

Hepatic cytokine BMP9 attenuates fatty liver by regulating the key lipid metabolism factor PPARα.
Abstract
Obesity drives the development of nonalcoholic fatty liver disease (NAFLD) characterized by hepatic steatosis. Several bone morphogenetic proteins (BMPs) except BMP9 were reported related to metabolic syndrome. This study demonstrates that liver cytokine BMP9 is decreased in the liver and serum of NAFLD model mice and patients. BMP9 knockdown induces lipid accumulation in Hepa 1-6 cells. BMP9–knockout mice exhibit hepatosteatosis due to down-regulated peroxisome proliferator–activated receptor α (PPARα) expression and reduced fatty acid oxidation. In vitro, recombinant BMP9 treatment attenuates triglyceride accumulation by enhancing PPARα promoter activity via the activation of p-smad. PPARα-specific antagonist GW6471 abolishes the effect of BMP9 knockdown. Furthermore, adeno-associated virus–mediated BMP9 overexpression in mouse liver markedly relieves liver steatosis and obesity-related metabolic syndrome. These findings indicate that BMP9 plays a critical role in regulating hepatic lipid metabolism in a PPARα-dependent manner and may provide a previously unknown insight into NAFLD therapeutic approaches.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a common metabolic disorder in obese individuals (1). Fatty liver is one of the major features of metabolic syndrome, and its development is associated with the dysregulation of systemic lipid and glucose homeostasis (2). Therefore, insulin resistance, inflammatory response, and hepatic steatosis are interconnected pathological events that are often observed in individuals with obesity (3). Generally, hepatic steatosis occurs when triglycerides (TGs) accumulate aberrantly in the liver due to an imbalance in fatty acid oxidation and de novo lipogenesis (4).
Bone morphogenetic protein 9 (BMP9), also known as growth differentiation factor 2 (GDF2), belongs to the BMP superfamily. BMP9 was reported to mainly express in the liver (5, 6). BMPs consist of 15 members (BMP1 to BMP15) that can function as signaling ligands in bone development, osteoblast differentiation, and angiogenesis (7). Recently, several BMP members were reported to be involved in metabolic diseases. BMP2 alters energy storage and adipogenesis in adipose tissue (8). BMP4 promotes a brown to white-like adipocyte shift that controls adipogenic commitment and differentiation (9). BMP7 exerts an anti-obesity effect by activating a full complement of brown adipogenesis genes. BMP8B facilitates obesity resistance by increasing brown adipose tissue (BAT) thermogenesis (10). These findings indicate that BMPs are potential metabolic regulatory genes playing roles in adipose tissue. We previously demonstrated the function of BMP9 in liver fibrosis and identified that BMP9 is highly expressed and secreted in liver cells (11). Emerging evidence indicates that BMP9 may play a critical role in adipose metabolic processes, glucose homeostasis, metabolic syndrome, and insulin resistance (12–14). However, the overall metabolic regulatory roles of BMP9 in mouse liver are not fully understood.
Here, we identified a robust decrease in BMP9 expression in the livers of obese mice and NAFLD patients with metabolic disorders. BMP9–knockout (KO) mice displayed fatty liver and obesity-related metabolic complications. BMP9 deletion leads to dysfunction of liver TG accumulation via down-regulation of the expression of peroxisome proliferator–activated receptor α (PPARα), a master of lipid metabolism (15, 16). Both recombinant BMP9 protein and adeno-associated virus (AAV)–BMP9 could reverse hepatic steatosis in vitro and in vivo. We provide a previously unidentified insight into BMP9-mediated maintenance of hepatic lipid homeostasis. BMP9 could be a potential target for fatty liver disease therapy.
RESULTS
BMP9 expression is decreased in metabolic syndrome
BMP2, BMP4, BMP7, and BMP8 have been reported to be involved in metabolic diseases and energy expenditure (8, 10, 17, 18). However, the relationship between BMP9 and metabolic syndrome remains unclear. To explore the role of BMP9 in metabolic diseases, we first examined the expression of BMP9 in mice fed a high-fat diet (HFD), a common method to generate fatty liver. Decreased BMP9 mRNA (Fig. 1A) and protein (Fig. 1B) expression in HFD-fed mouse livers was detected. Correspondingly, enzyme-linked immunosorbent assay (ELISA) results showed that serum BMP9 protein levels were also reduced, compared with normal chow diet (CD) mice (Fig. 1C). At this early stage, the liver of HFD mice displayed a notable liver steatosis (fig. S1A), but very few liver fibrosis (fig. S1B) was observed without fibrosis-specific diet induction.
Fig. 1. BMP9 expression is decreased in multiple metabolic disorder models.
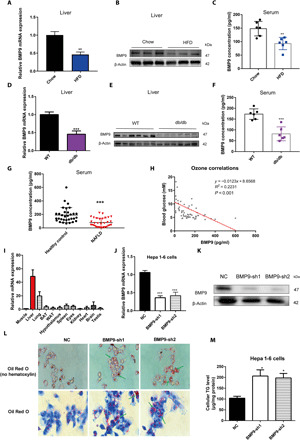
(A) Hepatic BMP9 mRNA, (B) BMP9 protein levels, and (C) serum BMP9 protein concentration were down-regulated in HFD mice. Eight-week-old C57BL/6 mice were fed an HFD for 8 weeks (n = 6). (D) Hepatic BMP9 mRNA, (E) BMP9 protein levels, and (F) serum BMP9 protein concentration were down-regulated in db/db mice. Eight-week-old db/db mice were fed a CD for 8 weeks (n = 6). (G) Serum BMP9 protein level in NAFLD patients. (H) Pearson’s R2 and P values for blood glucose levels versus BMP9 protein levels in human serum (n = 32). (I) Relative BMP9 mRNA expression level in multiple WT C57BL/6 mouse tissues (n = 6). (J) Knockdown of BMP9 mRNA in Hepa 1-6 cells by shRNA. NC, negative control. (K) BMP9 protein levels in BMP9-shRNA Hepa 1-6 cells. (L) Oil Red O staining showed cellular lipid accumulation in BMP9-shRNA Hepa 1-6 cells. (M) Cellular TG level. *P < 0.05, **P < 0.01, and ***P < 0.001.
To test whether the decrease of BMP9 in the liver represents a common feature of metabolic diseases, we checked the expression of BMP9 in db/db mice, a classic monogenic obesity mouse model accompanied by fatty liver (fig. S1, C and D). BMP9 mRNA (Fig. 1D) and BMP9 protein (Fig. 1E) levels were notably decreased in the livers of obese mice compared with wild-type (WT) mice. In addition, serum BMP9 protein (Fig. 1F) levels were also decreased in db/db mice.
Metabolic syndromes comprise fatty liver, obesity, hyperglycemia, dysfunctional liver glucose metabolism, and lipid metabolism. The liver fat content in HFD and db/db mice showed a distinct elevation (fig. S1, E and F). There was a distinct negative correlation between hepatic TG level and BMP9 in these two mouse models (fig. S1G). Markedly, serum BMP9 protein levels were also reduced in NAFLD patients with abnormal blood glucose levels (Fig. 1G). Furthermore, a significant correlation between blood glucose and serum BMP9 protein levels was observed (Fig. 1H). Clinical characteristics of all subjects, including abnormal liver fat content, were collected and analyzed in detail (table S1).
