

SUMMARY
The mammalian SWitch/Sucrose Non-Fermentable (SWI/SNF) chromatin-remodeling BAF (BRG1/BRM-associated factor) complex plays an essential role in developmental and pathological processes. We show that the deletion of Baf155, which encodes a subunit of the BAF complex, in the Tie2(+) lineage (Baf155 (CKO) leads to defects in yolk sac myeloid and definitive erythroid (EryD) lineage differentiation from erythromyeloid progenitors (EMPs). The chromatin of myeloid gene loci in Baf155 CKO EMPs is mostly inaccessible and enriched mainly by the ETS binding motif. BAF155 interacts with PU.1 and is recruited to PU.1 target gene loci together with p300 and KDM6a. Treatment of Baf155 CKO embryos with GSK126, an H3K27me2/3 methyltransferase EZH2 inhibitor, rescues myeloid lineage gene expression. This study uncovers indispensable BAF-mediated chromatin remodeling of myeloid gene loci at the EMP stage. Future studies exploiting epigenetics in the generation and application of EMP derivatives for tissue repair, regeneration, and disease are warranted.
Graphical Abstract
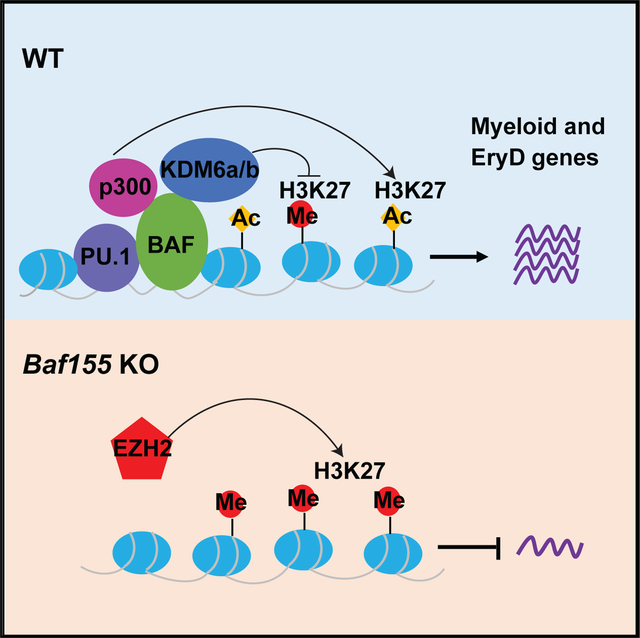
In Brief
The mammalian chromatin-remodeling BAF (BRG1/BRM-associated factor) complex has an essential role in developmental and pathological processes. Wu et al. show that BAF-mediated chromatin remodeling and activation of the myeloid and definitive erythroid transcriptional program at the EMP stage is critical for myeloid and definitive erythroid lineage development.
INTRODUCTION
The mammalian hematopoietic system is established from multiple embryonic origins. The first tissue to produce blood cells is the yolk sac, which generates primitive erythroid (EryP) cells and megakaryocytes (MegPs) presumably from bipotential MegP erythroid progenitors (pMEPs) (Tober et al., 2007). Although EryPs, which express embryonic globin genes, can be detected as early as embryonic day 7.25 (E7.25) in the blood islands of the yolk sac, mature MegP cells are not detected until later, around E9.5, in the yolk sac (Tober et al., 2007). The primary function of EryP cells is to provide developing embryos with oxygen and nutrients to accommodate the rapid growth of the embryo. Until recently, hematopoietic stem cells (HSCs) originating from the hemogenic endothelium of the aorta-gonad-mesonephros (AGM) have been thought to initiate definitive hematopoiesis. However, this paradigm has been challenged by the identification of erythromyeloid progenitors (EMPs), which generate definitive erythroid (EryD) and myeloid cell lineages before the establishment of HSCs (Hoeffel and Ginhoux, 2018; McGrath et al., 2015; Ginhoux et al., 2010; Schulz et al., 2012; Hashimoto et al., 2013; Gomez Perdiguero et al., 2015). EMPs develop transiently from the Tie2-expressing hemogenic endothelium of the yolk sac around E8.5–E10.5, migrate to the fetal liver, expand, and provide embryos with EryD and myeloid cells in fetal life (Chen et al., 2011; McGrath et al., 2015; Gomez Perdiguero et al., 2015; Palis, 2016). EMPs are ultimately replaced by HSCs, which generate a full spectrum of blood cells, including lymphoid cell lineages. The importance of EMPs was realized by the finding that tissue-resident macrophages originate from EMPs through monocyte intermediates, although microglia are believed to originate from yolk sac macrophages (Ginhoux et al., 2010). Despite the critical establishment of the cellular origin of tissue-resident macrophages from EMPs, few studies have examined the molecular mechanisms that regulate myeloid and EryD differentiation from EMPs.
In eukaryotes, DNA is packaged into nucleosomes and subsequent higher-order chromatin structures. As a result, DNA is not easily accessible for transcriptional machinery. Two fundamental mechanisms that allow cells to respond to signals and trigger gene expression are chromatin remodeling and histone modification. Transcription factors and other proteins are believed to gain access to nucleosome-bound DNA using ATP-dependent chromatin-modifying and remodeling enzymes, such as the SWI/SNF complex family (Hargreaves and Crabtree, 2011; Kadoch and Crabtree, 2015). SWI/SNF complexes, by utilizing energy derived from ATP hydrolysis, destabilize histone-DNA interactions and create an open chromatin state. SWI/SNF complexes are composed of one core ATPase (Brg1 or Brm) and distinct paralogous BRG1/BRM-associated factor (BAF) subunit family members that can interact with cofactors, including transcription factors. Combinatorial assembly of alternative families of subunits confers functional specificity to BAF complexes in different tissues and cell types (Wu et al., 2009). A recent study showing that BAF60c, a subunit of BAF critical for heart development (Lickert et al., 2004), together with GATA4 and TBX5, can reprogram mesoderm to cardiomyocytes in the mouse embryo (Takeuchi and Bruneau, 2009) highlights the fact that SWI/SNF-mediated chromatin remodeling is integral to the lineage-specific transcriptional network. Notably, enforced expression of just one component of the multi-protein BAF complex, BAF60c, in that study was sufficient to confer lineage-specific gene regulation.
Although chromatin-remodeling proteins are critical for establishing cell lineage specification in mammalian development, functional details of how these proteins affect specific cell lineage development are still lacking. Gene knockout studies have demonstrated that BRG1 ATPase and BAF155 are required for blood and vascular development. Particularly, Brg1- or Baf155-deficient embryos die around implantation (Bultman et al., 2000; Kim et al., 2001). The peri-implantation lethality of Baf155-deficient embryos can be extended to mid-gestation by Baf155 transgene expression (Baf155−/−; Tg+). However, these animals display severe blood vessel formation defects in the yolk sac around E10.5 (Han et al., 2008). Moreover, Tie2-Cre; Brg1f/f mice are embryonic lethal because of apoptosis of EryP and lack of embryonic globin gene expression (Griffin et al., 2008). Angiogenesis is also defective in these animals. While these studies demonstrate the critical role of BAF-mediated chromatin remodeling in EryP and angiogenesis, it remains unclear whether BAF-mediated chromatin remodeling is subsequently required for the yolk sac hematopoiesis. In this study, we determined BAF-mediated chromatin remodeling requirements from hemogenic endothelium to EMPs and myeloid and EryD lineage differentiation by deleting Baf155 in the Tie2 lineage. Our data demonstrate that BAF155-mediated chromatin remodeling of myeloid and EryD gene loci at the EMP stage is critical for activation of the myeloid and EryD transcriptional program as well as myeloid and EryD lineage differentiation.
RESULTS
Baf155 CKO Mice Are Embryonic Lethal, Showing Defects in Myeloid and EryD Lineage Development
To assess the role of chromatin remodeling in hematopoietic lineage development downstream of the yolk sac hemogenic endothelium, we first generated Tie2-Cre;Baf155f/+ mice from matings between Baf155f/f (Choi et al., 2012) and Tie2-Cre mice, which have been shown to target the yolk sac hemogenic endothelium (Chen et al., 2011; Gomez Perdiguero et al., 2015). We also observed effective tdTomato expression in EryP cells, endothelial cells, myeloid cells, and microglia in Tie2-Cre;R-osa-loxp-stop-loxp-tdTomato yolk sac and brain (Figure S1A). Tie2-Cre;Baf155f/+ mice were then crossed with Baf155f/f mice to generate Tie2-Cre;Baf155f/f mice (hereafter called Baf155 CKO). We found no live Baf155 CKO mice at weaning (Table S1). We found no Baf155 CKO embryos with abnormal morphology up to E9.5. However, at E10.5, some Baf155 CKO embryos were smaller compared with littermate controls and displayed occasional hemorrhage around the distal end of the tail and yolk sac (8 of 49), suggesting abnormal vessel formation. This is consistent with previous findings that Brg1 or Baf155 deficiency leads to angiogenesis defects (Griffin et al., 2008; Han et al., 2008). All Baf155 CKO embryos showed severe growth retardation and seemingly abnormal gross morphology at E13.5 (data not shown).
We assessed whether the embryonic lethality in Baf155 CKO mice was due to hematopoietic defects. Ter119+ erythroid cells from E9.5 and E10.5 yolk sacs, predominantly EryP cells at this stage, were present at similar levels in wild-type and Baf155 CKO yolk sacs (Figures 1A and S1B). CD45−CD31+ endothelial cells were also present similarly in wild-type and CKO yolk sacs (Figures 1A and S1B). Notably, there was a significant reduction in myeloid cells, CD45+CD11b+, in Baf155 CKO yolk sacs (Figures 1A and S1B). Yolk sac macrophages, CD45+F4/80+, as well as microglia of the brain, CD45+CX3CR1+CD11b+, were also reduced significantly in Baf155 CKO embryos (Figures 1A, S1B, and S1C). Although EryP progenitors from wild-type littermate control and Baf155 CKO yolk sacs were present similarly, myeloid progenitors and BFU-Es were decreased significantly in Baf155 CKO yolk sacs (Figure 1B). Corroborating these data, expression of myeloid lineage genes, including Irf8, Cx3cr1, and Fcgr3 (CD16), was reduced significantly in Baf155 CKO yolk sacs (Figures 1C and S1D). However, genes expressed in the EryP lineage, Klf1, Hbb-bh1, and Hbz, were detected similarly in wild-type and CKO yolk sacs (Figure 1C). Genes expressed in EMPs, cKit, Gata2, Tal1/Scl, and Myb, were also detected similarly in E9.5 wild-type (WT) and Baf155 CKO yolk sacs (Figure 1C). GP1bb (CD42c), Mpl, and Itga2b (CD41), genes expressed in megakaryocytes, were also expressed similarly in WT and CKO yolk sacs (Figure 1C). These results suggest that Baf155 deficiency leads to selective defects in myeloid and EryD lineage development.
Figure 1. Baf155 CKO Mice Show Defects in Myeloid and EryD Lineage Development.
