

Abstract
Interferons (IFNs) are well known as mediators of the antimicrobial response but also serve as important immunomodulatory cytokines in autoimmune and autoinflammatory diseases. An increasingly critical role for IFNs in evolution of skin inflammation in these patients has been recognized. IFNs are produced not only by infiltrating immune but also resident skin cells, with increased baseline IFN production priming for inflammatory cell activation, immune response amplification and development of skin lesions. The IFN response differs by cell type and host factors and may be modified by other inflammatory pathway activation specific to individual diseases, leading to differing clinical phenotypes. Understanding the contribution of IFNs to skin and systemic disease pathogenesis is key to development of new therapeutics and improved patient outcomes. In this review, we summarize the immunomodulatory role of IFNs in skin, with a focus on type I, and provide insight into IFN dysregulation in autoimmune and autoinflammatory diseases.
Intro/Background
The skin comprises a critical physical and chemical barrier, and resident skin cells and resident and migratory immune cells secrete immunomodulatory proteins to protect us from colonization and invasion by foreign microorganisms. Interferons (IFNs) are one important class of signaling proteins secreted to combat potential infection. While IFNs can serve a protective role, they also contribute to the pathogenesis of autoimmune and autoinflammatory diseases. Pathognomonic skin lesions frequently herald systemic autoimmune disease onset and represent key features that assist in diagnosis. Critically, an important role for type I IFNs in cutaneous and systemic disease pathogenesis has been recognized.
Types and role of interferons
There are three main classes of IFNs, with type I IFNs representing the largest class. Type I IFNs in humans encompass 17 members, including 13 IFNα subtypes, IFNβ, IFNω, IFNε and IFNκ(1). While most cell types produce IFNβ, the primary producers of IFNα are hematopoietic cells(2). There exists only a single type II interferon, IFNγ, which is produced predominantly by T and NK cells, and also assists in the antiviral immune response(3). The third class of IFNs, type III IFNs, is comprised of 4 IFNλ subtypes (IFNλ 1, 2, 3 or 4), with receptor expression mostly restricted to epithelial cells, myeloid cell subsets and neuronal cells(4). IFNλ is structurally similar to interleukin-10 family cytokines and has similar signaling effects to type I IFNs, exhibiting a role in the antimicrobial response of epithelial cells(5).
Type I interferons serve an important immunomodulatory role in healthy skin, leading to promotion of antigen presentation and NK cell activation through shaping the innate immune response, activation and augmentation of the adaptive immune system and induction of an antimicrobial state(1, 2). Type I IFNs also perform an anti-proliferative and tumor immune surveillance role. Type II IFNs can specifically inhibit keratinocyte proliferation(6). Type III IFNs are induced by nucleic acid signaling but their overall function in the skin requires additional study.
Downstream signaling of type I interferons
All type I IFNs bind to the same heterodimeric transmembrane receptor, the IFNα/β receptor (IFNAR), composed of IFNAR1 and 2; however, binding affinity and tissue-specific receptor expression can influence biological activity of the type I IFNs(7–10). The IFNAR is found on nearly all nucleated cells. Binding of type I IFNs to the IFNAR leads to activation of the Janus activated kinase-signal transducer and activation of transcription (JAK-STAT) pathway(11). IFNAR1 is associated with tyrosine kinase 2 (TYK2), and IFNAR2 is associated with JAK1. Once JAK1 and TYK2 are activated, they phosphorylate tyrosine residues on cytoplasmic tails of the IFNAR, which serve as binding sites for the Src-homology-2 (SH2) domain of STAT proteins 1–6(12, 13). Relative STAT expression also influences specific STAT activation. Classically, a phosphorylated STAT1 and STAT2 dimer translocates to the nucleus, associates with interferon response factor 9 (IRF9), resulting in formation of the IFN-stimulated gene factor 3 (ISGF3) complex(14). ISGF3 then binds to interferon-stimulated response elements (ISREs), resulting in activation of interferon-stimulated genes (ISGs) (Figure 1).
Figure 1.
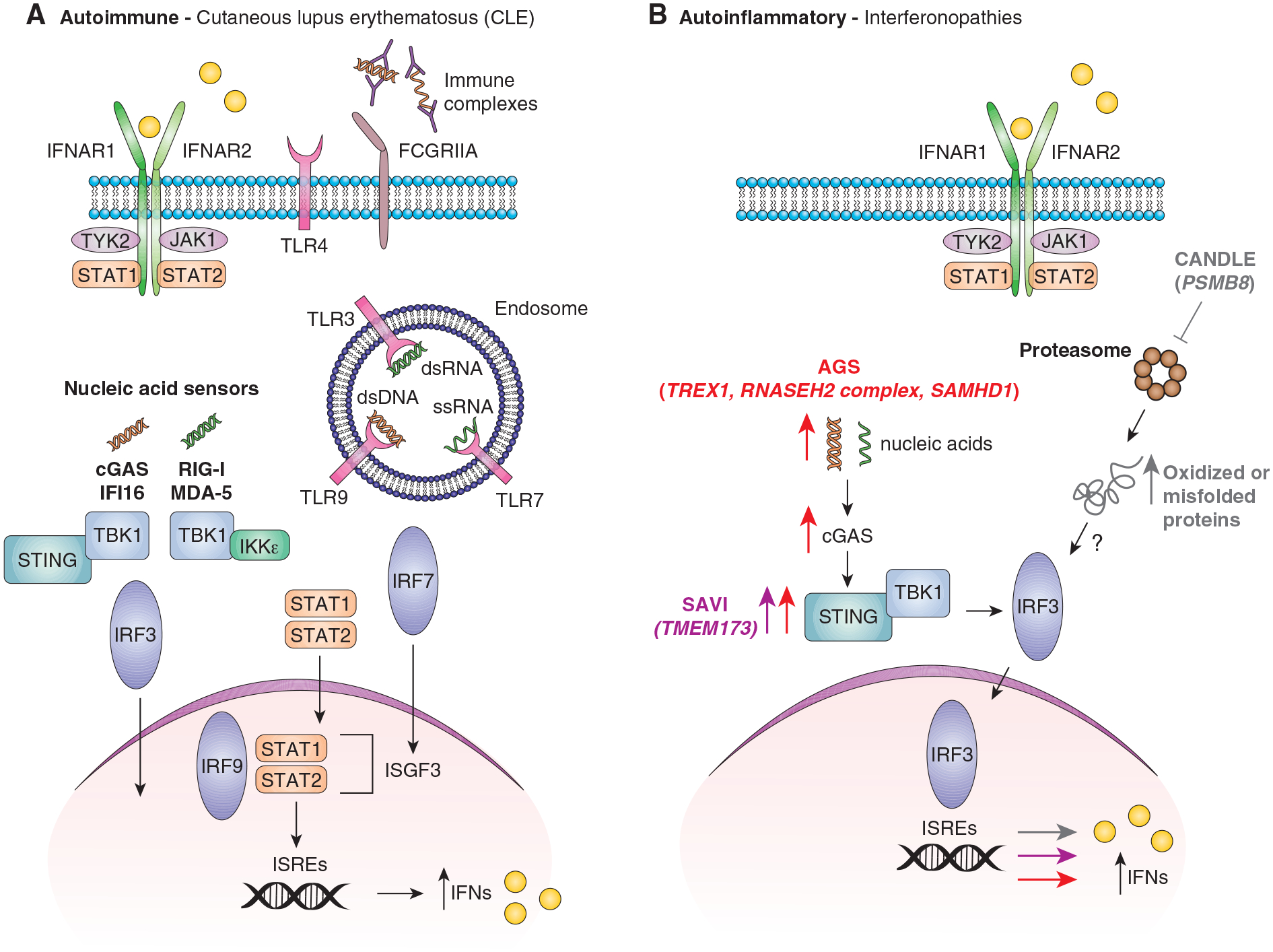
Pathways of interferon dysregulation in skin lesions of patients with autoimmune and autoinflammatory diseases. Cutaneous lupus erythematosus (CLE) is shown as an example in A. B represents pathways dysfunctional in Aicardi-Goutieres Syndrome (AGS) in red, Chronic Atypical Neutrophilic Dermatosis With Lipodystrophy and Elevated Temperature (CANDLE) in grey and STING-associated vasculopathy with onset in infancy (SAVI) in pink. dsDNA=double stranded DNA, dsRNA=double stranded RNA, ssRNA=single stranded RNA, IC=immune complex.
Localization of interferons in skin
In healthy resident skin cells, keratinocytes produce interferons at baseline, with minimal to no detectible production from fibroblasts or endothelial cells(15). This baseline interferon production is a result of chronic IFNκ production, with no apparent contribution from other type I IFNs(15, 16). IFNκ expression increases upon treatment with type I or II interferons(16), and chronic elevation of baseline IFNκ amplifies basal IFN responses(15). In inflammatory states, exposure to cytokines like TNF-α or antimicrobial peptides prime for additional type I IFN expression, particularly IFNβ(17).
Upon appropriate stimulation, many cell types are capable of type I IFN production. Immune cells, similar to keratinocytes, are poised to respond more rapidly to low levels of interferons. Plasmacytoid dendritic cells (pDCs) are able to rapidly secrete large amounts of IFNα and are known to accumulate in key tissues affected by inflammation in rheumatic disease. In autoimmune skin lesions, pDCs are recruited to the dermal-epidermal junction(18), contributing to interface dermatitis, a defining histopathologic feature in lupus and dermatomyositis. Immune complexes in cutaneous lupus lesions can induce type I IFN production in pDCs(19). Langerhans cells, a specialized subset of dendritic cells residing in skin, also produce interferons and release increased amounts of IFN-induced chemokines upon stimulation with Toll-like receptor (TLR) 3 agonist polyinosinic:polycytidylic acid (poly(I:C)) as compared to monocyte-derived DCs(20). Inflammatory monocytes have also been shown to be critical IFN producers in skin upon ultraviolet B (UVB) stimulation(21). Interestingly, IFNARKO mice demonstrate increased skin inflammation, suggesting a protective role for type I IFNs after UVB radiation in wild-type mice(21).