BMP9 was found to be expressed in multiple tissues in BMP9 WT mice but was highly expressed in the liver (Fig. 1I), which is consistent with previous reports showing that BMP9 originates in liver cells, especially hepatic stellate cells (HSCs) (11). Fatty liver is the abnormal accumulation of TGs in the liver (1, 4, 19). To study the role of BMP9 in hepatosteatosis in vitro, we used a short hairpin RNA (shRNA) lentivirus to knock down BMP9 in Hepa 1-6 cells and detected a successful knockdown of BMP9 at both mRNA and protein levels (Fig. 1, J and K). Oil Red O staining (Fig. 1L) showed abundant lipid droplet accumulation in BMP9 stable knockdown cells. Consistently, the cellular TG level (Fig. 1M) was markedly increased.
Thus, our results reveal a previously unknown feature of reduced BMP9 expression in obese mice and humans with NAFLD. Knockdown of BMP9 accelerates abnormal TG accumulation in liver cells in vitro.
BMP9-KO mice exhibit a distinct hepatic steatosis phenotype
To systematically assess the function of BMP9 in liver metabolic processes, we generated BMP9-KO mice by CRISPR-Cas9 system (Fig. 2A) (20). Genotyping and DNA sequencing results showed that 64 base pairs (bp) were deleted in BMP9-KO mice (Fig. 2, B and C). BMP9 is a secretory cytokine, and circulating serum BMP9 protein levels were significantly decreased in BMP9-KO mice (Fig. 2D). Moreover, compared with WT mice, BMP9 protein cannot be detected by Western blotting in the BMP9-KO mouse liver (Fig. 2E). These results show that BMP9 was markedly knocked out at both genomic and translational levels.
Fig. 2. BMP9-KO mice exhibit a distinct hepatic steatosis phenotype.
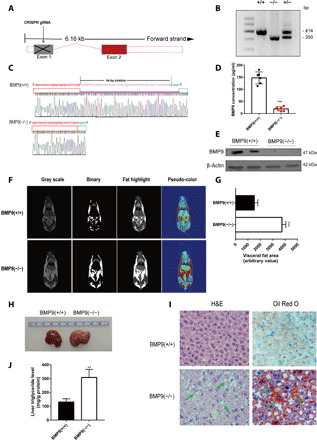
(A) Schematic diagram of BMP9 deletion process by targeting exon 1 via CRISPR guide RNA (gRNA). (B) Genotyping of BMP9 WT, KO, and heterozygous mice. (C) Gene sequencing results. (D) BMP9-KO mouse serum BMP9 protein concentrations (n = 6). (E) Hepatic BMP9 protein expression in BMP9-KO mice (n = 2). (F) NMR showed distribution of mice visceral fat. Photo credit: Z. Yang, Photographer Institution, Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine. (G) Quantification of visceral fat area (n = 3). (H) Representative image of a dysfunctional liver in 16-week-old BMP9 WT and KO mice fed a CD (n = 6). Photo credit: P. Li, Photographer Institution, Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine. (I) Liver H&E and Oil Red O staining (original magnification, ×400). (J) Liver TG level. **P < 0.01 and ***P < 0.001.
Furthermore, nuclear magnetic resonance (NMR) layer scanning results showed a marked fat accumulation in 16-week-old BMP9-KO mice (Fig. 2F), especially visceral fat (Fig. 2G). Upon dissecting the mice, we observed obvious steatosis and chylous particles in the liver. Liver size in BMP9-KO mouse was increased (Fig. 2H). Hematoxylin and eosin (H&E) and Oil Red O staining showed prominent liver steatosis in BMP9-KO mice (Fig. 2I), which was confirmed by the liver TG level analysis (Fig. 2J). To conclude, these results demonstrate that BMP9 ablation potently promotes lipid accumulation in mouse liver.
BMP9 ablation accelerates insulin resistance and obesity
NAFLD is often accompanied with insulin resistance (3, 16, 21). We further explored the impact of BMP9 in insulin resistance. Glucose tolerance test (GTT) (Fig. 3A) and insulin tolerance test (ITT) (Fig. 3B) results showed impaired glucose tolerance and insulin sensitivity in BMP9-KO mice. Consistently, fasted blood glucose concentration was much higher (Fig. 3C). We next proceeded to challenge the mice with an 8-week HFD treatment. HFD-fed mice showed a stronger impaired insulin sensitivity than CD-fed mice (Fig. 3, D and F).
Fig. 3. BMP9 ablation accelerates insulin resistance and obesity.
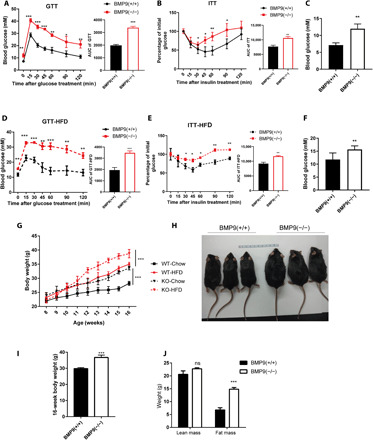
(A) GTT, (B) ITT, and (C) initial blood glucose comparison in CD-fed 16-week-old BMP9-KO mice (n = 6). (D) GTT, (E) ITT, and (F) initial blood glucose comparison in HFD-fed 16-week-old BMP9-KO mice (n = 6). (G) Weight gain of littermates fed a CD or HFD for 8 weeks (n = 6). (H) Representative photograph of CD-fed 16-week-old BMP9-KO mice (n = 3). Photo credit: P. Li, Photographer Institution, Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine. (I) Body weight (n = 6). (J) Comparison of fat mass and lean mass. *P < 0.05, **P < 0.01, and ***P < 0.001. ns, not significant.
Considering the critical role of BMP in growth and development (7), we measured body length and physical activity of BMP9-KO mice to exclude the effects of development on obesity formation (fig. S2, A to C). We further monitored body weights of WT and BMP9-KO mice fed a CD or HFD (Fig. 3G) and found that BMP9-KO mice both gradually exhibited higher body weight than WT mice in these two conditions. BMP9-KO mice exhibited a distinct physical appearance with a fatter body after approximately 16 weeks of CD feeding (Fig. 3, H and I). Notably, BMP9-KO mice had significantly increased body fat mass but no change in lean mass content (Fig. 3J). Moreover, after HFD feeding, BMP9-KO mice were more likely than WT mice to show body weight differences (Fig. 3G). As most researchers frequently did (21, 22), we performed GTT and ITT with precise doses of glucose and insulin, according to the timely body weight of each mouse. To further exclude the body weight difference impact and limitation on insulin resistance and glucose tolerance, we compared GTT and ITT data from WT-HFD and KO-CD mice to assess the effect of BMP9-KO on metabolic function because these mice were weight-matched. After body weight normalization, GTT results (fig. S2A) showed that KO mice exhibited a worse glucose clearance than WT-HFD mice. In addition, ITT results (fig. S2B) displayed an evenly contested insulin resistance between these two groups. These results indicated that BMP9 deficiency was the leading factor in metabolic processes.
