

Abstract
The generation of hematopoietic stem cells (HSCs) from induced pluripotent stem cells (iPSCs) is an active and promising area of research; however, generating engraftable HSCs remains a major obstacle. Ex vivo HSC derivation from renewable sources such as iPSCs offers an experimental tool for studying developmental hematopoiesis, disease modeling, and drug discovery, and yields tremendous therapeutic potential for malignant and nonmalignant hematological disorders. Although initial attempts mostly recapitulated yolk sac primitive/definitive hematopoiesis with inability to engraft, recent advances suggest the feasibility of engraftable HSC derivation from iPSCs utilizing ectopic transcription factor expression. Strategic development for de novo HSC generation includes further investigations of HSC ontogeny, and elucidation of critical signaling pathways, epigenetic modulations, HSC and iPSC microenvironment, and cell‐cell interactions that contribute to stem cell biology and function.
Keywords: embryo, hematopoiesis, hematopoietic differentiation, hemogenic endothelium, induced pluripotent stem cells, reprogramming
A potential future clinical setting for patient‐specific hematopoietic stem cell (HSC) or blood product generation from induced pluripotent stem cells (iPSCs).
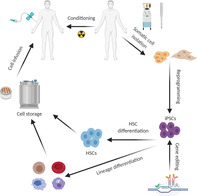
Significance statement.
Patient‐specific hematopoietic stem cells (HSCs) from induced pluripotent stem cells (iPSCs) offer possibility for the treatment of hematological diseases, particularly when no other options are available. This review highlights the current advances in HSC derivation from PSCs and discusses the obstacles that need to be overcome for future autologous PSC‐derived HSC transplantation as a therapeutic modality. The feasibility of HSC generation from transcription factor engineered PSCs has been demonstrated in laboratory conditions and is suggestive of clinically relevant application.
1. INTRODUCTION
Hematopoietic stem cells (HSCs) are immature multipotent adult stem cells that give rise to all mature blood cell lineages, demonstrate long‐term engraftment, and are able to hierarchically reconstitute the entire hematopoietic system in a conditioned recipient after infusion. Allogeneic HSC transplantation (AHSCT) with immunocompatible donors has been successfully used for more than 60 years for the treatment of various malignant and nonmalignant diseases. However, challenges including host‐dependent variability of response to therapy, scarce availability of human leukocyte antigen (HLA)‐matched donors, limited ex vivo proliferation and expansion of engraftable HSCs, and transplant‐related morbidity and mortality restrict broad use of this approach. 1 , 2 , 3 The paucity of HLA‐matched donors, especially for ethnic minorities who are underrepresented in the donor registries worldwide, are a major hurdle for curative options for patients with malignant and nonmalignant hematologic disorders. 4 , 5 In sickle cell disease (SCD), for instance, HLA‐matched sibling AHSCT demonstrates high overall survival and event‐free survival 6 ; however, greater than 85% of the patients lack an HLA‐matched sibling donor and alternative HSC sources such as unrelated, cord blood, or mismatched AHSCT are more variable in their success. 7 AHSCT is limited by donor choice, graft rejection, and graft‐vs‐host disease (GVHD); therefore, autologous HSCT after genetic engineering and correction of the pathologic genotype is an attractive option for patients that would theoretically be available to all.
Striking advances in gene therapy and genome editing tools, particularly lentiviral‐based gene therapies and CRISPR/Cas9 editing, hold tremendous power to cure hematological diseases. Chimeric antigen receptor‐T cell therapies are emerging as powerful treatments in hematologic malignancies, whereas autologous HSCT after gene correction is an ideal curative strategy in nonmalignant hemoglobinopathies where allogenic graft‐vs‐tumor effect does not apply. Initial results from ongoing gene therapy trials in hemoglobinopathies are encouraging 3 , 8 ; however, a lack of long‐term follow‐up data from gene addition studies, the generally low efficiency of genome editing tools, and cost concerns raise safety, applicability, and practicality concerns that must be addressed before there is widespread clinical use. Simultaneous research into alternative strategies is, therefore, important and necessary to continue in pursuit of disease‐modifying and curative strategies in blood disorders.
Ex vivo culture of embryonic stem cells (ESCs) that can produce all cell types in the adult body was established 40 years ago 9 and has provided an important understanding of developmental biology. Differentiated cells can be reprogrammed to an embryonic‐like state with the groundbreaking discovery in 2006 of inducing pluripotent stem cells (PSCs) from mouse stromal cells after introducing four transcription factors (OCT4, SOX2, KLF4, and c‐Myc) using integrating viral vectors under ES cell culture conditions. 10 This discovery brought the possibility of using this modality to produce patient‐specific production of adult stem/mature cells or HSCs in the context of SCD that can be produced and corrected, for an efficient autologous stem cell transplantation modality. This system was quickly adapted for human cells, 11 and various starting cells and manufacturing techniques have been introduced (reviewed in Reference 12). Although distinct gene expression signature is a reality for induced PSCs (iPSCs) (reviewed in Reference 13), and there is donor age‐ and epigenetic‐signature‐dependent variability in the iPSCs (reviewed in References 14, 15, respectively), iPSC models serve as a powerful tool to unravel the progression of malignant diseases, screen potential drug candidates for particular diseases, and have significant potential therapeutic applications in multiple hematologic and nonhematologic disorders. 16
Considering the remarkable therapeutic value of HSCs for regenerative medicine applications and their limited availability, significant experimental efforts have been devoted to generating true HSCs from PSCs. The simultaneous research into genome editing techniques offers a clinically relevant, therapeutic bridge when combined with iPSC generated HSCs, particularly in nonmalignant hematologic disorders where HLA‐matched sibling donors are lacking, GVHD is detrimental, and the generation of definitive HSCs from patient‐derived iPSCs has the potential to be curative when combined with genetic correction. While there remains significant potential, attempts to demonstrate engraftable HSC generation from PSCs have been largely unsuccessful ex vivo without teratoma formation or exogenous transcription factor expression (Table 1). Here, we review the dynamic regulations of developmental hematopoiesis as required for the establishment of bona fide HSC derivation protocols from PSCs, and discuss the unmet scientific needs and challenges required to overcome in order to translate this technology into successful therapeutic application in human diseases.
