

Genomes of 150 coral colonies reveal evolutionary processes related to past climatic change on the Great Barrier Reef.
Abstract
Genetic signatures caused by demographic and adaptive processes during past climatic shifts can inform predictions of species’ responses to anthropogenic climate change. To identify these signatures in Acropora tenuis, a reef-building coral threatened by global warming, we first assembled the genome from long reads and then used shallow whole-genome resequencing of 150 colonies from the central inshore Great Barrier Reef to inform population genomic analyses. We identify population structure in the host that reflects a Pleistocene split, whereas photosymbiont differences between reefs most likely reflect contemporary (Holocene) conditions. Signatures of selection in the host were associated with genes linked to diverse processes including osmotic regulation, skeletal development, and the establishment and maintenance of symbiosis. Our results suggest that adaptation to post-glacial climate change in A. tenuis has involved selection on many genes, while differences in symbiont specificity between reefs appear to be unrelated to host population structure.
INTRODUCTION
Coastal marine ecosystems worldwide are threatened by the effects of anthropogenic climate change including warming (1), acidification, altered patterns of freshwater input, and changing species interactions because of range shifts (2). Coral reefs are especially vulnerable because their structural integrity depends on the symbiosis between corals and photosymbionts [family Symbiodiniaceae (3)], but this relationship breaks down under stress, leading to bleaching and widespread mortality during marine heat waves (4). The projected severity of these impacts therefore depends on the ability of coral holobionts [coral, photosymbionts, and associated bacteria (5)] to adapt fast enough to maintain the integrity of reef ecosystems (6). A key factor that might improve the adaptive outlook for corals is the existence of stress-tolerant populations that could fuel adaptation in conspecifics through natural or assisted gene flow (7, 8), but the genetic basis of this stress tolerance is not yet well understood.
Although several studies have identified genetic correlates of the thermal stress response or divergent loci that characterize thermal resilience (9–11), little is known about how coral populations evolve when faced with the other stresses that are likely to accompany climate change. One way to identify the genetic factors involved in adaptive responses of corals across a range of stressors is to examine signatures of selection caused by past climatic change, but this is challenging because selection can be difficult to distinguish from demographic factors such as population bottlenecks. Here, we address this issue using dense allele frequency information from whole-genome sequencing to detect signatures of selection while using explicit demographic modeling to calculate a neutral distribution and control for false positives. An additional benefit to the whole-genome sequencing approach used here is that it also permits simultaneous genotyping of the dominant photosymbiont in each coral colony.
We apply our approach to identify signatures of selection and profile symbiont communities in Acropora tenuis populations from inshore reefs of the Great Barrier Reef (GBR). This species is found throughout the Indo-Pacific and is widespread across the GBR, where it is common on both inshore and offshore reefs (12). The species has also become an important model for research into water-quality stress (13), adaptive changes in symbiont composition (14), and the feasibility of assisted evolution approaches (15, 16).
The reefs sampled in this study were recolonized between 14 and 6 thousand years (ka) ago when rising sea levels resulted in marine flooding of the GBR shelf (17). This event changed the environment available for coral growth markedly, shifting from a narrow line of fringing reefs at the edge of the shelf to the distinct outer and inner reef characteristic of the GBR today (17, 18). On the basis of the limit of detection for selective sweeps, we argue that loci with signatures of selection in our study most likely reflect evolutionary responses to events since the last glacial maximum, a period that includes major increases in global temperature and CO2 and region-specific changes due to an increase in estuarine and shallow-water influence on water quality. Water quality on the GBR is a major driver of coral biodiversity and depends on many factors, some of which, such as the frequency of exposure to flood plumes, vary substantially between inshore locations (19–21). To examine these local factors, we also investigated differences between inshore sites with contrasting water quality due to relative proximity to large river systems and find that they host distinct populations of photosymbionts.
RESULTS
De novo assembly of the A. tenuis genome
To maximize our ability to model demographic history and detect selective sweeps, we generated a highly contiguous and complete reference genome for A. tenuis based on PacBio long-read sequencing. The resulting assembly of 487 Mb in 614 contigs with an N50 of 2.84 Mb and largest contig of 13.5 Mb represents a considerable improvement in contiguity compared with most previously published coral genomes (table S1) including a recent assembly from the same species (22). More than 80% of the genome assembled into contigs larger than 1 Mb, allowing us to minimize artifacts arising from ambiguous alignments due to unmerged haplotypes or misassemblies at the ends of contigs. Large genomic fragments also provide the necessary length to capture recent selective sweeps that can distort allele frequencies over centimorgan-sized regions (23).
Annotation of this genome assembly with Illumina RNA sequencing data resulted in 30,327 gene models of which more than 90% (27,825) belonged to one of 14,812 ortholog gene family groups shared with other cnidarian genomes. Of these gene families, 97% (14,403) were shared with other published Acropora genomes (Acropora millepora and Acropora digitifera). BUSCO analysis identified a near complete 91.8% set of core metazoan genes including a small fraction that were duplicated (2.6%) or fragmented (1.8%). A genome size estimate of 470 Mb obtained from kmer analysis of short-read data with sga.preqc (24) was close to the final assembly size, confirming the high completeness of the assembly and suggesting that there are few artificial duplications remaining after purging of unmerged haplotypes.
The completeness of our long-read assembly allowed us to resolve a substantially larger proportion of interspersed repeats (41.7%) compared with the published acroporid assemblies based on short-read data [A. digitifera (25) (30.2%), A. millepora (26) (31.6%), A. tenuis (22) (34.3%)]. The only other available coral genome with comparable repeat resolution is a recently released long-read assembly for A. millepora (27), which identified a very similar proportion (40.7%) of interspersed repeats to our A. tenuis assembly. This increased ability to resolve repeats is reflected in a substantially higher proportion of unclassified elements in long-read assemblies (20.8% A. tenuis; 21.2% A. millepora) compared with short-read assemblies (16.4% A. digitifera; 16.5% A. millepora), but the overall composition of known repeat classes was remarkably similar between Acropora genomes (fig. S1).
As an additional check on the accuracy of our A. tenuis assembly, we compared its high-level genomic organization with that of the congeneric species, A. millepora for which a chromosome-scale linkage map is available (27). Macrosynteny analysis between the two Acropora genomes revealed a high degree of collinearity and limited rearrangements between linkage groups (Fig. 1A) that most likely reflects their small evolutionary distance [approximately 15 Ma ago (22)]. For comparison, we also examined synteny between A. millepora and the much more distantly related anthozoan, Nematostella vectensis [greater than 500 Ma ago (28)]. Despite this long divergence time, the number and basic structure of linkage groups was remarkably well maintained, but with near complete rearrangement within groups (Fig. 1B).
Fig. 1. Macrosynteny conservation between A. tenuis and other cnidarian genomes.

Dots represent blocks of collinear groups of genes and margin lines represent boundaries between contigs (top) or A. millepora linkage groups (right). In (A), very few A. tenuis scaffolds map across A. millepora chromosomes (a single example is shown with arrows). In (B), the blocks along the diagonal reflect ancient conserved linkage groups that roughly correspond to chromosomes in A. millepora.
Population structure
To investigate recent evolutionary and demographic processes in A. tenuis, we performed shallow (approx. 3× depth) whole-genome sequencing on 150 colonies from five reefs in the central inshore GBR (n = 30 per reef). Individuals were selected randomly from a larger pool of samples after first excluding likely clones based on preliminary DArT (diversity arrays technology) sequencing (see Methods). Two samples from Magnetic Island were strong outliers in population structure analyses (fig. S2) and excluded from subsequent analyses resulting in a slightly reduced sample size (N = 28) for this location. Single-nucleotide polymorphism (SNP) calling based on the remaining 148 individuals resulted in a total of 14.3 million high quality biallelic SNPs. Reefs were chosen from opposite ends of two plume influence gradients, Burdekin in the south and Wet Tropics in the north (Fig. 2A). These gradients capture long-term frequency of exposure to riverine flood plume waters based on aerial and satellite monitoring data (19, 20) and predictive modeling (see Methods for full details and rationale). Although other factors are likely to also be involved, flood plume exposure is a strong driver of water quality (29). For the Burdekin gradient, two sites with high river influence (“plume;” Magnetic Island and Pandora Reef) and one site with low riverine influence (“marine;” Pelorus Island) were sampled, whereas for the Wet Tropics gradient, we sampled one plume (Dunk Island) and one marine site (Fitzroy Island). The choice of marine and plume sites reflects coastal topology and depth profiles that have changed slightly (sea level decline by 1 to 1.5 m) over the past 6 ka ago compared with the Holocene deglaciation period (20 to 6 ka ago) where sea levels increased by more than 100 m (18).