Muscle, fat, and liver are the major target organs of insulin. BMP9 has been reported to potently induce brown adipogenesis by reducing the sizes of white adipocytes (12). We detected body temperature of BMP9-KO mice, but no obvious changes were observed (fig. S3B). Histomorphology results showed that BAT and white adipose tissue (WAT) were undifferentiated (fig. S3H). A previous study revealed that BMP8B regulates central actions in the hypothalamus (10) and that BMP7 reverses obesity and regulates appetite through the central mammalian target of rapamycin (mTOR) pathway (23, 24), leading us to test whether BMP9 has a similar function in the hypothalamus to regulate food intake. However, food intake (fig. S3F) did not show significant change between WT and KO mice, and very few genes were changed in the hypothalamus (fig. S3G). These data indicate that BMP9 most likely functions in peripheral tissues, especially in the liver.
In summary, these results demonstrate that BMP9 deletion disturbs insulin sensitivity and promotes mouse genetic obesity and exacerbates dietary obesity. BMP9 plays a key role in modulating both glucose homeostasis and lipid metabolism in obese mice. The potential abnormal metabolic target organ is likely to be the liver.
BMP9 regulates liver lipid metabolism via PPARα signaling
We further investigated the underlying mechanisms that BMP9 acts in liver TG accumulation. TG disturbances can be caused by absorption or digestive disorders. Fecal TG excretion results showed that the digestive system of BMP9-KO mice functioned normally (fig. S3E). BMP9-KO and WT mouse livers were collected for gene microarray analysis. Volcano plot results showed that expression of many genes was altered by BMP9-KO (Fig. 4A). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed enrichment of differentially expressed genes in multiple pathways. BMP9 belongs to the transforming growth factor–β (TGF-β) family, which was shown in enriched signaling pathways after BMP9 ablation. In addition, several metabolic pathways were altered, which suggests liver metabolic process dysregulation. PPAR signaling pathway was one of the most enriched (Fig. 4B).
Fig. 4. BMP9 regulates liver lipid metabolism via PPARα signaling.
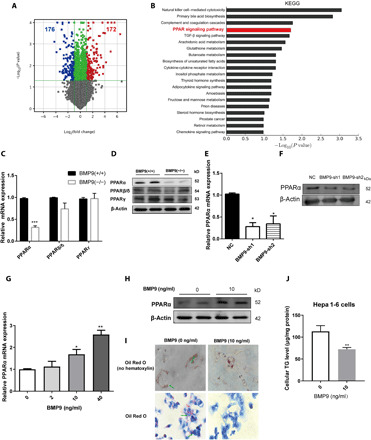
(A) Volcano plots: Differentially expressed genes (blue, down; red, up) in BMP9-KO mice livers (n = 3). (B) KEGG pathway analysis. PPAR signaling pathway is highlighted (n = 3). mRNA (C) and protein (D) expression of PPARs in the BMP9-KO mouse livers (n = 3). mRNA (E) and protein (F) expression of PPARα in BMP9 knockdown Hepa 1-6 cells. mRNA (G) and protein (H) expression of PPARα in BMP9 knockdown Hepa 1-6 cells treated with mouse recombinant BMP9 protein for 24 hours. (I) Cellular lipid droplets were visualized by Oil Red O staining. (J) Cellular TG levels. *P < 0.05, **P < 0.01, and ***P < 0.001.
PPARs constitute a subfamily of nuclear receptors with three current members: PPARα, PPARγ, and PPARδ (16). PPARα mRNA (Fig. 4C) and protein (Fig. 4D) expression was markedly down-regulated in the BMP9-KO liver. However, PPARβ/δ and PPARγ showed no obvious expression changes. PPARα is reported to be highly expressed in the liver, where it promotes fatty acid oxidation, gluconeogenesis, and ketogenesis (16, 25). Furthermore, to clarify the potential status of PPARα in hepatocytes, we measured PPARα expression at both mRNA (Fig. 4E) and protein (Fig. 4F) levels in Hepa 1-6 BMP9 shRNA stable cells in vitro. These results indicate that BMP9 deficiency leads to an apparent down-regulation of PPARα expression.
Previous studies showed that PPARα expression was reduced in type 2 diabetes mouse model ob/ob and db/db mice (26, 27). To confirm that BMP9 alters expression of PPARα, we next performed a set of rescue experiments in vitro. mRNA expression of PPARα in BMP9-knockdown Hepa 1-6 cells was altered by mouse recombinant BMP9 protein treatment in a dose-dependent manner. mRNA and protein expression of PPARα was notably up-regulated by BMP9 treatment (10 ng/ml) (Fig. 4, G and H). Moreover, Oil Red O staining (Fig. 4I) showed a visible reduction in lipid droplet formation after BMP9 treatment. Consistently, cellular TG level (Fig. 4J) was markedly decreased. Together, these data indicate a possible correlation between PPARα and BMP9, suggesting a potential positive regulatory effect on liver lipid metabolism.
BMP9 promotes fatty acid oxidation gene expression by up-regulating PPARα transcriptional activity
According to the findings regarding the role of PPARα in liver lipid metabolism, we next explored the molecular mechanism underlying the exacerbation of hepatic TG accumulation upon BMP9 deletion. On the basis of BMP9-KO liver gene microarray data, we performed a further Gene Ontology (GO) analysis to identify the top enriched biological processes (Fig. 5A). In addition to BMP signaling pathway itself, lipid metabolic process, fatty acid metabolic process, and regulation of metabolic process were among the top enriched pathways. These data confirm that BMP9-KO affects hepatic lipid metabolism. Generally, abnormal hepatic TG storage is caused by a disruption of homeostasis in fatty acid oxidation and de novo lipogenesis. The combination of GO-enriched pathway and KEGG-enriched top signaling pathway analysis (Fig. 4B) led us to focus on PPAR signaling pathway. PPARα target genes are principally associated with lipid metabolism processes, including fatty acid oxidation, fatty acid transportation, and lipogenesis (Fig. 5B). To verify microarray data, we confirmed expression of PPARα target genes in BMP9-KO mouse livers. Numerous fatty acid oxidation genes were down-regulated, including ACOX1, CYP4a12b, and SCPX (Fig. 5C).
Fig. 5. BMP9 promotes fatty acid oxidation gene expression by up-regulating PPARα transcriptional activity.
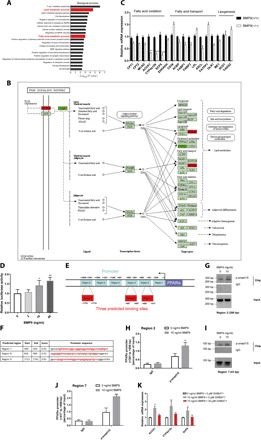
(A) GO analysis of BMP9-KO mouse liver microarray. Lipid metabolic processes and fatty acid metabolic processes are highlighted (n = 3). (B) PPAR signaling pathway map showing multiple lipid metabolism process–related genes. (C) mRNA expression of fatty acid oxidation, fatty acid transport, and lipogenesis genes in BMP9-KO mouse livers (n = 3). (D) Luciferase reporter activity was measured in BMP9 knockdown Hepa 1-6 cells treated with mouse recombinant BMP9 protein for 24 hours. (E) Schematic diagram of the PPARα promoter region. (F) Three 50-bp predicted TF binding site scores and sequences. Red sequences show the actual polymerase chain reaction (PCR) products. Agarose electrophoresis (G) and quantitative reverse transcription PCR (qRT-PCR) (H) showed an increased p-smad 1/5 ChIP product in region 2 after BMP9 treatment (10 ng/ml). Agarose electrophoresis (I) and qRT-PCR (J) showed an increased p-smad 1/5 ChIP product in predicted TF binding region 7. (K) mRNA expression of representative fatty acid oxidation genes after BMP9 and GW6471 treatment. *P < 0.05, **P < 0.01, and ***P < 0.001.