TABLE 1.
Selected reports for engraftable hematopoietic stem and progenitor cells (HSPCs) derived from PSCs
| Cellular source | Approach | Engraftment | Reference |
|---|---|---|---|
| HoxB4 engineered mouse yolk sac embryonic cells | Monolayer culture on OP9 cells | Myeloid biased and low lymphocyte engraftment (primary and secondary recipients, 10‐20 wk) | 17 |
| Human ESCs or iPSCs | Subcutaneous transplantation of PSCs with or without OP9 feeder cells and cytokines to derive HSPCs from PSCs induced teratomas | B and T cells and myeloid engraftment (primary and secondary recipients, 4‐12 wk) | 18, 19 |
| Human ESCs | Monolayer culture on stromal cells derived from mouse aorta‐gonad‐mesonephros (AGM) region | Myeloid and lymphoid engraftment (primary and secondary recipients, 8‐12 wk) | 20 |
| Human ESCs‐ and monkey iPSCs‐derived CD34+ cells | Embryoid body formation followed by sorting of CD34+ cells cultured on E4ORF1 engineered primary endothelial cells from human umbilical cords | Myeloid, lymphoid, and erythroid engraftment (primary and secondary recipients, 12‐16 wk) | 21 |
| HOXA9, ERG, RORA, SOX4 and MYB engineered CD34+CD45+ from human iPSCs | Embryoid Body formation followed by sorting of CD34+CD45+ cells to transduce with the transcription factors | Erythroid and myeloid engraftment (primary recipient, 4‐5 wk) | 22 |
| ERG, HOXA5, HOXA9, HOXA10, LCOR, RUNX1 and SPI1 engineered hemogenic endothelium cells derived from human ESCs and iPSCs | Embryoid body formation followed by sorting of hemogenic endothelium cell to transduce with the transcription factors | B and T cells and myeloid (primary and secondary recipient, 12‐16 wk) | 23 |
| MLL‐AF4 engineered HSPCs derived from human iPSCs | Monolayer differentiation followed by a collection of HSPCs to transduce with the transcription factor | B and T cells and myeloid (primary and secondary recipient, 8 wk) | 24 |
| Inducible Runx1 and Hoxa9 engineered mouse iPSCs and ESCs | Embryoid body formation followed by sorting of inducible hemogenic endothelium cells to culture on OP9‐DL1 cells | T cells (primary and secondary recipients, 4‐6 wk) | 25 |
| Mouse PSCs | Embryoid body formation followed by hematopoietic specification in 3D hydrogel | Myeloid and B cells (primary recipient, 3 wk) | 26 |
| Gfi1b, c‐Fos, and Gata2 Mouse iPSCs cells | Subcutaneous transplantation of engineered iPSCs to derive HSPCs from iPSCs induced teratomas | B and T cells and myeloid engraftment (primary and secondary recipients, 16 wk) | 27 |
2. DEVELOPMENTAL HEMATOPOIESIS: WAVES OF PRIMITIVE AND DEFINITIVE HEMATOPOIESIS ARE NECESSARY FOR LIFE‐LONG FUNCTIONAL HEMATOPOIESIS
Our understanding of mammalian hematopoiesis has primarily been derived from small animal models given ethical concerns surrounding research with human fetal tissue. Our current understanding of developmental hematopoiesis involves tightly regulated sequential events occurring in multiple, partially overlapping but spatiotemporally separated waves (Figure 1). The first wave, which appears in the yolk sac, lacks lymphoid potential but produces transitory primitive blood cells (erythrocytes, megakaryocytes, and macrophages) to provide the growth needs and first innate defense mechanism for the embryo. 28 , 29 , 30 The second wave also emerges in the yolk sac and generates lympho‐myeloid progenitors (LMPs) and erythro‐myeloid progenitors (EMPs) which subsequently migrate to the fetal liver as a distinct source of hematopoietic progenitors that precede HSC detection. 31 , 32 Although yolk‐sac progenitor cells cannot reconstitute hematopoiesis as HSCs do, they remain functional after birth and most likely throughout the lifespan as a common origin for tissue macrophages. 33 The first true HSCs are generated by the definitive third wave of hematopoiesis. This complex process involves generation of the aorta‐gonad‐mesonephros (AGM) region, a potent hematopoietic site within the mammalian embryo body, and the first place from which HSCs emerge. HSCs first appear in the dorsal aorta of AGM, with high frequencies of HSCs emerging from the vitelline and umbilical arteries suggesting a close relationship between the developing hematopoietic and major arterial vascular system of the vertebrate embryo. 34 , 35 After the specification of the HSCs, they localize the fetal liver via circulation where they propagate before they seed bone marrow that is capable of lifelong support of HSC self‐renewal. 36
FIGURE 1.
