

Abstract
Motivation
Microbial communities drive matter and energy transformations integral to global biogeochemical cycles, yet many taxonomic groups facilitating these processes remain poorly represented in biological sequence databases. Due to this missing information, taxonomic assignment of sequences from environmental genomes remains inaccurate.
Results
We present the Tree-based Sensitive and Accurate Phylogenetic Profiler (TreeSAPP) software for functionally and taxonomically classifying genes, reactions and pathways from genomes of cultivated and uncultivated microorganisms using reference packages representing coding sequences mediating multiple globally relevant biogeochemical cycles. TreeSAPP uses linear regression of evolutionary distance on taxonomic rank to improve classifications, assigning both closely related and divergent query sequences at the appropriate taxonomic rank. TreeSAPP is able to provide quantitative functional and taxonomic classifications for both assembled and unassembled sequences and files supporting interactive tree of life visualizations.
Availability and implementation
TreeSAPP was developed in Python 3 as an open-source Python package and is available on GitHub at https://github.com/hallamlab/TreeSAPP.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
We live in a world dominated by prokaryotic (archaea and bacteria) microorganisms. The collective properties of this unseen majority have an enormous impact on the world, driving matter and energy transformations through networks of metabolite exchange (Canfield et al., 2010; Falkowski et al., 2008; Hurwitz and Sullivan, 2013). Over geological time, these interactions have fundamentally transformed the surface chemistry of the earth and continue to shape elemental fluxes between atmospheric, terrestrial and aquatic compartments of the biosphere. A versatile set of functional genes that evolved early in the history of life are responsible for driving biogeochemical processes through largely defined metabolic pathways (Falkowski et al., 2008). Genes encoding diagnostic steps in these pathways (e.g. functional anchors) can be assembled into a phylogenetic framework that provides information on the distribution, abundance and taxonomic origin of environmental sequences. However, charting the diversity and distribution patterns of functional and taxonomic anchor genes is limited by our inability to quantitatively resolve microbial communities within a standard taxonomic hierarchy.
Despite a plethora of published software (Boyd et al., 2018; Buchfink et al., 2015; Darling et al., 2014), taxonomic assignment remains an unsolved problem (Peabody et al., 2015). Many factors must be considered when interpreting taxonomic profiles but perhaps the most vexing are legacy misclassifications in biological sequence databases, which are particularly difficult to predict and fix (Kozlov et al., 2016; Merchant et al., 2014; Nasko et al., 2018). The potential for misclassification is exacerbated by the recent inclusion of composite metagenome-assembled genomes (MAGs) in such databases (Shaiber and Eren, 2019). Moreover, despite enormous progress in high-throughput cultivation (Cross et al., 2019; Nichols et al., 2010) the majority of microorganisms from natural and engineered environments remain to be isolated in a laboratory setting (Rappé and Giovannoni, 2003; Solden et al., 2016; Steen et al., 2019). This ‘cultivation gap’ poses a formidable issue for taxonomic assignment as many sequences derived from the environment are distantly related to representatives in sequence databases that underpin taxonomic classification software. Single-cell amplified genomes (SAGs) have the potential to bridge the gap by linking individual, environmental genotypes to specific taxonomic labels (Rinke et al., 2013). However, their utility in annotation pipelines has yet to be realized.
Common methods for taxonomic classification include pairwise sequence alignment (Altschul et al., 1990; Buchfink et al., 2015), k-mer matching (Kim et al., 2016; Ondov et al., 2016) and phylogenetic placement (Barbera et al., 2019; Berger and Stamatakis, 2011; Matsen et al., 2010; Stark et al., 2010). These methods rely on indexed files from either custom, curated sequence datasets or massive repositories. Distance between query and reference sequences can be estimated using sequence similarity, evolutionary distance or derivatives thereof. Current taxonomic assignment methods do not factor these measures into classification, instead using either a lowest common ancestor (LCA), best-hit or ensemble approach to yield a single taxonomic label (Hanson et al., 2016; Huson et al., 2007; Konwar et al., 2013). This frequently leads to over-classification in the presence of unrepresented taxa. Phylogenetic placement methods are well-equipped to handle gene-centric assignment because the branch length distances between query and related reference sequences serve as a coordinate system when estimating taxonomic relationships (Ciccarelli et al., 2006). Applications that calibrate taxonomic rank thresholds to a phylogeny’s branch lengths have been developed (Parks et al., 2018; Wu et al., 2013). As part of the Genome Taxonomy Database (GTDB), nodes in reference phylogenies are calibrated by their relative evolutionary distance (RED) values (Chaumeil et al., 2019). However, the GTDB toolkit is not intended for gene-centric taxonomic assignment but for classifying genomes by placing concatenated single-copy marker genes sequences into pre-computed reference trees. Alternatively GraftM can use pplacer for phylogenetic placement of query sequences into gene trees but stops short of using evolutionary distance to correct for over-classification and annotates query sequences using one reference package at a time (Boyd et al., 2018).
Here, we present the Tree-based Sensitive and Accurate Phylogenetic Profiler (TreeSAPP), a gene-centric functional and taxonomic classification tool able to classify proteins and assembled or unassembled nucleotide sequences. Fragments per Kilobase per Million reads values can be calculated and used in all outputs for genomic or transcriptomic data with short-read sequences in FASTQ format. By using RAxML’s evolutionary placement algorithm (EPA), TreeSAPP leverages the evolutionary distance to reference sequences (EDR), a sum of the distal and pendant lengths as well as the mean length from the placement edge to all descendent leaf tips, correlated with taxonomic rank to recommend optimal taxonomic ranks for more accurate classifications. Structural and metabolic feature information can be annotated in the interactive tree of life (iTOL) and exported to allow for improved functional annotation of query sequences (Letunic and Bork, 2016). In addition to a classification table, placement outputs are compatible with iTOL for easy creation of publication-quality figures (Fig. 1).