Fig. 2. Population structure of coral host and dominant (Cladocopium) photosymbionts.
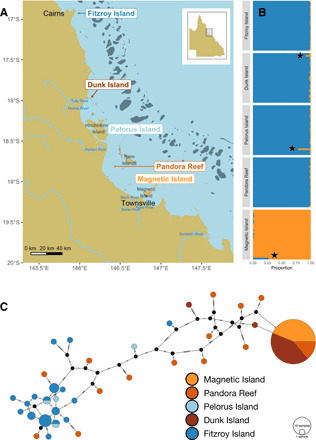
Plots (A) and (C) use the same color scheme (bottom right) that shows plume sites in shades of orange and marine sites in shades of blue. (A) Sampling locations in the central inshore GBR, Australia (N = 30 at each site except Magnetic Island, where two genetic outliers were discarded leading to N = 28). Major river systems are labeled and affect sites northward because of the direction of residual inshore current. (B) Coral host admixture proportions based on PCAngsd analysis for each of the five sampling locations. The northern population is shown in dark blue, while Magnetic island is in orange. Samples indicated with a black star represent likely recent hybrids. (C) Mitochondrial haplotype network based on the dominant strain of the symbiont Cladocopium in each sample. Substitutions separating nodes are represented by cross bars.
Superimposed on the water-quality gradients in our study design, we observed strong coral population structure separating Magnetic Island from the four reefs to its north. Analysis of population structure with PCAngsd showed these as two clear clusters (fig. S2) and admixture analysis showed near complete assignment to either of these clusters for all but three individuals (Fig. 2B). We refer to these clusters as north (all reefs other than Magnetic Island) and Magnetic Island. Genome-wide average Fst values between Magnetic Island and northern reefs were much larger (0.19) than between reefs within the northern cluster (0.008). We also observed evidence of recent gene flow between Magnetic Island and northern reefs with three colonies showing mixed ancestry (Fig. 2B). We also checked to determine whether this population structure was captured by differences in mitochondrial haplotypes between colonies but found that, in contrast to the nuclear data, mitochondrial haplotypes were completely unstructured (fig. S3). This most likely reflects the relatively recent divergence (see below) combined with a slow mutation rate that is a well-documented feature of anthozoan mitochondrial genomes (30).
Photosymbiont differences between reefs
The availability of complete genomes from five major genera within the Symbiodiniaceae allowed us to identify [using kraken (31)] between 100 and 300 k reads of symbiont [Symbiodiniaceae (3)] origin in each sample (fig. S4). For almost all samples, the dominant symbiont genus was Cladocopium [formerly Clade C (3)], with a single sample showing a high proportion of Durusdinium [formerly Clade D (3)]. Although the numbers of symbiont reads were insufficient to permit genotyping on nuclear loci, these did provide sufficient depth (at least 5×) for determination of the dominant mitochondrial haplotype in each sample. The resulting haplotype network appeared to be structured according to location, with most of samples from plume (Magnetic Island, Dunk Island, and Pandora Reef) sharing a single haplotype. Samples from marine locations (Fitzroy Island, Pelorus Island) did not include this haplotype but instead were highly diverse, forming a sparse network of divergent types. A test for genetic structuring using analysis of molecular variance confirmed that differentiation by reef was significant (P < 1 × 10−5), but it was not possible to link this unambiguously with the water quality designations from our study design (plume versus marine) (P = 0.1). Notably, the structuring of Symbiodiniaceae populations by reef followed a biogeographical pattern that was clearly distinct to that of hosts. Although the results point toward long-term frequency of plume exposure as one potential driver of these differences, we cannot rule out other factors such as thermal history or acute differences in plume exposure. Nevertheless, the observation of clear structuring by reef suggests that local conditions play a role in determining the symbiont community associated with A. tenuis.
Demographic history
To model demographic history and interpret and control for error in selection analyses, the complementary approaches MSMC (Multiple Sequentially Markovian Coalescent) and dadi were used, with combined results from both suggesting that the Magnetic Island and northern populations diverged between 600 and 270 ka ago (Fig. 3, A and B). Estimates using dadi are based on the best-fitting model (“isolation_asym_mig”), which allows for a period of isolation followed by secondary contact with asymmetric migration (see table S2 for full parameter lists of all models). An independent estimate based on divergence of population size trajectories inferred by MSMC suggests a slightly older split, with first divergence occurring at around 600 ka ago and becoming clearly distinct by around 300 ka ago. The overall demographic history inferred by MSMC features a peak in Ne around 1 Ma ago seen in other estimates from coral genomes (6, 22, 27, 32) and a more recent second peak that was also observed in an A. tenuis individual sampled from Okinawa, Japan (22). Although these features could represent changes in census population size, it is also possible that high values of Ne in the past could be because of contemporary or historical population structure (33).
Fig. 3. Population genomic statistics and models of demographic history.
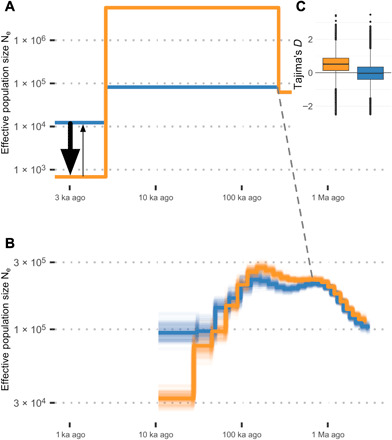
Orange represents Magnetic Island and blue represents northern populations in all component figures. Dashed line connects the point of divergence between populations. (A) Changes in effective population size and estimated gene flow for the best-fitting dadi model. Relative gene flow is shown using arrows with larger flow from north to south indicated by arrow size. (B) Changes in effective population size estimated using MSMC from high coverage (~20×) sequencing of a single colony from Magnetic Island (MI) and a colony from Fitzroy Island (FI). (C) (inset top right) Genome wide estimates of Tajima’s D calculated using PCAngsd. The scales for both time and Ne are dependent on values of the mutation rate (μ = 1.86 × 10−8 per bp per generation; see Methods) and generation time (5 years). Changes in these estimates would lead to a rescaling of axes but not the overall shape of demographic profiles.
Since it incorporates data from many individuals, the dadi approach is capable of more recent demographic inference and shows a recent decline in both A. tenuis populations. This is particularly pronounced in the case of the Magnetic Island population, which all dadi models (fig. S5) and MSMC estimate as having larger historical and much smaller contemporary effective population sizes.
Genome-wide estimates of Tajima’s D are sensitive to demographic history and were consistent with the main demographic inferences from dadi and MSMC analyses (Fig. 3C). In particular, a high value for Magnetic Island is consistent with a recent or ongoing bottleneck for that population (34), whereas values close to zero for northern reefs are consistent with minimal bottleneck effects there. Demographic models that included asymmetric migration provided a better fit to the two-dimensional (2D) site frequency spectrum (SFS) than those without migration or with symmetric migration (Fig. 3 and table S2). Across all of these models, migration coefficients were larger for the southward direction. Although this is against the predominant direction of inshore current flow (35) and predictions based on biophysical modeling (36), it is consistent with studies that have shown southward migration to be the predominant direction of gene flow for A. tenuis and A. millepora on the GBR (6, 36) including for inshore populations. This disconnect between biophysical modeling (which reflects prevailing, but variable, northward inshore currents) and apparent gene flow could reflect differences in effective population size. Demographic modeling and genome-wide Tajima’s D suggest a strong bottleneck and small effective population size for Magnetic Island, and since migration coefficients in dadi are measured as a proportion of individuals at the destination, the small size of the Magnetic Island population could skew outcomes toward southward flow. Thus, relative sizes of migration coefficients in our study most likely reflect differences in effective population size rather than absolute numbers of migrants.
Signatures of selection
A major challenge when identifying genomic sites under selection is that demographic history and migration can produce confounding signals (37). To estimate false-positive rates from these processes, SweepFinder 2 was used in combination with coalescent simulations under a range of demographic models (all dadi and MSMC models) to generate neutral data from which composite likelihood ratio (CLR) statistics could be calculated. Using this data as a control, we were able to calculate an empirical false discovery rate for a given CLR threshold and neutral model (see Methods). For northern reefs, we found that, irrespective of the neutral model used, it was always possible to find a CLR threshold that would control false positives to less than 10% (fig. S6). This was not the case for Magnetic Island, however, where its extreme demographic history (strong bottleneck) combined with small sample size meant that distinguishing neutral from selective processes was highly model-dependent and uniformly required a higher CLR threshold. We therefore focused our attention on the northern reefs where a CLR threshold of 100 was sufficient to control the false discovery rate to less than 10% under the best-fitting model and almost all other demographic models. In support of their status as putative sites under positive selection, the 98 loci passing this threshold also tended to have extremely low values of Tajima’s D (Fig. 4) and were often associated with regions of high differentiation (high pairwise Fst) between Magnetic Island and northern reefs (Fig. 4). Despite this relationship, a search for loci under selection based on Fst alone [using Bayescan 2 v2.1; (38)] failed to identify any outliers when considering Magnetic Island versus the northern cluster or when comparing between reefs within the northern cluster. This result likely reflects the existence of gene flow (Fig. 3) between reefs in our study that reduces power of the Fst outlier approach to detect sites under selection.