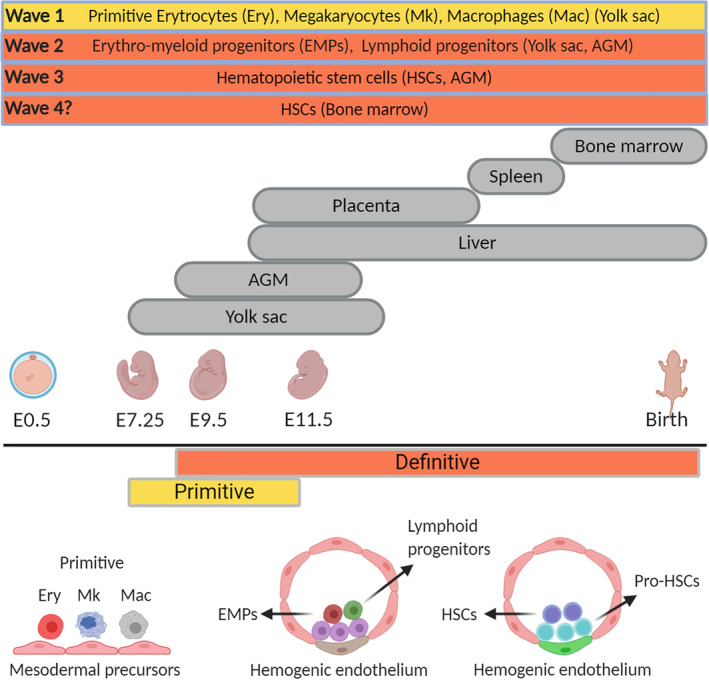
Schematic representations of mouse embryonic hematopoietic development. AGM, aorta‐gonad‐mesonephros
The terminology of “primitive” and “definitive” embryonic blood cells does not refer to engraftment potential, rather is dependent on the characteristics of the cell. The first primitive embryonic erythroblasts, detected at embryonic day 7.25 to 8.5 (E7.25‐E8.5) in the mouse embryo 37 and at 3 to 4 weeks in the human embryo, 38 mostly express embryonic globins, are nucleated, and larger compared to their definitive counterparts. In the mouse, these nucleated cells continue to divide and enter the embryo proper after the onset of cardiac contractions at E8.25. 39 Circulating primitive erythrocytes lose the ability to proliferate and eventually enucleate, remaining in the neonatal mouse circulation several days after birth. 40 Similarly, primitive megakaryocytes are detected in the yolk sac of the mouse embryo at E7.25 and are smaller in size compared to adult megakaryocytes with limited capacity to endoreplicate (reviewed in Reference 41). Compared to adult platelets, fetal platelets are relatively larger, have small alpha granules, respond to collagen inefficiently, and display low coagulation potential. 29 , 42 Primitive macrophages, first detected in the yolk sac after E9.5, 29 mature rapidly without peroxidase activity as otherwise observed in adult canonical monocyte‐derived macrophages from the bone marrow. 43
Although it is not known that whether definitive hematopoiesis emerges as a continuation of primitive hematopoiesis, increasing pluripotency in later hematopoietic waves is a hallmark of the developing embryo. The transient second wave in the yolk sac produces definitive LMPs that can differentiate into B and T cells but lack engraftment potential, 31 and EMPs that colonize the fetal liver as progenitors of erythrocytes, macrophages, megakaryocytes and neutrophils. 32 These early fetal liver erythroblasts express the definitive erythroid‐specific transcriptional modifiers c‐myb, Sox6, and Bcl11a, and produce erythrocytes that enucleate similar to adult RBCs; however, express γ‐globin with low levels of ε‐globin in contrast to adult RBCs expressing β‐globin and negligible levels of γ‐globin. 44 EMP‐derived megakaryocytes display relatively lower ploidy and generate fewer platelets compared to the HSC‐derived megakaryocytes. 45 Intriguingly, EMP‐derived macrophages become tissue‐resident macrophages that continue to remain in the adult body with a self‐renewal capacity similar to ESCs. 46
Definitive HSCs with extreme proliferation capacity give rise to all blood cell lineages and reconstitute the blood system of the adult body, and are detected at around E11 in the AGM region, and thereafter in the placenta and extraembryonic tissues. 34 , 47 , 48 Shortly after emerging in the vascular region of the embryo, HSCs migrate to the fetal liver where they produce blood lineages necessary to support the growing embryo, then initiate migration to the bone marrow shortly before birth to supply life‐long hematopoiesis. 49 This derivation of true HSCs capable of supporting lifelong hematopoiesis begins with strict dependence on the niche environment given stromal cell support from AGM allows for the maintenance, expansion, and multilineage potential of murine and human HSCs. 50 , 51 Specifically for definitive HSC‐derived RBCs, two major properties are required; efficient enucleation and activation of developmentally regulated machinery, referred to as globin switching, to switch the globin profile from fetal to adult globin after birth. 52
Recently, a previously unappreciated transient fourth wave of definitive HSC generation has been reported arising from hemogenic endothelium (HE) cells in the bone marrow in chickens and mice in the late fetus and young adult stage, 53 indicating HSCs can be generated de novo after developmental stages. It is well established that HSCs are generated from a transient subset of specialized endothelial cells termed hemogenic, present in the yolk sac, placenta, and aorta, through an endothelial‐to‐hematopoietic transition (EHT). HSC generation via EHT was thought to be restricted to the early stages of development; however, the discovery of bone marrow HE revealed that HSCs can be generated past embryonic stages. This transient wave might support the neonatal bone marrow environment until fetal liver amplified HSCs take up their adult‐type niches.
Definitive hematopoietic cells are derived through a process that does not involve cell division but instead results from a strong bending and rounding up of a small specialized form of endothelial cells with hemogenic potential (HE cells). 54 The process by which HE cells emerge from the aortic ventral wall into the subaortic space is called EHT. Many transcription factors (such as Runx1, Sox17, Scl, Gfi1/Gfi1b, and Gata2) and signaling factors (including Hedgehog, Bpm4, Notch ligands, and Wnt) are associated with either HE specification or EHT. 55 The Gata2 transcription factor is essential for HSC generation and function; therefore, recent index‐sorting of Gata2‐expressing intra‐aortic hematopoietic cluster cells combined with single‐cell RNA‐seq revealed a unique transcriptome for definitive HSCs with specific expression levels of CD31, Gata2, cKit, and CD27 from endothelium within the aortic clusters. 56 Further, Runx1 increases during HE specification, 53 while a Yes‐activated protein‐mediated mechanical signaling by way of a Rho‐GTPase‐mediated mechanism was shown to result in the commitment of HE to HSCs. 57 Here, although hemodynamic forces seem critical for in vivo HSC formation, the exact molecular mechanism is not known and adaptation of biomechanical forces to ex vivo differentiation protocols for successful de novo HSC generation is lacking. A recent study demonstrated primitive vascular endothelial cells (at E8) follow a two‐step fate choice to become HSC‐primed HE cells; specification into arterial phenotype followed by a subsequent hemogenic conversion. 58 This transition from pre‐HE to HE stage was further demonstrated through trajectory analyses and genetic perturbation experiments. 59 Here, two distinct subpopulations from intra‐arterial clusters, namely precursors of HSCs and CD45+ lympho‐myeloid‐biased progenitors, were reported, although it is not known whether these subpopulations differentiate from an equivalent or distinct population within the HE.
Understanding the complexity of developmental hematopoiesis and the connection among the hematopoietic waves aides in the establishment of ex vivo differentiation protocols, with definitive HSC production dependent on cell‐cell interactions, close regulation of growth factors, and identifying the various biomechanical interactions within the AGM. Further clarification of these complex interactions is required for the establishment of more sophisticated ex vivo protocols to derive HSCs from PSCs. For any therapeutic potential, however, definitive HSCs must have long‐term self‐renewal and engraftment capabilities.