Fig. 1.
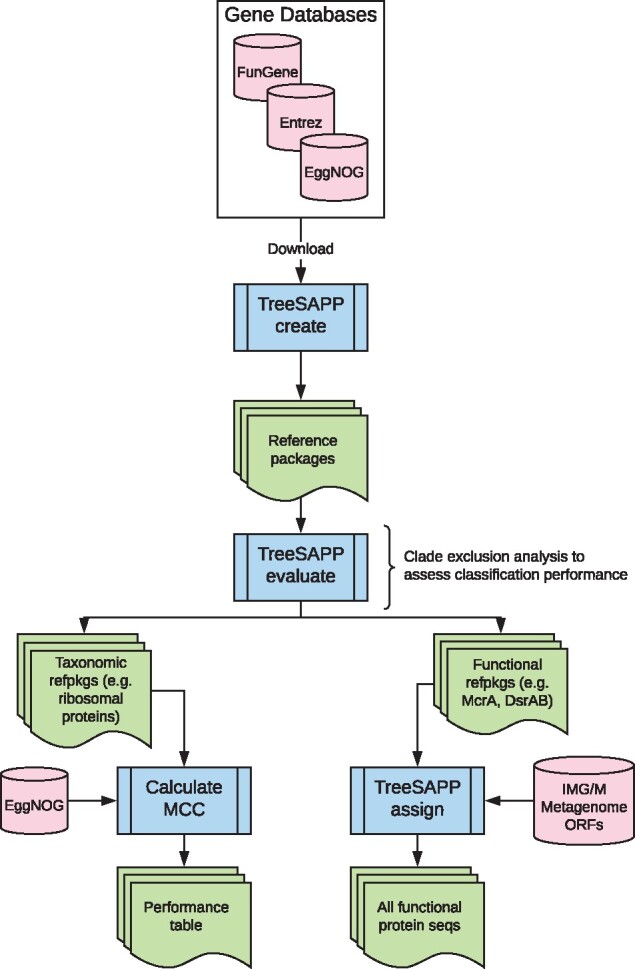
The workflow of the current study. Sequences for building reference packages were sourced from the NCBI and FunGene databases. Sequences were downloaded from EggNOG for validating reference packages and benchmarking TreeSAPP against GraftM. IMG/M metagenomes were used to explore the global diversity of Mcr
2 Materials and methods
TreeSAPP was developed in Python 3. It uses the Environment for Tree Exploration Python package for many tree manipulation operations (Huerta-Cepas et al., 2016b). The multiprocessing Python package is used to run executable processes in parallel unless it is more efficient to use the software’s native parallelization capabilities. BioPython is used for downloading lineage information from Entrez’s taxonomy database for each reference sequence when building new reference packages (Cock et al., 2009). Sequences for the reference packages described in this manuscript were primarily downloaded from the FunGene repository as this resource provides curated sequences for many of the functional anchor genes involved in major biogeochemical cycles (Fish et al., 2013) (Supplementary Table S1). Additionally, to provide the most comprehensive reference trees, newly published sequences that were not included in FunGene 9.6 were included from GenBank and the Joint Genome Institute’s Integrated Microbial Genomes and Microbiomes (IMG/M) (Borrel et al., 2019; Hua et al., 2019; McKay et al., 2019; Seitz et al., 2019; Wang et al., 2019). All benchmarking was performed on a server with a 20 physical core (40 virtual) Intel Xeon CPU (E5-2650 v3), 264 GB of RAM and a 5 TB HDD. The operating system was Red Hat Enterprise Linux Server release 7.5 (Maipo).
2.1 Building reference packages with TreeSAPP create
TreeSAPP’s classification workflow requires a multiple sequence alignment (MSA), profile hidden Markov model (HMM), taxonomic lineages and phylogenetic tree for all reference sequences (Supplementary Fig. S1). Together these files constitute a reference package, built using treesapp create, and allow for rapid query sequence filtering, phylogenetic placement and taxonomic classification. Construction began with removing truncated sequences using either a provided HMM or a database’s filter prior to downloading. An empirically determined threshold of 60% profile HMM coverage was required of candidate reference sequences to balance inclusivity and profile quality. NCBI taxonomic lineages were then downloaded for each candidate reference sequence with a valid accession. Lineage information of unaccessioned sequences can be provided via either a table or the FASTA file using a custom header format. Optional taxonomy-based filtering was performed (e.g. to remove viral or eukaryotic sequences). This set can then be clustered using USEARCH at a specified proportional similarity (Edgar, 2010). Non-homologous (i.e. outlier or mis-annotated) sequences were identified by OD-Seq and removed (Jehl et al., 2015). These automatically curated sequences were used to generate the reference MSA with MAFFT’s -‘auto’ algorithm (Katoh and Standley, 2013). The resulting MSA was used by HMMER’s ‘hmmbuild’ module to build a new profile HMM (Eddy, 1998). Before tree construction, the MSA was optionally trimmed using BMGE with the least conserved matrix, BLOSUM30 for proteins or PAM100 otherwise, to decrease the runtime required for phylogeny inference by removing non-conserved positions (Criscuolo and Gribaldo, 2010).
By default, RAxML is used for phylogenetic inference, automatically using the optimal substitution model (invoked with the -PROTGAMMAAUTO flag) and the minimum number of bootstraps necessary (using -‘autoMR’) (Pattengale et al., 2010; Stamatakis, 2006). Yet, in light of cursory taxonomic classification performance results indicating little difference between RAxML and FastTree (Supplementary Fig. S6) with a drastic difference in compute time, FastTree is available as well (Price et al., 2010). In this case, a bootstrapped tree will not be generated and the LG amino acid substitution model is used unless the user specifies differently (Le and Gascuel, 2008). To assign taxonomy at the most appropriate rank given a placement’s distance, a linear correlation of taxonomic rank with EDR is estimated for each reference package. Briefly, all sequences from a taxonomic group were removed from the reference tree before clustered sequences from the initial input FASTA file were mapped back to the tree using EPA. The placement distances were calculated and recorded before the next iteration involving a different taxon until all possible taxa have been exhaustively placed. Outliers are then removed from these data and rarefied to improve normality across the ranks before a linear model is fit.
