 000 | 30% | 30%–70% | mlp | merge2 | 1 | 0.694 | 0.819 |
| 10 000 | 30% | 20%–80% | mlp | merge2 | 1 | 0.671 | 0.803 |
| 10 000 | 30% | 10%–90% | mlp | merge3 | 1 | 0.6 | 0.750 |
| 10 000 | 30% | 5%–95% | mlp | merge3 | 1 | 0.459 | 0.629 |
| 7000 | 30% | 50%–50% | mlp | merge3 | 0.95 | 0.906 | 0.928 |
| 7000 | 30% | 40%–60% | mlp | merge3 | 1 | 0.824 | 0.903 |
| 7000 | 30% | 30%–70% | mlp | merge2 | 1 | 0.741 | 0.851 |
| 7000 | 30% | 20%–80% | mlp | merge3 | 1 | 0.741 | 0.851 |
| 7000 | 30% | 10%–90% | lsvc | merge3 | 1 | 0.553 | 0.712 |
| 7000 | 30% | 5%–95% | mlp | merge3 | 1 | 0.635 | 0.777 |
| 5000 | 30% | 50%–50% | logit | merge3 | 0.908 | 0.812 | 0.857 |
| 5000 | 30% | 40%–60% | logit | merge3 | 0.985 | 0.788 | 0.876 |
| 5000 | 30% | 30%–70% | logit | merge3 | 1 | 0.753 | 0.859 |
| 5000 | 30% | 20%–80% | mlp | merge3 | 0.985 | 0.776 | 0.868 |
| 5000 | 30% | 10%–90% | mlp | merge3 | 0.984 | 0.729 | 0.838 |
| 5000 | 30% | 5%–95% | mlp | merge3 | 1 | 0.706 | 0.828 |
We selected the best-performing test datasets (10 000-amino acid sliding window) to carry an evaluation using 5-fold CV classification models based on the best-performing training set (50%–50%). The predicted BGC candidates obtained with CV classification models were also processed with TOUCAN post-processing methods succ and merge, in the same manner as the models presented in Table 2. The best performance results obtained with the 10 000-amino acid sliding window test data among all 5-fold CV classification models are shown in Table 3.
Table 3.
TOUCAN best performances for the completely balanced (50% positive, 50% negative) CV models on A. niger test sets generated with a 10 000-amino acid sliding window
| Training | Sliding | Post- | |||||
|---|---|---|---|---|---|---|---|
| set | window | Overlap | Classifier | process | P | R | F-m |
| 50%–50% | 10 000 | 50% | svc | merge3 | 0.941 | 0.941 | 0.941 |
| 50%–50% | 10 000 | 30% | randomf | merge3 | 1 | 0.965 | 0.982 |
As shown in Table 3, the 5-fold CV classification models improved to a 0.982 F-m from the previously best 0.931 F-m achieved with models based on fixed training and validation splits. Performance results in Tables 2 and 3 show TOUCAN models’ discriminative power to identify candidate BGC regions from non-BGC regions. Our results also demonstrate TOUCAN models’ capacity of obtaining relevant BGC predictions on new or non-annotated genomes in test dataset instances generated solely based on sliding windows of fixed amino acid length. This aspect distinguishes TOUCAN from previous approaches that rely on gene models and other genomic annotations as input (14,15).
Performance comparison with DeepBGC
We compared the performance of three DeepBGC classification models using the 10 000-amino acid sliding window test datasets, which yield the best F-m with TOUCAN. As mentioned in the ‘Materials and Methods’ section, two out of the three DeepBGC models were trained using the best-performing constructed training dataset (50%–50% dataset in this case). The DeepBGC hyperparameters applied in this comparison are also listed in the ‘Materials and Methods’ section. As shown in (17), during validation phase the DeepBGC model trained using the original hyperparameters and the 50%–50% training dataset had early stopping at epoch 109, from the original total of 328 epochs, as applied in (15).
Table 4 shows P, R and F-m performances of the three DeepBGC models for the positive class on the test dataset with a 50% or 30% overlap. DeepBGC models built with original hyperparameters yielded high recall but very low precision, consequently leading to F-m metrics <0.3 for either models based on the 50%–50% training set or models based on DeepBGC original data. Models built with fungal optimized hyperparameters yielded a noticeable performance improvement, with a 0.627 F-m.