3. HSC GENERATION FROM PSCs: TRANSCRIPTION FACTORS SPECIFY THE GENERATION OF MULTIPOTENT PROGENITORS AND ENGRAFTABLE CELLS
Other than understanding and modeling developmental hematopoiesis, the long‐sought goal in the field of HSC generation from PSCs is to develop well‐established protocols that generate bona fide HSCs with long‐term engraftment potential. Early attempts could only recapitulate primitive first wave and inefficient definitive second wave hematopoiesis, similar to yolk sac hematopoiesis. 60 , 61 , 62 Since then, investigations using fetal stromal cells as feeder cells for PSCs, fine‐tuning growth factor supplementation, and developing tightly orchestrated differentiation protocols have allowed the generation of mostly phenotypically definitive PSC‐derived HSCs with multipotent differentiation potential but without engraftment ability. In general, RBC globin expression is a useful general assessment tool to define primitive/definitive characteristics of any HSC‐like cells from PSCs given differences in globin expression profiles. Alternatively, T lymphocyte potential has been shown to track the onset of definitive hematopoiesis in human pluripotent cultures. 63 , 64
Initially, RBCs derived from PSCs were reported to enucleate inefficiently and express mostly ε‐globin/γ‐globin with no or negligible levels of adult β‐globin. 65 , 66 Insights into developmental hematopoiesis and understanding of the regulatory mechanisms of globin switching allowed the derivation of relatively higher levels of β‐globin expressing RBCs from PSCs; however, engraftment has again not been demonstrated. 67 , 68 , 69 , 70 Although the improvement in β‐globin expression assumes successful activation of the definitive third wave‐like HSC generation from PSCs, single‐cell analysis in iPSC‐RBCs revealed that one cell can express all globin types to a certain degree. 71 This single‐cell analysis reflects a possibility of recapitulating engraftment deficient definitive yolk sac hematopoiesis for most cases rather than creation of a heterogeneous population with cells from distinct waves.
EMP‐RBCs and AGM‐RBCs in the embryo are phenotypically indistinguishable as both predominantly express γ‐globin; therefore, engraftment studies are the main hallmark for true HSC assessment. A plethora of the published protocols only generated HSC‐like cells with inefficient self‐renewal capacity without long‐term engraftment potential in conditioned immunodeficient mouse models, indicating a lack of definitive HSC generation. Although it is not the focus of this review, terminally differentiated blood cells from iPSCs could be used for transfusion purposes (although this topic is also debated), including generation of myeloid cells, RBCs (given normal oxygen transport for both fetal and adult hemoglobins), and megakaryocytes (for a deeper review, see Reference 72). Such therapeutic application would have implications to address issues surrounding resources and shortages, and would eliminate alloimmunization and transfusion‐transmitted infectious diseases from transfusion.
A long‐standing question has been whether inadequate engraftable HSC generation from PSCs is a technical or biological inefficiency. Ectopic expression of HoxB4 in mouse primitive yolk sac embryonic cells combined with culture on bone marrow stromal cells (OP9 cells) provided modest engraftment and multilineage differentiation in primary and secondary irradiated recipients with mostly myeloid bias and low lymphocyte (particularly T cells) reconstitution. 17 A follow‐up study demonstrated tetracycline‐inducible Cdx4 and HoxB4 engineered mouse ESC engraftment (with again low T‐cell chimerism), possibly due to activation of the Hox pathway. However, the results could not be confirmed for human ESCs. 73 On the other hand, subcutaneously transplanted human iPSCs into immunocompromised mice could provide long‐term repopulating intra‐teratoma HSC derivation, indicating the theoretical possibility of true HSC generation from PSCs under proper conditions without the need for ectopic gene expression. 18 , 19 Here, iPSC‐derived HSCs migrated from teratomas into the bone marrow, and resulted in multilineage and long‐term reconstitution of the hematolymphopoietic system in serial transfers. Ultimately, true engraftable HSCs from PSCs without teratoma formation would be required for any translatable clinical application.
Significant effort has been devoted to transcription factor screening for ex vivo hematopoiesis from PSCs on the basis of the finding that specific transcription factor expression (Hlf, Runx1t1, Pbx1, Lmo2, Zfp37, Prdm5, Meis1, and Mycn) in pro‐B cells and common myeloid progenitors were sufficient to derive serially transplantable HSCs with multilineage differentiation potential, 74 and that lineage‐restricted CD34+CD45+ myeloid precursors derived from iPSCs modified with five transcription factors (HOXA9, ERG, RORA, SOX4, and MYB) conferred short‐term engraftment with myeloid and erythroid lineages. 22 Such transcription factor screening studies provide proof‐of‐principle data for in vivo modeling using donor‐derived iPSCs for genetic blood diseases. In one such example, hematopoietic progenitors from Diamond‐Blackfan anemia (DBA) patient‐derived iPSCs engineered with aforementioned five transcription factors engrafted immunocompromised mice with impaired erythropoiesis 75 ; however, screening of small molecules identified autophagy inducer SMER28 as a potential therapeutic candidate for DBA that rescued impaired erythroid differentiation in chimeric mice. Additional transcription factor screening eventually resulted in the derivation of long‐term and serially transplantable HSCs from human iPSCs. In a separate study modeling hematopoietic disease in humanized mice for therapeutic strategies in genetic blood disorders, a larger cohort of transcription factors (ERG, HOXA5, HOXA9, HOXA10, LCOR, RUNX1, and SPI1) was sufficient to convert HE cells into HSCs that engrafted myeloid, B and T cells in primary and secondary mouse recipients (33 out of 76 mice engrafted, 9 multilineage reconstitution). 23 Here, characterization of differentiated cells revealed RBC enucleation potential and a hemoglobin profile similar to human umbilical cord blood, antigen‐specific responses in lymphocytes, and T‐cell receptor rearrangement, indicating recapitulation of developmental hematopoiesis for definitive HSC generation in ex vivo conditions. One of the rare human embryo studies recently reported that fetal liver HSPCs are specifically associated with HOX family transcription factors such as HOXA6 and HOXA10, confirming the importance of the HOX pathway for bona fide HSCs. 76 A recent report showed transient expression of a single transcription factor, MLL‐AF4, was sufficient to specify engraftable HSCs from PSCs; however, engrafted cells displayed a tendency for leukemic transformation during the long‐term follow‐up. 24 Here, the authors also tried to replicate five transcription factor engineering that was shown to result in myeloid and erythroid biased short‐term engraftment in mice, 22 and evaluate their potentially synergistic activity with MLL‐AF4 expression; however, their results revealed successful engraftment in only MLL‐AF4 engineered groups. These results indicate that complex transcription factor engineering approaches might be limited in their potential clinical use due to being hard to adapt in standard laboratory conditions, being time‐consuming and costly, and requires significant expertise.