2.2 Testing classification performance with TreeSAPP evaluate
TreeSAPP’s classification performance was evaluated using clade exclusion analysis (described below) and the binary classification metric, Matthews’ correlation coefficient (MCC) (Matthews, 1975 (Supplementary Equation S1). TreeSAPP was compared to GraftM and DIAMOND, as implemented in GraftM, using 15 taxonomic and 12 functional anchor reference packages (Supplementary Table S1). Functional anchor reference packages were only used during clade exclusion analysis as their representation in the MCC test data (EggNOG) was too limited and potentially a source of bias. To consistently compare between methods, all performance analyses relied on the NCBI’s taxonomic hierarchy for determining each sequence’s optimal taxonomic assignment: the highest resolution taxonomic classification with respect to the taxonomic composition of the reference package (i.e. the LCA between the query and most closely related reference). Under these conditions, ‘taxonomic distance’ is defined as the number of ranks separating the LCA of the optimal taxonomic assignment and the taxon assigned by the software (Supplementary Fig. S4). Given there are eight conventional taxonomic ranks used in the classification of Bacteria and Archaea, the maximum taxonomic distance is eight while a perfect classification has a taxonomic distance of zero.
Classification performance was measured utilizing 15 universal single-copy taxonomic anchor genes and the EggNOG database (v4.5.1) (Supplementary Table S1), with reference sequences from Bacteria and Archaea but excluding Eukaryotes to simplify the hierarchy. In addition to RecA, RadA and RpoB, nearly ubiquitous taxonomic anchor genes used by the GTDB were identified as being present in at least 90% of Archaea and Bacteria (Parks et al., 2018). Out of this set, 12 were selected that also had entries in the PFam database and were therefore easily accessed. Sequences that were either not in the PFam database or did not have an obvious corresponding orthologous group in EggNOG were omitted. For each taxonomic distance between zero and eight, sequence classifications matching their EggNOG annotations within the taxonomic distance threshold were counted as true positives. Sequences that were classified as taxa outside of the threshold or as the wrong gene were counted as false positives. EggNOG sequences that were orthologous to any of the 12 reference packages but were not classified counted as false negatives while all remaining sequences were true negatives. Classification performance was determined using MCC due to its ability to report reasonable values even with very different class sizes (Boughorbel et al., 2017).
Taxonomic classification accuracy was estimated by clade exclusion analysis using functional anchor reference packages for dissimilatory sulphite reductase alpha and beta subunits (DsrAB), methyl coenzyme-M reductase alpha, beta and gamma subunits (McrA, McrB and McrG), periplasmic nitrate reductase (NapA), NO-forming nitrite reductase (NirK and NirS), nitrogenase molybdenum–iron protein alpha chain(NifD), nitric oxide reductase subunit B (NorB), nitrite oxidoreductase subunits A and B (NxrA and NxrB) and a combined particulate methane monooxygenase and ammonia monooxygenase (PmoA/AmoA). Clade exclusion analysis measures classification performance in scenarios where the query sequences lack a close relative in the reference set, as is typical during taxonomic classification of metagenomes (Peabody et al., 2015) (Supplementary Fig. S3). The analysis required reference sequences with taxonomic lineages completely resolved to the rank evaluated. Sequences not resolved to the rank being evaluated (e.g. Archaea; Euryarchaeota; environmental samples for Class) were removed as their specific taxonomic relationship to other reference sequences was unknown. For each taxonomic rank tested, representative sequences were selected for each taxon (an arbitrary maximum of five so as to not introduce lineage-specific bias into the final estimate) and sequences belonging to that taxon were removed from the reference package. These representative sequences were then classified and compared to their optimal taxonomic assignment. Distances from their optimal taxonomic assignments were tabulated and reference sequences were returned to the reference package before the next taxon was tested. Only taxa that shared a common ancestor at the rank tested with one or more remaining reference sequences were evaluated. For example, evaluating the ability to classify at the rank of Class using sequences belonging to the order Methanosarcinales would require the reference package to contain other members of the parent taxonomic class Methanomicrobia that are not Methanosarcinales, such as Methanomicrobiales. This allows the software to optimally classify the sequences as Methanomicrobia.
2.3 Classifying query sequences with TreeSAPP assign
TreeSAPP begins by predicting open reading frames (ORFs) using Prodigal v2.6.3 if the inputs are nucleotide sequences, otherwise this step is skipped (Supplementary Fig. S2) (Hyatt et al., 2010). Resulting ORFs are conceptually translated into proteins that are then aligned to curated reference sequences using hmmalign, within the HMMER v3.1 package (Eddy, 1998). Homologous sequences are then extracted and mapped onto the reference MSA with hmmalign. Optionally, alignments are trimmed using BMGE to remove non-conserved positions from the alignment file and reduce computation time (Criscuolo and Gribaldo, 2010). BMGE uses the BLOSUM30 substitution matrix for protein sequences, as recommended by Tan et al. (2015). RAxML-EPA places the query sequences in the reference tree by finding the optimal phylogeny likelihood with the sequence inserted (Berger and Stamatakis, 2011). Query sequence placements are filtered by evolutionary distance and likelihood weight ratio prior to predicting taxonomy at the linear-model recommended rank. The complete set of ORF sequences predicted by Prodigal (both nucleotide and amino acid forms), a FASTA file containing only the classified ORFs and a classification table with taxonomy and abundance information are included as outputs. JPlace files, containing reference tree placement coordinates for all queries classified, are additionally provided for each reference package. Along with colour and style information files made for a specific phylogeny, these placements may be visualized in iTOL (Letunic and Bork, 2019).