Table 4.
Performance metrics of DeepBGC models for A. niger test sets generated with 10 000-amino acid sliding window
| Training | DeepBGC | Sliding | ||||
|---|---|---|---|---|---|---|
| dataset | model | window | Overlap | P | R | F-m |
| DeepBGC | Original | 10 000 | 50% | 0.114 | 1 | 0.205 |
| DeepBGC | Original | 10 000 | 30% | 0.159 | 1 | 0.274 |
| 50%–50% | Original | 10 000 | 50% | 0.075 | 1 | 0.140 |
| 50%–50% | Original | 10 000 | 30% | 0.105 | 1 | 0.191 |
| 50%–50% | Fungal | 10 000 | 50% | 0.464 | 0.765 | 0.578 |
| 50%–50% | Fungal | 10 000 | 30% | 0.580 | 0.682 | 0.627 |
For each of the three DeepBGC models, the test sets using a 30% overlap resulted in better performance than the ones using a 50% overlap. DeepBGC performance on predicting fungal BGCs shows high recall but very low precision, which consequently lead to F-m metrics <0.2. The most imbalanced models classified all test candidates as negative, which could be a sign of the model trying to optimize accuracy towards the majority class. Originally, DeepBGC was developed to predict bacterial BGCs, for which much more data are available compared to fungal BGCs. The larger amount of bacterial BGC data available benefits the development of supervised learning approaches. Fungal BGC data are more scarce, which makes it challenging to build robust classification models. Supervised learning approaches that fit bacteria may not be suitable to discover BGCs in fungi (17).
Performance comparison with fungiSMASH
We compared the performance of fungiSMASH on the same 10 000-amino acid sliding window test datasets used to compare with DeepBGC. The fungiSMASH parameters considered in this comparison are described in the ‘Materials and Methods’ section. fungiSMASH predictions are also assessed in terms of P, R and F-m, which are shown for the positive class in Table 5. fungiSMASH best performance yielded a 0.571 F-m when using a 50% overlap and 0.692 F-m when using a 30% overlap, both under relaxed strictness. As expected, loose strictness results in higher recall and lower precision, while a strict parameter results in higher precision but lower recall.
Table 5.
Performance metrics of fungiSMASH models for A. niger test sets generated with 10 000-amino acid sliding window
| fungiSMASH | Sliding | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| strictness | window | Overlap | P | R | F-m | Overlap | P | R | F-m |
| Relaxed (default) | 10 000 | 50% | 0.470 | 0.729 | 0.571 | 30% | 0.649 | 0.741 | 0.692 |
| Strict | 10 000 | 50% | 0.471 | 0.576 | 0.519 | 30% | 0.671 | 0.600 | 0.634 |
| Loose | 10 000 | 50% | 0.435 | 0.788 | 0.561 | 30% | 0.591 | 0.800 | 0.68 |
Similar to TOUCAN models, fungiSMASH seems to yield generally better performance on 30% overlap test candidates. fungiSMASH showed in general a more stable performance predicting fungal BGCs compared to DeepBGC. Apart from being based on a different approach than DeepBGC, fungiSMASH was developed focusing on fungal organisms. The difference in performance between the bacteria-focused approach of DeepBGC and the fungal-focused approach of fungiSMASH may be another indication that BGC discovery is a complex task, and can benefit from approaches built to target related organisms.
TOUCAN yields reproducible performance on Aspergillus nidulans
To assess TOUCAN reproducibility, we assessed the performance of its models in the A. nidulans genome. As in A. niger, A. nidulans is a species known as an important source of BGCs (3,19). Previous work on manual annotation of BGCs in Aspergilli (19) identified a total of 70 BGCs in A. nidulans, which are considered as gold standard for this analysis. To obtain candidate BGCs for testing, A. nidulans genome sequence was processed in the same manner as A. niger. Test candidate BGCs for A. nidulans were obtained by extracting genomic regions sequentially from its genome, using amino acid sliding windows of 10 000 amino acids that overlap by 30% and 50%. The analysis on A. nidulans used the best-performing model parameters previously established in A. niger: 50%–50% dataset, hyperparameter optimization and 5-fold CV.