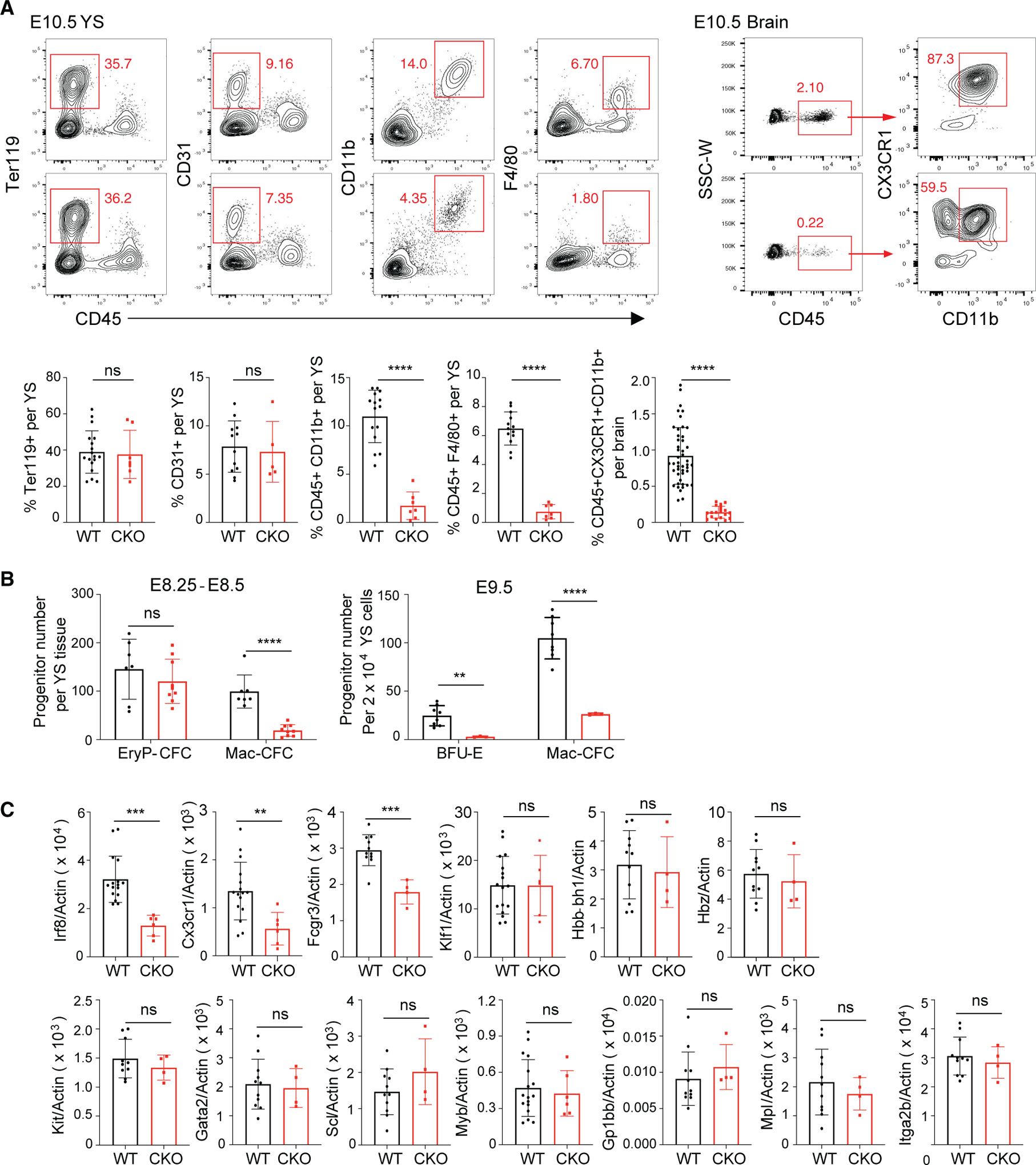
(A) A representative flow cytometry analysis of E10.5 yolk sacs (YS) EryP cells (CD45−Ter119+), ECs (CD45−CD31+), myeloid cells (CD45+CD11b+), macrophages (CD45+F4/80+), and brain microglia (CD45+CX3CR1+CD11b+) in wild-type (WT) and Baf155 CKO mice is shown in the top panel. The percentage of each population is shown in the bottom panel. At least 5 biological replicates in 4 independent experiments for either genotype were analyzed, each representing an individual YS. Data are presented as mean ± SD. Student’s t test; ns, not significant; ****p < 0.0001.
(B) Distribution of EryP (EryP-CFC), EryD (BFU-E), and macrophage (Mac-CFC) progenitors from E8.25–E8.5/E9.5 WT and Baf155 CKO YS cells. E8.25–E8.5 data from 7 WT and 9 Baf155 CKO biological replicates and E9.5 data from 8 WT and 3 Baf155 CKO biological replicates representing two independent experiments, with each replicate consisting of a single YS, are shown. Data are presented as mean ± SD. Student’s t test; **p < 0.005, ****p < 0.0001.
(C) qRT-PCR analysis of the indicated gene expression in E9.5 WT and Baf155 CKO YS cells is shown. Data are presented as mean ± SD. Student’s t test; **p < 0.005, ***p < 0.001.
See also Figure S1.
EMPs Develop in the Absence of Baf155
The selective myeloid and EryD lineage defects seen in Baf155 CKO yolk sacs might be due to defects in EMP generation. Alternatively, EMPs are generated, but their differentiation might be blocked. To differentiate these two possibilities, we first analyzed EMPs based on phenotypic markers in Baf155 CKO yolk sacs. EMPs are enriched in cKIT+CD41+CD16/32+ cells (McGrath et al., 2015). Because expression of Fcgr3 encoding CD16 was clearly decreased in Baf155 CKO yolk sacs (Figure 1C), we reasoned that inclusion of CD16/32 in the EMP analysis might be inadequate for measuring EMPs from Baf155 CKO yolk sacs. Indeed, the cKIT+CD16/32+CD41+ or CD16/32+CD41+ cell population was reduced greatly in Baf155 CKO yolk sacs (Figure S2). Because cKIT is critical for EMP development (Azzoni et al., 2018), and cKit expression was similar in WT and CKO yolk sacs (Figure 1C), we sorted cKIT+ cells and evaluated their myeloid and EryD potential compared with cKIT+CD41+CD16/32+ cells. There was an ~80% overlap between cKIT+ and cKIT+CD16/32+CD41+ cell populations (data not shown). Although cKIT+ and cKIT+CD41+CD16/32+ cells generated similar levels of BFU-Es, cKIT+ cells generated slightly more CFU-Es compared with cKIT+CD41+CD16/32+ cells, suggesting that cKIT+ cells also contain more committed erythroid progenitors (Figure 2A). Slightly more myeloid colonies were generated from cKIT+CD41+CD16/32+ cells compared with cKIT+ cells, suggesting that cKIT+CD41+CD16/32+ cells also enrich committed myeloid progenitors (Figure 2A). Importantly, cKIT+ cells were present at similar levels in WT and Baf155 CKO yolk sacs at E8.5–E10.5 (Figure 2B). However, cKIT+ cells from E10.5 Baf155 CKO yolk sacs generated significantly fewer myeloid or BFU-E colonies compared with controls (Figure 2C), suggesting that EMPs were generated in Baf155 CKO embryos but that Baf155 CKO EMPs have a block in myeloid and EryD differentiation. We assessed whether Baf155 inhibition in EMPs was sufficient to block myeloid and EryD differentiation. To this end, we knocked down Baf155 in EMPs by transfecting Baf155 esiRNA into sorted cKIT+ cells. We achieved more than 60% KD efficiency of Baf155 expression, whereas expression of irrelevant genes, such as Pu.1, was unaffected (Figure 2D). Notably, myeloid and EryD output from EMPs was reduced greatly by Baf155 knockdown (KD) (Figure 2E), indicating that acute deletion of Baf155 in EMPs is sufficient to inhibit myeloid and EryD lineage differentiation. These data collectively suggest that Baf155-mediated chromatin remodeling at the EMP stage is critical for efficient downstream myeloid and EryD lineage differentiation.
Figure 2. Baf155 Is Required for Myeloid and EryD Lineage Differentiation from EMPs.
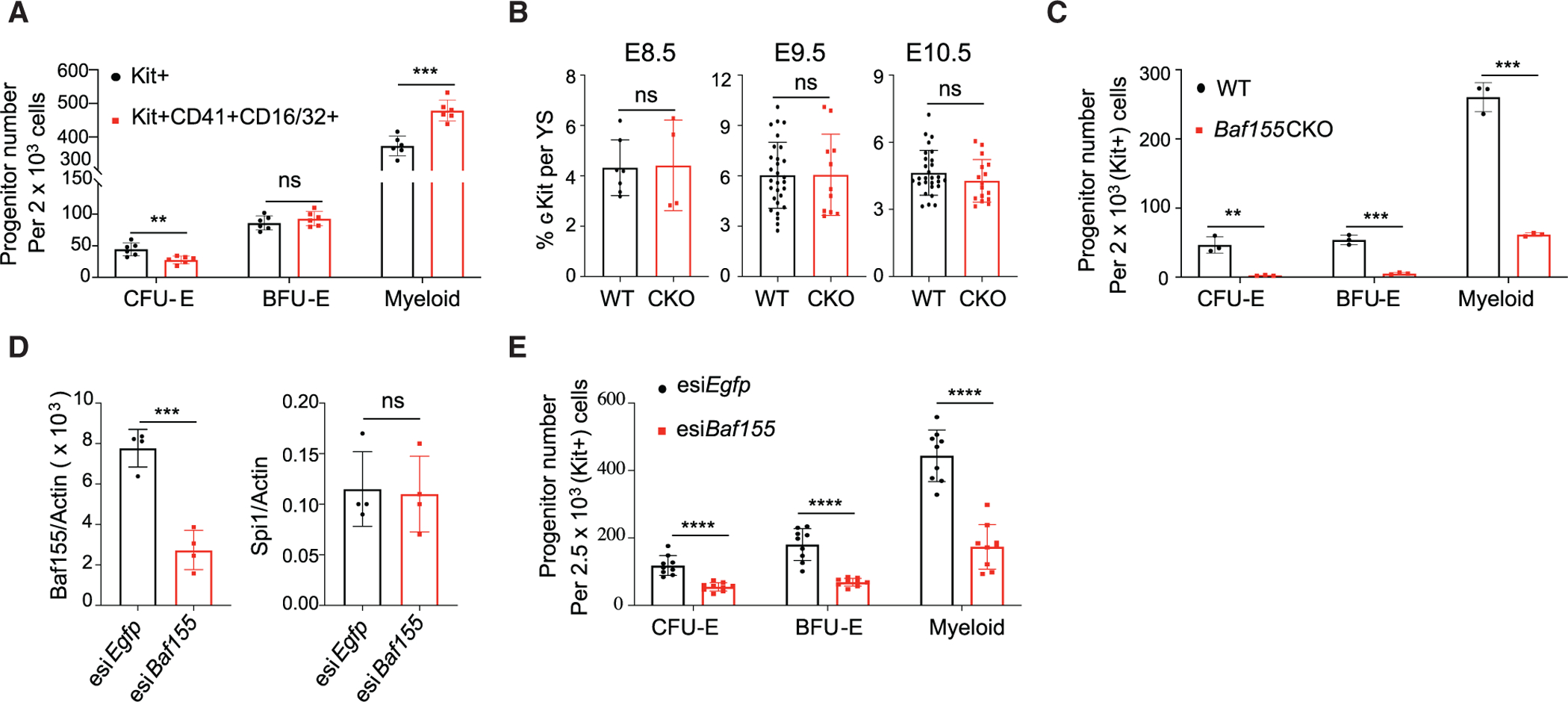
(A) Distribution of CFU-E, BFU-E, and myeloid colonies developing from KIT+ and KIT+CD41+CD16/32+ population from WT E10.5 YSs. Data are presented as mean ± SD. Student’s t test; **p < 0.005, ***p < 0.001; 6 biological replicates.