Fig. 4. Distribution of genome-wide Fst (Magnetic Island versus northern cluster) and Tajima’s D for all loci (red) and regions with CLR > 100 (blue) in the northern reefs population.
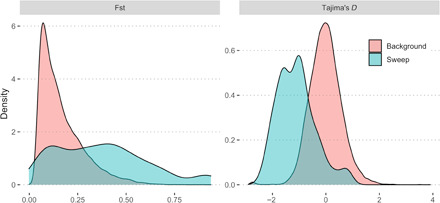
Horizontal axis shows the relevant statistical value (Fst or Tajima’s D) and vertical axis shows the relative abundance (as normalized density) of loci with the corresponding value.
Although SweepFinder 2 is designed to detect classical hard sweeps caused by positive selection, a similar signature can also be produced through persistent background selection (37). Since background selection involves removal of deleterious alleles, it should act most strongly on genes with highly conserved functions and is therefore more likely to act similarly in both Magnetic Island and northern populations. On the basis of this reasoning, 20 loci with low Fst values (<0.1) were removed to maintain our focus on genes under positive selection. Last, a further 7 loci with positive values of Tajima’s D were removed because these could represent genes under balancing selection (34).
Our final list of 71 regions affected by putative selective sweeps was spread throughout the genome (Fig. 5) and overlapped with 61 genes, of which 39 could be annotated based on homology or the presence of conserved domains. A complete list of sweep loci, along with associated genes and their functional groupings, is provided in Supplementary Information. Understanding the functional importance of these is challenging, since a high proportion of loci under selection involve genes that lack homologs in well-studied taxa. This is reflected in the results of Gene Ontology (GO) enrichment testing where we searched for GO terms over-represented in sweep genes using topGO (39). A single, relatively generic term (GO:0005509 calcium ion binding; P = 4.5 × 10−5) associated with epidermal growth factor (EGF) domain–containing genes and skeletal organic matrix proteins (SOMPs) was found to be enriched. While EGF domain–containing proteins are common in animal genomes, a specific class of these [here called EGF repeat proteins (EGFRs)] containing large tandem arrays of calcium-binding type EGF domains appeared to be under strong selection in A. tenuis. Two genes encoding EGFRs were associated with exceptionally strong sweep signals located adjacent to each other on a single scaffold of the A. tenuis genome. In addition, a further five EGFRs were located within 200 kb of these strong sweep signals (Fig. 5). Although the large tandem arrays of EGF domains in these proteins resemble those in the transforming growth factor–β binding proteins Fibrillin and LTBP, they lack the TB domains that characterize the latter class (40).
Fig. 5. Sites under selection.
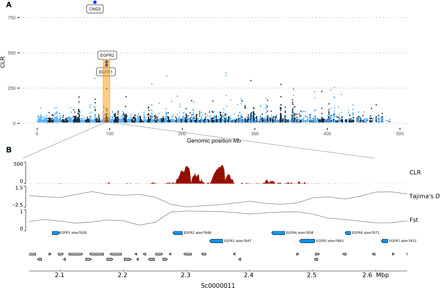
(A) Manhattan plot showing genome-wide distribution of the SweepFinder 2 CLR statistic. (B) Detailed view showing a region of the A. tenuis genome with evidence of strong selection on two genes encoding EGF repeats (EGFR1, EGFR2). These, and other EGF repeat-containing genes surrounding the sweep are numbered and shown in blue. All genes other than EGF repeat-containing genes are shown in gray.
DISCUSSION
The demographic modeling and population structure analysis presented here demonstrate that A. tenuis in the central inshore GBR comprises two populations that were genetically distinct (Fig. 2) before shelf flooding and are now experiencing a period of secondary contact (supported by the best-fitting dadi model). Signatures of selection detectable in one of these populations (northern cluster) implicate a diverse range of genes, but interpreting the adaptive function of these depends on the time frame (and associated environmental conditions) during which selection is thought to have occurred. We argue below that the most likely time frame corresponds to the period following the last glacial maximum and that the functional significance of sweeps is therefore best interpreted in the context of changes since that time. These changes include global shifts in temperature and atmospheric CO2 and local movement of populations from the paleo shelf-edge (17, 18) to the contemporary GBR inshore region where estuarine inputs and a large shallow-water shelf would have substantially affected water quality.
One of the characteristics of selective sweeps is that the signature of selection decays rapidly after fixation has occurred (41). Even under ideal conditions (no demographic change, classical hard sweep), the CLR has very little power to detect sweeps that originated more than 0.5 Ne generations ago and is best able to detect more recent sweeps (0.05 to 0.2 Ne generations ago) (37). Our best estimate of contemporary Ne for the northern population comes from dadi simulations (Fig. 3) and is around 10,000. If this is correct, it puts an upper bound on sweep ages at 25 ka ago with peak detectability between 2500 and 10 ka ago. This timing places the age of most sweeps during or after the period of global warming at the end of the last ice age. More recent changes, such as increases in sediment and nutrient flux since European settlement (42), could also produce signatures of selection but our data lack the resolution required to make inferences about this.
Global warming between 20 and 6 ka ago involved a temperature rise of over 3°C, an increase in atmospheric CO2 from 200 to 260 parts per million and over 100 m of sea level rise (18). In addition to these global changes, an especially rapid period of environmental change on the GBR occurred between 14 and 6 ka ago when sea levels were high enough to flood large regions of what is now the inshore shelf. This created coral habitat in the inshore region and increased sediment flux contributing to the death of some shelf-edge reefs during the period of sea level rise (18). Corals colonizing inshore shelf regions would likely have been subject to additional selective pressures arising from higher turbidity (18), regular influx of riverine sediment and nutrients (43, 44), and occasional periods of reduced salinity during severe flooding events (19, 20).
Major functional categories of genes implicated in selective sweeps in this study are consistent with the idea that modern inshore corals have experienced very different water quality conditions than their shelf-edge ancestors. Salinity is a crucial component of water quality especially in inshore environments where hypo-saline conditions can pose an acute and potentially lethal physiological challenge to corals. A recent gene expression study found that genes involved in solute transport, amino-acid metabolism, and protein recycling were major components of the response to osmotic stress in A. millepora (45). Many of these genes were implicated in selective sweeps in inshore corals in our study including homologs of the calcium and potassium ion transporters SERCA, CNG-3, CNGK1, TRPCG and GAT-3, EHD2 that regulates vesicle transport, and lysosomal α-mannosidase (Laman) that is involved in glycoprotein recycling. Notably, three of these (SERCA, GAT-3, and Laman) were very close homologs (97, 82, and 97% identity, respectively) to proteins differentially expressed in response to osmotic stress in A. millepora (45).
Genes potentially involved in skeletal growth and development comprised a high proportion (10 of 39) of those implicated in selective sweeps. Although the underlying mechanisms are not well understood, skeletal growth rates and morphology seem to be linked to water quality in A. tenuis. Marked differences between inshore sites have been observed along a water quality gradient (21), and a transplant experiment showed marked skeleton loss in colonies that were moved from a plume (Magnetic Island) to a marine (Pelorus Island) location (46). Related genes under positive selection in the northern population included 4 of the 36 SOMPs originally identified through proteomic analysis of A. millepora skeletal materials (47) and possibly EGF repeat proteins. EGF repeat proteins were associated with very strong signals of selection but their functional importance remains unclear. Proteins with various numbers of EGF-like domains have been reported as part of the skeletal organic matrix of both A. millepora (47) and A. digitifera (48), but developmental gene-expression data suggest that they may have a more general role in cell adhesion, which then leads to their incorporation in the skeletal organic matrix (48). Another potential role for these proteins is in the establishment and maintenance of symbiosis, with separate studies on Hydra (49) and corallimorpharians (50) both reporting increased expression in the symbiont-infected state compared to aposymbiotic controls.