Apparent changes in PPARα and PPARα target genes led us to investigate whether BMP9 plays a role in PPARα gene transcription. Using a PPARα promoter involving dual-luciferase reporter assay in Hepa 1-6 cells, BMP9 treatment (10 ng/ml) markedly enhanced PPARα promoter activity (Fig. 5D). BMP family molecules usually regulate gene expression by phosphorylating smad protein. As a transcription factor (TF), phosphorylated smad protein enters the nucleus and binds to the promoter region of the target gene to regulate gene transcription (7, 11, 28, 29). Consistent with the classic theory, positive regulation of pathway-restricted smad protein phosphorylation pathway was observed in GO biological process analysis (Fig. 5A), which was further confirmed in the protein level by in vivo and in vitro test (fig. S4, C and D). These data guided us to investigate the potential role of smad protein in PPARα transcriptional regulation after BMP9 treatment. As discussed above, BMP9 treatment increased PPARα promoter activity (Fig. 5D). To clarify whether phosphorylated smad 1/5 (p-smad 1/5) acts as a TF that binds to the PPARα promoter at specific sites, we divided the 1800-bp PPARα promoter into six 300-bp regions (Fig. 5E). We used an online promoter prediction tool (www.fruitfly.org/cgi-bin/seq_tools/promoter.pl) to generate potential PPARα promoter TF binding sites per 50 bp. There are three high-scoring potential TF binding sites according to the prediction results (Fig. 5F). We performed chromatin immunoprecipitation (ChIP) assay after BMP9 treatment (10 ng/ml) in Hepa 1-6 cells. There was notable enrichment in region 2, whereas no changes were observed in other regions (Fig. 5, G and H). To further verify this finding, we analyzed these three predicted TF binding sites. ChIP product in region 7, within region 2, was increased in the BMP9 treatment (10 ng/ml) group (Fig. 5, I and J). These results indicated the potential binding sites of TF p-smad at the PPARα promoter. To clarify whether BMP9 regulates PPARα transcriptional activity and consequently change the expression level of PPARα target genes, we used the PPARα-specific antagonist GW6471, as previously described (30), to abolish BMP9-mediated regulation of PPARα in Hepa 1-6 cells. Treatment with BMP9 (10 ng/ml) up-regulated expression of fatty acid oxidation genes downstream of PPARα. However, 50 μM GW6471 treatment effectively inhibited the effect of BMP9 treatment (Fig. 5K).
Fibroblast growth factor 21 (FGF21), a hormone predominantly secreted by the liver, has been reported to reverse hepatic steatosis, increase energy expenditure, and improve insulin sensitivity in obese mice (31). Moreover, recent study established a PPARα-FGF21 hormone axis that contributed to systemic metabolic regulation (22), including insulin resistance and abnormal energy expenditure (32, 33). We hypothesize that there would be a direct BMP9-PPARα-FGF21 regulation network in the liver. As expected, hepatic FGF21 mRNA (fig. S4A) and protein (fig. S4D) level was reduced after BMP9 ablation, which was consistent with a previous study (34). Meanwhile, the activity of downstream insulin signaling pathway proteins (p-Akt and p-IRβ) was decreased, which indicated a weakened insulin sensitivity (fig. S4D). Besides, reduced O2 consumption (fig. S4, G and H) and CO2 production (fig. S4, I and J) showed less energy expenditure in BMP9-KO mice, which was further confirmed by the decreased expression of adipocyte thermogenesis (UCP1) and fatty acid utilization (PGC-1) molecular marker in BAT and WAT (fig. S4, K and L). These results demonstrate that BMP9-PPARα-FGF21 deficiency contributes to the dysfunction of insulin resistance and energy expenditure, which leads to abnormal glucose homeostasis and weight gain.
In summary, these data indicate that PPARα is a major target of BMP9 in the liver. BMP9 activates the promoter region of PPARα through its downstream protein p-smad 1/5, which affects transcriptional regulation of PPARα. Subsequently, the expression of PPARα target genes, which are associated with fatty acid oxidation, energy expenditure, and insulin sensitivity, is altered.
AAV-mediated BMP9 overexpression attenuates hepatosteatosis and obesity complications
To explore whether overexpression of BMP9 in vivo could rescue metabolic syndrome phenotype in BMP9-KO mice, we fed the mice for approximately 2 months for short term and 4 months for long term after AAV delivery (Fig. 6A). AAV-meditated BMP9 overexpression was detected efficiently, particularly in the liver (Fig. 6B). The serotype 8 AAV vector, which exhibits a high affinity for the liver, is a promising vector for liver-directed gene therapy (35, 36). To evaluate the therapeutic effectiveness of AAV-BMP9, we collected the livers in the paired groups. The mRNA and protein levels of BMP9 and PPARα were increased in the livers of mice after 2 months of AAV-BMP9 treatment (Fig. 6, C and D). Notably, hepatic p-smad 1/5 level was elevated (fig. S4E) after AAV-BMP9 treatment. Circulating BMP9 levels were elevated in the serum (Fig. 6E). In addition, hepatic FGF21 (fig. S4, B and E) and serum FGF21 (fig. S4F) levels were notably increased. H&E and Oil Red O staining showed reduced liver steatosis in the AAV-BMP9 group (Fig. 6F), which was confirmed by the liver TG level test (Fig. 6G). In parallel, hyperglycemia was relieved (Fig. 6H). GTT analysis showed improved glucose clearance (Fig. 6I). Furthermore, ITT results showed increased insulin sensitivity (Fig. 6J). Insulin receptor signaling proteins (like p-IRβ and p-Akt) were greatly activated as BMP9 and FGF21 were rescued (fig. S4E), which was consistent with a previous study (37). These data show a marked glycemia recovery after AAV-BMP9 administration. However, no significant body weight loss was observed.
Fig. 6. AAV-mediated BMP9 overexpression attenuates insulin resistance and hepatosteatosis.