Table 6 shows TOUCAN best performance results among all six classifiers and post-processing methods for the A. nidulans 10 000-amino acid sliding window test sets. For comparison, we evaluated A. nidulans BGC predictions obtained with the best fungiSMASH and DeepBGC models on the same test sets, for which the results are also shown in Table 6. We observed that similar F-m performance metrics were achieved for A. nidulans and A. niger. TOUCAN and DeepBGC, both based on supervised learning, yielded the least F-m variation on the results obtained for the two Aspergillus species, suggesting that due to their generalization ability, supervised learning approaches may be a suitable approach to tackle BGC discovery.
Table 6.
Best performances per overlap of TOUCAN compared to fungiSMASH and DeepBGC for A. nidulans test sets generated with 10 000-amino acid sliding window
| Sliding | ||||||
|---|---|---|---|---|---|---|
| System | Model | window | Overlap | P | R | F-m |
| TOUCAN | lsvc + merge3 | 10 000 | 50% | 0.919 | 0.814 | 0.864 |
| TOUCAN | svc + merge3 | 10 000 | 30% | 0.953 | 0.871 | 0.910 |
| fungiSMASH | Relaxed (default) | 10 000 | 50% | 0.550 | 0.786 | 0.647 |
| fungiSMASH | Relaxed (default) | 10 000 | 30% | 0.775 | 0.786 | 0.780 |
| DeepBGC | 50%–50% fungal | 10 000 | 50% | 0.473 | 0.629 | 0.540 |
| DeepBGC | 50%–50% fungal | 10 000 | 30% | 0.631 | 0.586 | 0.607 |
TOUCAN TP predictions improve coverage of BGC genes
We compared TP predictions (BGC candidate predicted positives that have at least one gene matching a gold standard BGC) obtained from best-performing models in A. niger and A. nidulans for TOUCAN (0.982 F-m and 0.910 F-m, respectively) versus fungiSMASH (0.692 F-m and 0.780 F-m, respectively) and DeepBGC (0.620 F-m and 0.607 F-m, respectively). First, we analysed the distribution of clusterScore computed for each BGC candidate predicted positive. Figure 1 shows the clusterScore distribution in A. niger and A. nidulans TP predictions obtained with TOUCAN, DeepBGC and fungiSMASH best models.
Figure 1.
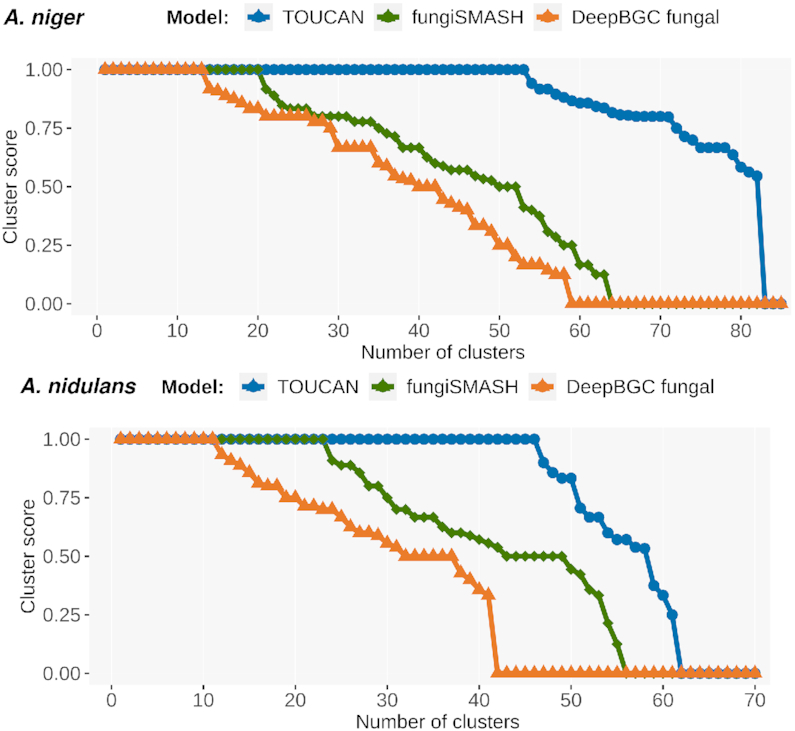
Distribution of clusterScore among TP predictions in A. niger and A. nidulans genomes. clusterScore distribution was computed for best-performing models of each system (A. niger: TOUCAN: 0.982 F-m, DeepBGC: 0.627 F-m, fungiSMASH: 0.692 F-m; A. nidulans: TOUCAN: 0.910 F-m, DeepBGC: 0.607 F-m, fungiSMASH: 0.780 F-m).