Triggers and regulation of IFN production in skin
Triggers for IFN production in skin can include ultraviolet radiation, infection, injury and cell death, all of which generate damage or pathogen-associated molecular patterns (DAMPs or PAMPs, respectively). Upon sensing of DAMPs or PAMPs by pattern recognition receptors (PRRs), IFN production is induced. The IFN response can differ depending on the underlying trigger, responding receptor and cell type, immune response and various host modifying factors. Keratinocytes express a wide range of PRRs, including toll-like receptors (TLRs) and cytoplasmic nucleic acid sensors, all with a unique ligand (PAMP or DAMP) preference (Table 1). Altered TLR and increased cytosolic nucleic acid sensor expression is noted in autoimmune skin diseases(22, 23), suggesting a general disease mechanism by which an environment with chronically elevated IFNs may modify IFN response mechanisms. UV radiation exposure to the skin of healthy volunteers and also mice has been shown to stimulate a striking cutaneous type I IFN response(21, 24). Which pathways sense and activate IFNs after UV exposure and how this differs in autoimmune disease are currently being investigated.
Table 1.
Toll-like receptors and cytosolic nucleic acid sensors in keratinocytes.
| Pattern recognition receptor (PRR) | Ligand | Location and Expression | Reference |
|---|---|---|---|
| Toll-like receptors (TLRs) | |||
| TLR1 | TLR2/1 heterodimer recognizes tri-acylated lipoproteins | Cell membrane, constitutively expressed, throughout the epidermis | (22, 136–138) |
| TLR2 | pathogen-derived lipoproteins (tri- or diacyl lipopeptides, lipoteichoic acid, peptidoglycan), fungal components | Cell membrane, constitutively expressed, throughout the epidermis | (22, 136, 138–140) |
| TLR3 | dsRNA, poly(I:C) | Intracellular membranes (Endosome/lysosome), constitutively expressed, basal layer of epidermis, expression increased upon exposure to IFNα + poly(I:C) | (23, 136, 138, 141) |
| TLR4 | Lipopolysaccharide (LPS) | Cell membrane | (136) |
| TLR5 | flagellin | Cell membrane, constitutively expressed, basal layer of epidermis | (22, 136, 138) |
| TLR6 | TLR2/6 heterodimer recognizes di-acylated lipoproteins | Cell membrane | (136, 137) |
| TLR7 | ssRNA | Endosome/lysosome, not expressed at baseline but treatment of keratinocytes with poly(I:C) can upregulate TLR7 expression | (142, 143) |
| TLR9 | dsDNA, chromatin-IgG complexes | Endosome/lysosome | (136, 137) |
| TLR10 | unknown | Constitutively expressed | (136, 138) |
| Cytosolic nucleic acid sensors | |||
| Protein kinase R (PKR) | dsRNA | Expression increased upon exposure to IFNα + poly(I:C) | (23) |
| Retinoic acid-inducible gene I (RIG-I) | ssRNA, dsRNA | Constitutively expressed, poly(I:C) leads to upregulation of type I IFNs while UVB has an opposing effect, expression increased upon exposure to IFNα + poly(I:C) | (23, 144, 145) |
| Melanoma differentiation associated gene 5 (MDA-5) | dsRNA | Expression increased upon exposure to IFNα + poly(I:C) | (23) |
| Interferon-γ-inducible protein 16 (IFI16) | dsDNA, ssDNA | Expression in upper epidermal layers in lesional skin of SLE patients | (146–148) |
| Cyclic GMP-AMP synthase (cGAS) | dsDNA | cGAS-stimulator of interferon genes (STING) pathway is activated by apoptosis-derived membrane vesicles from SLE patient sera | (146, 149) |
Multiple factors serve to modulate the cellular response to type I IFNs, including IFNAR downregulation, negative regulator and microRNA upregulation, differential STAT activation, cooperation of STATs with interferon regulatory factors (IRFs), post-translational modification and chromatin remodeling(2). As an example of potential host modifying factors, commensal microbial flora can serve as a rheostat of IFN responsiveness to viral infections in mice(25). Antibiotic-treated mice have been shown to demonstrate decreased IFN responsiveness after mucosal or systemic viral infection, and expression of IFN and IFN-stimulated genes (ISGs) is reduced in macrophages from antibiotic-treated mice(25). In human keratinocytes, treatment with interferons in vitro leads to decreased barrier gene expression and increased S. aureus adherence which may induce further IFN production(26).
IFN effects and involvement in pathogenesis of autoimmune skin disease
Cutaneous lupus erythematosus (CLE)
Cutaneous disease in SLE can be an isolated feature or associated with underlying systemic manifestations. Skin inflammation is present in the majority of patients and is often the first harbinger of disease onset or a disease flare, offering a crucial opportunity to potentially intervene even prior to onset of systemic inflammation. Multiple subtypes of cutaneous lupus exist, including acute CLE (ACLE), subacute CLE (SCLE), chronic CLE (CCLE, including discoid lupus) and intermittent CLE (ICLE or tumid lupus)(27). Cutaneous lupus lesions demonstrate a hallmark interface dermatitis, or inflammatory infiltrate bordering the dermoepidermal junction (DEJ), characterized by apoptotic keratinocytes, vacuolar changes, CD8+ lymphocytes and pDCs(28, 29). Even in SLE patients with no clinically apparent skin lesions, molecular signatures in non-lesional skin can still indicate an aberrant immune response. Both lesional and non-lesional skin from adults with lupus exhibit chronic upregulation of type I interferons(15, 30), and comparison of isolated CLE vs. systemic lupus associated CLE demonstrate similar gene expression profiles(31).
Although incompletely understood, the pathogenesis of CLE lesions is thought to be driven by IFNs (Figure 1A). CLE patients exhibit an elevated IFN signature in peripheral blood that correlates with clinical cutaneous disease activity as assessed by the Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI)(32, 33). IFNĸ production is increased at baseline in non-lesional SLE keratinocytes, leading to increased type I IFN responsiveness and UV light sensitivity(15, 34). Lupus keratinocytes also demonstrate a hypersensitive response to IFN stimulation, with a larger magnitude of change in ISG expression upon IFN treatment as compared to control keratinocytes(35). ISG expression, including Myxovirus resistance gene A (MxA), is upregulated at baseline in both the epidermis and dermis of lesional CLE skin(36). IFN-inducible CXCL9, CXCL10 and CXCL11 are 3 of the 5 most upregulated chemokines in lesional CLE skin, and their receptor (CXCR3) is among the top 3 most differentially regulated chemokine receptors(37, 38). MxA, CXCL9 and CXCL10 cutaneous expression patterns also differ by CLE subtype, suggesting that IFNs may have a role in directing differing clinical phenotypes(39).
Key genetic risk variants involved in IFN signaling pathways have also been described in SLE, including IRF5 and STAT4(40–42), but how each of these relates to skin disease hasn’t been delineated. SLE patients with these genetic risk variants have also been noted to have differences in disease phenotype, which may in part be explained through altered IFN signaling. As an example, SLE patients with high risk IRF5 genotypes were demonstrated to have elevated serum IFNα activity, with the highest levels observed in patients with anti-double-stranded DNA (dsDNA) or anti-RNA binding protein (RBP) autoantibodies(43). SLE patients with STAT4 risk alleles are diagnosed at a younger age and also more likely to have nephritis and anti-dsDNA autoantibodies(44, 45). Genetic risks for CLE have also been linked to IFN signaling as polymorphisms in IFNK(46) are associated with skin disease in African American and European ancestry females with SLE, and mutations in TREX1, a DNA exonuclease that when inhibited leads to accumulation of nucleic acids and increased IFN production, result in familial chilblain lupus (47, 48).
It is well known that UV radiation is a trigger for cutaneous inflammation and disease flare in SLE patients. UV radiation is known to amplify the IFN response to nucleic acids in keratinocytes, and mice lacking TREX1 develop UV-induced skin lesions(49). ISGs are increased in lupus-prone mice and human patients vs. healthy controls after UV radiation (50, 51) and this coincides with enhanced CD123+ dendritic cell and CD68+ macrophage recruitment in SLE skin after UV radiation (50). In C57BL/6J mice, UV radiation induces not only a type I IFN response in skin, but also a type I IFN response in peripheral blood and kidney tissue, suggesting a role for UV radiation and cutaneous IFNs in the initiation of systemic inflammation(24). Interestingly, this type I IFN response is more pronounced in female vs. male mice, lending insight into a potential mechanism by which females may be more susceptible to select autoimmune diseases such as SLE(24). In addition, IFNs repress UVB-mediated Treg induction in lupus-prone mice, which contributes to T cell activation(51). Importantly, persistence of IFN responses in CLE patients after UV exposure correlated with endothelial cell activation, likely contributing to leukocyte recruitment and development of clinical lesions(52). In pDCs, supernatant from UV-treated, apoptotic monocytes induces type I IFN production in combination with SLE total IgG (pooled from plasma of two patients), and both RNase and DNase treatment decrease type I IFN induction(53), suggesting that immune complexes predispose to inflammation following UVB.
Dermatomyositis
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by pathognomonic rash, muscle weakness and variable involvement of other organ systems, including the lungs, gastrointestinal tract and heart. In children, skin inflammation is the most common presenting symptom and most classically manifests as scaly, erythematous, raised lesions over the knuckles, or reddish purple discoloration of the upper eyelids with associated edema(54).
Similar to CLE, skin inflammation can be an important indicator of ongoing disease activity, photosensitivity is common, and lesions exhibit an interface dermatitis(55, 56). However, the pathophysiology of DM skin lesions is not as well understood. Type I IFN signaling is upregulated in DM and juvenile dermatomyositis (JDM) skin(57) as well as in muscle(58) and peripheral blood(59). The type I IFN signature in peripheral blood in DM and JDM has also been reported to correlate with disease activity(60, 61). Immunostaining of DM lesional skin demonstrates increased MxA staining in the epidermis, endothelial cells and inflammatory cell infiltrate(62, 63). CXCL10 expression is also higher in DM lesional skin, predominantly in the upper dermis in the presence of lymphocytic infiltrate and also in the epidermis near areas of interface dermatitis(62). In DM skin disease, similar to CLE, type I IFNs have been purported to lead to recruitment of CXCR3+ lymphocytes, with increased MxA staining correlating with higher numbers of CXCR3+ lymphocytes(62, 64). In anti-melanoma differentiation-associated 5 gene (MDA5) autoantibody-positive DM patients, MxA immunostaining in skin was distributed in blood vessels in the dermis, suggesting a role for IFNs in the vasculopathy that characterizes DM(65). Even non-lesional JDM skin has been described as altered, with increased numbers of pDCs and mast cells(66).