Structuring of symbiont genotypes according to local conditions on individual reefs and possibly to water quality (Fig. 2C) suggests that the ability to efficiently interact with specific symbionts may represent a key mode of adaptation of the coral holobiont. In support of this, we observed five genes associated with selective sweeps in the host genome that were implicated in the establishment and/or maintenance of symbiosis. All of these were homologous to genes observed to be differentially expressed in symbiotic versus aposymbiotic states in A. digitifera larvae (51), adult corallimorpharians (50), or Aiptasia (52), an anemone model for symbiosis. This group included genes involved in the cytoskeletal remodeling events required for symbiosome formation (MARE3 and CO1A2) and apoptotic regulators (A3Ar and S30BP).
Our results highlight two potential modes of adaptation for A. tenuis holobionts on the inshore GBR. Host genomes across the study area showed signatures of selection on a diverse range of genes while notable differences at a local scale were found in the photosymbiont association. The time frame for detection of signatures of selection in the host places them during a period of post-glacial climate change; however, it was not possible to specifically distinguish between changes related to inshore conditions and broader changes such as temperature or CO2 increase. Additional sampling in offshore locations could potentially resolve this question by providing an outgroup that has experienced the former (temperature/CO2 increase) without experiencing the latter (inshore conditions). Genetic structure in dominant photosymbionts seemed to be highly specific (single dominant mitochondrial haplotype) at some reefs and much less so (multiple divergent types) at others. While our study points toward water quality as a driver of these local changes, additional sampling locations coupled with more detailed long-term environmental data are required to understand the ecological basis for this genetic pattern.
The availability of a high-quality reference genome for A. tenuis and the use of whole-genome resequencing were crucial to our ability to confidently detect genomic regions under selection and to infer demographic history for the host. Although this approach has rarely been used so far in coral population genetics [although see (27, 53)], the moderate size of many coral genomes (~500 Mb) and continually falling sequencing costs means that whole-genome sequencing is now cost competitive with reduced representation approaches for population genomics. Our study demonstrates that the dense allele frequency information afforded by whole-genome approaches enables powerful inferences even with relatively shallow sequencing coverage per genome. As high-quality genome assemblies become available for a broader range of corals and their symbionts, we expect that this and related approaches will become a key tool in understanding the interaction between past climate change and the evolution of corals and coral reefs.
METHODS
Reference genome sequencing, assembly, and annotation
All genome sequencing was based on DNA extracted from sperm collected from a single A. tenuis colony collected on 10th of December 2014 from Orpheus Island. The species examined in this paper is widely referred to as A. tenuis on the GBR; however, we note that the taxonomy of this species is likely to change in light of recent molecular phylogenetic research indicating that traditional morphological taxonomy does not accurately reflect species boundaries or evolutionary relationships (54). The identity of the species is currently under examination using an integrated taxonomic approach, with preliminary analysis suggesting the species is most likely not A. tenuis (Dana 1846) but A. kenti (Brook 1892), a species described from the Torres Strait but synonymized with A. tenuis (Dana 1848) by Veron and Wallace (1984). Field images and a voucher specimen of the colony used in this study have been deposited in the Queensland Museum (QM registration number G335181). After taxonomic revisions are complete, this specimen will be compared to the holotypes of both A. kenti (Brook 1892) and A. tenuis (Dana 1848) and topotypes of specimens from both Fiji and the GBR to determine its actual identity.
An initial round of sequencing was performed on an Illumina HiSeq 2500 instrument using sheared and size selected DNA [~450 base pairs (bp)] to generate 88.6 million 250-bp paired-end reads. Further sequencing was then performed on a PacBio RSI instrument by Ramaciotti Center for genomics using 62 SMART Cells resulting in a total of 50.6 Gb of long read data (~108× genome coverage) with read N50 of 15Kb. In addition, to facilitate gene annotation, RNA was extracted from tissues collected from the same colony and sequenced on an illumina HiSeq 2500 instrument. All genome sequencing data are available under ENA accession PRJEB23295.
A draft reference genome was assembled purely from PacBio reads using the FALCON (version 0.7.3) assembler (55) followed by separation of haplotigs from the primary assembly with FALCON-unzip and error correction with Quiver (56). The resulting primary assembly was highly contiguous (N50 ~ 1320 Kb) but had a larger total size (629 Mb) than the haploid size of 470 Mb estimated by kmer analysis of Illumina data with sga.preqc (24). Mapping of transcript sequences to the draft genome assembly revealed the presence of numerous alternate haplotypes in the primary assembly that most likely accounts for the inflated assembly size. To address this issue, we used Haplomerger 2 to identify and merge alternate haplotypes. Last, small redundant scaffolds less than 2 kb in length were identified by BLAST and removed. This resulted in a final merged assembly with a primary size of 487 Mb and N50 of 2837 Kb. As any unmerged haplotypes that might remain should be located entirely on the shortest contigs, we used only the largest contigs (>1 Mb) for population genomic analyses. These large contigs accounted for 81% of the estimated genome size.
Protein-coding gene annotation was performed as previously described (57). Briefly, de novo and genome-guided transcriptome assembly was carried out using Trinity (58), followed by PSyTrans (https://github.com/sylvainforet/psytrans) to remove Symbiodinium transcripts. Transcripts were then assembled to the genome assembly using PASA (59), from which a set of likely ORFs were generated. On the basis of their protein coding ability and completeness, these ORFs were carefully assessed to produce a highly confident and nonredundant training gene set. This was used to train AUGUSTUS (60) and SNAP (61), and the resulting parameters were used by the corresponding program in the MAKER2 pipeline (62), from which an ab initio gene model was predicted. Last, putative transposable elements in the gene models were excluded based on transposonPSI (http://transposonpsi.sourceforge.net) and hhblits (63) search to transposon databases.
Functional annotation of gene models was performed as follows. For each gene in the A. tenuis genome, homologs with high quality functional annotations were identified using BLAST searches (E-value threshold of 1 × 10−5) of predicted proteins (BLASTp) and predicted transcripts (BLASTx) against the SwissProt (June 2018) database. Genes were also annotated by searching for conserved domains using InterProScan (64) (version 5.36-75). In addition to finding conserved domains, InterProScan results were also used to infer GO based on the conserved domains present.
Synteny analysis
To infer macro-syntenic linkages, one-to-one mutual best hit orthologs were computed against N. vectensis proteome (see section on orthology analysis for the list of all proteomes) using BLASTP. In total, 7053 orthologous groups containing representatives from all seven species were identified. Only scaffolds/chromosomes containing 10 or more such orthologous groups were considered. Macro-synteny plots were generated using a custom R script, where the synteny between A. millepora chromosomes to A. tenuis and N. vectensis scaffolds is represented by a dot for each orthologous gene family. The clustering of scaffolds for A. tenuis and N. vectensis was done by taking and sorting by the median position of the orthologous genes on A. millepora chromosomes.
Orthology analysis
Orthofinder (65) was used to search for orthologous groups of genes shared between gene sets from the following cnidarian genomes, A. digitifera (Genbank GCF_000222465.1), A. tenuis (http://aten.reefgenomics.org/), A. millepora (26), Fungia sp. (http://ffun.reefgenomics.org/), Galaxea fascicularis (http://gfas.reefgenomics.org/), N. vectensis (Genbank GCF_000209225.1), and Stylophora pistilata (Genbank GCF_002571385.1).
Sampling design and rationale for population genomics
Water quality is a key factor influencing the health of inshore coral reefs and is subject to intensive monitoring on the GBR (43). To capture adaptation in the A. tenuis holobiont related to water quality, we chose reef sites at contrasting ends of two water-quality gradients both of which are well characterized in terms of exposure to riverine flood plumes (19, 43). The southern gradient is primarily influenced by secondary plume waters from the Burdekin river that affects reefs to a diminishing extent heading northwards. Predicted numbers of wet-season days exposed are Magnetic Island >67%, Pandora Reef 33 to 67%, and Pelorus Island <33% (43). A similar trend is also seen in the number of occurrences of low salinity events (<30) between 2003 and 2010 which were Magnetic Island (8 of 8), Pandora Reef (6), and Pelorus Island (2). The northern gradient captures the wet tropics region where multiple rivers influence water quality. Here, the plume location, Dunk Island is highly exposed (>67% of wet-season days; 8 of 8 low salinity events between 2003 and 2010) to plume waters from the Tully and to a lesser extent the Herbert rivers (19), while Fitzroy Island receives flood waters from the Johnstone and Russell-Mulgrave river catchments much less often (lower end of 33 to 67% of wet-season days; 1 of 8 occurrence of low salinity between 2003 and 2010). The current location of rivers and coastal topology of these sites would have been greatly affected by sea level changes during the Holocene. Maximum rates of change (by flooded area) occurred between 10 and 14 ka ago (17), whereas much smaller sea level changes have occurred during the past 6 ka ago (decline by 1 to 1.5 m).