Ex vivo HSC derivation from PSCs offers an experimental tool for studying developmental hematopoiesis, disease modeling, and drug discovery, but engraftable HSCs are currently only possible with ectopic transcription factor expression or teratoma formation, thereby limiting clinical applications.
4. CHALLENGES AND OUTLOOK
iPSC‐derived cellular therapy has tremendous therapeutic potential for a wide breadth of disorders, ranging from regenerative needs to correction of hematologic disorders. Various diseases have been modeled in ex vivo conditions, now with the discovery of genome editing tools such as CRISPR/Cas9 contributing to the possibility of genetic correction of underlying disease mutations in patient‐derived iPSCs.
To date, ex vivo true HSC generation without genetic intervention remains theoretic and can only be achieved by ectopic expression of at least one transcription factor. MLL‐AF4 engineering has already been shown for possible leukemia transformation of long‐term engrafted iPSC‐HSCs, 24 and dysregulation of HOX genes is strongly associated with acute myeloid leukemia and acute lymphoid leukemia. 77 Therefore, to generate therapeutic‐grade HSCs from iPSCs, safety evaluation is required from both small and large animal models particularly if genetic manipulations are involved in order to avoid secondary mutations and possible carcinogenesis. For regenerative approaches, contamination of undifferentiated PSC during HSC generation causing teratoma formation is of concern. In addition to safety profiles, efficient purification/selection strategies are necessary to eliminate this possibility. To this end, it has been demonstrated that two drug‐inducible safeguards, which have separate functionalities, can eliminate undifferentiated PSCs to prevent teratoma formation and deplete all PSC‐derived cells in vivo if adverse events arise. 78
Although most of the current protocols can derive blood cells with a definitive phenotype, immunocompromised mice models are the only available tools to differentiate definitive yolk sac and AGM HSCs, and determine the functionality of HSC‐like cells derived from PSC. Mouse models are a costly and time‐consuming investment, and yet the success of these methods in larger animal models is eventually required for meaningful therapeutic translation. Establishment of ex vivo protocols with a proper HSC functionality evaluation would, therefore, enhance the speed of methodology screening, potentially reduce cost, and shorten the overall time for success in the animal model. To do such an evaluation, defining discrimination markers for human cells at different developmental stages is required. Currently, known cell surface markers to distinguish primitive/definitive hematopoiesis are similar; therefore, identifying globin profiles in RBCs and lymphoid potential of HSC‐like cells from PSCs are the generally applied methods. To improve this, for instance, a recent report demonstrated CD44 as a novel cell surface marker for HSC‐primed HE cells in the AGM region. 79 Besides, hepatic leukemia factor (Hlf) expression was recently reported as a discriminating factor for mouse liver HSCs from EMPs or hematopoietic clusters in the yolk sac (E9). 80 Although these findings are waiting to be confirmed for human, HSC‐specific reporter ES/iPS cell lines (ie, RUNX1c reporter lines 81 ) would be a versatile tool for ex vivo differentiation method optimizations. Devotion to the full characterization of progenitor blood cells, HSCs, and progenitors of HSCs would contribute to our understanding of engraftable HSCs while greatly reducing the need for animal engraftment studies in all protocol designs.
The microenvironment and interactions between various cell types play a critical role in true HSC derivation from PSCs given biological sufficiency of PSCs without a need for genetic manipulation shown in teratoma formation. 18 , 19 Currently, transcription factor engineered PSCs can compensate for the absence of microenvironmental signals; therefore, a detailed description of the crucial activated signaling pathways in fetal hematopoiesis would realize ex vivo bona fide HSC generation without genetic engineering where critical growth factors or small molecules could be provided in fine‐tuned differentiation protocols. Another critical consideration is the HSC niche in patients. Although the bone marrow microenvironment is difficult to recapitulate ex vivo, the establishment of nontoxic conditioning regimens that maintain critical function for homing and engraftment of iPSC‐HSCs to the bone marrow niche is important, for a less potent and supportive bone marrow environment of hematology patients (ie, SCD patients 82 ).
iPSC production is labor‐intensive; therefore, cost and maintenance of the generation and differentiation of patient‐specific iPSCs is a major concern, particularly for the developing countries. The high cost and labor necessary for clinical‐grade iPSC production requiring significant expertise would hamper the wide‐use of iPSC‐HSCs. To translate iPSC‐derived HSCs into clinical applications, cell products must be made under Good Manufacturing Practice (GMP)‐grade practices which further increases the cost. One possible approach to leverage the success of iPSC‐based regenerative therapies would be the generation of “universal” iPSCs by genetic inactivation of HLAs and overexpression of specific cell surface markers such as CD47 to mask them from immune cells. 83 A possible disadvantage of HLAs inactivation would be missing self‐response to natural killer cell (NK) activity. Knock‐in HLA‐E genes at the Beta‐2 Microglobulin locus in human iPSCs without surface expression of HLA‐A, B, or C were resistant to NK cell‐dependent lysis. 84 Another solution includes banking of particular iPSCs that serve the majority of the population. For instance, 12 iPSC lines retaining HLA‐C combined with HLA‐class II knockout are immune compatible and estimated to match >90% of the world's population, 85 proposing a drastic reduction in the costs and variability for PSC‐HSC therapy. However, HLA‐C expressing cells can still be detected by T‐cells, and suppression of other immune cells should also be considered for the long‐term survival of engrafted cells. In this manner, HLA‐A/‐B/‐C and CIITA molecules were simultaneously deleted in a recent study along with knock‐in of immune regulatory factors PD‐L1, HLA‐G, and CD47 to the safe harbor AAVS1 locus to evade immune surveillance by T cells, NK‐cells, and macrophages, respectively. 86 Although the upfront cost may ultimately be high for such therapies, the ability to potentially cure diseases that otherwise has high lifetime economic and quality‐of‐life costs may ultimately be advantageous in the long run. 87
5. CONCLUSION
Establishment of ex vivo protocols for iPSC derived HSCs contributes to the understanding of developmental hematopoiesis, is a methodology for high‐throughput drug screening and gene therapy development, and offers a promising potential therapeutic option for malignant and nonmalignant heritable blood disorders. Although the biological capacity of PSCs to generate HSCs have been presented through teratoma formation, ex vivo differentiation protocols without ectopic transcription factor expression have not been established and require further investigation. Further research must approximate all of the properties of true HSC generation, including but not limited by critical gene regulation, epigenetic modulations, microenvironment interactions, and signaling pathways in order to become a realistic, applicable, and ideally cost‐effective therapeutic option.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
S.D.: conception/design, manuscript writing, and final approval of the manuscript; A.L., J.F.T.: manuscript writing, final approval of the manuscript.