A recent analysis of two dermatomyositis skin microarray datasets revealed both a type I and type II IFN signature(67). Similarly, dermatomyositis muscle has been reported to have both a type I and II IFN signature, although the type I IFN signature may be somewhat more specific to DM versus other idiopathic inflammatory myopathies(68). IFNβ expression in the skin has been shown to correlate with ISGs(57), but whether IFNκ also plays a role in DM skin remains to be determined.
Scleroderma
Scleroderma is an autoimmune disorder with features of fibrosis, vasculopathy, and inflammation, contributing to pathogenesis at various stages of disease(69–71). In systemic sclerosis (SSc), the extent of skin involvement associates with prognosis, with lower survival in patients with a higher baseline skin score and improved survival in those patients with improvement in skin thickening(72).
IFN treatment has been known to trigger both systemic and localized scleroderma, leading to speculation on the role of IFNs in scleroderma pathogenesis. Localized scleroderma (LSc) has been described at IFNβ injection sites(73). SSc has also been reported in multiple sclerosis (MS) patients after IFNα and IFNβ treatment(74, 75). Immunostaining for MxA in lesional LSc biopsies shows expression that is most prominent in the deep dermis and subcutis near inflammatory infiltrates(76). CXCL10 staining is apparent in the dermal perivascular lymphoplasmacytic infiltrate(77). In pediatric LSc, bulk RNA sequencing of lesional skin confirms upregulation of IFNγ and ISGs, including CXCL9, CXCL10 and CXCL11, and LSc patients with more active skin lesions had higher IFN scores(78). A SSc skin microarray study also revealed ISGs as the top upstream transcriptional regulators, with IFNα and IFNγ as the top upstream activated cytokines(79). In this same study, approximately 75% of patients had a fibroinflammatory signature, which included gene expression scores of ISGs, that was found to correlate with the modified Rodnan skin score (mRSS)(79). Cutaneous expression of ISGs IFI44 and SIGLEC-1 has also been demonstrated to correlate with the mRSS(80). Interestingly, IFNκ was shown to be downregulated in SSc keratinocytes, suggesting that there may be different sources of type I IFNs based on cell type and individual autoimmune diseases(81).
Similar to gene expression studies in skin, an IFN signature in peripheral blood has been noted in both localized and systemic scleroderma patients(77, 82). As compared to the peripheral blood IFN signature in SLE, SSc patients were found to have upregulation of endothelial adhesion molecules, suggestive of the underlying vasculopathy that is central to the disease pathogenesis in SSc(82).
It has been suggested that IFN upregulation might play a role in the earlier stages of scleroderma pathogenesis. A study focused on patients with early SSc described that a type I IFN signature is still present in peripheral blood, despite the absence of clinical evidence of fibrosis(83). In fact, in early and non-fibrotic compared to fibrotic SSc patients, the IFN score was higher(83). pDCs have been shown to produce IFNα upon treatment with sera from SSc patients combined with necrotic material(84) in an FcgammaRII and RNA-dependent manner, suggesting a role for immune complexes(85). Indeed, TLR8 overexpression in a murine model of disease exacerbates fibrosis(86), and expression of ISGs also increases in skin and fibroblasts from SSc patients upon TLR3 stimulation(87). In SSc patients treated via hematopoietic stem cell transplantation (HSCT), there is a decrease in type I IFN expression in skin that correlates with decreased fibrosis and capillary regeneration(88).
Sjogren’s syndrome
Sjogren’s syndrome (SS) is characterized by inflammation of the lacrimal and salivary glands, resulting in exocrine dysfunction, with clinical features of keratoconjunctivitis sicca/xeropthalmia and xerostomia. SS can be both a primary disease or secondary to/associated with another underlying rheumatic disease and is associated with hypergammaglobulinemia and production of the classic autoantibodies SSA/Ro and SSB/La. Cutaneous manifestations in SS occur in up to 50% of patients and can include xerosis, angular cheilitis, eyelid dermatitis, pruritis, cutaneous vasculitis and skin lesions with histologic similarity to CLE(89, 90). Gene expression studies from both peripheral blood and salivary gland tissue highlight an IFN signature(91, 92), with a predominant type I IFN signature in peripheral blood and type II IFN signature in salivary gland tissue(92). Intriguingly, the type I IFN signature correlates with apoptotic gene expression(92), but whether this contributes to skin disease remains unknown. Monocytes from patients with primary SS also have a type I IFN signature in 55% of patients as compared to healthy controls(93). The importance of IFNs in SS is also reinforced by evidence in murine models, with SS mice that have a non-functional IFN receptor failing to develop clinical disease(94).
Psoriasis
Type I IFN activation has also been described in psoriasis and psoriatic keratinocytes. Genetic polymorphisms which lead to activation of cytosolic signaling pathways and IFN production are risk factors for psoriasis(95); indeed, DDX58 (RIG-I) activation is required for IL-23 activation and psoriasis in murine models(96). Type I IFNs and ISGs are significantly elevated in psoriatic plaques (97–100). A phase I trial of MEDI-545, an, anti-IFN-α monoclonal antibody was unable to show clinical benefit in patients with chronic psoriatic plaques, which may support the hypothesis that IFNs are involved in initiation of psoriasis but not in chronic plaque formation (101). Further work to understand how IFNs contribute to psoriatic development is required.
IFN effects and involvement in pathogenesis of autoinflammatory skin disease
Interferonopathies
The interferonopathies are autoinflammatory disorders characterized by overproduction of IFN due to mutations in genes involved in regulation of nucleic acid sensing. Through the study of interferonopathies, we have gained insight into the pathogenic role of interferons and underlying disease mechanisms driven by interferons. A spectrum of cutaneous manifestations are seen in the clinical presentation of interferonopathies especially vasculopathy (chilblain-like rash, microangiopathic vasculopathy, gangrene/ulcers/infarcts in acral areas) and skin eruptions of nodular erythema and violaceous plaques in cold-sensitive acral areas(102). Further, undifferentiated autoinflammatory disease patients with elevated IFN-response-gene scores more commonly had neutrophilic panniculitis(103). Further study has suggested that some disorders may favor NF-κB driven pathology over that mediated by interferons(103) but that IFN signature elevation is associated with erythematous, macular skin lesions and Gottron’s papules (skin lesions common in patients with DM). Understanding the balance between IFN-mediated and other inflammatory activation is an important goal for future research.
Aicardi-Goutieres Syndrome (AGS)
AGS patients were first described with progressive encephalopathy, basal ganglia calcifications, white matter hypodensities and persistent cerebrospinal fluid lymphocytosis(104). It was later noted that the most pathognomonic extra neurological symptom of AGS was the cutaneous finding of chilblain-like lesions on the digits and that these patients also had elevated IFNα in cerebrospinal fluid (CSF) and serum(105). Chilblain-like lesions are reported in approximately half of AGS patients, most often on the fingers and toes, but also other acral surfaces, including the ears(106).
Mutations in genes encoding the cellular nucleases TREX1(107), RNASEH2 complex(108), and SAMHD1(109) among others have been discovered in AGS patients. While these mutations lead to increased IFN generation, how these mutations directly lead to skin manifestations isn’t well understood. TREX1 encodes a 3’−5’ exonuclease that degrades ssDNA(110, 111), dsDNA(112), and ssRNA(113). Accumulation of nucleic acids causes a rise in IFN production in a cyclic GMP-AMP synthase (cGAS) and stimulator of IFN genes (STING) dependent manner, and deletion of TREX1 in keratinocytes raises ISG production in keratinocytes(114) (Figure 1B). However, mice with a dysfunctional TREX1 do not get spontaneous skin lesions(112). This suggests that triggers are needed for phenotype. Indeed, mice with dysfunctional TREX1 exhibit increased ear swelling and inflammation when injected with DNA, independent of its oxidation status (wild type mice develop lesions only from UV-oxidized DNA, which is resistant to TREX1 degradation)(115). Other mutations associated with TREX1 may also impact UVB sensitivity. Mutations in RNASEH2 can lead to defective repair of damaged RNA which increases the propensity for UVB-mediated damage and type I IFN production in response(116). Case reports have linked AGS with photosensitivity(117), but how individual mutations contribute remains to be determined. In C57BL/6J mice exposed to UV radiation, both the type I IFN response in skin and peripheral blood is primarily dependent on the cGAS-STING pathway in the early response phase at 6 hours post-radiation, lending insight into a potential role for cGAS-STING in the early type I IFN response and subsequent innate inflammatory cell recruitment(24).
CANDLE (Chronic Atypical Neutrophilic Dermatosis With Lipodystrophy and Elevated Temperature)
CANDLE is categorized as a proteasome-associated autoinflammatory syndrome (PRAAS) and is manifested by recurrent fevers, annular, purpuric rash, lipodystrophy and multisystem inflammation. Skin biopsies from CANDLE patients demonstrate mononuclear cell and neutrophilic infiltrate with dermal collagen degeneration(118). Mutations in the PSMB8 gene were initially found in 8/9 patients from a CANDLE cohort, accompanied by elevated serum levels of CXCL10 and IFN signaling as a top dysregulated pathway on whole blood gene expression analysis (pathway gene list including both type I and type II IFN-induced genes) (118). Additional mutations in genes involved in proteasome activity have since been identified that result in a CANDLE phenotype, including PSMB4, PSMA3, PSMB9 and POMP, which encodes a proteasome maturation protein(119). In patients with proteasome alterations other than in PSMB8, skin biopsies demonstrated increased ubiquitin-positive keratinocytes and ubiquitin-rich inclusions in keratinocytes. CANDLE patient keratinocytes showed impairment in proteasome assembly, and siRNA knockdown of patient proteasome mutations resulted in type I IFN induction(119). Indeed, in CANDLE, IFNs may participate in a feed-forward loop in which normal triggers of type I IFN production, such as UV light or infections, result in cellular stress and oxidized proteins that cannot be degraded, which results in further type I IFN production, upregulation of the immunoproteasome and subsequent inflammation (Figure 1B).