We observed that compared to the other tools, TOUCAN TP predictions more often present a clusterScore = 1, meaning that TOUCAN predictions better encompass genes matching gold standard BGCs, possibly as a result of TOUCAN merge post-processing. Although merge post-processing leads to more comprehensive predictions, it could result in overprediction of cluster boundaries. To mitigate, filtering methods could be applied to refine candidate cluster regions, and also as an opportunity to fine-tune TOUCAN predictions to specific genus or species of interest. One possible way to apply targeted filtering is to rely on manually curated annotations of relevant features, such as the annotated high and medium Pfam protein domains shown in the ‘Features’ section.
We also analysed the presence of backbone enzymes within genes of TP predictions. Backbone enzymes are considered as the BGC core (3), playing a key role in its biosynthesis and defining the BGC compound to be produced (19). We mapped the presence and absence of backbone genes among TOUCAN, DeepBGC and fungiSMASH best models’ TP predictions. Figure 2 shows backbone genes and product types found in A. niger and A. nidulans, respectively. Scores in Figure 2 (or the colour intensity) correspond to the clusterScore computed for the predicted BGC. Backbone enzyme genes were present in 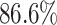 of all TOUCAN TP predictions for A. niger, versus
of all TOUCAN TP predictions for A. niger, versus 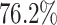 in fungiSMASH and
in fungiSMASH and 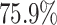 in DeepBGC. For A. nidulans, 93.5% of TOUCAN TP predictions found backbone enzymes, versus
in DeepBGC. For A. nidulans, 93.5% of TOUCAN TP predictions found backbone enzymes, versus 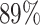 for fungiSMASH and
for fungiSMASH and 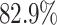 for DeepBGC.
for DeepBGC.
Figure 2.

Presence of backbone enzymes among positive predictions in A. niger and A. nidulans genomes. Each backbone enzyme is shown per the gene ID it is associated with and the clusterScore assigned to the candidate predicted BGC.
DISCUSSION
SMs are bioactive compounds that play a vital role in the production of various drugs. Discovery of novel fungal BGCs can potentially benefit human health. In this work, we presented TOUCAN, a supervised learning framework for fungal BGC discovery. We evaluated classification models based on fungal BGC datasets of various distributions, six classifiers, heterogeneous biological features and three post-processing methods. TOUCAN best-performing model achieved 0.982 F-m in A. niger and 0.910 F-m in A. nidulans, outperforming previous methods. The results obtained with TOUCAN models could indicate that standard supervised learning approaches are suitable to tackle BGC discovery. TOUCAN outperformance is possibly due to a combination of factors: combining feature types, evaluating the impact of different class distributions during training and post-processing candidate BGC predictions. merge post-processing can help identify regions that might have been missed, but in certain cases it may potentially lead to overestimation of predicted cluster boundaries.
The performance of TOUCAN models was compared to two BGC discovery state-of-the-art approaches: DeepBGC, based on deep learning, and fungiSMASH, based on probabilistic methods. TOUCAN models showed better F-m when predicting BGCs in A. niger and A. nidulans compared to DeepBGC and fungiSMASH. TOUCAN also yielded more comprehensive coverage of gold standard BGC genes within predicted clusters, and was able to identify backbone enzyme genes more often in its TP predictions compared to the other methods. The presence of backbone enzymes can be a crucial aspect in determining the presence of a BGC in a given genomic region. The results obtained by TOUCAN, as well as the performance of DeepBGC models, demonstrate the potential of exploring supervised learning approaches for BGC discovery, and relevance of developing BGC prediction tools focused on fungal organisms. Fungi were shown to be an important source for bioactive compounds (5,6) used in the pharmaceutical industry, but fungal BGC data available in open-access databases are scarce compared to bacteria. The availability of more annotated fungal BGCs is hence an important aspect to promote development and improvement of existing and new fungal BGC discovery approaches. Previous BGC discovery tools require curated data to identify candidate BGC regions in an organism, which may not be available or is expensive to obtain. Unlike previous approaches, TOUCAN is capable of outputting BGC predictions from amino acid sequences without requiring further data curation as input. This aspect can facilitate TOUCAN usage and its application on newly sequenced genomes, promoting the discovery of novel candidate BGC regions and potentially novel drugs, such as antibiotics, immunosuppressants and anticancer medications.