Demirci S, Leonard A, Tisdale JF. Hematopoietic stem cells from pluripotent stem cells: Clinical potential, challenges, and future perspectives. STEM CELLS Transl Med. 2020;9:1549–1557. 10.1002/sctm.20-0247
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Guilcher GM, Truong TH, Saraf SL, et al. Curative therapies: allogeneic hematopoietic cell transplantation from matched related donors using myeloablative, reduced intensity, and nonmyeloablative conditioning in sickle cell disease. Semin Hematol. 2018;55(2):87‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papa L, Djedaini M, Hoffman R. Ex vivo HSC expansion challenges the paradigm of unidirectional human hematopoiesis. Ann N Y Acad Sci. 2019;1466:39‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Demirci S, Uchida N, Tisdale JF. Gene therapy for sickle cell disease: an update. Cytotherapy. 2018;20(7):899‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dew A, Collins D, Artz A, et al. Paucity of HLA‐identical unrelated donors for African‐Americans with hematologic malignancies: the need for new donor options. Biol Blood Marrow Transplant. 2008;14(8):938‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Switzer GE, Bruce JG, Myaskovsky L, et al. Race and ethnicity in decisions about unrelated hematopoietic stem cell donation. Blood. 2013;121(8):1469‐1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem‐cell transplantation for sickle cell disease. N Engl J Med. 2009;361(24):2309‐2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leonard A, Tisdale J, Abraham A. Curative options for sickle cell disease: haploidentical stem cell transplantation or gene therapy? Br J Haematol. 2020;189(3):408‐423. [DOI] [PubMed] [Google Scholar]
- 8. Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion‐dependent β‐thalassemia. N Engl J Med. 2018;378(16):1479‐1493. [DOI] [PubMed] [Google Scholar]
- 9. Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292(5819):154‐156. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663‐676. [DOI] [PubMed] [Google Scholar]
- 11. Yu J, Vodyanik MA, Smuga‐Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917‐1920. [DOI] [PubMed] [Google Scholar]
- 12. González F, Boué S, Belmonte JCI. Methods for making induced pluripotent stem cells: reprogramming a la carte. Nat Rev Genet. 2011;12(4):231‐242. [DOI] [PubMed] [Google Scholar]
- 13. Narsinh KH, Plews J, Wu JC. Comparison of human induced pluripotent and embryonic stem cells: fraternal or identical twins? Mol Ther. 2011;19(4):635‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Strässler ET, Aalto‐Setälä K, Kiamehr M, Landmesser U, Kränkel N. Age is relative—impact of donor age on induced pluripotent stem cell‐derived cell functionality. Front Cardiovasc Med. 2018;5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liang G, Zhang Y. Genetic and epigenetic variations in iPSCs: potential causes and implications for application. Cell Stem Cell. 2013;13(2):149‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481(7381):295‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kyba M, Perlingeiro RC, Daley GQ. HoxB4 confers definitive lymphoid‐myeloid engraftment potential on embryonic stem cell and yolk sac hematopoietic progenitors. Cell. 2002;109(1):29‐37. [DOI] [PubMed] [Google Scholar]
- 18. Amabile G, Welner RS, Nombela‐Arrieta C, et al. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood. 2013;121(8):1255‐1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suzuki N, Yamazaki S, Yamaguchi T, et al. Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol Ther. 2013;21(7):1424‐1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ledran MH, Krassowska A, Armstrong L, et al. Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell. 2008;3(1):85‐98. [DOI] [PubMed] [Google Scholar]
- 21. Gori JL, Butler JM, Chan Y‐Y, et al. Vascular niche promotes hematopoietic multipotent progenitor formation from pluripotent stem cells. J Clin Invest. 2015;125(3):1243‐1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Doulatov S, Vo LT, Chou SS, et al. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage‐restricted precursors. Cell Stem Cell. 2013;13(4):459‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sugimura R, Jha DK, Han A, et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature. 2017;545(7655):432‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tan Y‐T, Ye L, Xie F, et al. Respecifying human iPSC‐derived blood cells into highly engraftable hematopoietic stem and progenitor cells with a single factor. Proc Natl Acad Sci USA. 2018;115(9):2180‐2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guo R, Hu F, Weng Q, et al. Guiding T lymphopoiesis from pluripotent stem cells by defined transcription factors. Cell Res. 2019;30:21‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shan W, Wang B, Xu Y, et al. Generation of hematopoietic cells from mouse pluripotent stem cells in a 3D culture system of self‐assembling peptide hydrogel. J Cell Physiol. 2020;235(3):2080‐2090. [DOI] [PubMed] [Google Scholar]
- 27. Tsukada M, Ota Y, Wilkinson AC, et al. In vivo generation of engraftable murine hematopoietic stem cells by Gfi1b, c‐Fos, and Gata2 overexpression within teratoma. Stem Cell Rep. 2017;9(4):1024‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Herbomel P, Thisse B, Thisse C. Ontogeny and behaviour of early macrophages in the zebrafish embryo. Development. 1999;126(17):3735‐3745. [DOI] [PubMed] [Google Scholar]
- 29. Tober J, Koniski A, McGrath KE, et al. The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood. 2007;109(4):1433‐1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baron MH, Isern J, Fraser ST. The embryonic origins of erythropoiesis in mammals. Blood. 2012;119(21):4828‐4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin Y, Yoder MC, Yoshimoto M. Lymphoid progenitor emergence in the murine embryo and yolk sac precedes stem cell detection. Stem Cells Dev. 2014;23(11):1168‐1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McGrath KE, Frame JM, Fegan KH, et al. Distinct sources of hematopoietic progenitors emerge before HSCs and provide functional blood cells in the mammalian embryo. Cell Rep. 2015;11(12):1892‐1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perdiguero EG, Klapproth K, Schulz C, et al. Tissue‐resident macrophages originate from yolk‐sac‐derived erythro‐myeloid progenitors. Nature. 2015;518(7540):547‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gekas C, Dieterlen‐Lièvre F, Orkin SH, Mikkola HKA. The placenta is a niche for hematopoietic stem cells. Dev Cell. 2005;8(3):365‐375. [DOI] [PubMed] [Google Scholar]