SAVI (STING-associated vasculopathy with onset in infancy)
Another group of patients exhibiting lupus-like malar rash and vasculitic skin lesions in conjunction with interstitial lung disease have been described to harbor TMEM173 mutations, leading to gain-of-function in stimulator of interferon genes (STING) and subsequent IFN overproduction(120, 121) (Figure 1B). At baseline, SAVI patients have maximal upregulation of type I IFN and ISGs with constitutive STAT1 phosphorylation(121). Lesional skin from SAVI patients is characterized by vascular inflammation of capillaries and microthrombosis, and dermal fibroblasts from SAVI patients are hypersensitive to treatment with even low-dose cyclic GMP-AMP (cGAMP), resulting in increased IRF3 phosphorylation and type I IFN transcription(121).
Murine models of SAVI-associated mutations also develop profoundly elevated ISG signatures. However, systemic disease is independent of the type I IFN receptor, suggesting other inflammatory pathways or other types of IFNs contribute to disease, at least in mice(122). Mice harboring SAVI-associated mutations have not been reported to develop skin disease, so how the type I IFN pathways participate in SAVI-associated skin manifestations is not yet known.
Insight into disease mechanisms by targeting the IFN pathway
Anifrolumab
Anifrolumab is a monoclonal antibody that binds to subunit 1 of the type I IFN receptor (IFNAR1), thereby blocking type I IFN activity. Trials of anifrolumab for treatment of SLE have shown promise for improvement in CLE disease activity. In a phase IIb, randomized, double-blind, placebo-controlled study of anifrolumab in adults with moderate-to-severe SLE (MUSE trial), there was greater efficacy of anifrolumab in patients with a higher IFN signature, including improvement in skin disease activity as assessed by the Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K), CLASI and British Isles Lupus Assessment Group (BILAG) index(123, 124). Improvement in rash was only significantly improved as assessed by the BILAG in the low IFN signature subgroup(124). In the Treatment of Uncontrolled Lupus via the IFN Pathway (TULIP) trial II, there was ≥50% decrease in CLASI scores in half of the anifrolumab group compared to only 25% of the placebo group (p = 0.04)(125). In systemic scleroderma patients, anifrolumab treatment has also been shown to decrease type I ISG expression in patient skin biopsies collected 28 days after dosing with anifrolumab(126).
Janus Kinase (JAK) inhibitors
JAK inhibitors block one or multiple JAKs (JAK1, JAK2, JAK3, TYK2), which are tyrosine kinases that bind to a wide variety of cytokine receptors (including all three types of IFNs) and thereby affect the immune response(127, 128). CLE lesions have been shown to exhibit high expression of phospho-JAK2 similar to CXCL10 and MxA, and treatment of keratinocytes and a 3d epidermis model with ruxolitinib after poly(I:C) stimulation decreases type I ISG expression(129). Treatment of murine lupus with tofacitinib resulted in improvement of both systemic and cutaneous disease manifestations(130). In DM, skin disease has shown improvement after treatment with ruxolitinib, further supporting a role for IFNs in DM pathogenesis(131). Treatment of 18 interferonopathy patients with baricitinib led to a decrease in IFN scores and clinical symptoms, with improvement in cutaneous disease also reported although not specifically scored(132). Similarly, treatment of cutaneous lesions in familial chilblain lupus with baricitinib leads to improvement in skin disease(133). Liu et al also demonstrated that treatment of SAVI patient T and B cells with JAK inhibitors blocks constitutive phosphorylation of STAT1(121).
Anti-BDCA2 antibody (BIIB059)
BIIB059 is a humanized monoclonal antibody that binds blood DC antigen 2 (BDCA2), a C-type lectin and pDC specific receptor. BIIB059 is believed to inhibit TLR-induced type I IFN and other inflammatory mediator production. In CLE, BIIB059 has been shown to reduce skin inflammation(134). In a randomized, double-blind, placebo-controlled trial of BIIB059 in SLE patients with active skin disease, BIIB059 decreased expression of MxA and IFITM3 and also CD45+ cellular infiltrate in skin biopsies four weeks after treatment and additionally improved CLASI scores(134). BIIB059 has also been described to reduce IFNα production from pDCs of CLE patients after stimulation with TLR agonists, providing an additive therapeutic benefit to hydroxychloroquine(135).
Conclusions
Overproduction of type I IFNs is a unifying theme amongst many autoimmune and autoinflammatory patients with skin manifestations. In SLE/CLE, this contributes to inflammatory cell activation and photosensitivity, a mechanism which likely extends to other diseases, possibly the autoinflammatory diseases as well. Further research is needed to understand the ways in which interferons drive disease and to identify which patients will benefit most from targeting of IFNs.
Acknowledgments
JMK was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award numbers R01-AR071384 (J.M.K.), the A. Alfred Taubman Medical Research Institute (J.M.K.), the Parfet Emerging Scholar Award (J.M.K.), the Rheumatology Research Foundation (J.M.K), and the Doris Duke Charitable Foundation through a Physician Scientist Development award to JMK. JLT was supported by a K12 Child Health Research Center Career Development Award to the University of Michigan Department of Pediatrics (K12 HD028820-28), a MICHR Pathway to First Grant Award and a Cure JM Foundation Research Grant.
References
- 1.Gibbert K, Schlaak JF, Yang D, and Dittmer U. 2013. IFN-alpha subtypes: distinct biological activities in anti-viral therapy. Br J Pharmacol 168: 1048–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ivashkiv LB, and Donlin LT. 2014. Regulation of type I interferon responses. Nat Rev Immunol 14: 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schroder K, Hertzog PJ, Ravasi T, and Hume DA. 2004. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75: 163–189. [DOI] [PubMed] [Google Scholar]
- 4.Hemann EA, Gale M Jr., and Savan R. 2017. Interferon Lambda Genetics and Biology in Regulation of Viral Control. Front Immunol 8: 1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gad HH, Dellgren C, Hamming OJ, Vends S, Paludan SR, and Hartmann R. 2009. Interferon-lambda is functionally an interferon but structurally related to the interleukin-10 family. J Biol Chem 284: 20869–20875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saunders NA, and Jetten AM. 1994. Control of growth regulatory and differentiation-specific genes in human epidermal keratinocytes by interferon gamma. Antagonism by retinoic acid and transforming growth factor beta 1. J Biol Chem 269: 2016–2022. [PubMed] [Google Scholar]
- 7.Behrendt R, Schumann T, Gerbaulet A, Nguyen LA, Schubert N, Alexopoulou D, Berka U, Lienenklaus S, Peschke K, Gibbert K, Wittmann S, Lindemann D, Weiss S, Dahl A, Naumann R, Dittmer U, Kim B, Mueller W, Gramberg T, and Roers A. 2013. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell Rep 4: 689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cull VS, Tilbrook PA, Bartlett EJ, Brekalo NL, and James CM. 2003. Type I interferon differential therapy for erythroleukemia: specificity of STAT activation. Blood 101: 2727–2735. [DOI] [PubMed] [Google Scholar]
- 9.Moll HP, Maier T, Zommer A, Lavoie T, and Brostjan C. 2011. The differential activity of interferon-alpha subtypes is consistent among distinct target genes and cell types. Cytokine 53: 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schreiber G 2017. The molecular basis for differential type I interferon signaling. J Biol Chem 292: 7285–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, and Melero I. 2011. Direct effects of type I interferons on cells of the immune system. Clin Cancer Res 17: 2619–2627. [DOI] [PubMed] [Google Scholar]
- 12.Levy DE, and Darnell JE Jr. 2002. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3: 651–662. [DOI] [PubMed] [Google Scholar]
- 13.Kalliolias GD, and Ivashkiv LB. 2010. Overview of the biology of type I interferons. Arthritis Res Ther 12 Suppl 1: S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hile GA, Gudjonsson JE, and Kahlenberg JM. 2018. The influence of interferon on healthy and diseased skin. Cytokine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, Berthier CC, Swindell WR, Patrick MT, Shao S, Tsou PS, Uppala R, Beamer MA, Srivastava A, Bielas SL, Harms PW, Getsios S, Elder JT, Voorhees JJ, Gudjonsson JE, and Kahlenberg JM. 2018. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis 77: 1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LaFleur DW, Nardelli B, Tsareva T, Mather D, Feng P, Semenuk M, Taylor K, Buergin M, Chinchilla D, Roshke V, Chen G, Ruben SM, Pitha PM, Coleman TA, and Moore PA. 2001. Interferon-kappa, a novel type I interferon expressed in human keratinocytes. J Biol Chem 276: 39765–39771. [DOI] [PubMed] [Google Scholar]
- 17.Zhang LJ, Sen GL, Ward NL, Johnston A, Chun K, Chen Y, Adase C, Sanford JA, Gao N, Chensee M, Sato E, Fritz Y, Baliwag J, Williams MR, Hata T, and Gallo RL. 2016. Antimicrobial Peptide LL37 and MAVS Signaling Drive Interferon-beta Production by Epidermal Keratinocytes during Skin Injury. Immunity 45: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macal M, Tam MA, Hesser C, Di Domizio J, Leger P, Gilliet M, and Zuniga EI. 2016. CD28 Deficiency Enhances Type I IFN Production by Murine Plasmacytoid Dendritic Cells. J Immunol 196: 1900–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, and Luster AD. 2005. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 115: 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Renn CN, Sanchez DJ, Ochoa MT, Legaspi AJ, Oh CK, Liu PT, Krutzik SR, Sieling PA, Cheng G, and Modlin RL. 2006. TLR activation of Langerhans cell-like dendritic cells triggers an antiviral immune response. J Immunol 177: 298–305. [DOI] [PubMed] [Google Scholar]