(B) Flow cytometry analysis of the cKIT+ population per YS from WT and Baf155 CKO embryos on the indicated embryonic day. E8.5 data (mean ± SD) are from 2 independent experiments. E9.5 and E10.5 data (mean ± SD) are from 6 independent experiments, each representing a single YS. Data are presented as mean ± SD. Student’s t test.
(C) CFU-E, BFU-E, and myeloid colonies from cKIT+ cells from WT (n = 3) and Baf155 CKO (n = 3) YSs. Data are from 2 independent experiments, with each replicate consisting of a single or 2 pooled YSs of the same genotype. Data are presented as mean ± SD. Student’s t test; **p < 0.01, ***p < 0.001.
(D) qRT-PCR analysis of the indicated gene expression in cKIT+ cells from E10.5 WT YSs transfected with esiRNA against Baf155 or Egfp. Data are presented as mean ± SD. Student’s t test; ***p < 0.001; four biological replicates from 3 independent experiments.
(E) CFU-E, BFU-E, and myeloid colonies from cKIT+ cells from E10.5 WT YSs transfected with esiRNA against Baf155 or Egfp. Data are presented as mean ± SD. Student’s t test; ***p < 0.001. Nine biological replicates from 3 independent experiments.
See also Figure S2.
Single-Cell RNA Sequencing Reveals Myeloid Lineage Differentiation Defects of Baf155-Deficient EMPs
To better understand the myeloid lineage differentiation block in Baf155 CKO embryos, we subjected yolk sacs from WT and Baf155 CKO mice to single-cell RNA sequencing (scRNA-seq). After filtering out low-quality cells, 722 WT and 791 Baf155 CKO yolk sac cells were chosen for further analysis. t-stochastic neighbor embedding (t-SNE) was used to visualize the populations. WT yolk sac cells were clustered into 7 populations based on similarities of the transcriptome (Figure S3A). High expression of Kdr, Cdh5, and Pecam1 highlights cluster 5 to be an endothelial cell population (Figure S3B). Two distinct erythroid cell populations were visible based on the erythroid lineage markers Gata1, Klf1, and EpoR (Figure S3C). Expression of Hbb-y, Hbb-x, and Hbb-bh1 (embryonic β-globin genes) separated cluster 2 as primitive and cluster 4 as an EryD cell population (Figure S3D). We also identified Gm15915, Ccl17, Muc13, and Gdf3 to be expressed in an EryD-specific manner (Figures S3E and S3I). Expression of the mature myeloid lineage genes Trem2, Emr1, Cx3cr1, Csf1r, Irf8, and Cd68 identified cluster 6 as a myeloid cell population (Figures S3F and S3J). Enriched expression of cKit and Itga2b, encoding CD41, and Cd34 identified cluster 0 as EMPs (Figure S3G). Expression of Pu.1 (Spi1), a critical factor for myeloid lineage development, expression was high in EMPs, and its expression was detected continuously in the myeloid lineage arm (Figures S3G and S3K). Bcl11a, Myb, Scl (Tal1), and Gata2 expression was detected in EMPs, and their expression was detected continuously in the EryD cell cluster (Figure S3K). It is also notable that Baf155 and Cd34 expression was high in the EMP population (Figure S3M). As we reported recently (Zhao and Choi, 2019), clusters 1 and 3 represent smooth muscle cells and pericytes, based on Hand1, Hand2, Acta2, Tbx20, and Cd248 expression (Figures S3H and S3L). Desmin expression separated pericytes (cluster 3) from smooth muscle cells (cluster 1; Figure S3L).
When we overlaid WT and Baf155 CKO scRNA-seq data, we observed that the transcriptomes of endothelial cells (ECs; Figure 3A, cluster 7), EryP cells (Figure 3A, clusters 1 and 6), and smooth muscle cells and pericytes (Figure 3A, clusters 2, 4, 5, and 8) overlapped each other, indicating that Baf155 deficiency did not affect their overall transcriptome (Figures 3A–3D). However, clusters 0 and 3, both expressing EMP genes (Figures 3E and 3F), showed clear separation between WT and Baf155 CKO yolk sac cells (Figures 3B–3D). Baf155 expression was clearly absent in cluster 3, indicating that Baf155 was deleted effectively in this population (Figure 3B). Strikingly, a population with the mature myeloid gene signature (cluster 9) was readily visible in WT yolk sacs but absent in Baf155 CKO yolk sacs (Figures 3E and 3G). A population with the EryD gene signature was also reduced greatly in Baf155 CKO yolk sacs (Figures 3E and 3H). Intriguingly, although EryD cells were reduced greatly, the megakaryocytic lineage gene signature was high in the presumptive Baf155 CKO EryD cell population (Figure 3I). This suggests that the megakaryocytic lineage may be the default pathway in EryD and megakaryocytic lineage choice and that chromatin remodeling is also critical for EryD and megakaryocyte lineage bifurcation. Additionally, endothelial genes were still expressed in the Baf155 CKO EMP cell population (Figure 3J). These data collectively suggest that chromatin remodeling is needed at the EMP stage for further differentiation into myeloid and EryD lineages to occur. The endothelial gene program is sustained in EMPs when subsequent differentiation is blocked. Alternatively, termination of the endothelial gene program might require chromatin remodeling.
Figure 3. scRNA-Seq Data Reveal Myeloid and EryD Differentiation Defects from Baf155-Deficient EMPs.

(A) t-SNE projection of all cells, showing 10 different clusters.
(B) Baf155/Smarcc1 expression in all YSs, showing the absence of Baf155 expression in the CKO EMP cell population.
(C) An overlay of scRNA-seq data between WT and Baf155 CKO YSs.
(D) Percentage of cells in each cluster from WT versus Baf155 CKO YSs.
(E) Heatmap showing differentially expressed genes in each cluster.
(F) The EMP signature genes Gata2, Tal1, cKit, Cd34, and Pu.1/Spi1 are similarly expressed in WT and Baf155 CKO EMPs.
(G) A cell population with myeloid lineage signature gene expression (Irf8, Maf, Csf1r, Cx3cr1, and Trem2) is absent in Baf155 CKO YSs.
(H) A cell population with EryD lineage signature genes expression (Gdf3, Muc13, Ccl17, and Gm15915) is significantly lower in Baf155 CKO YSs.
(I) A population with elevated megakaryocyte lineage signature gene expression is increased in Baf155 CKO YS cells.
(J) Endothelial lineage signature genes are still expressed in the Baf155-deficient EMP cell population.
See also Figure S3.
Chromatin Accessibility of Myeloid and EryD Gene Loci Is Reduced Greatly in Baf155 CKO EMPs
So far, the data suggested that Baf155 deficiency leads to myeloid and EryD differentiation block from EMPs. To better understand the myeloid lineage differentiation defect in Baf155-deficient EMPs, we sorted cKIT+ cells, EMP enriched, from WT and Baf155 CKO yolk sacs (Figure S4A) and assessed genome-wide chromatin accessibility by assay for transposase-accessible chromatin using sequencing (ATAC-seq; Buenrostro et al., 2013). We identified 103,043 and 143,021 accessible chromatin regions in Baf155 CKO and WT cKIT+ cells, respectively. Among these ATAC-seq peaks, 7,422 regions were more accessible in WT than in Baf155 CKO cells, whereas only 56 regions were more accessible in Baf155 CKO cells than in WT cells (Figures 4A and 4B). The differentially accessible genomic regions (DARs) were enriched for the binding motifs of transcription factors (TFs) such as PU.1, IRF8, AP-1, and CEBP, whereas 13,213 unaffected accessible genomic regions, commonly open in CKO and WT EMPs (fold change < 1.1 and p > .05), were enriched for the different TF binding motifs, such as CTCF (Figures 4B, 4C, and S4B). Of the 7,422 DARs specific to WT EMPs, 3,679 peaks contained PU.1 motifs, whereas 1,472 of 13,213 unaffected peaks contained PU.1 motifs. This suggests that loss of Baf155 leads to a closed chromatin structure at selective genomic regions. The data also suggest potential interplay between ETS factors and BAF-mediated chromatin remodeling in activating the myeloid lineage program.
Figure 4. Baf155 CKO EMPs Have Reduced Chromatin Accessibility at the Myeloid and EryD Gene Loci.
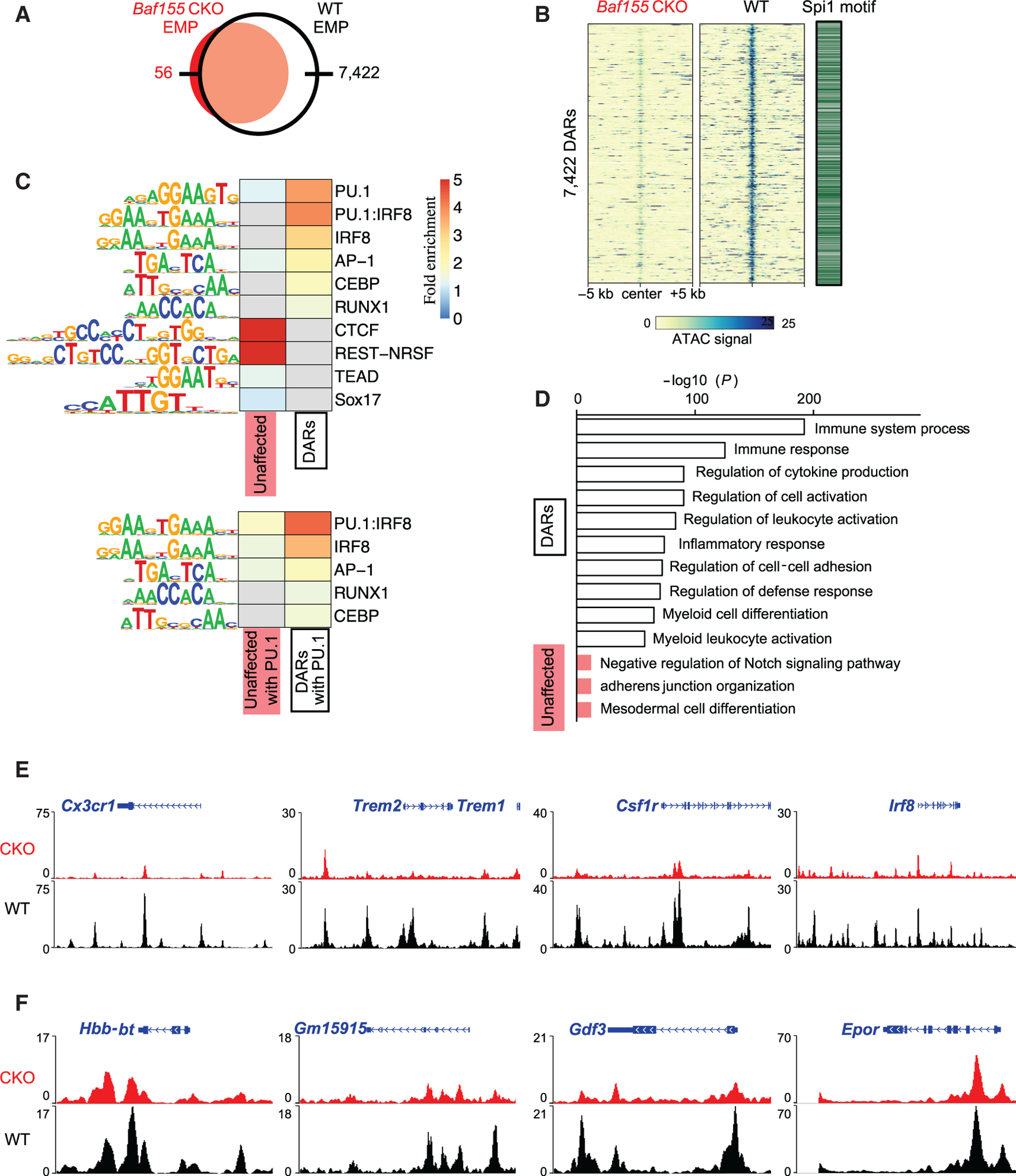
(A) A Venn diagram of the numbers of ATAC-seq peaks found in Baf155 CKO and WT EMPs.