Sample collection, sequencing, and variant calling
Samples were collected in early 2015 from a total of 267 adult corals taken from five inshore locations on the central GBR (see Fig. 2) including Magnetic Island and four northern locations, Fitzroy Island, Dunk Island, Pandora Reef, and Pelorus Island. These samples were subjected to preliminary sequencing with DArTseq, and analysis of this data was used to identify a total of 43 likely clones and 8 likely species misidentifications. After excluding these samples, a total of 30 individuals from each site were randomly selected for whole genome sequencing. Samples were taken by cutting a branch and then placing it in ethanol for storage. Most samples were collected on a voyage of the RV Cape Ferguson in February [Fitzroy Island, 20 February 2015; Dunk Island, 21 February 2015; Pandora Reef, 22 February 2015; and Magnetic Island (Geoffrey Bay), 23 February 2015]. Half of the samples from Pelorus Island were collected on 22 February 2015 with the remaining 15 samples collected from nearby Orpheus Island (Pioneer Bay) on 24 February 2015. Sequencing libraries were prepared separately for each sample and then used to generate four pools for sequencing across four flow cells of a HiSeq 2500 instrument (Ramaciotti Center for Genomics) to obtain a total of 1.2 billion 100-bp paired-end reads. With the exception of two samples that were chosen for high (~20×) coverage sequencing (one from Fitzroy Island and one from Magnetic Island), libraries were pooled evenly so as to obtain approximately 3× coverage per sample. All population sequencing data are available under ENA study accession number PRJEB37470.
Raw reads for all samples were preprocessed and mapped against the A. tenuis genome using the GATK (Genome Analysis Toolkit) best-practices workflow as follows. Reads passing quality checks were converted to unmapped bam files with sample, lane, and flowcell tags added as appropriate. Adapters were marked using Picard (v2.2.1; http://broadinstitute.github.io/picard), mapping was performed using bwa mem [v0.7.13; (66)], and polymerase chain reaction duplicates were marked using Picard (v2.2.1). Mapping rates (proportion of reads mapping to at least one host genomic location) were at least 86% for all samples, with more than 85% of samples having rates higher than 92% (fig. S7). Mapping rates were slightly lower on average for samples from Magnetic Island, most likely reflecting the fact that the reference genome is based on an individual from the northern cluster (Pelorus Island). Variants were then called using Freebayes [v1.0.2-16; (67)] with haplotype calling turned off and ignoring reads with mapping quality less than 30 or sites with read quality less than 20. The two higher (~20×) coverage samples were downsampled to 3× using samtools to ensure parity with other samples before variant calling.
To ensure that only high-quality variant sites and high-quality genotype calls were used for population genomic analyses, we performed the following basic filtering steps on raw variant calls from Freebayes. In cases where additional filtering was required, it is described under the relevant analysis section. Sites with Phred scaled site quality scores less than 30 were removed. Only biallelic sites were retained. Sites where the mean read depth per individual was less than one third or greater than twice the genome-wide average (2.7×) after mapping were removed. Genotypes with Phred scaled genotype quality score less than 20 were set to missing, and sites with more than 50% missing genotypes were removed. Sites with highly unbalanced allele read counts (Freebayes allele balance Phred scaled probability of >20) were removed. The mdust program was used to mark low complexity regions, and sites within these were removed. SNP sites within 10 bp of an indel were removed. Indel sites with length greater than 50 bp were removed.
Population structure
To determine whether samples could be divided into genetically distinct clusters, we used the program PCAngsd (0.973) (68) that has been designed specifically to infer population structure and admixture from low-coverage data. As input to PCAngsd, we used genotype likelihoods at all variant sites after quality filtering (see above) and converted these to Beagle format with ANGSD (0.920) (69). Individual samples were plotted as a function of the top two principal components based on an eigendecomposition of the genotype relationship matrix calculated by PCAngsd (fig. S2). This revealed two distinct clusters corresponding to the Magnetic Island and northern populations that is consistent with the optimal cluster number of 2, also inferred by PCAngsd. Admixture proportions for each sample are shown in Fig. 2 and show nearly complete assignment to either northern or Magnetic Island populations for most samples. Three samples (MI-1-1, PI-1-16, and DI-2-4) showed mixed ancestry and were likely to be recent North/Magnetic Island hybrids, while principal components analysis revealed two other samples (MI-2-9 and MI-1-16) from Magnetic Island that did not appear to belong to either population cluster. We excluded all potential hybrids and these two outlying individuals from selective sweep and population demographic analyses. Outlying individuals were excluded on the basis that they could represent migrants from a third (poorly sampled) population or species misidentifications. Hybrids were excluded to avoid potential bias resulting from assignment to one of the two source populations.
Demographic history with PSMC′
Changes in effective population size through time were inferred on the basis of the genome-wide distribution of heterozygous sites in two individual colonies sequenced to high coverage (Fitzroy Island FI-1-3, 18×; Magnetic Island MI-1-4, 20×) using PSMC′ implemented in the software MSMC (v2.1.2) (70). To avoid biases due to differences in callability of heterozygous sites across the genome, we only used these two high coverage samples and also used the snpable (http://lh3lh3.users.sourceforge.net/snpable.shtml) program to mask all regions of the genome where it was not possible to obtain unambiguous read mappings. Variant calling was performed for samples sequenced to high (~20×) coverage using samtools (1.7) and bcftools (1.9), as these generate outputs in the format required by the bamCaller python script included as part of the msmc-tools package (https://github.com/stschiff/msmc-tools). This script supplements the callability mask (see above) with an additional mask to avoid regions where read coverage is too low (less than half the genome-wide mean) or too high (double the mean), indicating a problematic region such as a collapsed repeat. As a final quality control measure, only scaffolds larger than 1 Mb (~80% of the genome) were included in MSMC analyses.
A distribution of MSMC estimates was obtained by generating 100 bootstrap runs, each of which involved recombining a random subsampling of 500-kb-sized chunks to produce 20 large scaffolds of length 10 Mb. Raw outputs from MSMC were converted to real values using a mutation rate of 1.86 × 10−8 events per base per generation and a generation time of 5 years that is consistent with the fast growth rate and relatively high turnover of Acroporid corals.
The neutral mutation rate used in the calculation above was estimated by exploiting the equivalence between mutation and substitution rates for well-separated lineages. The well-studied congeneric species A. digitifera was chosen for this comparison because its divergence time from A. tenuis has been estimated at 15 Ma (22) on the basis of fossil-calibrated phylogenomic analyses. This value is sufficient to allow near complete lineage separation but small enough that the number of multiply mutated sites should be small. First, 5583 one-to-one orthologs shared by A. digitifera, A. tenuis, and A. millepora were identified using ProteinOrtho [v5.16b (71)]. Amino acid sequences for A. tenuis and A. digitifera for these orthologs were then aligned with MAFFT [7.407 (72)] and converted to codon alignments using pal2nal [v14 (73)]. Synonymous substitution rates, dS was estimated for each ortholog pair using codeml from the PAML suite [4.9i (74)]. Since dS is a ratio, its values follow a log-normal distribution, and we therefore chose the central value as the mean of log(dS). Last, the neutral mutation rate, μ was estimated using a divergence time (T) of 15.5 Ma ago, a generation time (G) of 5 years, and the formula μ = G × dS/(2 T). The generation time of 5 years is based on an age of first reproduction of 3 years (75) combined with high growth rate and mechanical vulnerability in large colonies.
Demographic history with dadi
Historical population sizes inferred under PSMC′ are made under the assumption of a single population without migration and have limited power to resolve changes in the recent past. To address these issues, we used the dadi framework (76) to fit a variety of more complex demographic models (table S2) to the joint SFS between Magnetic Island and northern populations. A total of six demographic models were chosen to explore the timing and completeness of separation, population size changes, and possible secondary contact between the northern and Magnetic Island populations. To explore the relationship between model fit and complexity, we included models ranging from a very simple island model without migration (3 parameters) to a complex model allowing for population sizes before and after population split and asymmetric migration (10 parameters).
To prepare inputs for dadi, we created a python script, vcf2dadi (https://github.com/iracooke/atenuis_wgs_pub/blob/master/bin/vcf2dadi.py), to calculate allele frequencies at each site based on observed read counts for each allele encoded in the Freebayes generated vcf file. The script rejects sites with fewer than 50% called genotypes and calculates allele frequencies by using the ratio of total observations for each allele to infer the number of alleles of each type present among called samples. This method takes advantage of the high overall read depth across all samples to accurately calculate allele frequencies for each population without using potentially biased low-coverage genotype calls. Before running the vcf2dadi script, further filtering was performed on the vcf file to remove sites with minor allele count less than 2 and to thin sites to a physical separation distance of at least 1000 bp. This produced a final dataset consisting of 266k SNPs in dadi format. These were imported to dadi and converted to a folded 2D SFS and projected down from the original sample size of 56 × 240 alleles to 45 × 180. This resulted in a final dataset that included 15.5k segregating sites.