3 Results
Many taxonomic assignment software methods are evaluated by a mock community of known, but severely limited diversity. So while these analyses are useful, their inherent oversimplification prohibits their performance estimates from being extensible to natural and engineered microbial communities. This is especially perplexing for gene-centric annotation software, where the total diversity of query sequences is reduced compared to whole-genome binning and profiling tools. Efforts are being made to provide well-designed mock communities for metagenome analysis, including taxonomic assignment, though they are still limited in their diversity (Sczyrba et al., 2017). Therefore, we decided to compare the taxonomic classification performance of TreeSAPP to GraftM and DIAMOND using the large, but still well curated, EggNOG database (v4.5) (Huerta-Cepas et al., 2016a) comprised of 2031 organisms for universal taxonomic anchor genes. Additionally, we used treesapp evaluate to simulate reference packages that do not represent taxa from Species to Class-level sequence divergence to determine how each tool classifies well and poorly represented query sequences. Finally, TreeSAPP was used to profile all metagenome-derived proteins in the IMG/M database.
3.1 Performance
Classification and runtime performance of TreeSAPP were benchmarked against GraftM with the default hmmsearch search strategy and pplacer placement method as well as DIAMOND-mode as a proxy for an alignment-based search and classification strategy (Boyd et al., 2018; Buchfink et al., 2015). Classifying the EggNOG database (version 4.5.0) (Huerta-Cepas et al., 2016a) with 12 single-copy taxonomic anchors revealed TreeSAPP’s overall classification performance was, with little exception, preferable to that of either a pairwise alignment strategy or GraftM (Fig. 2 and Supplementary Table S2). This is due in large part to TreeSAPP’s evolutionary distance-based filtering thresholds that favourably remove false positives without increasing false negatives, thereby increasing precision; GraftM and DIAMOND accrued over 3000 more false positives than TreeSAPP. Notably, this performance was achieved only using RAxML version 8.2.12 as we found pendant length distances were estimated accurately beginning with this version. Based on the performance of TreeSAPP using alignment trimming but assigning taxonomy to queries only by LCA (TreeSAPP-BMGE-Raw), adjusting a query’s assigned taxonomic rank by its EDR is worthwhile. However, relative to DIAMOND and GraftM, TreeSAPP’s recall was not as strong, mostly stemming from the initial HMM search, missing 1775 EggNOG sequences versus 561 and 789 for GraftM and DIAMOND at the most relaxed taxonomic rank distance allowed, respectively (Supplementary Fig. S5 and Table S2). The vast majority of false negatives were accounted for by Eukaryotic sequences and this is sensible given the reference packages were built using only sequences from Bacteria and Archaea. Only 7.1% of unclassified sequences were bacterial or archaeal in origin. Still, 5420 Eukaryotic sequences were correctly classified suggesting TreeSAPP is able to accurately identify very distantly related homologous sequences. False negatives were unevenly distributed across the tested reference packages with over 60% from just 3 (minimum =1 from Ribosomal S3Ae family, maximum =653 from Ribosomal protein L1p/L10e family, median =23).
Fig. 2.

Classification performance of TreeSAPP, GraftM and DIAMOND as evaluated by the MCC. TreeSAPP was run both with (TreeSAPP-BMGE) and without MSA trimming using BMGE. TreeSAPP-BMGE-Raw represents the classification performance of TreeSAPP with BMGE but without the linear-model-based rank recommendation. Distance from optimal rank is the accepted taxonomic distance in order for a classified sequence to be considered a true positive. Sequences that failed to meet the distance from optimal rank were included in the MCC calculation as false positives
Clade exclusion analysis was used to dually determine the accuracy of these methods using functional anchors with less congruent phylogenetic and taxonomic relations as compared to the ribosomal proteins, and how these methods perform when databases lack reference sequences that are closely related to query sequences. Query sequences were downloaded from FunGene version 9.6 and clustered at 99% similarity with USEARCH (Edgar, 2010). EggNOG was not used in this case as its taxonomic breadth of 2031 organisms, many of which do not contain any of the functional markers, was deemed inadequate for reference package construction. Using treesapp evaluate, iterative clade exclusion analyses were performed for every testable taxon and each reference package was independently analysed. Classification performance was variable, but not considerably so, across the reference packages tested. McrA tended to yield the best assignments while PmoA/AmoA and DsrAB consistently performed poorly. GraftM and TreeSAPP both perform much better than DIAMOND when classifying divergent sequences (Class-, Order- and Family-level relations to closest relative in reference set) but comparable when query sequences were similar to the reference set (Fig. 3). Most of TreeSAPP’s classifications were <2 taxonomic ranks away from the optimal assignment on average, regardless of sequence divergence, and sequences were classified with approximately equivalent accuracy across taxonomic ranks. Since false negatives were unavailable for this analysis the F1 score, the harmonic mean of precision and recall, was used to compare across all reference packages (Supplementary Fig. S7). The difference in each of the methods’ ability to handle distantly related sequences is emphasized in these values. All did comparably well when classifying queries closely related to reference sequences (F1 scores between 0.71 and 0.92) but varied significantly for distantly related queries (between 0.04 and 0.8).
Fig. 3.