(B) ATAC-seq signals over 10-kb regions centered on the differentially accessible regions (DARs) with reduced signals in Baf155 CKO EMPs compared with the WT (left) and the presence of the Spi1/Pu.1 motif in the DARs (right).
(C) Heatmaps of HOMER known TF motif fold enrichment in the DARs and unaffected accessible regions. Gray cells indicate no enrichment found (p > 0.05).
(D) Enriched Gene Ontology (GO) terms and their binomial p values from analyzing the DARs with reduced signals in Baf155 CKO EMPs (white) and the unaffected peaks (red) using GREAT.
(E) Epigenome browser views of representative myeloid gene loci.
(F) Epigenome browser views of representative erythroid gene loci.
See also Figure S4.
The genes associated with selective genomic regions with loss of chromatin accessibility in Baf155 CKO EMPs were enriched for biological functions related to immune responses, including inflammatory response and myeloid leukocyte activation, whereas genes with unaffected accessible regions were enriched for biological functions different from those (Figure 4D). Genes under the inflammatory response and myeloid leukocyte activation categories mainly include myeloid genes such as Cx3cr1, Trem2, Csf1r, Irf8, Emr1, and Fcgr3 (CD16). Importantly, chromatin of these gene loci was largely inaccessible in Baf155 CKO EMPs (Figures 4E and S4C), explaining the block of myeloid lineage differentiation. In contrast, EC gene loci, Kdr, Cdh5, and Esam, were similarly accessible in WT and Baf155 CKO EMPs (Figure S4D). EMP gene loci, cKit, Itga2b (Cd41), and Cd34, were also readily accessible in WT and Baf155 CKO EMPs (Figure S4E). Importantly, although chromatin of erythroid lineage genes that were commonly expressed in EryP cells was readily accessible, chromatin of the erythroid lineage genes that were specifically upregulated in EryD cells, Hbb-bt, Gm15915, and Gdf3, were not (Figure 4F). These data suggest that BAF155-mediated chromatin remodeling of myeloid and EryD gene loci at the EMP stage is necessary for subsequent myeloid and EryD lineage differentiation.
BAF155 Is Recruited to PU.1 Target Gene Loci
PU.1 is a master ETS factor critical for myeloid lineage development. PU.1 regulates its own expression (Chen et al., 1995), although the mechanisms of this autoregulation have not been elucidated clearly. Because reduced accessible regions in Baf155 CKO EMPs are represented predominantly by the ETS binding motif, we assessed whether PU.1 requires the BAF complex for activating the myeloid lineage program. Intriguingly, Pu.1 expression itself was diminished in CKO yolk sacs (Figure 5A). Chromatin of the Pu.1 locus was less accessible in CKO EMPs (Figure 5A). Because Baf155 KD in EMPs could block myeloid and EryD differentiation without affecting Pu.1 expression (Figures 2D and 2E), we reasoned that diminished expression of Pu.1 and its target genes in Baf155 CKO yolk sacs could be due to deficiency of BAF-mediated chromatin remodeling at Pu.1 and its target gene loci. We first assessed whether BAF155 is recruited to PU.1 target genes. We selected 7 genomic regions that contain the ETS motif and are differentially accessible in WT and Baf155 CKO yolk sacs. These include genomic regions that are associated with the Cx3cr1, Trem2, Csf1r, Irf8, and Cd68 genes (Figure 5B; Table S3). We also selected 2 unaffected ETS regions, UER1 and UER2, from the 1,472 peaks that contain the PU.1 motif but whose chromatin accessibility is unaffected by Baf155 deficiency (Figure 5B; Table S3). Although a significant mean enrichment was observed for PU.1 and BAF155 binding at these PU.1 target gene loci, only BAF155 binding, not PU.1, was enriched at UER1 and UER2 (Figure 5B). We next determined whether BAF155 can form a complex with PU.1. Specifically, we generated a mouse embryonic fibroblast (MEF) line that expresses Flag-Baf155 and Pu.1 or HA-Pu.1. Cells were then subjected to immunoprecipitation using an antibody against the FLAG tag, followed by PU.1 or hemagglutinin (HA) immunoblot. PU.1 was co-immunoprecipitated with BAF155 (Figure 5C). Conversely, when the anti-HA antibody was used for immunoprecipitation, BAF155 was co-immunoprecipitated with PU.1 (Figure S5A). As reported previously (Alver et al., 2017; Narayanan et al., 2015), we additionally found BRG1, p300, and KDM6a (UTX) to be co-immunoprecipitated with BAF155, suggesting that PU.1 activates its target genes by forming a transcriptional complex with BAF, p300, and KDM6a. Because p300 and KDM6a mainly target H3K27, we postulated that PU.1 target gene loci remain methylated at H3K27 sites in the absence of BAF155, leading to repression of PU.1 target gene expression. Indeed, although inaccessible regions in Baf155 CKO yolk sacs showed higher H3K27me3 levels compared with WT yolk sacs, H3K27me3 levels were similar at UER1 and UER2 in WT and CKO yolk sacs (Figure S5B). If this were truly the case, then we would expect that inhibition of EZH2, the catalytic subunit of Polycomb repressive complex 2 (PRC2), which methylates H3K27 (Laugesen et al., 2019), might rescue PU.1 target gene expression in Baf155 CKO yolk sacs. Thus, we set up matings between Tie2-Cre;Baf155f/+ (father) and Baf155f/f (mother) mice and injected GSK-126 (an EZH2 inhibitor) intraperitoneally into the mother at E8. E9.5 yolk sacs were collected and subjected to qRT-PCR. Although vehicle treatment did not affect myeloid lineage gene expression, indicating that the GSK-126 effect would be specific (Figure S5C), expression of many PU.1 target genes was rescued in Baf155 CKO yolk sacs when EZH2 was inhibited (Figure 5D). Pu.1/Spi1 expression was also rescued, suggesting the BAF-mediated remodeling mechanism of PU.1 autoregulation. It is worth noting that GSK-126 treatment led to baseline elevation of expression of some genes, including Irf8, Itgam, Gata2, Scl, and cKit (Figures 5D and S5D), suggesting that these genes are normally repressed by the EZH2-mediated mechanism.
Figure 5. BAF155 Interacts with PU.1 and Is Recruited to Its Target Genes.
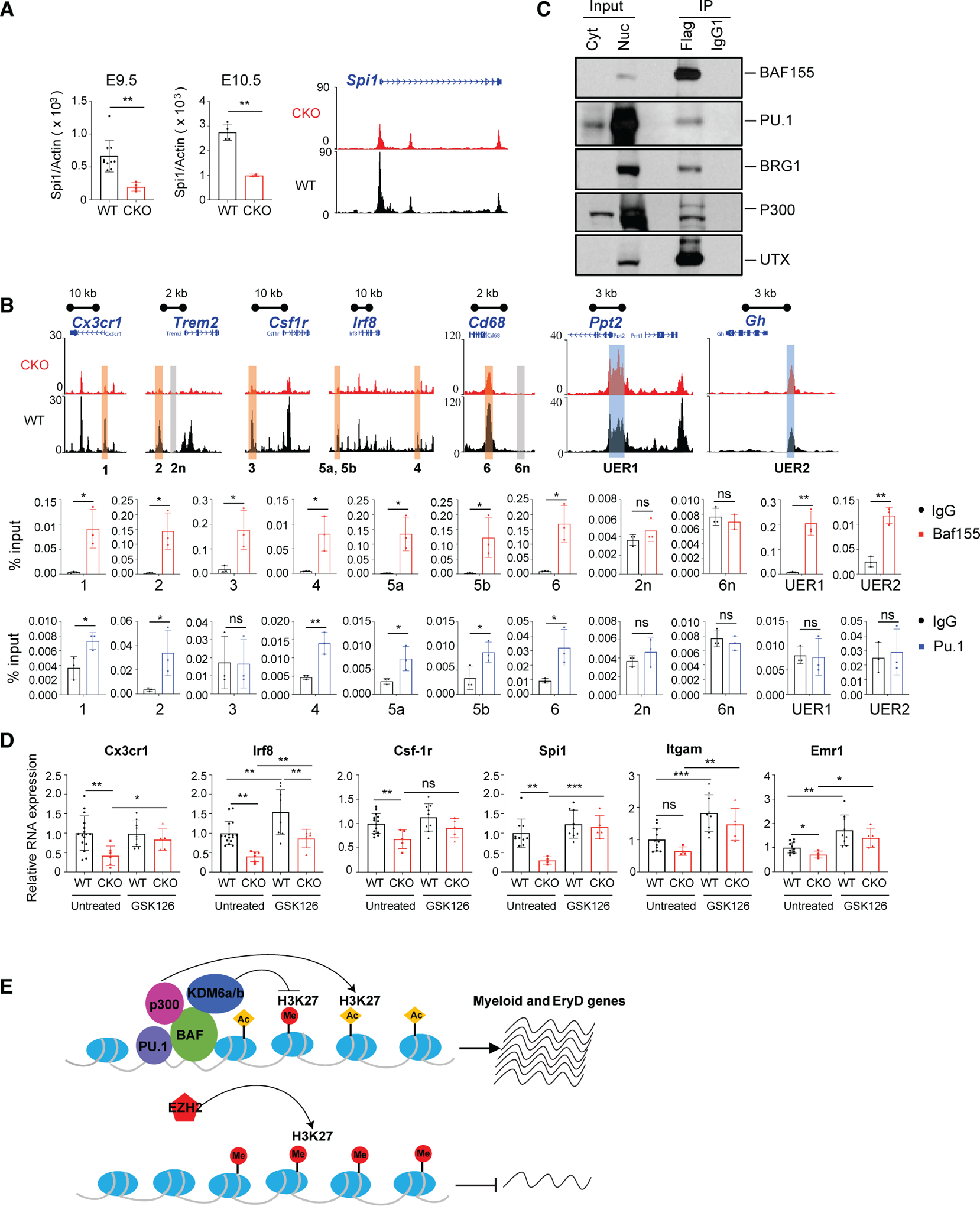
(A) qRT-PCR analysis of Pu.1 expression (top) and epigenome browser view of the Spi1/Pu.1 locus (bottom) from WT and Baf155 CKO YSs. Data are from at least two biological replicates for either genotype, with each replicate consisting of an individual YS. Data are presented as mean ± SD. Student’s t test; **p < 0.01.