All dadi models were optimized in a hierarchical manner to minimize the possibility of obtaining best-fit parameters corresponding to a local optimum. All models used generic starting parameters to begin 10 independent runs, where each run involved four successive rounds with the best-fit parameters from the previous round undergoing perturbation before being used as input for the next round.
Signatures of selection
A genome-wide search for selective sweeps was performed using SweepFinder 2 (37) that calculates a test statistic for each site in the genome that reflects the likelihood of observing SNP data under the assumption of a sweep compared with a null model where no sweep has occurred. To prepare inputs for SweepFinder 2, we used the same vcf2dadi script described under the section “demographic history with dadi,” with an option to generate outputs in SweepFinder format. The site frequencies generated by this script were then used to generate a combined frequency spectrum across the entire genome that SweepFinder 2 uses as an empirical neutral model. Each population was then scanned for sweeps by calculating the SweepFinder test statistic on a genome-wide grid every 1000 bp. To avoid treating adjacent loci as independent sweeps, we then ran another python script sf2gff.py (https://github.com/iracooke/atenuis_wgs_pub/blob/master/bin/sf2gff.py) that identifies contiguous sweep regions and converts them to gff format. In this script, a sweep region is deemed to start when the SweepFinder statistic rises above a value of 10 and ends when it falls below this value. Within the region, the maximum value of the test statistic is taken to represent the strength of the sweep. This resulted in 5315 putative sweeps that were then subjected to significance testing (see below).
Since the SweepFinder test statistic is a composite likelihood, it requires an appropriate null model to determine a significance threshold. To address this problem and also to control for potential false positives arising from demographic factors, we used the program, ms (77) to generate simulated allele frequency data for all demographic scenarios modeled using dadi and the demographic history inferred by MSMC analyses. Neutral model data simulated with ms was subjected to SweepFinder 2 analysis to calculate a null distribution of the CLR statistic for each demographic model. At a given threshold value, T, of the CLR, it is then possible to calculate the false discovery rate as the ratio of the number of loci with (CLR > T) in the neutral model versus the Magnetic Island or northern population data, respectively. For the northern population, a threshold CLR value of 100 or greater resulted in a false discovery rate of less than 10% under any of the demographic models except for “sym_mig_size,” which was not the best-fitting model and predicted a deep bottleneck followed by population expansion. For Magnetic Island, no such threshold could be found indicating that the influence of neutral factors was too large to allow genuine sweeps to be detected. For this reason, we did not proceed further with SweepFinder analyses using the Magnetic Island data. For the northern population, all sweeps with a CLR value greater than a conservative significance threshold of 100 were identified, and BEDTools [2.7.1 (78)] was used to find genes that lay within 1000 bp of these.
In addition to searching for regions under selection with SweepFinder 2, we also used Bayescan (38) (version 2.1) to look for regions of high differentiation between populations (Fst outliers) as indicators of positive selection. This was performed separately for two sets of samples; first, between all four northern reefs, treating each reef as a separate sub-population, and second, between northern reefs as a whole and Magnetic Island. This separation between analyses was necessary because Bayescan is not designed to deal with hierarchical population structure.
To ensure that only independent loci were provided to Bayescan 2, we first thinned data to ensure a physical distance of at least 10 kb. This resulted in a total of 27,109 sites available for analysis across all populations. The same allele frequency data used for SweepFinder 2 analyses was then converted to Bayescan format using an awk script. Bayescan was run with prior odds set to 1000 and a burn-in length of 200,000 for both analyses. Convergence was confirmed with the Geweke diagnostic implemented in the R package coda. Statistical analysis to search for outlier loci at a false discovery rate of 20% was performed using the plot_bayescan function distributed with Bayescan.
Folded versus unfolded frequency spectra
We used the folded SFS for both dadi and SweepFinder 2 analyses. Although both methods can benefit from provision of an unfolded SFS, this requires an accurate determination of the ancestral state for each allele. This is a particular challenge for A. tenuis because other Acropora species that could potentially be used as an outgroup (A. digitifera and A. millepora) are separated by a relatively large evolutionary distance [15 Ma (22)]. With these distantly related outgroup taxa, there is a high likelihood that the outgroup sequence will not represent the ancestral state but will instead capture independently fixed mutations in each lineage. We therefore decided to conduct all analyses with folded SFS in this study, which reduces power but avoids the risk of false inferences because of incorrect ancestral assignment.
Population genetic statistics, Fst, and Tajima’s D
To avoid biases because of inaccurate genotype calls in low-coverage data, we calculated Fst and Tajima’s D using ANGSD (69) (version 0.938) that calculates these parameters without calling genotypes. All calculations were performed on the basis of the 14 million variant sites identified after quality filtering on Freebayes calls (see section on variant calling).
Mitochondrial genomes
Host consensus mitochondrial genome sequences were obtained for each sample as follows. Reads were mapped against the published A. tenuis mitochondrial genome sequence (Genbank accession AF338425) using bwa mem (v0.7.17). Mapped mitochondrial reads for all samples constituted at least 25× coverage and were used to call a consensus sequence for each sample using bcftools (v1.9). Full-length consensus mitochondrial genome sequences for all samples were used to build a TCS type haplotype network and visualized using the software, PopArt (79) (v1.7).
Mitochondrial genome sequences for photosymbionts were obtained in a similar way. Reads previously identified as not mapping to the host genome were mapped against the Cladocopium goreaui mitochondrial sequences available from the reefgenomics website (http://symbs.reefgenomics.org/download/). Mapped reads were then used to call a consensus sequence using bcftools in the same manner as for host mitogenomes. Overall mitogenome coverage was much lower and more variable for symbiont genomes than for host genomes reflecting a larger mitogenome sequence (55 kb) and reduced number of reads. We therefore removed samples with fewer than 2500 reads (~5× coverage) to avoid obtaining consensus calls from very low coverage. The resulting mitogenome sequences from 107 remaining samples were loaded into Geneious Prime (2019.2.3), and only sites with no ambiguous bases for any sample were retained. The resulting 6170 bp segment was used to construct a TC haplotype network shown in Fig. 2 using PopArt (79) (v1.7).