- 35. de Bruijn MF, Speck NA, Peeters MC, Dzierzak E. Definitive hematopoietic stem cells first develop within the major arterial regions of the mouse embryo. EMBO J. 2000;19(11):2465‐2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Christensen JL, Wright DE, Wagers AJ, et al. Circulation and chemotaxis of fetal hematopoietic stem cells. PLoS Biol. 2004;2(3):368‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Palis J, Robertson S, Kennedy M, Wall C, Keller G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development. 1999;126(22):5073‐5084. [DOI] [PubMed] [Google Scholar]
- 38. Van Handel B, Prashad SL, Hassanzadeh‐Kiabi N, et al. The first trimester human placenta is a site for terminal maturation of primitive erythroid cells. Blood. 2010;116(17):3321‐3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ji RP, Phoon CK, Aristizábal O, et al. Onset of cardiac function during early mouse embryogenesis coincides with entry of primitive erythroblasts into the embryo proper. Circ Res. 2003;92(2):133‐135. [DOI] [PubMed] [Google Scholar]
- 40. Kingsley PD, Malik J, Fantauzzo KA, Palis J. Yolk sac‐derived primitive erythroblasts enucleate during mammalian embryogenesis. Blood. 2004;104(1):19‐25. [DOI] [PubMed] [Google Scholar]
- 41. Palis J. Hematopoietic stem cell‐independent hematopoiesis: emergence of erythroid, megakaryocyte, and myeloid potential in the mammalian embryo. FEBS Lett. 2016;590(22):3965‐3974. [DOI] [PubMed] [Google Scholar]
- 42. De Cuyper IM, Meinders M, Van De Vijver E, et al. A novel flow cytometry‐based platelet aggregation assay. Blood. 2013;121(10):e70‐e80. [DOI] [PubMed] [Google Scholar]
- 43. Takahashi K, Yamamura F, Naito M. Differentiation, maturation, and proliferation of macrophages in the mouse yolk sac: a light‐microscopic, enzyme‐cytochemical, immunohistochemical, and ultrastructural study. J Leukoc Biol. 1989;45(2):87‐96. [DOI] [PubMed] [Google Scholar]
- 44. McGrath KE, Frame JM, Fromm GJ, et al. A transient definitive erythroid lineage with unique regulation of the β‐globin locus in the mammalian embryo. Blood. 2011;117(17):4600‐4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Alarcon PA, Graeve JL. Analysis of megakaryocyte ploidy in fetal bone marrow biopsies using a new adaptation of the feulgen technique to measure DNA content and estimate megakaryocyte ploidy from biopsy specimens. Pediatr Res. 1996;39(1):166‐170. [DOI] [PubMed] [Google Scholar]
- 46. Soucie EL, Weng Z, Geirsdóttir L, et al. Lineage‐specific enhancers activate self‐renewal genes in macrophages and embryonic stem cells. Science. 2016;351(6274):aad5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gordon‐Keylock S, Sobiesiak M, Rybtsov S, Moore K, Medvinsky A. Mouse extraembryonic arterial vessels harbor precursors capable of maturing into definitive HSCs. Blood. 2013;122(14):2338‐2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Taoudi S, Gonneau C, Moore K, et al. Extensive hematopoietic stem cell generation in the AGM region via maturation of VE‐cadherin+ CD45+ pre‐definitive HSCs. Cell Stem Cell. 2008;3(1):99‐108. [DOI] [PubMed] [Google Scholar]
- 49. Crisan M, Dzierzak E. The many faces of hematopoietic stem cell heterogeneity. Development. 2016;143(24):4571‐4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Durand C, Robin C, Bollerot K, Baron MH, Ottersbach K, Dzierzak E. Embryonic stromal clones reveal developmental regulators of definitive hematopoietic stem cells. Proc Natl Acad Sci USA. 2007;104(52):20838‐20843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oostendorp RA, Harvey KN, Kusadasi N, et al. Stromal cell lines from mouse aorta‐gonads‐mesonephros subregions are potent supporters of hematopoietic stem cell activity. Blood. 2002;99(4):1183‐1189. [DOI] [PubMed] [Google Scholar]
- 52. Demirci S, Tisdale JF. Definitive erythropoiesis from pluripotent stem cells: recent advances and perspectives. Cell Bio Transl Med. 2018;3:1‐13. [DOI] [PubMed] [Google Scholar]
- 53. Yvernogeau L, Gautier R, Petit L, et al. In vivo generation of haematopoietic stem/progenitor cells from bone marrow‐derived haemogenic endothelium. Nat Cell Biol. 2019;21(11):1334‐1345. [DOI] [PubMed] [Google Scholar]
- 54. Kissa K, Herbomel P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature. 2010;464(7285):112‐115. [DOI] [PubMed] [Google Scholar]
- 55. Blaser BW, Zon LI. Making HSCs in vitro: don't forget the hemogenic endothelium. Blood. 2018;132(13):1372‐1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vink CS, Calero‐Nieto FJ, Wang X, et al. Iterative single‐cell analyses define the transcriptome of the first functional hematopoietic stem cells. Cell Rep. 2020;31(6):107627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lundin V, Sugden WW, Theodore LN, et al. YAP regulates hematopoietic stem cell formation in response to the biomechanical forces of blood flow. Dev Cell. 2020;52(4):446‐460.e445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hou S, Li Z, Zheng X, et al. Embryonic endothelial evolution towards first hematopoietic stem cells revealed by single‐cell transcriptomic and functional analyses. Cell Res. 2020;30:376‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhu Q, Gao P, Tober J, et al. Developmental trajectory of pre‐hematopoietic stem cell formation from endothelium. Blood. 2020. 10.1182/blood.2020004801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zambidis ET, Peault B, Park TS, Bunz F, Civin CI. Hematopoietic differentiation of human embryonic stem cells progresses through sequential hematoendothelial, primitive, and definitive stages resembling human yolk sac development. Blood. 2005;106(3):860‐870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kaufman DS, Hanson ET, Lewis RL, Auerbach R, Thomson JA. Hematopoietic colony‐forming cells derived from human embryonic stem cells. Proc Natl Acad Sci USA. 2001;98(19):10716‐10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chadwick K, Wang L, Li L, et al. Cytokines and BMP‐4 promote hematopoietic differentiation of human embryonic stem cells. Blood. 2003;102(3):906‐915. [DOI] [PubMed] [Google Scholar]