(B) Epigenome browser views of selected myeloid and negative control genomic regions and unaffected ETS regions between WT and Baf155 CKO YS EMPs (top panel). Also shown is ChIP-qPCR showing enrichment of PU.1 and BAF155 binding at selected myeloid gene loci (highlighted regions in the top panel, 1–6), negative control gene loci (highlighted regions in the top panel, 2n and 6n), and unaffected ETS regions (highlighted regions in the top panel, UER1 and UER2) in E10.5 WT YSs (bottom panel). qPCR primers and genomic locations are provided in Table S2. Data are presented as mean ± SD; n = 3.
(C) Cytoplasmic (Cyt) and nuclear (Nuc) protein input and anti-FLAG and isotype control (IgG1) immunoprecipitation of nuclear extracts from MEFs over-expressing FLAG-Baf155 and Pu.1. Shown are immunoblots for BAF155, PU.1, BRG1, P300, and UTX (KDM6a).
(D) qRT-PCR analysis of Cx3cr1, Irf8, Csf-1r, Pu.1/Spi1, Itgam, and Emr1 gene expression in E9.5 WT and Baf155 CKO YSs with or without GSK126 treatment. Gene expression was normalized to the untreated WT mean value. Data are from at least four biological replicates for either genotype, with each replicate consisting of an individual YS. Data are presented as mean ± SD. Student’s t test; *p < 0.05, **p < 0.01, ***p < 0.001.
(E) A model showing BAF-mediated chromatin remodeling in PU.1 transcriptional gene activation.
See also Figure S5.
DISCUSSION
Chromatin remodeling by the mammalian BAF complex is required for the development of multiple lineages during embryogenesis. Brg1 deletion within the Tie2(+) lineage leads to defective yolk sac angiogenesis and primitive erythropoiesis. EryP cell defects in these mice are due to increased apoptosis and lack of embryonic α- and β-globin gene expression (Griffin et al., 2008). We show that Baf155 deletion within the Tie2(+) lineage also causes angiogenesis defects, as evidenced by hemorrhage in some Baf155 CKO mice. However, we found that embryonic globin gene expression was similar, and EryPs and their progenitors were present at similar levels in WT and Baf155 CKO yolk sacs. Unexpectedly, Baf155 CKO mice display defective yolk sac myelopoiesis and definitive erythropoiesis. We attribute the phenotype difference between the two mice, Tie2-Cre; Brg1f/f and Tie2-Cre; Baf155f/f, to the nature of the deleted gene. Brg1 encodes for an ATPase that is the core of the BAF complex, whereas Baf155 encodes a BAF structural component. Presumably, Baf155 deletion might have delayed the phenotype’s manifestation to reflect the hematopoietic lineage hierarchy; i.e., the EC and EryP lineages arise before EMPs, which generate myeloid and EryD lineages. Intriguingly, we observed that EC genes were still expressed in Baf155-deficient EMPs, suggesting that the extinction of previous lineage genes is necessary for new lineage establishment. Alternatively, the previous lineage gene loci are still accessible in the absence of the next lineage gene loci’s active chromatin remodeling. Collectively, these data establish that the BAF complex has a critical role in myeloid and EryD lineage differentiation from EMPs by remodeling the chromatin of the myeloid and EryD lineage gene loci.
Although chromatin remodeling is critical for developing many different lineages, it is still unclear whether and how the specificity of the BAF chromatin remodeling complex of the target genes is achieved. Our data demonstrate that BAF-mediated chromatin remodeling of myeloid and EryD lineage genes at the EMP stage is necessary for downstream myeloid and EryD lineage development. We found that DARs in WT and Baf155-deficient EMPs are enriched predominantly for the ETS motif. Moreover, BAF155 interacted with PU.1 and was recruited to PU.1 target gene loci. This strongly argues that the BAF complex’s target gene specificity is achieved by ETS TFs. Consistent with this idea, recent studies have shown that AP-1 and ETS motifs are enriched in enhancer regions sensitive to Smarcb1 loss (Alver et al., 2017). Moreover, TMPRSS2-ERG, a fusion gene product from a chromosomal translocation in prostate cancer, interacts with the BAF complex in an ETS-dependent manner (Sandoval et al., 2018), indicating that the BAF complex is required for ERG-mediated prostate oncogenesis. The BAF complex has been shown to interact with p300 and acetylates H3K27 (Alver et al., 2017). The BAF complex also interacts with KDM6a/6b and demethylates H3K27 (Narayanan et al., 2015). We also observed that BAF155 forms a complex with p300 and KDM6a. Our data show that DARs in Baf155 CKO EMPs included mostly PU.1 target genes and displayed higher H3K27me3 levels. EZH2 inhibitor treatment could rescue some of the PU.1 target gene expression in Baf155 CKO yolk sacs. These data suggest that PU.1 activates its target genes by forming a complex with BAF, p300, and KDM6a/6b and triggering H3K27 acetylation/demethylation of the target genes (Figure 5E). In the absence of the BAF complex, PU.1 target loci are occupied by EZH2, suppressing PU.1 target gene expression (Figure 5E).
Reduced Pu.1 expression leads to acute myeloid leukemia (Will et al., 2015; Steidl et al., 2007). Moreover, Pu.1 activation in hematopoietic stem and progenitor cells can lead to myeloid lineage skewing and deregulated hematopoiesis in chronic inflammatory conditions (Pietras et al., 2016; Etzrodt et al., 2019). Methylation of BAF155 at the R1064 residue by coactivator-associated arginine methyltransferase 1 (CARM1; also known as PRMT4) is critical for tumor progression and metastasis (Wang et al., 2014). Although CARM1 is essential for myeloid leukemogenesis, it is dispensable for normal hematopoiesis (Greenblatt et al., 2018). UTX (KDM6a) suppresses myeloid leukemogenesis partially by repressing an ETS-mediated transcriptional program (Gozdecka et al., 2018). Kdm6b is required for fetus-derived T-ALL and adult-derived AML (Mallaney et al., 2019). These studies collectively suggest that controlling Pu.1 expression and its activity might be critical for managing cancer and chronic inflammatory diseases. Intriguingly, ETS factors can regulate Baf155 expression (Ahn et al., 2005), supporting an interplay between ETS TFs and BAF expression and function. Future studies delineating the crosstalk between ETS factors and BAF and interaction among BAF, p300, PU.1, and KDM6a/6b and PU.1 target gene expression will be critical for further understanding myeloid lineage and leukemia development. Future applications of the epigenetics involving PU.1 and BAF155 expression and function for tissue repair, regeneration, and diseases are warranted.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author and Lead Contact, Dr. Kyunghee Choi (kchoi@wustl.edu).
Materials Availability
This study did not generate any unique reagents.
Data and Code Availability
The ATAC-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE144243 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144243).
The accession number for the scRNA-Seq data reported in this paper is GEO: GSE159381.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Tie2-Cre;Baf155 CKO mice were obtained by first crossing Tie2-Cre (Stock No: 4128, Jackson Labs) males (2–3 months old) to Baf155f/f (Choi et al., 2012) females (2–3 months old) to generate Tie2-cre; Baf155f/+ mice. Next, timed matings using Tie2-Cre; Baf155f/+ males (2–8 months old) and Baf155f/f females (2–3 months old) were set up in the evening and females checked for vaginal plugs the following morning (12pm = E0.5). Females were separated from males and housed in the animal barrier until the desired time point. Females were euthanized using CO2 asphyxiation and uteri removed for embryo collection. Embryos and collected tissue were kept on ice in PBS with 10% FBS until processed for analysis. Wild-type (WT) littermates were used as controls. Animal husbandry, generation, and handling were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Washington University School of Medicine in St. Louis.
Cell culture and transduction
The Mouse Embryonic Fibroblast (MEF) cell line has been previously described (Lybarger et al., 2003). MEF and MEF-derived stable cell lines were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM) (2440046, GIBCO) supplemented with 10% (v/v) Fetal bovine serum (FBS) (S12450, Atlanta Biologicals), and 100U/ml penicillin-streptomycin (15140122, GIBCO). MEF cells were transduced with Flag-Baf155-IRES-GFP lentiviral and Pu.1-IRES-mCherry retroviral particles. Hexadimethrine bromide (8 μg/ml) (H9268, Sigma) was added during transductions to increase viral particle uptake. Transduced cells were sorted twice to ensure greater than 90% purity. When HA-Pu.1 lentivirus was used, MEF-Flag-Baf155 cells were transduced with HA-Pu.1 lentiviral particles and selected with 1μg/ml Blasticidin S Hydrochloride for 2 weeks. The overexpression efficiency of target proteins was confirmed by western blot.
METHOD DETAILS
Genotyping
The following primers were used to obtain genotype information for breeders and embryos:
Baf155 - TGTCATCCATGAGGAGTGGTC3′ (F); 5′GGTAGCTCACAAATGCCTGT3′ (R); WT = 400 bp; Floxed = 450 bp. Cre −5′ACCAGAGACGGAAATCCATCG3′ (F); 5′CCACGACCA AGTGACAGCAATG3′ (R); Cre = 390 bp.
Lentiviral and retroviral particle production
Baf155 lentiviral plasmid DNA, pRRL_CAGpN-Flag-Baf155-IRES-GFP, was a gift from Jerry Crabtree (Addgene, plasmid# 2456). MISSION pLKO.1-puro-Ubc-TurboGFP (Sigma, SHC014) was used as a transduction efficiency control. The Pu.1 lentiviral plasmid was constructed by adding a HA tag at the Pu.1 N-terminal and inserting it into the CSII-EF-MCS-IRES2-bsr lentiviral backbone. Lentiviral packaging plasmid psPAX2 (Addgene, plasmid# 12260) and VSV-G envelope expressing plasmid pMD2.G (Addgene, plasmid# 12259) were gifts from Didier Trono. For cloning purposes, viral plasmid DNA was transformed using One Shot Stbl3 Chemically Competent E.Coli (C737303, ThermoFisher). Lentiviral particles were produced using the 293FT cell line (R70007, ThermoFisher), which was maintained in high glucose Dulbecco’s Modified Eagle Medium (DMEM) (11965092, GIBCO), 10% FBS, 200 mM L-Gluta-mine (35050061, GIBCO), 10 mM MEM Non-Essential Amino Acids (25–025-Cl, Corning), 100 mM MEM Sodium Pyruvate (25–000-Cl, Corning), and 500 μg/ml Geneticin (10131–035, GIBCO). Cells were transfected with lentiviral DNA using the calcium phosphate method. Sixteen hours after transfection, media was replaced and cells were incubated at 37°C in 5% CO2 for an additional 48 hours. Virus titer was determined by QuickTiter Lentivirus Associated HIV p24 Titer Kit (Cell Biolabs, INC). Pu.1-IRES-mCherry retroviral plasmid DNA was a gift from Ellen Rothenberg (Addgene, plasmid# 80140). Platinum-E (Plat-E) retroviral packaging cell line was used to generate the Pu.1 retrovirus using the calcium phosphate method. Cells were maintained in high glucose DMEM supplemented with 10% FBS, 1 μg/ml puromycin (P8833, Sigma), 10 μg/ml Blasticidin S Hydrochloride (B12200, Research Products International Corp), and 100 U/ml penicillin-streptomycin. Media was replaced the following morning and virus harvested 48 hours after transfection.
esiRNA transfection
E10.5 WT yolk sac (YS) cKIT+ cells (1×104) were plated in 100 μL maturation media (IMDM, 20% FBS, 1% interleukin-3 (IL-3) super-natant, 10 ng/ml murine stem cell factor (PeproTech), 10 ng/ml M-CSF (PeproTech), 10 ng/ml GM-CSF (PeproTech), 10 ng/ml IL-6, 10 ng/ml IL-11 (R&D Systems) and 2 U/ml erythropoietin (PeproTech) in a 96-well plate and transfected with 300 ng esiRNA against either Baf155 (Sigma, EMU012611) or Egfp (Sigma, EHUEGFP) with 2 μL lipofectamine 3000 (Thermofisher). Cells were cultured in a 37°C incubator with 5% CO2 for 36–48 hours and then subjected to either RNA extraction or re-plating in methylcellulose (MethoCult 3434, Stem Cell Technologies).