Supplementary Material
Acknowledgments
We would like to thank A. Bellantuono, C. Granados-Cifuentes, and K. Dougan for early access to the Durusdinium trenchii genome. AIMS staff J. Davidson, P. Costello, and K. Morrow are thanked for the help in coral collections and L. Peplow for the help with DNA extractions. G. Torda collected the colony used for genome assembly and T. Bridge provided advice on taxonomy. Funding: This project was supported by a Queensland Government DSITIA Accelerate Partnerships award to the University of Queensland on behalf of the Australian Institute of Marine Science (AIMS), the Australian National University, Bioplatforms Australia, the Great Barrier Reef Foundation, the Great Barrier Reef Marine Park Authority, and James Cook University (2014). This research/project was undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), which is supported by the Australian Government. The data used in this project were funded by the Great Barrier Reef Foundation’s Resilient Coral Reefs Successfully Adapting to Climate Change research and development program in collaboration with the Australian Government, Bioplatforms Australia through the National Collaborative Research Infrastructure Strategy (NCRIS), Rio Tinto, and a family foundation. Genome sequencing was supported by the Reef Future Genomics (ReFuGe) 2020 Consortium organized by the Great Barrier Reef Foundation. Author contributions: I.C., S.F., P.B., D.G.B., M.J.H.v.O., M.A.R., and D.J.M. designed the study. I.C. wrote the manuscript with assistance from D.J.M. and feedback from all authors. H.Y., S.F., O.S., and J.Z. assembled the genome and performed comparative genomic analysis. I.C. performed host population genomic analyses with assistance from P.B., J.M.S., and M.A.F. Symbiont population genetic analysis was performed by I.C. using genomic resources provided by M.R.-L. and with assistance from P.B. Samples were collected and prepared for sequencing by S.C.B. and D.G.B. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The genome assembly and gene annotations for A. tenuis are available for download at http://aten.reefgenomics.org/. Sequencing data used for genome assembly are available on the European Nucleotide Archive, ENA accession PRJEB23295. Population genomic resequencing data are available under ENA accession PRJEB37470. Detailed methods including all scripts and commands used to perform population genomic analyses are provided at https://github.com/iracooke/atenuis_wgs_pub. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/48/eabc6318/DC1
REFERENCES AND NOTES
- 1.Hughes T. P., Barnes M. L., Bellwood D. R., Cinner J. E., Cumming G. S., Jackson J. B. C., Kleypas J., van de Leemput I. A., Lough J. M., Morrison T. H., Palumbi S. R., van Nes E. H., Scheffer M., Coral reefs in the Anthropocene. Nature 546, 82–90 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Doney S. C., Ruckelshaus M., Duffy J. E., Barry J. P., Chan F., English C. A., Galindo H. M., Grebmeier J. M., Hollowed A. B., Knowlton N., Polovina J., Rabalais N. N., Sydeman W. J., Talley L. D., Climate change impacts on marine ecosystems. Ann. Rev. Mar. Sci. 4, 11–37 (2012). [DOI] [PubMed] [Google Scholar]
- 3.LaJeunesse T. C., Parkinson J. E., Gabrielson P. W., Jeong H. J., Reimer J. D., Voolstra C. R., Santos S. R., Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral endosymbionts. Curr. Biol. 28, 2570–2580.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Hughes T. P., Kerry J. T., Álvarez-Noriega M., Álvarez-Romero J. G., Anderson K. D., Baird A. H., Babcock R. C., Beger M., Bellwood D. R., Berkelmans R., Bridge T. C., Butler I. R., Byrne M., Cantin N. E., Comeau S., Connolly S. R., Cumming G. S., Dalton S. J., Diaz-Pulido G., Eakin C. M., Figueira W. F., Gilmour J. P., Harrison H. B., Heron S. F., Hoey A. S., Hobbs J.-P. A., Hoogenboom M. O., Kennedy E. V., Kuo C.-Y., Lough J. M., Lowe R. J., Liu G., McCulloch M. T., Malcolm H. A., McWilliam M. J., Pandolfi J. M., Pears R. J., Pratchett M. S., Schoepf V., Simpson T., Skirving W. J., Sommer B., Torda G., Wachenfeld D. R., Willis B. L., Wilson S. K., Global warming and recurrent mass bleaching of corals. Nature 543, 373–377 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Robbins S. J., Singleton C. M., Chan C. X., Messer L. F., Geers A. U., Ying H., Baker A., Bell S. C., Morrow K. M., Ragan M. A., Miller D. J., Forêt S.; ReFuGe2020 Consortium, Voolstra C. R., Tyson G. W., Bourne D. G., A genomic view of the reef-building coral Porites lutea and its microbial symbionts. Nat. Microbiol. 4, 2090–2100 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Matz M. V., Treml E. A., Aglyamova G. V., Bay L. K., Potential and limits for rapid genetic adaptation to warming in a Great Barrier Reef coral. PLOS Genet. 14, e1007220 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anthony K., Bay L. K., Costanza R., Firn J., Gunn J., Harrison P., Heyward A., Lundgren P., Mead D., Moore T., Mumby P. J., van Oppen M. J. H., Robertson J., Runge M. C., Suggett D. J., Schaffelke B., Wachenfeld D., Walshe T., New interventions are needed to save coral reefs. Nat. Ecol. Evol. 1, 1420–1422 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Schoepf V., Carrion S. A., Pfeifer S. M., Naugle M., Dugal L., Bruyn J., McCulloch M. T., Stress-resistant corals may not acclimatize to ocean warming but maintain heat tolerance under cooler temperatures. Nat. Commun. 10, 4031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kenkel C. D., Matz M. V., Gene expression plasticity as a mechanism of coral adaptation to a variable environment. Nat. Ecol. Evol. 1, 14 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Bay R. A., Palumbi S. R., Multilocus adaptation associated with heat resistance in reef-building corals. Curr. Biol. 24, 2952–2956 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Dixon G. B., Davies S. W., Aglyamova G. A., Meyer E., Bay L. K., Matz M. V., Genomic determinants of coral heat tolerance across latitudes. Science 348, 1460–1462 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Lukoschek V., Riginos C., van Oppen M. J. H., Congruent patterns of connectivity can inform management for broadcast spawning corals on the Great Barrier Reef. Mol. Ecol. 25, 3065–3080 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Humanes A., Noonan S. H. C., Willis B. L., Fabricius K. E., Negri A. P., Cumulative effects of nutrient enrichment and elevated temperature compromise the early life history stages of the coral Acropora tenuis. PLOS ONE 11, e0161616 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ulstrup K. E., Van Oppen M. J. H., Geographic and habitat partitioning of genetically distinct zooxanthellae (Symbiodinium) in Acropora corals on the Great Barrier Reef. Mol. Ecol. 12, 3477–3484 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Quigley K. M., Bay L. K., van Oppen M. J. H., The active spread of adaptive variation for reef resilience. Ecol. Evol. 9, 11122–11135 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chakravarti L. J., Beltran V. H., van Oppen M. J. H., Rapid thermal adaptation in photosymbionts of reef-building corals. Glob. Chang. Biol. 23, 4675–4688 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Hinestrosa G., Webster J. M., Beaman R. J., Spatio-temporal patterns in the postglacial flooding of the Great Barrier Reef shelf, Australia. Cont. Shelf Res. 173, 13–26 (2019). [Google Scholar]
- 18.Webster J. M., Braga J. C., Humblet M., Potts D. C., Iryu Y., Yokoyama Y., Fujita K., Bourillot R., Esat T. M., Fallon S., Thompson W. G., Thomas A. L., Kan H., McGregor H. V., Hinestrosa G., Obrochta S. P., Lougheed B. C., Response of the Great Barrier Reef to sea-level and environmental changes over the past 30,000 years. Nat. Geosci. 11, 426–432 (2018). [Google Scholar]
- 19.Devlin M., Schaffelke B., Spatial extent of riverine flood plumes and exposure of marine ecosystems in the Tully coastal region, Great Barrier Reef. Mar. Freshwater Res. 60, 1109–1122 (2009). [Google Scholar]
- 20.Schroeder T., Devlin M. J., Brando V. E., Dekker A. G., Brodie J. E., Clementson L. A., McKinna L., Inter-annual variability of wet season freshwater plume extent into the Great Barrier Reef lagoon based on satellite coastal ocean colour observations. Mar. Pollut. Bull. 65, 210–223 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Rocker M. M., Francis D. S., Fabricius K. E., Willis B. L., Bay L. K., Variation in the health and biochemical condition of the coral Acropora tenuis along two water quality gradients on the Great Barrier Reef, Australia. Mar. Pollut. Bull. 119, 106–119 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Mao Y., Economo E. P., Satoh N., The roles of introgression and climate change in the rise to dominance of Acropora corals. Curr. Biol. 28, 3373–3382.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Nielsen R., Hellmann I., Hubisz M., Bustamante C., Clark A. G., Recent and ongoing selection in the human genome. Nat. Rev. Genet. 8, 857–868 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simpson J. T., Exploring genome characteristics and sequence quality without a reference. Bioinformatics 30, 1228–1235 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shinzato C., Shoguchi E., Kawashima T., Hamada M., Hisata K., Tanaka M., Fujie M., Fujiwara M., Koyanagi R., Ikuta T., Fujiyama A., Miller D. J., Satoh N., Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 476, 320–323 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Ying H., Hayward D. C., Cooke I., Wang W., Moya A., Siemering K. R., Sprungala S., Ball E. E., Forêt S., Miller D. J., The whole-genome sequence of the coral Acropora millepora. Genome Biol. Evol. 11, 1374–1379 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuller Z. L., Mocellin V. J. L., Morris L. A., Cantin N., Shepherd J., Sarre L., Peng J., Liao Y., Pickrell J., Andolfatto P., Matz M., Bay L. K., Przeworski M., Population genetics of the coral Acropora millepora: Toward genomic prediction of bleaching. Science 369, eaba4674 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Zapata F., Goetz F. E., Smith S. A., Howison M., Siebert S., Church S. H., Sanders S. M., Ames C. L., McFadden C. S., France S. C., Daly M., Collins A. G., Haddock S. H. D., Dunn C. W., Cartwright P., Phylogenomic analyses support traditional relationships within cnidaria. PLOS ONE 10, e0139068 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Devlin M. J., Petus C., Da Silva E., Tracey D., Wolff N. H., Waterhouse J., Brodie J., Water quality and river plume monitoring in the Great Barrier Reef: An overview of methods based on ocean colour satellite data. Remote Sens. (Basel) 7, 12909–12941 (2015). [Google Scholar]
- 30.Shearer T. L., Van Oppen M. J. H., Romano S. L., Wörheide G., Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol. Ecol. 11, 2475–2487 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Wood D. E., Salzberg S. L., Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prada C., Hanna B., Budd A. F., Woodley C. M., Schmutz J., Grimwood J., Iglesias-Prieto R., Pandolfi J. M., Levitan D., Johnson K. G., Knowlton N., Kitano H., DeGiorgio M., Medina M., Empty niches after extinctions increase population sizes of modern corals. Curr. Biol. 26, 3190–3194 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Mazet O., Rodríguez W., Grusea S., Boitard S., Chikhi L., On the importance of being structured: Instantaneous coalescence rates and human evolution–lessons for ancestral population size inference? Heredity 116, 362–371 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maruyama T., Fuerst P. A., Population bottlenecks and nonequilibrium models in population genetics. II. Number of alleles in a small population that was formed by a recent bottleneck. Genetics 111, 675–689 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.C. Thomas, thesis, Ecole Polytechnique de Louvain (2015). [Google Scholar]
- 36.Riginos C., Hock K., Matias A. M., Mumby P. J., van Oppen M. J. H., Lukoschek V., Asymmetric dispersal is a critical element of concordance between biophysical dispersal models and spatial genetic structure in Great Barrier Reef corals. Divers. Distrib. 25, 1684–1696 (2019). [Google Scholar]
- 37.Huber C. D., DeGiorgio M., Hellmann I., Nielsen R., Detecting recent selective sweeps while controlling for mutation rate and background selection. Mol. Ecol. 25, 142–156 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foll M., Gaggiotti O., A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 180, 977–993 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexa A., Rahnenführer J., Lengauer T., Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Robertson I., Jensen S., Handford P., TB domain proteins: Evolutionary insights into the multifaceted roles of fibrillins and LTBPs. Biochem. J. 433, 263–276 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Przeworski M., The signature of positive selection at randomly chosen loci. Genetics 160, 1179–1189 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De’ath G., Fabricius K. E., Sweatman H., Puotinen M., The 27–year decline of coral cover on the Great Barrier Reef and its causes. Proc. Natl. Acad. Sci. U.S.A. 109, 17995–17999 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.A. Thompson, C. Lønborg, P. Costello, J. Davidson, M. Logan, M. Furnas, K. Gunn, M. Liddy, M. Skuza, S. Uthicke, M. Wright, I. Zagorskis, B. Schaffelke, “Marine Monitoring Program. Annual Report for Inshore water quality and coral reef monitoring 2013–2014” (Australian Institute of Marine Science, 2014); http://elibrary.gbrmpa.gov.au/jspui/bitstream/11017/2975/5/MMP_AIMS_Inshore_WQ_Coral_Monitoring_2013-2014%20v2.pdf.