- 63. Ditadi A, Sturgeon CM, Tober J, et al. Human definitive haemogenic endothelium and arterial vascular endothelium represent distinct lineages. Nat Cell Biol. 2015;17(5):580‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kennedy M, Awong G, Sturgeon CM, et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2(6):1722‐1735. [DOI] [PubMed] [Google Scholar]
- 65. Lapillonne H, Kobari L, Mazurier C, et al. Red blood cell generation from human induced pluripotent stem cells: perspectives for transfusion medicine. Haematologica. 2010;95(10):1651‐1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kobari L, Yates F, Oudrhiri N, et al. Human induced pluripotent stem cells can reach complete terminal maturation: in vivo and in vitro evidence in the erythropoietic differentiation model. Haematologica. 2012;97(12):1795‐1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Haro‐Mora JJ, Uchida N, Demirci S, Wang Q, Zou J, Tisdale JF. Biallelic correction of sickle cell disease‐derived iPSCs confirmed at the protein level through serum‐free iPS‐sac/erythroid differentiation. Stem Cells Translational Medicine. 2020;9(5):590‐602.32034898 [Google Scholar]
- 68. Uchida N, Haro‐Mora JJ, Fujita A, et al. Efficient generation of β‐globin‐expressing erythroid cells using stromal cell‐derived induced pluripotent stem cells from patients with sickle cell disease. Stem Cells. 2017;35(3):586‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bernecker C, Ackermann M, Lachmann N, et al. Enhanced ex vivo generation of erythroid cells from human induced pluripotent stem cells in a simplified cell culture system with low cytokine support. Stem Cells Dev. 2019;28(23):1540‐1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ruiz JP, Chen G, Mora JJH, et al. Robust generation of erythroid and multilineage hematopoietic progenitors from human iPSCs using a scalable monolayer culture system. Stem Cell Res. 2019;41:101600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vanuytsel K, Matte T, Leung A, et al. Induced pluripotent stem cell‐based mapping of β‐globin expression throughout human erythropoietic development. Blood Adv. 2018;2(15):1998‐2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hansen M, von Lindern M, van den Akker E, Varga E. Human‐induced pluripotent stem cell‐derived blood products: state of the art and future directions. FEBS Lett. 2019;593(23):3288‐3303. [DOI] [PubMed] [Google Scholar]
- 73. Wang L, Menendez P, Shojaei F, et al. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J Exp Med. 2005;201(10):1603‐1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Riddell J, Gazit R, Garrison BS, et al. Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell. 2014;157(3):549‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Doulatov S, Vo LT, Macari ER, et al. Drug discovery for Diamond‐Blackfan anemia using reprogrammed hematopoietic progenitors. Sci Transl Med. 2017;9(376):eaah5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bian Z, Gong Y, Huang T, et al. Deciphering human macrophage development at single‐cell resolution. Nature. 2020;582:571‐576. [DOI] [PubMed] [Google Scholar]
- 77. Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia. 2013;27(5):1000‐1008. [DOI] [PubMed] [Google Scholar]
- 78. Martin RM, Fowler JL, Cromer MK, et al. Improving the safety of human pluripotent stem cell therapies using genome‐edited orthogonal safeguards. Nat Commun. 2020;11(1):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Oatley M, Bölükbası ÖV, Svensson V, et al. Single‐cell transcriptomics identifies CD44 as a marker and regulator of endothelial to haematopoietic transition. Nat Commun. 2020;11(1):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yokomizo T, Watanabe N, Umemoto T, et al. Hlf marks the developmental pathway for hematopoietic stem cells but not for erythro‐myeloid progenitors. J Exp Med. 2019;216(7):1599‐1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ferrell PI, Xi J, Ma C, Adlakha M, Kaufman DS. The RUNX1+ 24 enhancer and P1 promoter identify a unique subpopulation of hematopoietic progenitor cells derived from human pluripotent stem cells. Stem Cells. 2015;33(4):1130‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Leonard A, Bonifacino A, Dominical VM, et al. Bone marrow characterization in sickle cell disease: inflammation and stress erythropoiesis lead to suboptimal CD34 recovery. Br J Haematol. 2019;186(2):286‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Deuse T, Hu X, Gravina A, et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat Biotechnol. 2019;37(3):252‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gornalusse GG, Hirata RK, Funk SE, et al. HLA‐E‐expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol. 2017;35(8):765‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Xu H, Wang B, Ono M, et al. Targeted disruption of HLA genes via CRISPR‐Cas9 generates iPSCs with enhanced immune compatibility. Cell Stem Cell. 2019;24(4):566‐578.e567. [DOI] [PubMed] [Google Scholar]
- 86. Han X, Wang M, Duan S, et al. Generation of hypoimmunogenic human pluripotent stem cells. Proc Natl Acad Sci USA. 2019;116(21):10441‐10446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Leonard A, Tisdale JF. Stem cell transplantation in sickle cell disease: therapeutic potential and challenges faced. Expert Rev Hematol. 2018;11(7):547‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.