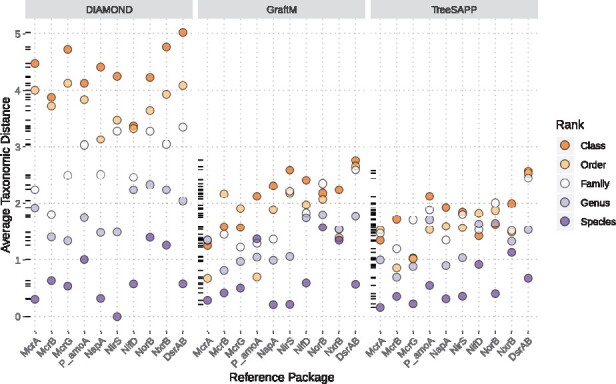
Average taxonomic distance across all taxa evaluated for 12 functional anchors. Colours correspond to the taxonomic rank evaluated by clade exclusion analysis and serve as a proxy for sequence divergence. Dashes along the y-axis show the distribution of points on a single plane. P_amoA is a reference package with sequences containing both PmoA and AmoA
Clade exclusion analysis was used again to confirm the results observed by classifying the taxonomic anchor genes in EggNOG. The mean distances and F1 scores were consistent with the functional anchor genes, with a few reference packages performing poorly (Supplementary Figs S8 and S9). This could not be completed for DIAMOND and GraftM as one of its dependencies for building reference packages, Taxtastic (https://github.com/fhcrc/taxtastic), would frequently timeout.
Classification and JPlace files can be used for purposes beyond taxonomic and functional profiling; TreeSAPP can also discover genes, reactions and pathways associated with organismal genomes, SAGs or MAGs. To evaluate this aspect of the TreeSAPP pipeline, we developed a use case for McrA, because this reference package performed the best during benchmarking. The mcrA gene along with mcrB and mcrG encode the holoenzyme mediating the terminal step in biological methane production, though it is also capable of binding and activating methane in the anaerobic oxidation of methane and more recently may be used in the oxidation of short-chain alkanes (Laso-Pérez et al., 2016). The McrA phylogeny has been shown to be fairly congruent with both small subunit ribosomal RNA (SSU or 16S rRNA) gene and concatenated marker gene phylogenies, with limited recorded instances of lateral gene transfer, and is therefore more likely to perform well as a taxonomic anchor (Evans et al., 2019; Springer et al., 1995). Moreover, methane-metabolism pathway information has been provided for each known clade and expected to be conserved, making accurate metabolic inferences directly from phylogenetic placement possible (Evans et al., 2019).
3.2 Global McrA survey
TreeSAPP was used to find and classify all McrABG sequences from the Joint Genome Institute’s IMG/M database image from January 10, 2017. There were 7.715×109 (∼1.2 TB) of putative amino acid sequences included in this analysis and 14 919 McrA, 11 825 McrB and 8609 McrG sequences were taxonomically classified. A total of 816 metagenomes were found to contain at least 1 of the 3 Mcr subunits. Sequences shorter than 84 AA, corresponding to the first quantile of length-sorted sequences, were removed to mitigate incorrect classifications leaving 11 256 McrA. About 58% (6533) of the McrA sequences were classified at the rank of Genus or Species and only 3.6% were classified as Archaea (Supplementary Fig. S10). Methanomicrobia and Methanobacteria were the most sampled classes with 5505 and 1572, respectively. Methanoculleus, Methanobrevibacter, Methanobacterium and Methanoregula were the most common genera accounting for 2891 sequences. Of all the McrA sequences classified 4251 sequences were resolved to at least Phylum (to reduce the chance they were false positives) and no further than Family. Even though 2764 of these were classified to the NCBI’s Phylum rank as either ‘environmental samples’ or ‘metagenomes’, they can still be considered novel with respect to cultured archaea. These novel sequences were from 432 metagenomes, primarily represented by wetland and hydrothermal vent communities. Strikingly, 89% of the 806 McrA sequences from hydrothermal vent metagenomes were considered novel. While the number of uniquely novel sequences was not determined, updating the reference tree after clustering all sequences at 97% similarity added 412 leaves, an increase of 180%.
Additionally, the McrA tree was annotated with methane-metabolism pathway information for each known clade (Borrel et al., 2014, 2019; Evans et al., 2019; Whitman et al., 2006) (Supplementary Fig. S11) and this was used to provide metabolic labels for an additional 10 839 sequences. A metabolic label was not assigned to sequences placed at a node where descendants possess multiple metabolic labels. CO2-dependent hydrogenotrophy was the most common metabolism by a large margin and methylotrophy was the least common. There were no signs of mutual exclusion among metabolisms or between metabolisms and broad ecosystem categories but some trends were identified for specific ecosystem sub-types. Hydrothermal vents, oil seeps, thermal springs and an asphaltene lake harboured relatively abundant McrA associated with short-chain alkane-oxidizing and anaerobic methanotrophic Archaea. Only thermal springs also hosted an abundant methanogenic population. McrA associated with CH3-dependent hydrogenotrophic methanogenesis were most prevalent in digestive systems and other environments rich in organic matter. The most aceticlastic McrA were found in soil and freshwater environments as well as anaerobic digesters.
Together, using TreeSAPP’s phylogenetically derived taxonomic classifications, these metagenomic data indicate that there are plenty of novel methanogenic and short-chain alkane-oxidizing Archaeal lineages that remain to be described.
4 Discussion
TreeSAPP is a functional and taxonomic annotation software that uses phylogenetic placement for accurate classifications. It is readily able to classify sequences derived from organismal genomes, environmental genomes (SAGs, MAGs) and metagenomes—even those that are distantly related to reference genomes present in contemporary databases. Moreover, it capitalizes on the phylogenetic framework to transitively assign taxonomic and functional feature information. In cases of complex evolutionary histories, internalizing these features in the reference package, by annotating clades, ensures that distantly related genes are not mis-annotated, thereby reducing false discovery. TreeSAPP was designed to integrate with iTOL as well as biological sequence databases so sequences can be easily linked to their respective taxonomic lineages. We are looking to expand support for databases beyond EggNOG and Entrez so users can more easily create reference packages. Moreover, to keep reference packages as current as possible, query sequences that meet profile HMM proportion thresholds and are deemed sufficiently divergent from current reference sequences are used to readily rebuild reference MSA, profile HMM and tree using treesapp update. We do not plan to regularly update all reference packages centrally. Rather, in their current state, these are meant to be used as mutable objects that users can update as required to suit their unique efforts (Fig. 4).