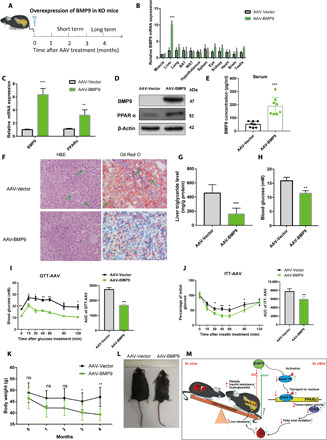
(A) Schematic diagram of AAV-BMP9 administration in BMP9-KO mice for 2 or 4 months. (B) mRNA expression of BMP9 in multiple mouse tissues after 2-month AAV-BMP9 treatment. (C) mRNA expression of BMP9 and PPARα in BMP9-KO mouse livers (n = 3). (D) Expression of BMP9 and PPARα proteins in BMP9-KO mouse livers. (E) Serum BMP9 protein concentrations (n = 6). (F) H&E and Oil Red O staining: Hepatic steatosis. Original magnification, ×200. (G) Liver TG level. (H) Initial blood glucose, (I) GTT, and (J) ITT (n = 6). (K) Body weight loss during 4 months of AAV-BMP9 treatment (n = 6). (L) Representative photograph of AAV-BMP9 mice (n = 6). Photo credit: Z. Yang, Photographer Institution, Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine. (M) Schematic overview. *P < 0.05, **P < 0.01, and ***P < 0.001.
For long-term AAV treatment, there was a significant body weight loss in the AAV-BMP9 group compared with the AAV vector group by the end of the continuous mouse body weight monitoring period (Fig. 6K). Attenuation of obesity by AAV-BMP9 was also observed (Fig. 6L).
In short, administration of AAV vector encoding BMP9 in the liver specifically caused the sustained reversion of hepatic steatosis, insulin resistance, and obesity. Our study demonstrates that liver BMP9 could correlate with liver steatosis via regulation of PPARα-mediated liver lipid metabolism.
DISCUSSION
To conclude, we revealed a previously unknown role of BMP9 in lipid homeostasis by regulating key lipid metabolism gene PPARα in fatty liver (Fig. 6M). Loss of BMP9 decreases phosphorylation of the downstream TF smad 1/5, which further resulted in a reduction in PPARα promoter activity and PPARα expression. PPARα reduction in the liver led to a decrease in fatty acid oxidation gene expression, thereby aggravating TG accumulation and expediting hepatic steatosis, obesity, insulin resistance, and hyperglycemia. Correspondingly, in vivo AAV-mediated BMP9 overexpression in mice or in vitro recombinant BMP9 protein treatment in hepatocytes rescued the above metabolic disorder consequences. Our studies uncover a critical function of BMP9 in NAFLD and provide insight into the understanding of liver lipid metabolism.
To our knowledge, no previous studies have used BMP9-KO mice to investigate obesity and liver lipid metabolism. Some studies have shown that ablation of genes, including DB-related gene, OB gene, and FAT-related gene, causes monogenic obesity (38), which are widely used as metabolic models. However, it is uncommon for a single gene mutation to result in spontaneous obesity. Most of the reported obesity susceptibility genes display an inconspicuous obesity phenotype unless HFD treatment is used (39). In this study, we provide direct evidence that BMP9 ablation displayed an obvious obesity tendency even under CD-feeding conditions. Moreover, the metabolic phenotypes were exacerbated when BMP9-KO mice were in an HFD feeding. These results show that BMP9 deficiency strongly induces obesity and fatty liver.
It is quite interesting that BMP9 plays dual roles in hepatic steatosis and fibrosis. In this study, BMP9 deficiency and significant hepatic steatosis were simultaneously observed in both NAFLD mouse model of HFD mice and db/db mice. However, mice exhibit no evident difference in liver fibrosis while lack of BMP9. Previous studies found that BMP9 accelerated hepatic fibrosis in mice especially induced fibrosis with carbon tetrachloride (CCl4) (11, 40), which suggested that BMP9 acted as a liver fibrosis risk factor. At the same time, a large number of studies supported the idea that BMP9 could have a promising therapeutic potential in suppressing obesity, reducing blood glucose concentration, and especially activating the expression of key lipid metabolism enzymes (12, 13, 34, 41). According to the “multiple hit model” (42), NAFLD is characterized by the presence of simple hepatic steatosis, followed by nonalcoholic steatohepatitis (NASH), fibrosis, and even cirrhosis (43). Briefly, fibrosis is the progressive form of steatosis (44). Liver fibrosis occurrence usually relies on specific diet inductions, like methionine/choline deficient diet (MCD) diet, NASH diet, CCl4, or mineral oil challenge (45). In addition, it is possible that the dual role of BMP9 in hepatic steatosis and fibrosis is due to the different target cells and biological processes. In other words, BMP9 attenuates hepatic steatosis by improving TG accumulation in hepatocytes by regulating PPARα. While BMP9 promotes fibrosis by activating HSCs, it is common that one gene or cytokine has dual or even contradictory function in steatosis and fibrosis. Similar studies and conclusions were found in one team and published contemporaneously. Gα12 ablation exacerbates liver steatosis by suppressing mitochondrial respiration (46). Gα12 overexpression also contributes to liver fibrosis by promoting autophagy in HSCs (47). It would be interesting to further investigate whether BMP9 activates the autophagic process and increases the fibrogenic response through the breakdown of lipid droplets in HSCs.
PPARα-dependent transcription is tightly regulated to dictate lipid handling. In hepatocytes, B cell lymphoma 6 (BCL6) has been shown to negatively regulate oxidative metabolism in a PPARα-dependent manner (48). In cardiac myocyte cells, Kruppel-like factor 5 (KLF5) transcriptionally regulates PPARα and cardiac energetics. KLF5 and c-Jun have opposite effects on PPARα gene expression and compete for binding to the PPARα promoter. In KLF5-KO mice, expression of cardiac PPARα and its downstream fatty acid oxidation–related target genes is decreased, causing sepsis and heart failure (27). Recently, smad 1/5/8 deficiency was reported to contribute to liver injury (49). Here, we demonstrate that p-smad acts as an unreported PPARα TF, influencing its promoter activity, thus regulating expression of PPARα in the liver. We clarified a previously unknown BMP-involved liver lipid metabolism mechanism that BMP9 regulates fatty acid oxidation via PPARα, rather than lipogenesis through sterol regulatory element–binding protein–1c (SREBP-1c) (41). The BMP9-smad-PPARα axis may play a vital role in biological processes, with the exception of liver lipid metabolism.
In the past few years, AAV-mediated gene therapy has become an efficient strategy to fight obesity and metabolic diseases (50). We used AAV8-mediated BMP9 overexpression in metabolic disease mice. Obesity, insulin resistance, and fatty liver were significantly attenuated in the treated mice. Our results provide previously unreported insights into fatty liver and metabolic complications in the future.
As previously reported, lipid inflammation promotes the initiation of obesity and multiple metabolic diseases (51). Natural killer (NK) cells protect against obesity and metabolic disorder through regulatory cytokine production in the liver (52–54). Recently, studies found a positive immunity function by influencing BMP6/Smad activity in the liver (55). Here, NK cell–mediated cytotoxicity signaling pathway was affected in BMP9-KO mouse livers (Fig. 4C), which indicates potential NK cell population changes or functional changes in hepatocytes. Whether BMP9 ablation disrupts the immunological behavior of NK cells requires further investigation.