Hematopoietic progenitor assays
Methylcellulose colony-forming assays were performed using MethoCult 3434 (Stem Cell Technologies). E10.5 WT YS were pooled and sorted for either cKIT+ or cKIT+CD41+CD16/32+ populations. Sorted cells were mixed in methylcellulose (2,000/ml) and plated in triplicate using 35mm Petri-dishes. Cultures were maintained in a humidified incubator at 37°C, 5% CO2. CFU-E colonies were counted after 2–3 days of culture. Primitive erythroid, definitive erythroid (BFU-E), macrophage, and granulocyte/macrophage colonies were counted following 5–7 days of culture.
In vivo GSK-126 treatment
GSK-126 (HY-13470, MedChem) was dissolved in SBE-β-CD (HY-17031, MedChem) at a final concentration of 20 mg/ml. Pregnant mice (E8) were injected intraperitoneally with equal volumes of either SBE-β-CD (vehicle) or GSK126 (100mg/kg). At E9.5, pregnant females were euthanized using CO2 asphyxiation and the uteri removed for embryo collection and dissection.
Nuclear extract preparation
Transduced MEF cells expressing Baf155 and Pu.1 were treated with DSP (Dithiobis [succinimidyl propionate], Thermo Scientific), a membrane permeable cleavable crosslinker, before subjected to nuclear extraction. Cells were detached using 0.25% trypsin-EDTA, washed twice with phosphate-buffered saline (PBS), resuspended in PBS containing 1mM DSP at approximately 1×107/ml, and incubated at room temperature (RT) for 30 min. The crosslink reaction was stopped by adding 1M Tris-HCl (pH7.5) at a final concentration of 20mM for 10min. DSP treated cells after wash were then incubated in hypotonic buffer (25mM HEPES (pH7.6), 25mM KCl, 5mM MgCl2, 0.05mM EDTA, 0.1% NP40, 5% Glycerol, 1mM PMSF) on ice for 10min, vigorously vortexed for 10 s, and centrifuged at 13,000 xg for 1 min. Supernatant, primarily containing soluble cytosolic protein, was collected for immunoblot while the pellet containing nuclei was resuspended in nuclear extraction buffer (10mM HEPES (pH7.6), 100mM KCl, 3mM MgCl2, 0.1mM EDTA, 5% lycerol supplemented with 1mM PMSF and 1X protease inhibitor cocktail (complete mini Roche, 11836170001, Sigma)), incubated on ice for 15min, and followed by 3×10s of sonication at 50% amplitude. Insoluble proteins and debris were removed from the nuclear extract by high-speed centrifugation (10 min at 18,000 g).
Immunoprecipitation
Nuclear extract was mixed 1:1 with IP buffer (10 mM Tris-HCl (pH 7.5), 200 mM NaCl, 1 mM EDTA, 0.2% Tween 20, 1X protease inhibitor cocktail, 20mM Iodoacetamide) and precleared by Sepharose 4B (4B200, Sigma). Immunoprecipitation was performed by incubating precleared nuclear extract with antibody-bound beads (anti-Flag M2 affinity gel, anti-HA (clone HA-7) affinity gel, or Protein A as a negative control (Sigma)) at 4°C overnight. Precipitates were washed 4 times with washing buffer (10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1% Tween 20, 20 mM Iodoacetamide) and eluted in 1X LDS buffer (Invitrogen). DSP was cleaved by adding 5% beta- mercaptoethanol in LDS loading buffer at 100°C for 10 min.
Western blotting
Western blotting was conducted following standard protocols. Primary antibodies used for western blotting are listed in key resources table. Secondary antibodies were horseradish peroxidase-conjugated mouse anti-rabbit IgG light chain and mouse IgG kappa chain binding protein (Santa Cruz Biotechnology, 1:10,000 dilution). Membranes were developed with ECL chemiluminescence substrate (ThermoFisher) and visualized using photographic film.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CDD45-BV421 (Clone 30-F11) | BioLegend | Cat#103134; RRID:AB_2562559 |
| Anti-mouse CDD45-APC (Clone 30-F11) | BioLegend | Cat#103112; RRID:AB_312977 |
| Anti-mouse TER119-PE (Clone TER-119) | BioLegend | Cat#116208; RRID:AB_313709 |
| Anti-mouse TER119-APC-Cy7 (Clone TER-119) | BioLegend | Cat#116223; RRID:AB_2137788 |
| Anti-mouse CD31-Biotin (Clone MEC13.3) | BioLegend | Cat#102504; RRID:AB_312911 |
| Anti-mouse CD31-FITC (Clone MEC13.3) | BD Biosciences | Cat#553372; RRID:AB_394818 |
| Anti-mouse/human CD11b-BV421 (Clone M1/70) | BioLegend | Cat#101235; RRID:AB_10897942 |
| Anti-mouse/human CD11b-Biotin (Clone M1/70) | BioLegend | Cat#101204; RRID:AB_312787 |
| Anti-mouse F4/80-PE (Clone BM8) | BioLegend | Cat#123110; RRID:AB_893486 |
| Anti-mouse F4/80-APC (Clone BM8) | BioLegend | Cat#123116; RRID:AB_893481 |
| Anti-mouse CX3CR1-PE (Clone SA011F11) | BioLegend | Cat#149006; RRID:AB_2564315 |
| Anti-mouse CD117(c-Kit)-PE (Clone 2B8) | BioLegend | Cat#105807; RRID:AB_313216 |
| Anti-mouse CD117(c-Kit)-APC (Clone 2B8) | BioLegend | Cat#105812; RRID:AB_313221 |
| Anti-mouse CD41-APC (Clone MWReg30) | BioLegend | Cat#133914; RRID:AB_11125581 |
| Anti-mouse CD16/32-BV421 (Clone 93) | BioLegend | Cat#101331; RRID:AB_2562188 |
| Anti-mouse CD16/32-PE (Clone 2.4G2) | BD Biosciences | Cat#553145; RRID:AB_394660 |
| Purified anti-mouse CD16/32 (Clone 93) | BioLegend | Cat# 101302; RRID:AB_312801 |
| BV421-Streptavidin | BioLegend | Cat#405225 (No RRID number available) |
| BV605-Streptavidin | BD Biosciences | Cat#563260 (No RRID number available) |
| SMARCC1/BAF155 (D7F8S) Rabbit mAb antibody | Cell Signaling Tech. | Cat#11956; RRID:AB_2797776 |
| PU.1 (9G7) Rabbit mAb antibody | Cell Signaling Tech. | Cat# 2258; RRID:AB_2186909 |
| p300 (D8Z4E) Rabbit mAb antibody | Cell Signaling Tech. | Cat# 86377; RRID:AB_2800077 |
| Brg1 (D1Q7F) Rabbit mAb antibody | Cell Signaling Tech. | Cat# 49360; RRID:AB_2728743 |
| UTX (D3Q1I) Rabbit mAb antibody | Cell Signaling Tech. | Cat# 33510; RRID:AB_2721244 |
| HA-Tag(C29F4) Rabbit mAb antibody | Cell Signaling Tech. | Cat# 3724; RRID:AB_1549585 |
| Rabbit IgG | Cell Signaling Tech. | Cat#2729; RRID:AB_1031062 |
| Rabbit Anti-Histone H3, trimethyl (Lys27) Polyclonal antibody | Millipore | Cat# 07–449; RRID:AB_310624 |
| Monoclonal ANTI-FLAG® M2 antibody | Sigma-Aldrich | Cat# F3165; RRID:AB_259529 |
| EZview Red ANTI-FLAG® M2 Affinity Gel | Sigma-Aldrich | Cat# F2426; RRID:AB_2616449 |
| EZview Red Anti-HA Affinity Gel | Sigma-Aldrich | Cat# E6779; RRID:AB_10109562 |
| Bacterial and Virus Strains | ||
| One Shot Stbl3 Chemically Competent E.Coli | Thermo Fisher Scientific | Cat#C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | GIBCO | Cat#11965092 |
| FBS | Atlanta Biologicals | Cat#S12450 |
| L-Glutamine | GIBCO | Cat#35050061 |
| MEM Non-Essential Amino Acids | Corning | Cat#25–025-Cl |
| MEM Sodium Pyruvate | Corning | Cat#25–000-Cl |
| Geneticin | GIBCO | Cat#10131–035 |
| Puromycin | Sigma | Cat#P8833 |
| Blasticidin S Hydrochloride | Research Products International Corp | Cat#B12200 |
| penicillin-streptomycin | GIBCO | Cat# 15140122 |
| IMDM | GIBCO | Cat#2440046 |
| Hexadimethrine bromide | Sigma | Cat#H9268 |
| Interleukin-3 (IL-3) supernatant | This paper | N/A |
| Murine stem cell factor | PeproTech | Cat#250–03 |
| M-CSF | PeproTech | Cat#315–02 |
| GM-CSF | PeproTech | Cat#315–03 |
| IL-6 | PeproTech | Cat#216–16 |
| IL-11 | R&D Systems | Cat#418-ML |
| Erythropoietin (EPO) | PeproTech | Cat#100–64 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat#L3000–001 |
| esiBaf155 | Sigma | Cat#EMU012611–50UG |
| esiEGFP | Sigma | Cat#EHUEGFP-50UG |
| MethoCult3434 | Stem Cell Technologies | Cat#M3434 |
| GSK-126 | MedChem | Cat#HY-13470 |
| SBE-β-CD | MedChem | Cat# HY-17031 |
| DSP (Dithiobis (succinimidyl propionate)) | Thermo Fisher Scientific | Cat#22585 |
| 0.25% trypsin-EDTA | GIBCO | Cat#25200–056 |
| Protease inhibitor cocktail | Sigma | Cat#11836170001 |
| Tween 20 | Sigma | Cat#P9416 |
| Iodoacetamide | Sigma | Cat#I6125 |
| Sepharose 4B | Sigma | Cat#4B200 |
| LDS buffer | Invitrogen | Cat#NP0007 |
| Beta – mercaptoethanol | Sigma | Cat#444203 |
| ECL chemiluminescence substrate | Thermo Fisher Scientific | Cat#32106 |
| Collagenase type IV | Worthington | Cat#LS004188 |
| Deoxyribonuclease I | Worthington | Cat#LS002139 |
| 0.25% Collagenase | Stem Cell Technologies | Cat#07902 |
| ProteinA-Sepharose® 4 | Sigma-Aldrich | Cat#P9424 |
| Critical Commercial Assays | ||
| QuickTiter Lentivirus Associated HIV p24 Titer Kit | Cell Biolabs, INC | Cat#VPK-107 |
| SimpleChIP® Plus Enzymatic Chromatin IP Kit | Cell Signaling Tech. | Cat#9005 |
| RNeasy Micro/Mini Kit | QIAGEN | Cat#74004/74106 |
| qScript cDNA SuperMix | Quanta | Cat#101414–106 |
| DNA Clean and Concentrator 5 | Zymo Research | Cat#D4014 |
| AMPure XP beads | Beckman Coulter | N/A |
| Deposited Data | ||
| ATAC-seq data | This paper | GEO: GSE144243 |
| GRCm10/mm10 | UCSC genome browser | http://hgdownload.soe.ucsc.edu/goldenPath/mm10/bigZips/ |
| scRNA-seq data | This paper | GEO: GSE159381 |
| scRNA-seq data of WT cells | Zhao and Choi, 2019 | GEO: GSE130146 |
| Experimental Models: Cell Lines | ||
| 293FT cell line | ThermoFisher | Cat#R70007 |