- 21.Sontheimer C, Liggitt D, and Elkon KB. 2017. Ultraviolet B Irradiation Causes Stimulator of Interferon Genes-Dependent Production of Protective Type I Interferon in Mouse Skin by Recruited Inflammatory Monocytes. Arthritis & rheumatology (Hoboken, N.J.) 69: 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baker BS, Ovigne JM, Powles AV, Corcoran S, and Fry L. 2003. Normal keratinocytes express Toll-like receptors (TLRs) 1, 2 and 5: modulation of TLR expression in chronic plaque psoriasis. Br J Dermatol 148: 670–679. [DOI] [PubMed] [Google Scholar]
- 23.Prens EP, Kant M, van Dijk G, van der Wel LI, Mourits S, and van der Fits L. 2008. IFN-alpha enhances poly-IC responses in human keratinocytes by inducing expression of cytosolic innate RNA receptors: relevance for psoriasis. J Invest Dermatol 128: 932–938. [DOI] [PubMed] [Google Scholar]
- 24.Skopelja-Gardner S, An J, Tai J, Tanaka L, Sun X, Hermanson P, Baum R, Kawasumi M, Green R, Gale M Jr., Kalus A, Werth VP, and Elkon KB. 2020. The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent. Sci Rep 10: 7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, Wherry EJ, and Artis D. 2012. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37: 158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sirobhushanam S, Parsa N, Reed TJ, Berthier CC, Sarkar MK, Hile GA, Tsoi LC, Banfield J, Dobry C, Horswill AR, Gudjonsson JE, and Kahlenberg JM. 2019. Staphylococcus aureus Colonization Is Increased on Lupus Skin Lesions and Is Promoted by IFN-Mediated Barrier Disruption. J Invest Dermatol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wenzel J 2019. Cutaneous lupus erythematosus: new insights into pathogenesis and therapeutic strategies. Nat Rev Rheumatol 15: 519–532. [DOI] [PubMed] [Google Scholar]
- 28.Crowson AN, Magro CM, and Mihm MC Jr. 2008. Interface dermatitis. Arch Pathol Lab Med 132: 652–666. [DOI] [PubMed] [Google Scholar]
- 29.Wenzel J, and Tuting T. 2008. An IFN-associated cytotoxic cellular immune response against viral, self-, or tumor antigens is a common pathogenetic feature in “interface dermatitis”. The Journal of investigative dermatology 128: 2392–2402. [DOI] [PubMed] [Google Scholar]
- 30.Der E, Ranabothu S, Suryawanshi H, Akat KM, Clancy R, Morozov P, Kustagi M, Czuppa M, Izmirly P, Belmont HM, Wang T, Jordan N, Bornkamp N, Nwaukoni J, Martinez J, Goilav B, Buyon JP, Tuschl T, and Putterman C. 2017. Single cell RNA sequencing to dissect the molecular heterogeneity in lupus nephritis. JCI insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berthier CC, Tsoi LC, Reed TJ, Stannard JN, Myers EM, Namas R, Xing X, Lazar S, Lowe L, Kretzler M, Gudjonsson JE, and Kahlenberg JM. 2019. Molecular Profiling of Cutaneous Lupus Lesions Identifies Subgroups Distinct from Clinical Phenotypes. J Clin Med 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Braunstein I, Klein R, Okawa J, and Werth VP. 2012. The interferon-regulated gene signature is elevated in subacute cutaneous lupus erythematosus and discoid lupus erythematosus and correlates with the cutaneous lupus area and severity index score. Br J Dermatol 166: 971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Albrecht J, Taylor L, Berlin JA, Dulay S, Ang G, Fakharzadeh S, Kantor J, Kim E, Militello G, McGinnis K, Richardson S, Treat J, Vittorio C, Van Voorhees A, and Werth VP. 2005. The CLASI (Cutaneous Lupus Erythematosus Disease Area and Severity Index): an outcome instrument for cutaneous lupus erythematosus. J Invest Dermatol 125: 889–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stannard JN, Reed TJ, Myers E, Lowe L, Sarkar MK, Xing X, Gudjonsson JE, and Kahlenberg JM. 2017. Lupus Skin Is Primed for IL-6 Inflammatory Responses through a Keratinocyte-Mediated Autocrine Type I Interferon Loop. J Invest Dermatol 137: 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsoi LC, Hile GA, Berthier CC, Sarkar MK, Reed TJ, Liu J, Uppala R, Patrick M, Raja K, Xing X, Xing E, He K, Gudjonsson JE, and Kahlenberg JM. 2019. Hypersensitive IFN Responses in Lupus Keratinocytes Reveal Key Mechanistic Determinants in Cutaneous Lupus. J Immunol 202: 2121–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wenzel J, Uerlich M, Haller O, Bieber T, and Tueting T. 2005. Enhanced type I interferon signaling and recruitment of chemokine receptor CXCR3-expressing lymphocytes into the skin following treatment with the TLR7-agonist imiquimod. J Cutan Pathol 32: 257–262. [DOI] [PubMed] [Google Scholar]
- 37.Meller S, Winterberg F, Gilliet M, Muller A, Lauceviciute I, Rieker J, Neumann NJ, Kubitza R, Gombert M, Bunemann E, Wiesner U, Franken-Kunkel P, Kanzler H, Dieu-Nosjean MC, Amara A, Ruzicka T, Lehmann P, Zlotnik A, and Homey B. 2005. Ultraviolet radiation-induced injury, chemokines, and leukocyte recruitment: An amplification cycle triggering cutaneous lupus erythematosus. Arthritis Rheum 52: 1504–1516. [DOI] [PubMed] [Google Scholar]
- 38.Zahn S, Rehkamper C, Ferring-Schmitt S, Bieber T, Tuting T, and Wenzel J. 2011. Interferon-alpha stimulates TRAIL expression in human keratinocytes and peripheral blood mononuclear cells: implications for the pathogenesis of cutaneous lupus erythematosus. Br J Dermatol 165: 1118–1123. [DOI] [PubMed] [Google Scholar]
- 39.Wenzel J, Zahn S, Mikus S, Wiechert A, Bieber T, and Tuting T. 2007. The expression pattern of interferon-inducible proteins reflects the characteristic histological distribution of infiltrating immune cells in different cutaneous lupus erythematosus subsets. Br J Dermatol 157: 752–757. [DOI] [PubMed] [Google Scholar]
- 40.Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G, Jonsen A, Rantapaa-Dahlqvist S, Moller B, Kere J, Koskenmies S, Widen E, Eloranta ML, Julkunen H, Kristjansdottir H, Steinsson K, Alm G, Ronnblom L, and Syvanen AC. 2005. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet 76: 528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, Ortmann WA, Koeuth T, Gonzalez Escribano MF, Argentine, G. Spanish Collaborative, Pons-Estel B, Petri M, Daly M, Gregersen PK, Martin J, Altshuler D, Behrens TW, and Alarcon-Riquelme ME. 2006. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet 38: 550–555. [DOI] [PubMed] [Google Scholar]
- 42.Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, Li W, Masters SL, Booty MG, Carulli JP, Padyukov L, Alfredsson L, Klareskog L, Chen WV, Amos CI, Criswell LA, Seldin MF, Kastner DL, and Gregersen PK. 2007. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 357: 977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, and Crow MK. 2008. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum 58: 2481–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sigurdsson S, Nordmark G, Garnier S, Grundberg E, Kwan T, Nilsson O, Eloranta ML, Gunnarsson I, Svenungsson E, Sturfelt G, Bengtsson AA, Jonsen A, Truedsson L, Rantapaa-Dahlqvist S, Eriksson C, Alm G, Goring HH, Pastinen T, Syvanen AC, and Ronnblom L. 2008. A risk haplotype of STAT4 for systemic lupus erythematosus is over-expressed, correlates with anti-dsDNA and shows additive effects with two risk alleles of IRF5. Hum Mol Genet 17: 2868–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor KE, Remmers EF, Lee AT, Ortmann WA, Plenge RM, Tian C, Chung SA, Nititham J, Hom G, Kao AH, Demirci FY, Kamboh MI, Petri M, Manzi S, Kastner DL, Seldin MF, Gregersen PK, Behrens TW, and Criswell LA. 2008. Specificity of the STAT4 genetic association for severe disease manifestations of systemic lupus erythematosus. PLoS Genet 4: e1000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harley IT, Niewold TB, Stormont RM, Kaufman KM, Glenn SB, Franek BS, Kelly JA, Kilpatrick JR, Hutchings D, Divers J, Bruner GR, Edberg JC, McGwin G Jr., Petri MA, Ramsey-Goldman R, Reveille JD, Vila-Perez LM, Merrill JT, Gilkeson GS, Vyse TJ, Alarcon-Riquelme ME, Cho SK, Jacob CO, Alarcon GS, Moser KL, Gaffney PM, Kimberly RP, Bae SC, Langefeld CD, Harley JB, Guthridge JM, and James JA. 2010. The role of genetic variation near interferon-kappa in systemic lupus erythematosus. Journal of biomedicine & biotechnology 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, Pfeiffer C, Hollis T, Gahr M, Perrino FW, Lieberman J, and Hubner N. 2007. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med (Berl) 85: 531–537. [DOI] [PubMed] [Google Scholar]
- 48.Lee-Kirsch MA, Gong M, Schulz H, Ruschendorf F, Stein A, Pfeiffer C, Ballarini A, Gahr M, Hubner N, and Linne M. 2006. Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am J Hum Genet 79: 731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scholtissek B, Zahn S, Maier J, Klaeschen S, Braegelmann C, Hoelzel M, Bieber T, Barchet W, and Wenzel J. 2017. Immunostimulatory Endogenous Nucleic Acids Drive the Lesional Inflammation in Cutaneous Lupus Erythematosus. The Journal of investigative dermatology 137: 1484–1492. [DOI] [PubMed] [Google Scholar]
- 50.Zahn S, Graef M, Patsinakidis N, Landmann A, Surber C, Wenzel J, and Kuhn A. 2014. Ultraviolet light protection by a sunscreen prevents interferon-driven skin inflammation in cutaneous lupus erythematosus. Exp Dermatol 23: 516–518. [DOI] [PubMed] [Google Scholar]