Further studies will be required to test the functional receptors of BMP9 participating hepatic steatosis process in hepatocyte. Generally, mature BMP signaling depends on the secreted ligands and the type I and II BMP receptors in target cells (7). Ligand-receptor complexes recruit and phosphorylate the downstream intracellular transcriptional factors, which subsequently regulate gene expression. In this study, we confirmed that p-smad 1/5 was the downstream of BMP9 in both knockdown and overexpression strategies. However, BMP9-activated receptors remain unclear. To date, type I and II BMP receptors are the only known transmembrane cell surface receptors in humans with serine/threonine kinase activity. As the upstream of p-smad 1/5, the potential type I BMP receptors include activin receptor type 1 (ALK1), ALK2, ALK3, and ALK6. Moreover, ACVR2A, ACVR2B, BMPR2, and AMHR2 are considered as type II BMP receptor candidates of BMP9 (7). To some extent, ALK1 was the most well-recognized receptor of BMP9, especially in endothelial cells (56). Moreover, the co-receptor endoglin may also act as the potential functional receptor for BMP9 signaling (57).
In conclusion, our studies uncover a previously unreported and critical factor regulating hepatic TG homeostasis and the mechanism of hepatosteatosis in NAFLD mice and humans. We identified BMP9 as a critical regulatory cytokine involved in the progression of fatty liver. Thus, BMP9 might be a potential therapeutic target for NAFLD and obesity complications.
MATERIALS AND METHODS
Clinical samples
Clinical samples were collected from the Clinical Laboratory of the Ninth People’s Hospital, affiliated to Shanghai Jiao Tong University School of Medicine. These included 32 normal healthy control samples and 32 samples from patients with endocrine and metabolic diseases. Clinical characteristics of subjects were collected and measured as previously described, and more than 5% liver fat content abnormal accumulation was defined as NAFLD (42, 44, 58). Liver fat content was measured by magnetic resonance spectroscopy. Concentration of BMP9 in serum was detected by a BMP9 commercial ELISA kit (RayBiotech, ELH-BMP9). This research was performed in accordance with the World Medical Association Declaration of Helsinki. All the studies were approved by the Ethics Committee of Shanghai Jiao Tong University.
Animal experiments
Male C57BL/6 and db/db mice aged 8 to 12 weeks were purchased from the Shanghai Laboratory Animal Company (Shanghai). BMP9-KO mice were generated by CRISPR-Cas9 on a C57BL/6 background, and KO mice were compared to WT littermates. All animal procedures were approved by the Shanghai Jiao Tong University Animal Care and Use Committee.
Cell culture
Mouse Hepa 1-6 cells (SCSP-512) were provided by Stem Cell Bank, Chinese Academy of Sciences. Hepa 1-6 cells were cultured in complete Dulbecco’s modified Eagle’s medium (Gibco, 10938) supplemented with 10% (v/v) fetal bovine serum (Gibco, 16140071) and 1% (v/v) antibiotics (penicillin/streptomycin; Gibco, 10378016) in a humidified incubator with 5% CO2 at 37°C.
Gene microarray analysis
Three independent 16-week-old littermate WT and BMP9-KO mouse livers and hypothalami were collected and frozen in liquid nitrogen for microarray analysis. Differentially expressed genes were then identified through the fold change (FC), and the P value was calculated by t test. The threshold for up- and down-regulated genes was FC ≥ 2.0 and P ≤ 0.05.
ChIP analysis
The PPARα promoter was divided into six 300-bp regions. Furthermore, we investigated the precise TF binding sites of the PPARα promoter. ChIP assays were performed using a ChIP kit (Merck Millipore, 17-371).
Statistical analysis
Statistical analyses were performed using Student’s two-tailed t test (GraphPad Prism 7). Data were represented as the mean ± SEM of n independent experiments. Area under the curve (AUC) was calculated to access GTT and ITT sensitivity by “total area” and “standard error.” Values of P < 0.05 were considered significant (*P < 0.05, **P < 0.01, and ***P < 0.001).
Supplementary Material
Acknowledgments
We thank all the NAFLD patients enrolled in our study and wish them good health. Funding: The authors acknowledge the financial support from the National Natural Science Foundation of China (grants U1932135 and 81772875), the National Key Research and Development Plan (2018YFC1106100), the Science and Technology Commission of Shanghai (17DZ2260100 and 19JC1410200), the National Natural Science Foundation of China Youth Fund (81802919), and the Shanghai Yang Fan Youth Project (20YF1442300). Author contributions: X.F. and S.G. designed and supervised the study. Z.Y. drafted the manuscript and performed animal and molecular experiments. P.L. constructed CRISPR BMP9-KO mice. Q.S. contributed to some animal feeding experiments. Y.W. and J.H. helped with clinical sample collection. X.F., R.J., and S.G. revised the draft. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/48/eabc5022/DC1
REFERENCES AND NOTES
- 1.Lu Y., Liu X., Jiao Y., Xiong X., Wang E., Wang X., Zhang Z., Zhang H., Pan L., Guan Y., Cai D., Ning G., Li X., Periostin promotes liver steatosis and hypertriglyceridemia through downregulation of PPARα. J. Clin. Invest. 124, 3501–3513 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heidenreich S., Witte N., Weber P., Goehring I., Tolkachov A., von Loeffelholz C., Döcke S., Bauer M., Stockmann M., Pfeiffer A. F. H., Birkenfeld A. L., Pietzke M., Kempa S., Muenzner M., Schupp M., Retinol saturase coordinates liver metabolism by regulating ChREBP activity. Nat. Commun. 8, 384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang P. X., Zhang X. J., Luo P., Jiang X., Zhang P., Guo J., Zhao G. N., Zhu X., Zhang Y., Yang S., Li H., Hepatocyte TRAF3 promotes liver steatosis and systemic insulin resistance through targeting TAK1-dependent signalling. Nat. Commun. 7, 10592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fabbrini E., Sullivan S., Klein S., Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 51, 679–689 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bidart M., Ricard N., Levet S., Samson M., Mallet C., David L., Subileau M., Tillet E., Feige J. J., Bailly S., BMP9 is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell. Mol. Life Sci. 69, 313–324 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller A. F., Harvey S. A., Thies R. S., Olson M. S., Bone morphogenetic protein-9. An autocrine/paracrine cytokine in the liver. J. Biol. Chem. 275, 17937–17945 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Salazar V. S., Gamer L. W., Rosen V., BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 12, 203–221 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Guiu-Jurado E., Unthan M., Böhler N., Kern M., Landgraf K., Dietrich A., Schleinitz D., Ruschke K., Klöting N., Faßhauer M., Tönjes A., Stumvoll M., Körner A., Kovacs P., Blüher M., Bone morphogenetic protein 2 (BMP2) may contribute to partition of energy storage into visceral and subcutaneous fat depots. Obesity (Silver Spring) 24, 2092–2100 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Modica S., Straub L. G., Balaz M., Sun W., Varga L., Stefanicka P., Profant M., Simon E., Neubauer H., Ukropcova B., Ukropec J., Wolfrum C., Bmp4 promotes a brown to white-like adipocyte shift. Cell Rep. 16, 2243–2258 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Whittle A. J., Carobbio S., Martins L., Slawik M., Hondares E., Vázquez M. J., Morgan D., Csikasz R. I., Gallego R., Rodriguez-Cuenca S., Dale M., Virtue S., Villarroya F., Cannon B., Rahmouni K., López M., Vidal-Puig A., BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell 149, 871–885 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li P., Li Y., Zhu L., Yang Z., He J., Wang L., Shang Q., Pan H., Wang H., Ma X., Li B., Fan X., Ge S., Jia R., Zhang H., Targeting secreted cytokine BMP9 gates the attenuation of hepatic fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 709–720 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Kuo M. M.