| Platinum-E (Plat-E) retroviral packaging cell line | Cell Biolabs, INC | Cat#RV-101 |
| Mouse Embryonic Fibroblast (MEF) | Hansen, T.H. Washington University in St. Louis; Lybarger et al., 2003 | N/A |
| MEF-Flag-Baf155-IRES-GFP+Pu.1-IRES-mCherry | This paper | N/A |
| MEF-Flag-Baf155+HA-PU.1 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BI6/J Wild Type | Jackson Laboratories | Stock No:000664 |
| Tie2-Cre | Jackson Laboratories | Stock No:004128 |
| Baf155f/f | Rho Hyun Seong; Choi et al., 2012 | N/A |
| Oligonucleotides | ||
| See Tables S2 and S3 for a list of oligonucleotide sequences | N/A | |
| Recombinant DNA | ||
| pRRL_CAGpN-Flag-Baf155-IRES-GFP | Addgene | Cat#24561; RRID:Addgene_24561 |
| CSII-EF-MCS-IRES2-bsr-PU.1-HA | This paper | N/A |
| psPAX2 | Addgene | Cat#12260; RRID:Addgene_12260 |
| pMD2.G | Addgene | Cat#12259; RRID:Addgene_12259 |
| Pu.1-IRES-mCherry retroviral plasmid DNA | Addgene | Cat#80140; RRID:Addgene_80140 |
| pLKO.1-puro-Ubc-TurboGFP | Sigma | Cat#SHC014; (No RRID number available) |
| Software and Algorithms | ||
| FlowJo software version 10.5.3 | TreeStar Inc. | https://www.flowjo.com |
| Graphpad Prism version 8.4.3 | Graphpad Software, LLC. | https://www.graphpad.com/scientific-software/prism/ |
| Cutadapt version 1.11 | Martin, 2011 | https://github.com/marcelm/cutadapt/ |
| Bowtie 2 version 2.3.4.1 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS2 version 2.1.1 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| WashU Epigenome Browser | Zhou et al., 2011 | https://epigenomegateway.wustl.edu/ |
| DiffBind version 2.6.6 | Ross-Innes et al., 2012 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| deepTools | Ramírez et al., 2016 | https://github.com/deeptools/deepTools |
| GREAT version 4.0.4 | McLean et al., 2010 | http://great.stanford.edu/public/htmI/index.php |
| HOMER version 4.8 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/index.html |
| Cell Ranger Single Cell Software Suite (v2.0.1) | Cell Ranger | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Seurat version 2.3.4 | Seurat | https://satijalab.org/seurat/ |
| Other | ||
| Fisherbrand Model 120 Sonic Dismembrator | Fisher scientific | N/A |
Chromatin immunoprecipitation (ChIP) coupled with quantitative real-time PCR (ChIP-qPCR)
Chromatin immunoprecipitation (ChIP) was performed according to the manufacture’s protocol (ChIP kit, 9005, Cell Signaling Technologies) with the following modification: 25 mg cross-linked YS tissue was used per preparation. After cell lysis, nuclei extracts were digested by adding 0.5 μL Micrococcal Nuclease per IP prep and incubating for 20 min at 37°C with frequent mixing to digest DNA to a size of approximately 150–900 bp. Digestion was stopped by adding 10 μL 0.5 M EDTA and samples placed on ice for 2 min. Nuclei was pelleted and resuspend in 100 μL ChIP buffer. Nuclear lysates were further subjected to sonication to break nuclear membrane using a 120 Sonic Dismembrator (Fisher Scientific) at 4°C for 3 cycles, cycling ON for 10 s and OFF for 30 s at 50% amplitude. Approximately 10 μg of digested, cross-linked chromatin and 5 μg of antibody (BAF155, Cell Signaling Technologies, 11956; PU.1, Cell Signaling Technologies, 2258; H3K27me3, Millipore,07–449) were used per immunoprecipitation. IP samples were incubated overnight at 4°C with rotation, followed by 30 μL of protein G Magnetic Beads per IP reaction, and incubated for an additional 2 hr at 4°C with rotation. After elution of chromatin from the antibody/protein G magnetic beads, reverse cross-link performed by adding 6 μL 5 M NaCl and 2 μL proteinase K per IP, and incubating for 6 hr at 65°C. Immunoprecipitated DNA fragments were isolated using spin columns provided by the kit and subjected to qPCR with appropriate primers indicated in Table S2. Rabbit IgG (Cell Signaling Technologies, 2729) was used as a negative control. Quantitative PCR was performed in triplicate from 3 independent experiments, and data were normalized to input values.
Tissue processing for flow cytometry
YS were collected between E8.25–10.5, and brain rudiments collected between E9.5–10.5. To obtain single-cell suspension, tissues were incubated in Hank’s balanced salt solution (HBSS) containing 0.2 mg/ml collagenase type IV (Worthington), 100 U/ml deoxyribonuclease I (Worthington) and 5% FBS at 37°C for 1 h with tubes inverted every 5 to 10 min. Tissues were further dissociated by gently passing through a 20G needle 5 to 10 times. Cells were pelleted and resuspend in 0.3–1 mL IMDM media with 10% FBS. Cells were then passed through a 70 μm cell strainer and counted for viability.
Flow cytometry and cell sorting
Single cell suspensions were centrifuged at 400 g for 5 min, resuspended in 200 μL staining buffer (1X PBS, 1% BAS, 2 mM EDTA), placed in 5ml round-bottom tubes, and immunolabeled for FACS analysis. Before immunostaining, cell suspensions were pre-incubated with diluted (1:50) purified anti-CD16/32 (clone: 93, Biolegend, 101302) for 10 min on ice to block non-specific binding to Fcreceptors. Next, antibodies were added and incubated for 40 min on ice. Where appropriate, cells were further incubated with streptavidin conjugates for 20 min. All antibodies used can be found in key resources table. All FACS analyses were carried out on LSR Fortessa or Fortessa X-20 (BD Biosciences). Cell sorting was performed on FACS Aria II (BD Biosciences) sorter using 85 μm nozzle. All data were analyzed using FlowJo10 software (Tree Star).
Quantitative real-time reverse transcription PCR (qRT-PCR)
Total RNA from YS was prepared with RNeasy Micro/Mini Kit (QIAGEN), and reverse-transcribed into cDNA with qScript cDNA SuperMix (101414–106, Quanta) according to the manufacturer’s protocol. Gene expression was measured by quantitative real-time PCR with primers indicated in Table S3. Gene expression levels were normalized to β-actin.
Single-cell RNA sequencing
An equal number of E9.5 and E10.5 WT YS were combined and dissociated with 0.25% collagenase at 37°C for 30 minutes. Cells were briefly stored at 80°C in 90% FBS and 10% DMSO. Cells were thawed, washed with PBS, and stained with TER-119 antibody. Dead cells and TER-119+ cells were excluded by sorting to enrich live non-erythroid cells. Single cell suspension at 300 cells/μL in PBS were subjected to Chromium 10x Genomics library construction and HiSeq2500 sequencing (The Genome Technology Access Center, Washington University in St. Louis).
ATAC-seq library generation, sequencing, and mapping
For ATAC-seq library generation, approximately 50,000 cKIT+ cells were isolated from WT and Baf155 CKO YS using FACS sorter as described above. ATAC-seq libraries were generated following the Omni-ATAC protocol (Corces et al., 2017) with the following modification: Cells were harvested by centrifuging at 500 g for 5 minutes at 4°C. Supernatant was carefully aspirated and cells were washed once with cold PBS. Cell pellets were lysed in 100 μl of ATAC-seq RSB (10 mM Tris pH 7.4, 10 mM NaCl, 3 mM MgCl2) containing 0.1% NP40, 0.1% Tween-20, and 0.01% Digitonin by pipetting up and down and incubating on ice for 3 minutes. Next, 1 mL of ATAC-seq RSB containing 0.1% Tween-20 was added and mixed with the lysis reaction. Nuclei were pelleted by centrifuging at 800 g for 5 minutes at 4°C. Supernatant was carefully removed, and nuclear pellets were resuspended in 20 μl 2x TD buffer (20 mM Tris pH 7.6, 10 mM MgCl2, 20% Dimethyl Formamide). Nuclei were counted using trypan blue. Approximately 50,000 nuclei were transferred to 25 μL of 2x TD buffer. 25 μl of transposition mix (2.5 μl transposase (100 nM final), 16.5 μl PBS, 0.5 μl 1% digitonin, 0.5 μl 10% Tween-20, and 5 μl H2O) was then added to the nuclei. Transposition reactions were mixed and incubated at 37°C for 30 min gently tapping every 10 min to mix. Reactions were cleaned up with Zymo DNA Clean and Concentrator 5 columns. ATAC-seq library was prepared by amplifying the DNA for 9 cycles on a thermal cycler. The PCR reaction was purified with AMPure XP beads using double size selection following the manufacture’s protocol, in which 27.5 μl beads (0.55x sample volume) and 50 μl beads (1.5x sample volume) were used based on 50 μl PCR reaction. ATAC-seq libraries were quantitated by Qubit assays. Paired-end ATAC-seq libraries were sequenced on an Illumina NextSeq 500 machine. The reads were de-multiplexed by using sample-specific index sequences. Nextera adaptor sequences were trimmed by using cutadapt (Martin, 2011) version 1.11. The trimmed reads were mapped to the mouse genome sequence by using bowtie2 (Langmead and Salzberg, 2012) version 2.3.4.1 with the following parameters: −local −k 4 −X 2000 −mm. Secondary alignment, multiply mapped reads, and PCR duplicated reads were removed from the total aligned reads.