- 51.Wolf SJ, Estadt SN, Theros J, Moore T, Ellis J, Liu J, Reed TJ, Jacob CO, Gudjonsson JE, and Kahlenberg JM. 2019. Ultraviolet light induces increased T cell activation in lupus-prone mice via type I IFN-dependent inhibition of T regulatory cells. J Autoimmun 103: 102291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reefman E, Kuiper H, Limburg PC, Kallenberg CG, and Bijl M. 2008. Type I interferons are involved in the development of ultraviolet B-induced inflammatory skin lesions in systemic lupus erythaematosus patients. Ann Rheum Dis 67: 11–18. [DOI] [PubMed] [Google Scholar]
- 53.Lovgren T, Eloranta ML, Bave U, Alm GV, and Ronnblom L. 2004. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum 50: 1861–1872. [DOI] [PubMed] [Google Scholar]
- 54.Huber AM 2018. Juvenile Idiopathic Inflammatory Myopathies. Pediatr Clin North Am 65: 739–756. [DOI] [PubMed] [Google Scholar]
- 55.Stringer E, Singh-Grewal D, and Feldman BM. 2008. Predicting the course of juvenile dermatomyositis: significance of early clinical and laboratory features. Arthritis Rheum 58: 3585–3592. [DOI] [PubMed] [Google Scholar]
- 56.Christen-Zaech S, Seshadri R, Sundberg J, Paller AS, and Pachman LM. 2008. Persistent association of nailfold capillaroscopy changes and skin involvement over thirty-six months with duration of untreated disease in patients with juvenile dermatomyositis. Arthritis Rheum 58: 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wong D, Kea B, Pesich R, Higgs BW, Zhu W, Brown P, Yao Y, and Fiorentino D. 2012. Interferon and biologic signatures in dermatomyositis skin: specificity and heterogeneity across diseases. PLoS One 7: e29161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tezak Z, Hoffman EP, Lutz JL, Fedczyna TO, Stephan D, Bremer EG, Krasnoselska-Riz I, Kumar A, and Pachman LM. 2002. Gene expression profiling in DQA1*0501+ children with untreated dermatomyositis: a novel model of pathogenesis. J Immunol 168: 4154–4163. [DOI] [PubMed] [Google Scholar]
- 59.O’Connor KA, Abbott KA, Sabin B, Kuroda M, and Pachman LM. 2006. MxA gene expression in juvenile dermatomyositis peripheral blood mononuclear cells: association with muscle involvement. Clin Immunol 120: 319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Greenberg SA, Higgs BW, Morehouse C, Walsh RJ, Kong SW, Brohawn P, Zhu W, Amato A, Salajegheh M, White B, Kiener PA, Jallal B, and Yao Y. 2012. Relationship between disease activity and type 1 interferon- and other cytokine-inducible gene expression in blood in dermatomyositis and polymyositis. Genes Immun 13: 207–213. [DOI] [PubMed] [Google Scholar]
- 61.Bilgic H, Ytterberg SR, Amin S, McNallan KT, Wilson JC, Koeuth T, Ellingson S, Newman B, Bauer JW, Peterson EJ, Baechler EC, and Reed AM. 2009. Interleukin-6 and type I interferon-regulated genes and chemokines mark disease activity in dermatomyositis. Arthritis Rheum 60: 3436–3446. [DOI] [PubMed] [Google Scholar]
- 62.Wenzel J, Schmidt R, Proelss J, Zahn S, Bieber T, and Tuting T. 2006. Type I interferon-associated skin recruitment of CXCR3+ lymphocytes in dermatomyositis. Clin Exp Dermatol 31: 576–582. [DOI] [PubMed] [Google Scholar]
- 63.Magro CM, Segal JP, Crowson AN, and Chadwick P. 2010. The phenotypic profile of dermatomyositis and lupus erythematosus: a comparative analysis. J Cutan Pathol 37: 659–671. [DOI] [PubMed] [Google Scholar]
- 64.Caproni M, Torchia D, Cardinali C, Volpi W, Del Bianco E, D’Agata A, and Fabbri P. 2004. Infiltrating cells, related cytokines and chemokine receptors in lesional skin of patients with dermatomyositis. Br J Dermatol 151: 784–791. [DOI] [PubMed] [Google Scholar]
- 65.Ono N, Kai K, Maruyama A, Sakai M, Sadanaga Y, Koarada S, Inoue T, and Tada Y. 2020. The relationship between type 1 IFN and vasculopathy in anti-MDA5 antibody-positive dermatomyositis patients. Rheumatology (Oxford) 59: 918. [DOI] [PubMed] [Google Scholar]
- 66.Shrestha S, Wershil B, Sarwark JF, Niewold TB, Philipp T, and Pachman LM. 2010. Lesional and nonlesional skin from patients with untreated juvenile dermatomyositis displays increased numbers of mast cells and mature plasmacytoid dendritic cells. Arthritis and rheumatism 62: 2813–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neely J, Rychkov D, Paranjpe M, Waterfield M, Kim S, and Sirota M. 2019. Gene Expression Meta-Analysis Reveals Concordance in Gene Activation, Pathway, and Cell-Type Enrichment in Dermatomyositis Target Tissues. ACR Open Rheumatol 1: 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pinal-Fernandez I, Casal-Dominguez M, Derfoul A, Pak K, Plotz P, Miller FW, Milisenda JC, Grau-Junyent JM, Selva-O’Callaghan A, Paik J, Albayda J, Christopher-Stine L, Lloyd TE, Corse AM, and Mammen AL. 2019. Identification of distinctive interferon gene signatures in different types of myositis. Neurology 93: e1193–e1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Denton CP, and Khanna D. 2017. Systemic sclerosis. Lancet 390: 1685–1699. [DOI] [PubMed] [Google Scholar]
- 70.Varga J, and Abraham D. 2007. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 117: 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stevens AM, Torok KS, Li SC, Taber SF, Lu TT, and Zulian F. 2019. Immunopathogenesis of Juvenile Systemic Sclerosis. Front Immunol 10: 1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Steen VD, and Medsger TA Jr. 2001. Improvement in skin thickening in systemic sclerosis associated with improved survival. Arthritis Rheum 44: 2828–2835. [DOI] [PubMed] [Google Scholar]
- 73.Bezalel SA, Strober BE, and Ferenczi K. 2015. Interferon beta-1a-induced morphea. JAAD Case Rep 1: 15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hugle T, Gratzl S, Daikeler T, Frey D, Tyndall A, and Walker UA. 2009. Sclerosing skin disorders in association with multiple sclerosis. Coincidence, underlying autoimmune pathology or interferon induced? Ann Rheum Dis 68: 47–50. [DOI] [PubMed] [Google Scholar]
- 75.Powell A, Myles ML, and Yacyshyn E. 2008. The development of systemic sclerosis in a female patient with multiple sclerosis following beta interferon treatment. Clin Rheumatol 27: 1467–1468. [DOI] [PubMed] [Google Scholar]
- 76.Ghoreishi M, Vera Kellet C, and Dutz JP. 2012. Type 1 IFN-induced protein MxA and plasmacytoid dendritic cells in lesions of morphea. Exp Dermatol 21: 417–419. [DOI] [PubMed] [Google Scholar]
- 77.Magee KE, Kelsey CE, Kurzinski KL, Ho J, Mlakar LR, Feghali-Bostwick CA, and Torok KS. 2013. Interferon-gamma inducible protein-10 as a potential biomarker in localized scleroderma. Arthritis Res Ther 15: R188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Torok KS, Li SC, Jacobe HM, Taber SF, Stevens AM, Zulian F, and Lu TT. 2019. Immunopathogenesis of Pediatric Localized Scleroderma. Front Immunol 10: 908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Assassi S, Swindell WR, Wu M, Tan FD, Khanna D, Furst DE, Tashkin DP, Jahan-Tigh RR, Mayes MD, Gudjonsson JE, and Chang JT. 2015. Dissecting the heterogeneity of skin gene expression patterns in systemic sclerosis. Arthritis Rheumatol 67: 3016–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Farina G, Lafyatis D, Lemaire R, and Lafyatis R. 2010. A four-gene biomarker predicts skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum 62: 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McCoy SS, Reed TJ, Berthier CC, Tsou PS, Liu J, Gudjonsson JE, Khanna D, and Kahlenberg JM. 2017. Scleroderma keratinocytes promote fibroblast activation independent of transforming growth factor beta. Rheumatology (Oxford) 56: 1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tan FK, Zhou X, Mayes MD, Gourh P, Guo X, Marcum C, Jin L, and Arnett FC Jr. 2006. Signatures of differentially regulated interferon gene expression and vasculotrophism in the peripheral blood cells of systemic sclerosis patients. Rheumatology (Oxford) 45: 694–702. [DOI] [PubMed] [Google Scholar]
- 83.Brkic Z, van Bon L, Cossu M, van Helden-Meeuwsen CG, Vonk MC, Knaapen H, van den Berg W, Dalm VA, Van Daele PL, Severino A, Maria NI, Guillen S, Dik WA, Beretta L, Versnel MA, and Radstake T. 2016. The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann Rheum Dis 75: 1567–1573. [DOI] [PubMed] [Google Scholar]
- 84.Eloranta ML, Franck-Larsson K, Lovgren T, Kalamajski S, Ronnblom A, Rubin K, Alm GV, and Ronnblom L. 2010. Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis 69: 1396–1402. [DOI] [PubMed] [Google Scholar]
- 85.Kim D, Peck A, Santer D, Patole P, Schwartz SM, Molitor JA, Arnett FC, and Elkon KB. 2008. Induction of interferon-alpha by scleroderma sera containing autoantibodies to topoisomerase I: association of higher interferon-alpha activity with lung fibrosis. Arthritis Rheum 58: 2163–2173. [DOI] [PubMed] [Google Scholar]
- 86.Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, Gordon JK, and Barrat FJ. 2018. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Farina GA, York MR, Di Marzio M, Collins CA, Meller S, Homey B, Rifkin IR, Marshak-Rothstein A, Radstake TR, and Lafyatis R. 2010. Poly(I:C) drives type I IFN- and TGFbeta-mediated inflammation and dermal fibrosis simulating altered gene expression in systemic sclerosis. J Invest Dermatol 130: 2583–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ciechomska M, and Skalska U. 2018. Targeting interferons as a strategy for systemic sclerosis treatment. Immunol Lett 195: 45–54. [DOI] [PubMed] [Google Scholar]