Fig. 4.

Phylogenetic and metabolic analysis of IMG metagenome-derived McrA sequences. (A) All predicted metagenome-derived McrA sequences (14 919) from IMG/M (as of January 10, 2017) were classified using TreeSAPP and visualized in iTOL. The tree shown here contains 228 reference McrA sequences including most newly described lineages from the “divergent McrA” clade hypothesized to be involved in oxidizing higher alkanes. A version of the tree with leaf labels is available as Supplementary Figure S12. (B) Proportion of sequences assigned at each taxonomic rank. (C) Putative methanogenesis and methanotrophic metabolisms supported in each ecosystem category as inferred by their placement on the reference McrA tree. Sequences that mapped deeply and converge across multiple annotated metabolisms were omitted
4.1 Performance
Classification performance analyses indicate that TreeSAPP was better at both identifying and assigning taxonomy to query protein sequences than DIAMOND and GraftM, especially when query sequences were novel with respect to the reference sequences. The MCC values generated using EggNOG showed TreeSAPP’s taxonomic classifications were better than both GraftM and DIAMOND, though this was only achieved with taxonomic rank recommendation from linear models. Even still, it is important to point out that the pairwise alignment performance of DIAMOND shown here is not extensible to pairwise alignment in general because databases tend to be more comprehensive than reference packages used in this study. Unfortunately, we were unable to compare our classification results to those of the critical assessment of metagenome interpretation project since their performance summaries were based on classifications of DNA sequences (Sczyrba et al., 2017). A recent meta-analysis of taxonomic classification tools by Ye et al. (2019), though not directly comparable, produced F1 scores similar to our analyses for DIAMOND at the Genus and Species ranks (ranging from 0.1 to 0.4, Supplementary Fig. S7) indicating that both GraftM and TreeSAPP would outperform their tested ‘DNA-to-protein’ classifiers (tools that classify DNA sequences using protein databases).
Through these analyses, we also found classification precision to vary by reference package. All classifiers struggled to accurately assign taxonomy with the DsrAB reference package [included reductive and oxidative forms of DsrA and DsrB, and validated with sequences from Müller et al. (2015)]. DsrAB is a composite reference package containing the homologous subunits DsrA and DsrB, anaerobic sulphite reductase subunit C and a nitrite and sulphite reductase clade of Euryarchaeota. Including homologous but functionally diverse genes in a single phylogeny can safeguard from erroneous functional attributions by performing a second classification with additional tree decoration information, as implemented in treesapp layer, but may result in less accurate taxonomic assignments (Supplementary Methods). More research into building reference packages for complex gene families is needed.
4.2 Application
TreeSAPP was used to classify all metagenome-derived McrABG subunits in IMG/M. It identified several thousand novel sequences (i.e. not represented by either a Genus or Species) with respect to our contemporary, metabolically annotated reference McrA tree. Specifically, Gulf of California hydrothermal vent samples contained diverse methanotrophic and short-chain alkane-oxidizing Archaea, of which nearly 90% do not have a Genus-level representative. We also estimated the relative abundance of McrA associated with known methanogenic and alkanotrophic metabolisms, identifying CO2-dependent hydrogenotrophy as vastly more common than any other on a per-ORF basis. This use case illustrates the power of TreeSAPP in identifying new lineages and resolving quantitative functional differences between locations. This rich dataset is ripe for further diversity and correlation-based analyses to inform future sequencing and cultivation efforts.
4.3 Future development
In the process of developing and testing TreeSAPP several potential areas of improvement were recognized. TreeSAPP is slower and requires more RAM than GraftM (Supplementary Fig. S13) for a number of reasons. Intermediate files are written so runs can be restarted from checkpoints at the cost of more time spent performing I/O operations. TreeSAPP uses Prodigal for ORF prediction and conceptual translation (Hyatt et al., 2010), while GraftM employs the simple and significantly faster OrfM (Woodcroft et al., 2016). TreeSAPP uses one HMM for both search and profile multiple alignments. This is less sensitive than GraftM’s search-specific HMM where sequences were taxonomically deduplicated before building (data not shown). Adopting this strategy could be beneficial if it does not increase the false positive rate. Finally, RAxML’s EPA is used for phylogenetic placement, instead of the faster pplacer. While neither tool scales particularly well on a single compute node (Supplementary Fig. S14) future versions of TreeSAPP will see marked improvements in sequential and parallel computing efficiency by adopting the faster RAxML-NG and EPA-NG (Barbera et al., 2019; Kozlov et al., 2019).
Several developments in phylogenetic placement have been published recently that may lead to further improvements in efficiency and accuracy (Barbera et al., 2019; Czech et al., 2019). Among them is hierarchical phylogenetic placement, which involves placing query sequences onto a taxonomically broad and sparse backbone tree with subsequent placement onto high-resolution phylogenies based on the backbone edge position. This method has the potential to increase phylogenetic placement precision while reducing computational requirements (Czech et al., 2019). Moreover, new phylogenetically informed Bacterial and Archaeal taxonomic hierarchies are being introduced (Parks et al., 2018). Leveraging a principled taxonomic framework based on phylogenetic relationships will likely improve the taxonomic classifications of all phylogenetic methods.