QUANTIFICATION AND STATISTICAL ANALYSIS
scRNA-seq bioinformatics analyses
The sequenced reads were mapped to the GRCm38 assembly using Cell Range 2.0.1 (10× Genomics). Sample demultiplexing, bar-code processing, and single-cell 3′ counting was performed using the Cell Ranger Single-Cell Software Suite (10× Genomics). Cellranger count was used to align samples to the mm10 reference genome, quantify reads, and filter reads with a quality score below 30. The resultant files were input into Seurat for normalization across all samples and merging. The Seurat package in R was used for subsequent analysis (Butler et al., 2018). Cells with Mitochondrial content greater than 5 percent were removed for downstream analysis. Data were normalized using a scaling factor of 10,000, and nUMI was regressed with a negative binomial model. Principal component analysis was performed using the top 3000 most variable genes and t-SNE analysis was performed with the top 15 PCAs. Clustering was performed using the FindClusters function which works on K-nearest neighbor (KNN) graph model with the granularity ranging from 0.1–0.9 and selected 0.6 for the downstream clustering. For identifying the markers for each cluster, we performed differential expression of each cluster against all other clusters identifying negative and positive markers for that cluster
Identification of ATAC peaks
Filtered aligned ATAC-seq reads were used to map to the transposon insertion sites, and ATAC peaks were called from those insertion sites. First, ATAC-seq reads mapped to mitochondrial DNA were removed from the aligned reads. Both ends of the paired-end reads were then treated as two Tn5 insertion sites. Tn5 insertion sites were adjusted to reflect the actual binding center of transpo-sons as follows. All reads mapped to the + strand were offset by +4 bp, and all reads mapped to the – strand were offset by −5 bp. The ATAC peaks were identified from these insertion sites by using the MACS2 (Zhang et al., 2008) version 2.1.1 callpeak function with the following parameters: −g mm --keep-dup all −B --SPMR --nomodel --extsize 73 --shift −37 −p 0.01 --call-summits. The ATAC-seq signals were visualized on the WashU Epigenome Browser (Zhou et al., 2011) as fold change over background using bedGraph tracks generated by using the MACS2 bdgcmp function with the following parameter: −m FE.
Identification and analyses of DARs
To identify DARs, DiffBind (Ross-Innes et al., 2012) version 2.6.6 was used on the union set of ATAC peaks with the following parameters: minOverlap = 1, fragmentSize = 1, summits = 0. DARs were defined as the ATAC peaks with fold change > 2 and P-value < 0.05. Unaffected accessible regions were defined as the ATAC peaks that are present both wild-type and Baf155 CKO cells and that are also with fold change < 1.1 and P-value > 0.05 from DiffBind. Heatmaps of ATAC-seq signal levels of DARs along with their neighboring regions were plotted by using deepTools (Ramírez et al., 2016). Gene Ontology enrichment analysis on DARs and unaffected regions were performed using GREAT (McLean et al., 2010) version 4.0.4. Motif enrichment analysis on DARs was performed using HOMER (Heinz et al., 2010) version 4.8. HOMER scanned the sequences of DARs for known motifs, and calculated enrichment score P-values using a binomial test. The heatmap of the selected known motifs were plotted using fold enrichment against the background. HOMER also discovered de novo motifs with their best matches to a known motif in DARs.
Statistics
GraphPad Prism 8 software was used for performing statistical analysis and generating graphs/plots. Data are presented as mean with standard deviation for all the measurements. All experimental data were reliably reproduced in two or more individual biological replicates. Details of the statistical tests performed are given in the respective figure legends. p < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
Baf155 is required for yolk sac myeloid and definitive erythroid lineage development
BAF-mediated chromatin remodeling of myeloid gene loci occurs at the EMP stage
Inaccessible chromatin in Baf155-deficient EMPs is enriched by the ETS binding motif
BAF155 interacts with PU.1 and is recruited to PUx.1 target gene loci
ACKNOWLEDGMENTS
We thank the Choi lab members for constructive criticism and support. We also thank the Washington University Pathology FACS core. This work was supported by NIH grants R01HL55337 and R01HL149954 (to K.C) and the Edward Mallinckrodt Foundation (to K.C. and D.H.F.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108395.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahn J, Ko M, Lee K, Oh J, Jeon SH, and Seong RH (2005). Expression of SRG3, a core component of mouse SWI/SNF chromatin-remodeling complex, is regulated by cooperative interactions between Sp1/Sp3 and Ets transcription factors. Biochem. Biophys. Res. Commun 338, 1435–1446. [DOI] [PubMed] [Google Scholar]
- Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, Haswell JR, Park PJ, and Roberts CW (2017). The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun 8, 14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzoni E, Frontera V, McGrath KE, Harman J, Carrelha J, Nerlov C, Palis J, Jacobsen SEW, and de Bruijn MF (2018). Kit ligand has a critical role in mouse yolk sac and aorta-gonad-mesonephros hematopoiesis. EMBO Rep 19, e45477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G, and Magnuson T (2000). A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell 6, 1287–1295. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ray-Gallet D, Zhang P, Hetherington CJ, Gonzalez DA, Zhang DE, Moreau-Gachelin F, and Tenen DG (1995). PU.1 (Spi-1) autoregulates its expression in myeloid cells. Oncogene 11, 1549–1560. [PubMed] [Google Scholar]
- Chen MJ, Li Y, De Obaldia ME, Yang Q, Yzaguirre AD, Yamada-Inagawa T, Vink CS, Bhandoola A, Dzierzak E, and Speck NA (2011). Erythroid/myeloid progenitors and hematopoietic stem cells originate from distinct populations of endothelial cells. Cell Stem Cell 9, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Ko M, Jeon S, Jeon Y, Park K, Lee C, Lee H, and Seong RH (2012). The SWI/SNF-like BAF complex is essential for early B cell development. J. Immunol 188, 3791–3803. [DOI] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzrodt M, Ahmed N, Hoppe PS, Loeffler D, Skylaki S, Hilsenbeck O, Kokkaliaris KD, Kaltenbach HM, Stelling J, Nerlov C, and Schroeder T (2019). Inflammatory signals directly instruct PU.1 in HSCs via TNF. Blood 133, 816–819. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, and Rodewald HR (2015). Tissue-resident macrophages originate from yolk-sac-derived erythromyeloid progenitors. Nature 518, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozdecka M, Meduri E, Mazan M, Tzelepis K, Dudek M, Knights AJ, Pardo M, Yu L, Choudhary JS, Metzakopian E, et al. (2018). UTX-mediated enhancer and chromatin remodeling suppresses myeloid leukemogenesis through noncatalytic inverse regulation of ETS and GATA programs. Nat. Genet 50, 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt SM, Man N, Hamard PJ, Asai T, Karl D, Martinez C, Bilbao D, Stathias V, Jermakowicz AM, Duffort S, et al. (2018). CARM1 Is Essential for Myeloid Leukemogenesis but Dispensable for Normal Hematopoiesis. Cancer Cell 33, 1111–1127.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin CT, Brennan J, and Magnuson T (2008). The chromatin-remodeling enzyme BRG1 plays an essential role in primitive erythropoiesis and vascular development. Development 135, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Jeon S, Sohn DH, Lee C, Ahn S, Kim WK, Chung H, and Seong RH (2008). SRG3, a core component of mouse SWI/SNF complex, is essential for extra-embryonic vascular development. Dev. Biol 315, 136–146. [DOI] [PubMed] [Google Scholar]
- Hargreaves DC, and Crabtree GR (2011). ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 21, 396–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, et al. (2013). Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffel G, and Ginhoux F (2018). Fetal monocytes and the origins of tissue-resident macrophages. Cell. Immunol 330, 5–15. [DOI] [PubMed] [Google Scholar]
- Kadoch C, and Crabtree GR (2015). Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv 1, e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Huh SO, Choi H, Lee KS, Shin D, Lee C, Nam JS, Kim H, Chung H, Lee HW, et al. (2001). Srg3, a mouse homolog of yeast SWI3, is essential for early embryogenesis and involved in brain development. Mol. Cell. Biol 21, 7787–7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugesen A, Højfeldt JW, and Helin K (2019). Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 74, 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickert H, Takeuchi JK, Von Both I, Walls JR, McAuliffe F, Adamson SL, Henkelman RM, Wrana JL, Rossant J, and Bruneau BG (2004). Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature 432, 107–112. [DOI] [PubMed] [Google Scholar]
- Lybarger L, Wang X, Harris MR, Virgin HW 4th, and Hansen TH (2003). Virus subversion of the MHC class I peptide-loading complex. Immunity 18, 121–130. [DOI] [PubMed] [Google Scholar]
- Mallaney C, Ostrander EL, Celik H, Kramer AC, Martens A, Kothari A, Koh WK, Haussler E, Iwamori N, Gontarz P, et al. (2019). Kdm6b regulates context-dependent hematopoietic stem cell self-renewal and leukemogenesis. Leukemia 33, 2506–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 17, 10–12. [Google Scholar]
- McGrath KE, Frame JM, Fegan KH, Bowen JR, Conway SJ, Catherman SC, Kingsley PD, Koniski AD, and Palis J (2015). Distinct Sources of Hematopoietic Progenitors Emerge before HSCs and Provide Functional Blood Cells in the Mammalian Embryo. Cell Rep 11, 1892–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Pirouz M, Kerimoglu C, Pham L, Wagener RJ, Kiszka KA, Rosenbusch J, Seong RH, Kessel M, Fischer A, et al. (2015). Loss of BAF (mSWI/SNF) Complexes Causes Global Transcriptional and Chromatin State Changes in Forebrain Development. Cell Rep 13, 1842–1854. [DOI] [PubMed] [Google Scholar]
- Palis J (2016). Hematopoietic stem cell-independent hematopoiesis: emergence of erythroid, megakaryocyte, and myeloid potential in the mammalian embryo. FEBS Lett 590, 3965–3974. [DOI] [PubMed] [Google Scholar]
- Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. (2016). Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol 18, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44 (W1), W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval GJ, Pulice JL, Pakula H, Schenone M, Takeda DY, Pop M, Boulay G, Williamson KE, McBride MJ, Pan J, et al. (2018). Binding of TMPRSS2-ERG to BAF Chromatin Remodeling Complexes Mediates Prostate Oncogenesis. Mol. Cell 71, 554–566.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, et al. (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90. [DOI] [PubMed] [Google Scholar]
- Steidl U, Steidl C, Ebralidze A, Chapuy B, Han HJ, Will B, Rosenbauer F, Becker A, Wagner K, Koschmieder S, et al. (2007). A distal single nucleotide polymorphism alters long-range regulation of the PU.1 gene in acute myeloid leukemia. J. Clin. Invest 117, 2611–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi JK, and Bruneau BG (2009). Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature 459, 708–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tober J, Koniski A, McGrath KE, Vemishetti R, Emerson R, de Mesy-Bentley KK, Waugh R, and Palis J (2007). The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood 109, 1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhao Z, Meyer MB, Saha S, Yu M, Guo A, Wisinski KB, Huang W, Cai W, Pike JW, et al. (2014). CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell 25, 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will B, Vogler TO, Narayanagari S, Bartholdy B, Todorova TI, da Silva Ferreira M, Chen J, Yu Y, Mayer J, Barreyro L, et al. (2015). Minimal PU.1 reduction induces a preleukemic state and promotes development of acute myeloid leukemia. Nat. Med 21, 1172–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JI, Lessard J, and Crabtree GR (2009). Understanding the words of chromatin regulation. Cell 136, 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, and Choi K (2019). Single cell transcriptome dynamics from pluripotency to FLK1+ mesoderm. Development 146, dev182097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Maricque B, Xie M, Li D, Sundaram V, Martin EA, Koebbe BC, Nielsen C, Hirst M, Farnham P, et al. (2011). The Human Epigenome Browser at Washington University. Nat. Methods 8, 989–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ATAC-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE144243 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144243).
The accession number for the scRNA-Seq data reported in this paper is GEO: GSE159381.