- 44.Brodie J. E., Devlin M., Haynes D., Waterhouse J., Assessment of the eutrophication status of the Great Barrier Reef lagoon (Australia). Biogeochemistry 106, 281–302 (2011). [Google Scholar]
- 45.Aguilar C., Raina J.-B., Fôret S., Hayward D. C., Lapeyre B., Bourne D. G., Miller D. J., Transcriptomic analysis reveals protein homeostasis breakdown in the coral Acropora millepora during hypo-saline stress. BMC Genomics 20, 148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rocker M. M., Brandl S. J., Transplantation of corals into a new environment results in substantial skeletal loss in Acropora tenuis. Mar. Biodivers. 45, 321–326 (2015). [Google Scholar]
- 47.Ramos-Silva P., Kaandorp J., Huisman L., Marie B., Zanella-Cléon I., Guichard N., Miller D. J., Marin F., The skeletal proteome of the coral Acropora millepora: The evolution of calcification by co-option and domain shuffling. Mol. Biol. Evol. 30, 2099–2112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takeuchi T., Yamada L., Shinzato C., Sawada H., Satoh N., Stepwise evolution of coral biomineralization revealed with genome-wide proteomics and transcriptomics. PLOS ONE 11, e0156424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamada M., Schröder K., Bathia J., Kürn U., Fraune S., Khalturina M., Khalturin K., Shinzato C., Satoh N., Bosch T. C., Metabolic co-dependence drives the evolutionarily ancient Hydra-Chlorella symbiosis. Elife 7, e35122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin M.-F., Takahashi S., Forêt S., Davy S. K., Miller D. J., Transcriptomic analyses highlight the likely metabolic consequences of colonization of a cnidarian host by native or non-native Symbiodinium species. Biol. Open. 8, bio038281 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mohamed A. R., Cumbo V., Harii S., Shinzato C., Chan C. X., Ragan M. A., Bourne D. G., Willis B. L., Ball E. E., Satoh N., Miller D. J., The transcriptomic response of the coral Acropora digitifera to a competent Symbiodinium strain: The symbiosome as an arrested early phagosome. Mol. Ecol. 25, 3127–3141 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Oakley C. A., Ameismeier M. F., Peng L., Weis V. M., Grossman A. R., Davy S. K., Symbiosis induces widespread changes in the proteome of the model cnidarian Aiptasia. Cell. Microbiol. 18, 1009–1023 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Shinzato C., Mungpakdee S., Arakaki N., Satoh N., Genome-wide SNP analysis explains coral diversity and recovery in the Ryukyu Archipelago. Sci. Rep. 5, 18211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cowman P. F., Quattrini A. M., Bridge T. C. L., Watkins-Colwell G. J., Fadli N., Grinblat M., Roberts T. E., McFadden C. S., Miller D. J., Baird A. H., An enhanced target-enrichment bait set for Hexacorallia provides phylogenomic resolution of the staghorn corals (Acroporidae) and close relatives. Mol. Phylogenet. Evol. 153, 106944 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Chin C.-S., Peluso P., Sedlazeck F. J., Nattestad M., Concepcion G. T., Clum A., Dunn C., O’Malley R., Figueroa-Balderas R., Morales-Cruz A., Cramer G. R., Delledonne M., Luo C., Ecker J. R., Cantu D., Rank D. R., Schatz M. C., Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chin C.-S., Alexander D. H., Marks P., Klammer A. A., Drake J., Heiner C., Clum A., Copeland A., Huddleston J., Eichler E. E., Turner S. W., Korlach J., Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Ying H., Cooke I., Sprungala S., Wang W., Hayward D. C., Tang Y., Huttley G., Ball E. E., Forêt S., Miller D. J., Comparative genomics reveals the distinct evolutionary trajectories of the robust and complex coral lineages. Genome Biol. 19, 175 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q., Chen Z., Mauceli E., Hacohen N., Gnirke A., Rhind N., di Palma F., Birren B. W., Nusbaum C., Lindblad-Toh K., Friedman N., Regev A., Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haas B. J., Papanicolaou A., Yassour M., Grabherr M., Blood P. D., Bowden J., Couger M. B., Eccles D., Li B., Lieber M., MacManes M. D., Ott M., Orvis J., Pochet N., Strozzi F., Weeks N., Westerman R., William T., Dewey C. N., Henschel R., LeDuc R. D., Friedman N., Regev A., De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stanke M., Keller O., Gunduz I., Hayes A., Waack S., Morgenstern B., AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Korf I., Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Holt C., Yandell M., MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Remmert M., Biegert A., Hauser A., Söding J., HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9, 173–175 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G., Pesseat S., Quinn A. F., Sangrador-Vegas A., Scheremetjew M., Yong S.-Y., Lopez R., Hunter S., InterProScan 5: Genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Emms D. M., Kelly S., OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H., Durbin R., Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.E. Garrison, G. Marth, Haplotype-based variant detection from short-read sequencing. arXiv [q-bio.GN] (2012); http://arxiv.org/abs/1207.3907.
- 68.Meisner J., Albrechtsen A., Inferring population structure and admixture proportions in low depth NGS data. Genetics 210, 719–731 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Korneliussen T. S., Albrechtsen A., Nielsen R., ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics 15, 356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schiffels S., Durbin R., Inferring human population size and separation history from multiple genome sequences. Nat. Genet. 46, 919–925 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lechner M., Findeiß S., Steiner L., Marz M., Stadler P. F., Prohaska S. J., Proteinortho: Detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 12, 124 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Katoh K., Standley D. M., MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suyama M., Torrents D., Bork P., PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang Z., PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007). [DOI] [PubMed] [Google Scholar]
- 75.Madin J. S., Baird A. H., Dornelas M., Connolly S. R., Mechanical vulnerability explains size-dependent mortality of reef corals. Ecol. Lett. 17, 1008–1015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gutenkunst R. N., Hernandez R. D., Williamson S. H., Bustamante C. D., Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLOS Genet. 5, e1000695 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hudson R. R., Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18, 337–338 (2002). [DOI] [PubMed] [Google Scholar]
- 78.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Leigh J. W., Bryant D., popart : Full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116 (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/48/eabc6318/DC1