Modelling the relationship between taxonomic rank and EDR for rank recommendation benefits taxonomic classification performance (Fig. 2). It is most beneficial when classifying distantly related query sequences that map to either a leaf’s edge or a sparse clade resulting in a shallow (Species or Genus) LCA. However, generating the EDR data for training is currently a slow, iterative process. Moreover, a reliable model is not created if there is insufficient taxonomic redundancy within reference sequences (i.e. a single Order is representing a Class, so removing that Order removes the entire Class and next closest ancestor is at Phylum). Using distances directly from the tree, as was used in the GTDB-Tk (Chaumeil et al., 2019) with RED, would accelerate this process and benefit consistency.
5 Conclusion
We have developed a functional and taxonomic annotation software, TreeSAPP, with improved classification performance based on regression of evolutionary distances and taxonomic ranks to recommend more accurate taxonomic assignments. TreeSAPP is able to provide quantitative functional and taxonomic information for both assembled and unassembled sequences, classification tables and files supporting interactive iTOL visualizations. Using TreeSAPP, we explored the global distribution of McrA and recovered many new sequences associated with methane metabolizing archaea. With an expanded set of reference packages under development TreeSAPP will support community-driven taxonomic assignment and metabolic reconstruction on a truly global scale.
Supplementary Material
Acknowledgements
We would like to thank Tanya Woyke, Natalia Ivanova and Frederik Schulz at the Joint Genome Institute (JGI; Berkeley, California) and Daniel Udwary for providing metagenomic data from IMG/M and assistance with the National Energy Research Scientific Computing (NERSC) Center systems. Many thanks to Simonetta Gribaldo and Guillaume Borrel at the Institut Pasteur (Paris) for validating metabolic annotations and recommending BMGE. We also thank Kishori Konwar for formative conversations and preliminary code used to guide early TreeSAPP development cycles.
Funding
This work was performed under the auspices of the US Department of Energy (DOE) Joint Genome Institute, an Office of Science User Facility, supported by the Office of Science of the US Department of Energy under Contract DE-AC02-05CH11231 through the Facilities Integrating Collaborations for User Science (FICUS) initiative between JGI and NERSC; the Natural Sciences and Engineering Research Council of Canada; Genome British Columbia; Genome Canada; and Compute Canada through grants awarded to S.J.H. is a co-founder of Koonkie Inc., a bioinformatics consulting company that designs and provides scalable algorithmic and data analytics solutions in the cloud.
Conflict of Interest: none declared.
Contributor Information
Connor Morgan-Lang, Graduate Program in Bioinformatics, University of British Columbia, Genome Sciences Centre, Vancouver, British Columbia V5Z 4S6, Canada.
Ryan McLaughlin, Graduate Program in Bioinformatics, University of British Columbia, Genome Sciences Centre, Vancouver, British Columbia V5Z 4S6, Canada.
Zachary Armstrong, Genome Science and Technology Program, University of British Columbia, Vancouver, BC V6T 1Z4, Canada.
Grace Zhang, Department of Electrical and Computer Engineering, University of British Columbia, Vancouver BC V6T 1Z4, Canada.
Kevin Chan, Department of Electrical and Computer Engineering, University of British Columbia, Vancouver BC V6T 1Z4, Canada.
Steven J Hallam, Graduate Program in Bioinformatics, University of British Columbia, Genome Sciences Centre, Vancouver, British Columbia V5Z 4S6, Canada; Genome Science and Technology Program, University of British Columbia, Vancouver, BC V6T 1Z4, Canada; Department of Electrical and Computer Engineering, University of British Columbia, Vancouver BC V6T 1Z4, Canada; Department of Microbiology and Immunology, University of British Columbia, 2552-2350 Health Sciences Mall, Vancouver, British Columbia V6T 1Z3, Canada; ECOSCOPE Training Program, University of British Columbia, Vancouver, British Columbia V6T 1Z, Canada.
References
- Altschul S.F. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Barbera P. et al. (2019) EPA-ng: massively parallel evolutionary placement of genetic sequences. Syst. Biol., 68, 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger S.A., Stamatakis A. (2011) Aligning short reads to reference alignments and trees. Bioinformatics, 27, 2068–2075. [DOI] [PubMed] [Google Scholar]
- Borrel G. et al. (2014) Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genomics, 15, 679–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrel G. et al. (2019) Wide diversity of methane and short-chain alkane metabolisms in uncultured archaea. Nat. Microbiol., 4, 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boughorbel S. et al. (2017) Optimal classifier for imbalanced data using Matthews Correlation Coefficient metric. PLoS One, 12, e0177678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd J.A. et al. (2018) GraftM: a tool for scalable, phylogenetically informed classification of genes within metagenomes. Nucleic Acids Res., 46, e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B. et al. (2015) Fast and sensitive protein alignment using DIAMOND. Nat. Methods, 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Canfield D.E. et al. (2010) The evolution and future of earth’s nitrogen cycle. Science, 330, 192–196. [DOI] [PubMed] [Google Scholar]
- Chaumeil P.-A. et al. (2019) GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics, 36, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli F.D. et al. (2006) Toward automatic reconstruction of a highly resolved tree of life. Science, 311, 1283–1287. [DOI] [PubMed]
- Cock P.J. et al. (2009) Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo A., Gribaldo S. (2010) BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol., 10, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross K.L. et al. (2019) Targeted isolation and cultivation of uncultivated bacteria by reverse genomics. Nat. Biotechnol., 37, 1314–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech L. et al. (2019) Methods for automatic reference trees and multilevel phylogenetic placement. Bioinformatics, 35, 1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A.E. et al. (2014) PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ, 2, e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S.R. (1998) Profile hidden Markov models. Bioinformatics, 14, 755–763. [DOI] [PubMed] [Google Scholar]
- Edgar R.C. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Evans P.N. et al. (2019) An evolving view of methane metabolism in the Archaea. Nat. Rev. Microbiol., 17, 219–232. [DOI] [PubMed] [Google Scholar]
- Falkowski P.G. et al. (2008) The microbial engines that drive earth’s biogeochemical cycles. Science, 320, 1034–1039. [DOI] [PubMed] [Google Scholar]