DATA AVAILABILITY
TOUCAN source code as well as all datasets applied in our experiments are made publicly available at http://github.com/bioinfoUQAM/TOUCAN. TOUCAN source code is available under the MIT permissive software license. The datasets used in this work were obtained from open-access databases, which are available under the Creative Commons Attribution 4.0 international license.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge the bioinformatics teams at CSFG and UQAM for many inspiring discussions and comments on this manuscript; Amine Remita for helpful discussions and for providing comments on an earlier version of this manuscript; Diego Maupomé for helping with deep learning frameworks; and Antoine Briand for helping with web frameworks.
Contributor Information
Hayda Almeida, Departement d’Informatique, UQAM, Montréal, QC, H2X 3Y7, Canada; Laboratoire d’Algèbre, de Combinatoire, et d’Informatique Mathématique, Montréal, QC, H2X 3Y7, Canada; Centre for Structural and Functional Genomics, Concordia University, Montréal, QC, H4B 1R6, Canada.
Sylvester Palys, Centre for Structural and Functional Genomics, Concordia University, Montréal, QC, H4B 1R6, Canada.
Adrian Tsang, Departement d’Informatique, UQAM, Montréal, QC, H2X 3Y7, Canada; Centre for Structural and Functional Genomics, Concordia University, Montréal, QC, H4B 1R6, Canada.
Abdoulaye Baniré Diallo, Departement d’Informatique, UQAM, Montréal, QC, H2X 3Y7, Canada; Laboratoire d’Algèbre, de Combinatoire, et d’Informatique Mathématique, Montréal, QC, H2X 3Y7, Canada; Centre of Excellence in Research on Orphan Diseases—Courtois Foundation (CERMO-FC), Montréal, QC, H2X 3Y7, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Natural Sciences and Engineering Research Council of Canada (NSERC); Fonds de Recherche du Québec—Nature et Technologies (FRQNT).
Conflict of interest statement. None declared.
REFERENCES
- 1. Chavali A.K., Rhee S.Y. Bioinformatics tools for the identification of gene clusters that biosynthesize specialized metabolites. Brief. Bioinform. 2017; 19:1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kautsar S.A., Blin K., Shaw S., Navarro-Muñoz J.C., Terlouw B.R., van der Hooft J.J., Van Santen J.A., Tracanna V., Suarez Duran H.G., Pascal Andreu V. et al. MIBiG 2.0: a repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020; 48:D454–D458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kjærbølling I., Vesth T., Frisvad J.C., Nybo J.L., Theobald S., Kildgaard S., Petersen T.I., Kuo A., Sato A., Lyhne E.K. et al. A comparative genomics study of 23 Aspergillus species from section Flavi. Nat. Commun. 2020; 11:1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Keller N.P. Fungal secondary metabolism: regulation, function and drug discovery. Nat. Rev. Microbiol. 2019; 17:167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Macheleidt J., Mattern D.J., Fischer J., Netzker T., Weber J., Schroeckh V., Valiante V., Brakhage A.A. Regulation and role of fungal secondary metabolites. Annu. Rev. Genet. 2016; 50:371–392. [DOI] [PubMed] [Google Scholar]
- 6. de Vries R.P., Riley R., Wiebenga A., Aguilar-Osorio G., Amillis S., Uchima C.A., Anderluh G., Asadollahi M., Askin M., Barry K. et al. Comparative genomics reveals high biological diversity and specific adaptations in the industrially and medically important fungal genus Aspergillus. Genome Biol. 2017; 18:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takeda I., Umemura M., Koike H., Asai K., Machida M. Motif-independent prediction of a secondary metabolism gene cluster using comparative genomics: application to sequenced genomes of Aspergillus and ten other filamentous fungal species. DNA Res. 2014; 21:447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wolf T., Shelest V., Nath N., Shelest E. CASSIS and SMIPS: promoter-based prediction of secondary metabolite gene clusters in eukaryotic genomes. Bioinformatics. 