-C., Kim S., Tseng C. Y., Jeon Y. H., Choe S., Lee D. K., BMP-9 as a potent brown adipogenic inducer with anti-obesity capacity. Biomaterials 35, 3172–3179 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Chen C., Grzegorzewski K. J., Barash S., Zhao Q., Schneider H., Wang Q., Singh M., Pukac L., Bell A. C., Duan R., Coleman T., Duttaroy A., Cheng S., Hirsch J., Zhang L., Lazard Y., Fischer C., Barber M. C., Ma Z. D., Zhang Y. Q., Reavey P., Zhong L., Teng B., Sanyal I., Ruben S. M., Blondel O., Birse C. E., An integrated functional genomics screening program reveals a role for BMP-9 in glucose homeostasis. Nat. Biotechnol. 21, 294–301 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Xu X., Li X., Yang G., Li L., Hu W., Zhang L., Liu H., Zheng H., Tan M., Zhu D., Circulating bone morphogenetic protein-9 in relation to metabolic syndrome and insulin resistance. Sci. Rep. 7, 17529 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chakravarthy M. V., Pan Z., Zhu Y., Tordjman K., Schneider J. G., Coleman T., Turk J., Semenkovich C. F., “New” hepatic fat activates PPARα to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 1, 309–322 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Chakravarthy M. V., Lodhi I. J., Yin L., Malapaka R. R. V., Xu H. E., Turk J., Semenkovich C. F., Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell 138, 476–488 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gustafson B., Hammarstedt A., Hedjazifar S., Hoffmann J. M., Svensson P. A., Grimsby J., Rondinone C., Smith U., BMP4 and BMP antagonists regulate human white and beige adipogenesis. Diabetes 64, 1670–1681 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Tseng Y. H., Kokkotou E., Schulz T. J., Huang T. L., Winnay J. N., Taniguchi C. M., Tran T. T., Suzuki R., Espinoza D. O., Yamamoto Y., Ahrens M. J., Dudley A. T., Norris A. W., Kulkarni R. N., Kahn C. R., New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 454, 1000–1004 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badman M. K., Pissios P., Kennedy A. R., Koukos G., Flier J. S., Maratos-Flier E., Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Hsu P. D., Lander E. S., Zhang F., Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J., Liu R., Wang F., Hong J., Li X., Chen M., Ke Y., Zhang X., Ma Q., Wang R., Shi J., Cui B., Gu W., Zhang Y., Zhang Z., Wang W., Xia X., Liu M., Ning G., Ablation of LGR4 promotes energy expenditure by driving white-to-brown fat switch. Nat. Cell Biol. 15, 1455–1463 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Vernia S., Cavanagh-Kyros J., Garcia-Haro L., Sabio G., Barrett T., Jung D. Y., Kim J. K., Xu J., Shulha H. P., Garber M., Gao G., Davis R. J., The PPARα-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 20, 512–525 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Townsend K. L., Suzuki R., Huang T. L., Jing E., Schulz T. J., Lee K., Taniguchi C. M., Espinoza D. O., McDougall L. E., Zhang H., He T.-C., Kokkotou E., Tseng Y. H., Bone morphogenetic protein 7 (BMP7) reverses obesity and regulates appetite through a central mTOR pathway. FASEB J. 26, 2187–2196 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saini S., Duraisamy A. J., Bayen S., Vats P., Singh S. B., Role of BMP7 in appetite regulation, adipogenesis, and energy expenditure. Endocrine 48, 405–409 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Bernal-Mizrachi C., Weng S., Feng C., Finck B. N., Knutsen R. H., Leone T. C., Coleman T., Mecham R. P., Kelly D. P., Semenkovich C. F., Dexamethasone induction of hypertension and diabetes is PPAR-alpha dependent in LDL receptor-null mice. Nat. Med. 9, 1069–1075 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Buchanan J., Mazumder P. K., Hu P., Chakrabarti G., Roberts M. W., Yun U. J., Cooksey R. C., Litwin S. E., Abel E. D., Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146, 5341–5349 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Drosatos K., Pollak N. M., Pol C. J., Ntziachristos P., Willecke F., Valenti M.-C., Trent C. M., Hu Y., Guo S., Aifantis I., Goldberg I. J., Cardiac myocyte KLF5 regulates ppara expression and cardiac function. Circ. Res. 118, 241–253 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Derynck R., Zhang Y. E., Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425, 577–584 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Heldin C. H., Miyazono K., ten Dijke P., TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Xu H. E., Stanley T. B., Montana V. G., Lambert M. H., Shearer B. G., Cobb J. E., McKee D. D., Galardi C. M., Plunket K. D., Nolte R. T., Parks D. J., Moore J. T., Kliewer S. A., Willson T. M., Stimmel J. B., Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARα. Nature 415, 813–817 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Xu J., Lloyd D. J., Hale C., Stanislaus S., Chen M., Sivits G., Vonderfecht S., Hecht R., Li Y.-S., Lindberg R. A., Chen J.-L., Jung D. Y., Zhang Z., Ko H.-J., Kim J. K., Véniant M. M., Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 58, 250–259 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morita M., Siddiqui N., Katsumura S., Rouya C., Larsson O., Nagashima T., Hekmatnejad B., Takahashi A., Kiyonari H., Zang M., St-Arnaud R., Oike Y., Giguère V., Topisirovic I., Okada-Hatakeyama M., Yamamoto T., Sonenberg N., Hepatic posttranscriptional network comprised of CCR4-NOT deadenylase and FGF21 maintains systemic metabolic homeostasis. Proc. Natl. Acad. Sci. U.S.A. 116, 7973–7981 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y., Wong K., Giles A., Jiang J., Lee J. W., Adams A. C., Kharitonenkov A., Yang Q., Gao B., Guarente L., Zang M., Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology 146, 539–549.e7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim S., Choe S., Lee D. K., BMP-9 enhances fibroblast growth factor 21 expression and suppresses obesity. Biochim. Biophys. Acta 1862, 1237–1246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gan S. U., Fu Z., Sia K. C., Kon O. L., Calne R., Lee K. O., Development of a liver-specific Tet-off AAV8 vector for improved safety of insulin gene therapy for diabetes. J. Gene Med. 21, e3067 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tenney R. M., Bell C. L., Wilson J. M., AAV8 capsid variable regions at the two-fold symmetry axis contribute to