- 89.Soy M, and Piskin S. 2007. Cutaneous findings in patients with primary Sjogren’s syndrome. Clin Rheumatol 26: 1350–1352. [DOI] [PubMed] [Google Scholar]
- 90.Bernacchi E, Amato L, Parodi A, Cottoni F, Rubegni P, De Pita O, Papini M, Rebora A, Bombardieri S, and Fabbri P. 2004. Sjogren’s syndrome: a retrospective review of the cutaneous features of 93 patients by the Italian Group of Immunodermatology. Clin Exp Rheumatol 22: 55–62. [PubMed] [Google Scholar]
- 91.Hjelmervik TO, Petersen K, Jonassen I, Jonsson R, and Bolstad AI. 2005. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjogren’s syndrome patients from healthy control subjects. Arthritis Rheum 52: 1534–1544. [DOI] [PubMed] [Google Scholar]
- 92.Nezos A, Gravani F, Tassidou A, Kapsogeorgou EK, Voulgarelis M, Koutsilieris M, Crow MK, and Mavragani CP. 2015. Type I and II interferon signatures in Sjogren’s syndrome pathogenesis: Contributions in distinct clinical phenotypes and Sjogren’s related lymphomagenesis. J Autoimmun 63: 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brkic Z, Maria NI, van Helden-Meeuwsen CG, van de Merwe JP, van Daele PL, Dalm VA, Wildenberg ME, Beumer W, Drexhage HA, and Versnel MA. 2013. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjogren’s syndrome and association with disease activity and BAFF gene expression. Ann Rheum Dis 72: 728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nguyen CQ, and Peck AB. 2013. The Interferon-Signature of Sjogren’s Syndrome: How Unique Biomarkers Can Identify Underlying Inflammatory and Immunopathological Mechanisms of Specific Diseases. Front Immunol 4: 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ, Ruether A, Schreiber S, Weichenthal M, Gladman D, Rahman P, Schrodi SJ, Prahalad S, Guthery SL, Fischer J, Liao W, Kwok PY, Menter A, Lathrop GM, Wise CA, Begovich AB, Voorhees JJ, Elder JT, Krueger GG, Bowcock AM, and Abecasis GR. 2009. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet 41: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhu H, Lou F, Yin Q, Gao Y, Sun Y, Bai J, Xu Z, Liu Z, Cai W, Ke F, Zhang L, Zhou H, Wang H, Wang G, Chen X, Zhang H, Wang Z, Ginhoux F, Lu C, Su B, and Wang H. 2017. RIG-I antiviral signaling drives interleukin-23 production and psoriasis-like skin disease. EMBO Mol Med 9: 589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van der Fits L, van der Wel LI, Laman JD, Prens EP, and Verschuren MC. 2004. In psoriasis lesional skin the type I interferon signaling pathway is activated, whereas interferon-alpha sensitivity is unaltered. J Invest Dermatol 122: 51–60. [DOI] [PubMed] [Google Scholar]
- 98.Schmid P, Itin D Fau - Cox P, Cox GK Fau - McMaster D, McMaster MA Fau - Horisberger Gk, and Horisberger MA. The type I interferon system is locally activated in psoriatic lesions. [DOI] [PubMed]
- 99.Fah J, Pavlovic G Fau - Burg J, and Burg G. Expression of MxA protein in inflammatory dermatoses. [DOI] [PubMed]
- 100.Suomela S, Cao A Fau - Bowcock L, Bowcock U Fau - Saarialho-Kere A, and Saarialho-Kere U. Interferon alpha-inducible protein 27 (IFI27) is upregulated in psoriatic skin and certain epithelial cancers. [DOI] [PubMed]
- 101.Goldberg A, Geppert T, Schiopu E, Frech T, Hsu V, Simms RW, Peng SL, Yao Y, Elgeioushi N, Chang L, Wang B, and Yoo S. 2014. Dose-escalation of human anti-interferon-alpha receptor monoclonal antibody MEDI-546 in subjects with systemic sclerosis: a phase 1, multicenter, open label study. Arthritis Res Ther 16: R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sonmez HE, Karaaslan C, de Jesus AA, Batu ED, Anlar B, Sozeri B, Bilginer Y, Karaguzel D, Cagdas Ayvaz D, Tezcan I, Goldbach-Mansky R, and Ozen S. 2020. A clinical score to guide in decision making for monogenic type I IFNopathies. Pediatr Res 87: 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.de Jesus AA, Hou Y, Brooks S, Malle L, Biancotto A, Huang Y, Calvo KR, Marrero B, Moir S, Oler AJ, Deng Z, Montealegre Sanchez GA, Ahmed A, Allenspach E, Arabshahi B, Behrens E, Benseler S, Bezrodnik L, Bout-Tabaku S, Brescia AC, Brown D, Burnham JM, Caldirola MS, Carrasco R, Chan AY, Cimaz R, Dancey P, Dare J, DeGuzman M, Dimitriades V, Ferguson I, Ferguson P, Finn L, Gattorno M, Grom AA, Hanson EP, Hashkes PJ, Hedrich CM, Herzog R, Horneff G, Jerath R, Kessler E, Kim H, Kingsbury DJ, Laxer RM, Lee PY, Lee-Kirsch MA, Lewandowski L, Li S, Lilleby V, Mammadova V, Moorthy LN, Nasrullayeva G, O’Neill KM, Onel K, Ozen S, Pan N, Pillet P, Piotto DG, Punaro MG, Reiff A, Reinhardt A, Rider LG, Rivas-Chacon R, Ronis T, Rosen-Wolff A, Roth J, Ruth NM, Rygg M, Schmeling H, Schulert G, Scott C, Seminario G, Shulman A, Sivaraman V, Son MB, Stepanovskiy Y, Stringer E, Taber S, Terreri MT, Tifft C, Torgerson T, Tosi L, Van Royen-Kerkhof A, Wampler Muskardin T, Canna SW, and Goldbach-Mansky R. 2020. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J Clin Invest. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Aicardi J, and Goutieres F. 1984. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol 15: 49–54. [DOI] [PubMed] [Google Scholar]
- 105.Goutieres F, Aicardi J, Barth PG, and Lebon P. 1998. Aicardi-Goutieres syndrome: an update and results of interferon-alpha studies. Ann Neurol 44: 900–907. [DOI] [PubMed] [Google Scholar]
- 106.Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, Baxter P, Benko WS, Bergmann C, Bertini E, Biancheri R, Blair EM, Blau N, Bonthron DT, Briggs T, Brueton LA, Brunner HG, Burke CJ, Carr IM, Carvalho DR, Chandler KE, Christen HJ, Corry PC, Cowan FM, Cox H, D’Arrigo S, Dean J, De Laet C, De Praeter C, Dery C, Ferrie CD, Flintoff K, Frints SG, Garcia-Cazorla A, Gener B, Goizet C, Goutieres F, Green AJ, Guet A, Hamel BC, Hayward BE, Heiberg A, Hennekam RC, Husson M, Jackson AP, Jayatunga R, Jiang YH, Kant SG, Kao A, King MD, Kingston HM, Klepper J, van der Knaap MS, Kornberg AJ, Kotzot D, Kratzer W, Lacombe D, Lagae L, Landrieu PG, Lanzi G, Leitch A, Lim MJ, Livingston JH, Lourenco CM, Lyall EG, Lynch SA, Lyons MJ, Marom D, McClure JP, McWilliam R, Melancon SB, Mewasingh LD, Moutard ML, Nischal KK, Ostergaard JR, Prendiville J, Rasmussen M, Rogers RC, Roland D, Rosser EM, Rostasy K, Roubertie A, Sanchis A, Schiffmann R, Scholl-Burgi S, Seal S, Shalev SA, Corcoles CS, Sinha GP, Soler D, Spiegel R, Stephenson JB, Tacke U, Tan TY, Till M, Tolmie JL, Tomlin P, Vagnarelli F, Valente EM, Van Coster RN, Van der Aa N, Vanderver A, Vles JS, Voit T, Wassmer E, Weschke B, Whiteford ML, Willemsen MA, Zankl A, Zuberi SM, Orcesi S, Fazzi E, Lebon P, and Crow YJ. 2007. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet 81: 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, and Lindahl T. 2006. Mutations in the gene encoding the 3’−5’ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 38: 917–920. [DOI] [PubMed] [Google Scholar]
- 108.Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martinez-Frias ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JB, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, and Jackson AP. 2006. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet 38: 910–916. [DOI] [PubMed] [Google Scholar]
- 109.Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BC, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L, Landrieu PG, Lourenco CM, Marom D, McDermott MF, van der Merwe W, Orcesi S, Prendiville JS, Rasmussen M, Shalev SA, Soler DM, Shinawi M, Spiegel R, Tan TY, Vanderver A, Wakeling EL, Wassmer E, Whittaker E, Lebon P, Stetson DB, Bonthron DT, and Crow YJ. 2009. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 41: 829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yang YG, Lindahl T, and Barnes DE. 2007. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131: 873–886. [DOI] [PubMed] [Google Scholar]
- 111.Mazur DJ, and Perrino FW. 2001. Excision of 3’ termini by the Trex1 and TREX2 3’-->5’ exonucleases. Characterization of the recombinant proteins. J Biol Chem 276: 17022–17029. [DOI] [PubMed] [Google Scholar]
- 112.Grieves JL, Fye JM, Harvey S, Grayson JM, Hollis T, and Perrino FW. 2015. Exonuclease TREX1 degrades double-stranded DNA to prevent spontaneous lupus-like inflammatory disease. Proc Natl Acad Sci U S A 112: 5117–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yuan F, Dutta T, Wang L, Song L, Gu L, Qian L, Benitez A, Ning S, Malhotra A, Deutscher MP, and Zhang Y. 2015. Human DNA Exonuclease TREX1 Is Also an Exoribonuclease That Acts on Single-stranded RNA. J Biol Chem 290: 13344–13353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Peschke K, Achleitner M, Frenzel K, Gerbaulet A, Ada SR, Zeller N, Lienenklaus S, Lesche M, Poulet C, Naumann R, Dahl A, Ravens U, Gunther C, Muller W, Knobeloch KP, Prinz M, Roers A, and Behrendt R. 2016. Loss of Trex1 in Dendritic Cells Is Sufficient To Trigger Systemic Autoimmunity. J Immunol 197: 2157–2166. [DOI] [PubMed] [Google Scholar]
- 115.Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, Tuting T, Hartmann G, and Barchet W. 2013. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 39: 482–495. [DOI] [PubMed] [Google Scholar]
- 116.Gunther C, Kind B, Reijns MA, Berndt N, Martinez-Bueno M, Wolf C, Tungler V, Chara O, Lee YA, Hubner N, Bicknell L, Blum S, Krug C, Schmidt F, Kretschmer S, Koss S, Astell KR, Ramantani G, Bauerfeind A, Morris DL, Cunninghame Graham DS, Bubeck D, Leitch A, Ralston SH, Blackburn EA, Gahr M, Witte T, Vyse TJ, Melchers I, Mangold E, Nothen MM, Aringer M, Kuhn A, Luthke K, Unger L, Bley A, Lorenzi A, Isaacs JD, Alexopoulou D, Conrad K, Dahl A, Roers A, Alarcon-Riquelme ME, Jackson AP, and Lee-Kirsch MA. 2015. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J Clin Invest 125: 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Juern A, Robbins A, Galbraith S, and Drolet B. 2010. Aicardi-Goutieres syndrome: cutaneous, laboratory, and radiologic findings: a case report. Pediatr Dermatol 27: 82–85. [DOI] [PubMed] [Google Scholar]