- Fish J.A. et al. (2013) FunGene: the functional gene pipeline and repository. Front. Microbiol., 4, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson N.W. et al. (2016) LCA*: an entropy-based measure for taxonomic assignment within assembled metagenomes. Bioinformatics, 32, 3535–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Z-s. et al. (2019) Insights into the ecological roles and evolution of methyl-coenzyme M reductase-containing hot spring Archaea. Nat. Commun., 10, 4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J. et al. (2016. a) eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res., 44, D286–D293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J. et al. (2016. b) ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol., 33, 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz B.L. et al. (2013) Metabolic reprogramming by viruses in the sunlit and dark ocean. Genome Biol., 14, R123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D.H. et al. (2007) MEGAN analysis of metagenomic data. Genome Res., 17, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D. et al. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehl P. et al. (2015) OD-seq: outlier detection in multiple sequence alignments. BMC Bioinformatics, 16, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol., 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. et al. (2016) Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res., 26, 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konwar K.M. et al. (2013) MetaPathways: a modular pipeline for constructing pathway/genome databases from environmental sequence information. BMC Bioinformatics, 14, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov A.M. et al. (2016) Phylogeny-aware identification and correction of taxonomically mislabeled sequences. Nucleic Acids Res., 44, 5022–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov A.M. et al. (2019) RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics, 35, 4453–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laso-Pérez R. et al. (2016) Thermophilic archaea activate butane via alkyl-coenzyme M formation. Nature, 539, 396–401. [DOI] [PubMed] [Google Scholar]
- Le S.Q., Gascuel O. (2008) An improved general amino acid replacement matrix. Mol. Biol. Evol., 25, 1307–1320. [DOI] [PubMed] [Google Scholar]
- Letunic I., Bork P. (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res., 44, W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Bork P. (2019) Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res., 47, W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsen F.A. et al. (2010) pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics, 11, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews B.W. (1975) Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta, 405, 442–451. [DOI] [PubMed] [Google Scholar]
- McKay L.J. et al. (2019) Co-occurring genomic capacity for anaerobic methane and dissimilatory sulfur metabolisms discovered in the Korarchaeota. Nat. Microbiol., 4, 614–622. [DOI] [PubMed] [Google Scholar]
- Merchant S. et al. (2014) Unexpected cross-species contamination in genome sequencing projects. PeerJ, 2, e675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller A.L. et al. (2015) Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J., 9, 1152–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasko D.J. et al. (2018) RefSeq database growth influences the accuracy of k-mer-based lowest common ancestor species identification. Genome Biol., 19, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols D. et al. (2010) Use of ichip for high-throughput in situ cultivation of “uncultivable microbial species”. Appl. Environ. Microbiol., 76, 2445–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondov B.D. et al. (2016) Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol., 17, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks D.H. et al. (2018) A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol., 36, 996–1004. [DOI] [PubMed] [Google Scholar]
- Pattengale N.D. et al. (2010) How many bootstrap replicates are necessary? J. Comput. Biol., 17, 337–354. [DOI] [PubMed] [Google Scholar]
- Peabody M.A. et al. (2015) Evaluation of shotgun metagenomics sequence classification methods using in silico and in vitro simulated communities. BMC Bioinformatics, 16, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M.N. et al. (2010) FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One, 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappé M.S., Giovannoni S.J. (2003) The uncultured microbial majority. Annu. Rev. Microbiol., 57, 369–394. [DOI] [PubMed] [Google Scholar]
- Rinke C. et al. (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature, 499, 431–437. [DOI] [PubMed] [Google Scholar]
- Sczyrba A. et al. (2017) Critical Assessment of Metagenome Interpretation-a benchmark of metagenomics software. Nat. Methods, 14, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz K.W. et al. (2019) Asgard archaea capable of anaerobic hydrocarbon cycling. Nat. Commun., 10, 1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaiber A., Eren A.M. (2019) Composite metagenome-assembled genomes reduce the quality of public genome repositories. mBio, 10, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solden L. et al. (2016) The bright side of microbial dark matter: lessons learned from the uncultivated majority. Curr. Opin. Microbiol., 31, 217–226. [DOI] [PubMed] [Google Scholar]
- Springer E. et al. (1995) Partial gene sequences for the A subunit of methyl-coenzyme M reductase (mcrI) as a phylogenetic tool for the family Methanosarcinaceae. Int. J. Syst. Bacteriol., 45, 554–559. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22, 2688–2690. [DOI] [PubMed] [Google Scholar]
- Stark M. et al. (2010) MLTreeMap - accurate Maximum Likelihood placement of environmental DNA sequences into taxonomic and functional reference phylogenies. BMC Genomics, 11, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen A.D. et al. (2019) High proportions of bacteria and archaea across most biomes remain uncultured. ISME J., 13, 3126–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan G. et al. (2015) Current methods for automated filtering of multiple sequence alignments frequently worsen single-gene phylogenetic inference. Syst. Biol., 64, 778–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al. (2019) Expanding anaerobic alkane metabolism in the domain of Archaea. Nat. Microbiol., 4, 595–602. [DOI] [PubMed] [Google Scholar]
- Whitman W.B. et al. (2006) The methanogenic bacteria. In: Dworkin,M. et al. (eds), Prokaryotes. Springer, New York, NY, pp. 165–207. [Google Scholar]
- Woodcroft B.J. et al. (2016) OrfM: A fast open reading frame predictor for metagenomic data. Bioinformatics, 32, 2702–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D. et al. (2013) TreeOTU: operational taxonomic unit classification based on phylogenetic trees. Preprint at https://arxiv.org/abs/1308.6333. [Google Scholar]
- Ye S.H. et al. (2019) Benchmarking metagenomics tools for taxonomic classification. Cell, 178, 779–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.