high liver transduction by mediating nuclear entry and capsid uncoating. Virology 454-455, 227–236 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Pan Y., Wang B., Zheng J., Xiong R., Fan Z., Ye Y., Zhang S., Li Q., Gong F., Wu C., Lin Z., Li X., Pan X., Pancreatic fibroblast growth factor 21 protects against type 2 diabetes in mice by promoting insulin expression and secretion in a PI3K/Akt signaling-dependent manner. J. Cell. Mol. Med. 23, 1059–1071 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cool D. R., Normant E., Shen F. S., Chen H. C., Pannell L., Zhang Y., Loh Y. P., Carboxypeptidase E is a regulated secretory pathway sorting receptor: Genetic obliteration leads to endocrine disorders in Cpe(fat) mice. Cell 88, 73–83 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Liang N., Damdimopoulos A., Goñi S., Huang Z., Vedin L. L., Jakobsson T., Giudici M., Ahmed O., Pedrelli M., Barilla S., Alzaid F., Mendoza A., Schröder T., Kuiper R., Parini P., Hollenberg A., Lefebvre P., Francque S., van Gaal L., Staels B., Venteclef N., Treuter E., Fan R., Hepatocyte-specific loss of GPS2 in mice reduces non-alcoholic steatohepatitis via activation of PPARα. Nat. Commun. 10, 1684 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breitkopf-Heinlein K., Meyer C., König C., Gaitantzi H., Addante A., Thomas M., Wiercinska E., Cai C., Li Q., Wan F., Hellerbrand C., Valous N. A., Hahnel M., Ehlting C., Bode J. G., Müller-Bohl S., Klingmüller U., Altenöder J., Ilkavets I., Goumans M. J., Hawinkels L. J. A. C., Lee S. J., Wieland M., Mogler C., Ebert M. P., Herrera B., Augustin H., Sánchez A., Dooley S., ten Dijke P., BMP-9 interferes with liver regeneration and promotes liver fibrosis. Gut 66, 939–954 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Yang M., Liang Z., Yang M., Jia Y., Yang G., He Y., Li X., Gu H. F., Zheng H., Zhu Z., Li A. L., Role of bone morphogenetic protein-9 in the regulation of glucose and lipid metabolism. FASEB J. 33, 10077–10088 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Fang Y. L., Chen H., Wang C. L., Liang L., Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model”. World J. Gastroenterol. 24, 2974–2983 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jayakumar S., Middleton M. S., Lawitz E. J., Mantry P. S., Caldwell S. H., Arnold H., Mae Diehl A., Ghalib R., Elkhashab M., Abdelmalek M. F., Kowdley K. V., Stephen Djedjos C., Xu R., Han L., Mani Subramanian G., Myers R. P., Goodman Z. D., Afdhal N. H., Charlton M. R., Sirlin C. B., Loomba R., Longitudinal correlations between MRE, MRI-PDFF, and liver histology in patients with non-alcoholic steatohepatitis: Analysis of data from a phase II trial of selonsertib. J. Hepatol. 70, 133–141 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Chalasani N., Younossi Z., Lavine J. E., Diehl A. M., Brunt E. M., Cusi K., Charlton M., Sanyal A. J., The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 55, 2005–2023 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Guo L., Zhang P., Chen Z., Xia H., Li S., Zhang Y., Kobberup S., Zou W., Lin J. D., Hepatic neuregulin 4 signaling defines an endocrine checkpoint for steatosis-to-NASH progression. J. Clin. Invest. 127, 4449–4461 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim T. H., Yang Y. M., Han C. Y., Koo J. H., Oh H., Kim S. S., You B. H., Choi Y. H., Park T. S., Lee C. H., Kurose H., Noureddin M., Seki E., Wan Y. J. Y., Choi C. S., Kim S. G., Gα12 ablation exacerbates liver steatosis and obesity by suppressing USP22/SIRT1-regulated mitochondrial respiration. J. Clin. Invest. 128, 5587–5602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim K. M., Han C. Y., Kim J. Y., Cho S. S., Kim Y. S., Koo J. H., Lee J. M., Lim S. C., Kang K. W., Kim J. S., Hwang S. J., Ki S. H., Kim S. G., Gα12 overexpression induced by miR-16 dysregulation contributes to liver fibrosis by promoting autophagy in hepatic stellate cells. J. Hepatol. 68, 493–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sommars M. A., Ramachandran K., Senagolage M. D., Futtner C. R., Germain D. M., Allred A. L., Omura Y., Bederman I. R., Barish G. D., Dynamic repression by BCL6 controls the genome-wide liver response to fasting and steatosis. eLife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang C. Y., Xiao X., Bayer A., Xu Y., Dev S., Canali S., Nair A. V., Masia R., Babitt J. L., Ablation of Hepatocyte Smad1, Smad5, and Smad8 causes severe tissue iron loading and liver fibrosis in mice. Hepatology 70, 1986–2002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sponton C. H., Kajimura S., AAV-mediated gene therapy as a strategy to fight obesity and metabolic diseases. EMBO Mol. Med. 10, e9431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shan B., Wang X., Wu Y., Xu C., Xia Z., Dai J., Shao M., Zhao F., He S., Yang L., Zhang M., Nan F., Li J., Liu J., Liu J., Jia W., Qiu Y., Song B., Han J.-D. J., Rui L., Duan S.-Z., Liu Y., The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat. Immunol. 18, 519–529 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Lynch L., Nowak M., Varghese B., Clark J., Hogan A. E., Toxavidis V., Balk S. P., O’Shea D., O’Farrelly C., Exley M. A., Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity 37, 574–587 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lukens J. R., Kanneganti T. D., Fat chance: Not much against NKT cells. Immunity 37, 447–449 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Wensveen F. M., Jelenčić V., Valentić S., Šestan M., Wensveen T. T., Theurich S., Glasner A., Mendrila D., Štimac D., Wunderlich F. T., Brüning J. C., Mandelboim O., Polić B., NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat. Immunol. 16, 376–385 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Eddowes L. A., al-Hourani K., Ramamurthy N., Frankish J., Baddock H. T., Sandor C., Ryan J. D., Fusco D. N., Arezes J., Giannoulatou E., Boninsegna S., Chevaliez S., Owens B. M. J., Sun C. C., Fabris P., Giordani M. T., Martines D., Vukicevic S., Crowe J., Lin H. Y., Rehwinkel J., McHugh P. J., Binder M., Babitt J. L., Chung R. T., Lawless M. W., Armitage A. E., Webber C., Klenerman P., Drakesmith H., Antiviral activity of bone morphogenetic proteins and activins. Nat. Microbiol. 4, 339–351 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salmon R. M., Guo J., Wood J. H., Tong Z., Beech J. S., Lawera A., Yu M., Grainger D. J., Reckless J., Morrell N. W., Li W., Molecular basis of ALK1-mediated signalling by BMP9/BMP10 and their prodomain-bound forms. Nat. Commun. 11, 1621 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lawera A., Tong Z., Thorikay M., Redgrave R. E., Cai J., van Dinther M., Morrell N. W., Afink G. B., Charnock-Jones D. S., Arthur H. M., ten Dijke P., Li W., Role of soluble endoglin in BMP9 signaling. Proc. Natl. Acad. Sci. U.S.A. 116, 17800–17808 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang H. J., Zhang X.-F., Ma Z.-M., Pan L.-L., Chen Z., Han H.-W., Han C.-K., Zhuang X.-J., Lu Y., Li X.-J., Yang S.-Y., Li X.-Y., Irisin is inversely associated with intrahepatic triglyceride contents in obese adults. J. Hepatol. 59, 557–562 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/48/eabc5022/DC1