2016; 32:1138–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vesth T.C., Brandl J., Andersen M.R. FunGeneClusterS: predicting fungal gene clusters from genome and transcriptome data. Synth. Syst. Biotechnol. 2016; 1:122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Umemura M., Koike H., Nagano N., Ishii T., Kawano J., Yamane N., Kozone I., Horimoto K., Shin-ya K., Asai K. et al. MIDDAS-M: motif-independent de novo detection of secondary metabolite gene clusters through the integration of genome sequencing and transcriptome data. PLoS One. 2013; 8:e84028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blin K., Wolf T., Chevrette M.G., Lu X., Schwalen C.J., Kautsar S.A., Suarez Duran H.G., De Los Santos E.L., Kim H.U., Nave M. et al. antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017; 45:W36–W41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cimermancic P., Medema M.H., Claesen J., Kurita K., Brown L. C.W., Mavrommatis K., Pati A., Godfrey P.A., Koehrsen M., Clardy J. et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell. 2014; 158:412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khaldi N., Seifuddin F.T., Turner G., Haft D., Nierman W.C., Wolfe K.H., Fedorova N.D. SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet. Biol. 2010; 47:736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Agrawal P., Khater S., Gupta M., Sain N., Mohanty D. RiPPMiner: a bioinformatics resource for deciphering chemical structures of RiPPs based on prediction of cleavage and cross-links. Nucleic Acids Res. 2017; 45:W80–W88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hannigan G.D., Prihoda D., Palicka A., Soukup J., Klempir O., Rampula L., Durcak J., Wurst M., Kotowski J., Chang D. et al. A deep learning genome-mining strategy for biosynthetic gene cluster prediction. Nucleic Acids Res. 2019; 47:e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019; 47:D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Almeida H., Tsang A., Diallo A.B. Supporting supervised learning in fungal biosynthetic gene cluster discovery: new benchmark datasets. Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2019; Piscataway: IEEE; 1280–1287. [Google Scholar]
- 18. Kriventseva E.V., Kuznetsov D., Tegenfeldt F., Manni M., Dias R., Simão F.A., Zdobnov E.M. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2018; 47:D807–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Inglis D.O., Binkley J., Skrzypek M.S., Arnaud M.B., Cerqueira G.C., Shah P., Wymore F., Wortman J.R., Sherlock G. Comprehensive annotation of secondary metabolite biosynthetic genes and gene clusters of Aspergillus nidulans, A. fumigatus, A. niger and A. oryzae. BMC Microbiol. 2013; 13:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vinje H., Liland K.H., Almøy T., Snipen L. Comparing K-mer based methods for improved classification of 16S sequences. BMC Bioinformatics. 2015; 16: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang Y., Pedersen J.O. A comparative study on feature selection in text categorization. Proceedings of the International Conference on Machine Learning (ICML). 1997; 97:Nashville: 35. [Google Scholar]
- 22. UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rost B. Twilight zone of protein sequence alignments. Protein Eng. Des. Sel. 1999; 12:85–94. [DOI] [PubMed] [Google Scholar]
- 24. Skinnider M.A., Dejong C.A., Rees P.N., Johnston C.W., Li H., Webster A.L., Wyatt M.A., Magarvey N.A. Genomes to natural products prediction informatics for secondary metabolomes (PRISM). Nucleic Acids Res. 2015; 43:9645–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
TOUCAN source code as well as all datasets applied in our experiments are made publicly available at http://github.com/bioinfoUQAM/TOUCAN. TOUCAN source code is available under the MIT permissive software license. The datasets used in this work were obtained from open-access databases, which are available under the Creative Commons Attribution 4.0 international license.