- 118.Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, Kim PW, Sheikh A, Lee CC, Chen Y, Vera A, Zhang X, Goldbach-Mansky R, and Zlotogorski A. 2012. Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum 64: 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, Montealegre G, Biancotto A, Reinhardt A, de Jesus AA, Pelletier M, Tsai WL, Remmers EF, Kardava L, Hill S, Kim H, Lachmann HJ, Megarbane A, Chae JJ, Brady J, Castillo RD, Brown D, Casano AV, Gao L, Chapelle D, Huang Y, Stone D, Chen Y, Sotzny F, Lee CC, Kastner DL, Torrelo A, Zlotogorski A, Moir S, Gadina M, McCoy P, Wesley R, Rother KI, Hildebrand PW, Brogan P, Kruger E, Aksentijevich I, and Goldbach-Mansky R. 2016. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest 126: 795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, Goudin N, Fremond ML, Nitschke P, Molina TJ, Blanche S, Picard C, Rice GI, Crow YJ, Manel N, Fischer A, Bader-Meunier B, and Rieux-Laucat F. 2014. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest 124: 5516–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, Lee CR, DiMattia MA, Cowen EW, Gonzalez B, Palmer I, DiGiovanna JJ, Biancotto A, Kim H, Tsai WL, Trier AM, Huang Y, Stone DL, Hill S, Kim HJ, St Hilaire C, Gurprasad S, Plass N, Chapelle D, Horkayne-Szakaly I, Foell D, Barysenka A, Candotti F, Holland SM, Hughes JD, Mehmet H, Issekutz AC, Raffeld M, McElwee J, Fontana JR, Minniti CP, Moir S, Kastner DL, Gadina M, Steven AC, Wingfield PT, Brooks SR, Rosenzweig SD, Fleisher TA, Deng Z, Boehm M, Paller AS, and Goldbach-Mansky R. 2014. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371: 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Motwani M, Pawaria S, Bernier J, Moses S, Henry K, Fang T, Burkly L, Marshak-Rothstein A, and Fitzgerald KA. 2019. Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proc Natl Acad Sci U S A 116: 7941–7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, Illei GG, Drappa J, Wang L, Yoo S, and Investigators CDS. 2017. Anifrolumab, an Anti-Interferon-alpha Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol 69: 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Merrill JT, Furie R, Werth VP, Khamashta M, Drappa J, Wang L, Illei G, and Tummala R. 2018. Anifrolumab effects on rash and arthritis: impact of the type I interferon gene signature in the phase IIb MUSE study in patients with systemic lupus erythematosus. Lupus Sci Med 5: e000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, Bae SC, Brohawn PZ, Pineda L, Berglind A, Tummala R, and Investigators T-T. 2020. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med 382: 211–221. [DOI] [PubMed] [Google Scholar]
- 126.Guo X, Higgs BW, Bay-Jensen AC, Karsdal MA, Yao Y, Roskos LK, and White WI. 2015. Suppression of T Cell Activation and Collagen Accumulation by an Anti-IFNAR1 mAb, Anifrolumab, in Adult Patients with Systemic Sclerosis. J Invest Dermatol 135: 2402–2409. [DOI] [PubMed] [Google Scholar]
- 127.O’Shea JJ, and Gadina M. 2019. Selective Janus kinase inhibitors come of age. Nat Rev Rheumatol 15: 74–75. [DOI] [PubMed] [Google Scholar]
- 128.Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, and O’Shea JJ. 2017. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov 17: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Klaeschen AS, Wolf D, Brossart P, Bieber T, and Wenzel J. 2017. JAK inhibitor ruxolitinib inhibits the expression of cytokines characteristic of cutaneous lupus erythematosus. Exp Dermatol 26: 728–730. [DOI] [PubMed] [Google Scholar]
- 130.Furumoto Y, Smith CK, Blanco L, Zhao W, Brooks SR, Thacker SG, Abdalrahman Z, Sciume G, Tsai WL, Trier AM, Nunez L, Mast L, Hoffmann V, Remaley AT, O’Shea JJ, Kaplan MJ, and Gadina M. 2017. Tofacitinib Ameliorates Murine Lupus and Its Associated Vascular Dysfunction. Arthritis Rheumatol 69: 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ladislau L, Suarez-Calvet X, Toquet S, Landon-Cardinal O, Amelin D, Depp M, Rodero MP, Hathazi D, Duffy D, Bondet V, Preusse C, Bienvenu B, Rozenberg F, Roos A, Benjamim CF, Gallardo E, Illa I, Mouly V, Stenzel W, Butler-Browne G, Benveniste O, and Allenbach Y. 2018. JAK inhibitor improves type I interferon induced damage: proof of concept in dermatomyositis. Brain 141: 1609–1621. [DOI] [PubMed] [Google Scholar]
- 132.Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, Schalm S, Murias S, Dare JA, Brown D, Stone DL, Gao L, Klausmeier T, Foell D, de Jesus AA, Chapelle DC, Kim H, Dill S, Colbert RA, Failla L, Kost B, O’Brien M, Reynolds JC, Folio LR, Calvo KR, Paul SM, Weir N, Brofferio A, Soldatos A, Biancotto A, Cowen EW, Digiovanna JJ, Gadina M, Lipton AJ, Hadigan C, Holland SM, Fontana J, Alawad AS, Brown RJ, Rother KI, Heller T, Brooks KM, Kumar P, Brooks SR, Waldman M, Singh HK, Nickeleit V, Silk M, Prakash A, Janes JM, Ozen S, Wakim PG, Brogan PA, Macias WL, and Goldbach-Mansky R. 2018. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest 128: 3041–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zimmermann N, Wolf C, Schwenke R, Luth A, Schmidt F, Engel K, Lee-Kirsch MA, and Gunther C. 2019. Assessment of Clinical Response to Janus Kinase Inhibition in Patients With Familial Chilblain Lupus and TREX1 Mutation. JAMA Dermatol 155: 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Furie R, Werth VP, Merola JF, Stevenson L, Reynolds TL, Naik H, Wang W, Christmann R, Gardet A, Pellerin A, Hamann S, Auluck P, Barbey C, Gulati P, Rabah D, and Franchimont N. 2019. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Invest 129: 1359–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gardet A, Pellerin A, McCarl CA, Diwanji R, Wang W, Donaldson D, Franchimont N, Werth VP, and Rabah D. 2019. Effect of in vivo Hydroxychloroquine and ex vivo Anti-BDCA2 mAb Treatment on pDC IFNalpha Production From Patients Affected With Cutaneous Lupus Erythematosus. Front Immunol 10: 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lebre MC, van der Aar AM, van Baarsen L, van Capel TM, Schuitemaker JH, Kapsenberg ML, and de Jong EC. 2007. Human keratinocytes express functional Toll-like receptor 3, 4, 5, and 9. J Invest Dermatol 127: 331–341. [DOI] [PubMed] [Google Scholar]
- 137.Akira S, and Hemmi H. 2003. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett 85: 85–95. [DOI] [PubMed] [Google Scholar]
- 138.Kollisch G, Kalali BN, Voelcker V, Wallich R, Behrendt H, Ring J, Bauer S, Jakob T, Mempel M, and Ollert M. 2005. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology 114: 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Miller LS 2008. Toll-like receptors in skin. Adv Dermatol 24: 71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li M, Chen Q, Shen Y, and Liu W. 2009. Candida albicans phospholipomannan triggers inflammatory responses of human keratinocytes through Toll-like receptor 2. Exp Dermatol 18: 603–610. [DOI] [PubMed] [Google Scholar]
- 141.Olaru F, and Jensen LE. 2010. Chemokine expression by human keratinocyte cell lines after activation of Toll-like receptors. Exp Dermatol 19: e314–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kalali BN, Kollisch G, Mages J, Muller T, Bauer S, Wagner H, Ring J, Lang R, Mempel M, and Ollert M. 2008. Double-stranded RNA induces an antiviral defense status in epidermal keratinocytes through TLR3-, PKR-, and MDA5/RIG-I-mediated differential signaling. J Immunol 181: 2694–2704. [DOI] [PubMed] [Google Scholar]
- 143.Fukui A, Ohta K, Nishi H, Shigeishi H, Tobiume K, Takechi M, and Kamata N. 2013. Interleukin-8 and CXCL10 expression in oral keratinocytes and fibroblasts via Toll-like receptors. Microbiol Immunol 57: 198–206. [DOI] [PubMed] [Google Scholar]
- 144.Ohta K, Fukui A, Shigeishi H, Ishida Y, Nishi H, Tobiume K, Takechi M, and Kamata N. 2014. Expression and function of RIG-I in oral keratinocytes and fibroblasts. Cell Physiol Biochem 34: 1556–1565. [DOI] [PubMed] [Google Scholar]
- 145.Kimura K, Matsuzaki Y, Nishikawa Y, Kitamura H, Akasaka E, Rokunohe D, Nakano H, Imaizumi T, Satoh K, and Sawamura D. 2012. Characterization of retinoic acid-inducible gene-I (RIG-I) expression corresponding to viral infection and UVB in human keratinocytes. J Dermatol Sci 66: 64–70. [DOI] [PubMed] [Google Scholar]
- 146.Almine JF, O’Hare CA, Dunphy G, Haga IR, Naik RJ, Atrih A, Connolly DJ, Taylor J, Kelsall IR, Bowie AG, Beard PM, and Unterholzner L. 2017. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun 8: 14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Costa S, Borgogna C, Mondini M, De Andrea M, Meroni PL, Berti E, Gariglio M, and Landolfo S. 2011. Redistribution of the nuclear protein IFI16 into the cytoplasm of ultraviolet B-exposed keratinocytes as a mechanism of autoantigen processing. Br J Dermatol 164: 282–290. [DOI] [PubMed] [Google Scholar]
- 148.Demaria O, Di Domizio J, and Gilliet M. 2014. Immune sensing of nucleic acids in inflammatory skin diseases. Semin Immunopathol 36: 519–529. [DOI] [PubMed] [Google Scholar]
- 149.Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, Ueda M, Nishide M, Koyama S, Hayama Y, Kinehara Y, Hirano T, Shima Y, Narazaki M, and Kumanogoh A. 2018. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis 77: 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]