

Abstract
Following our discovery that silver(I) oxide-promoted glycosylation with glycosyl bromides can be greatly accelerated in the presence of catalytic TMSOTf or TfOH, reported herein is a new discovery that glycosyl chlorides are even more effective glycosyl donors under these reaction conditions. The developed reaction conditions work well with a variety of glycosyl chlorides. Both benzoylated and benzylated chlorides were successfully glycosidated, and these reaction conditions were effective in coupling substrates containing nitrogen and sulfur atoms. Another convenient feature of this glycosylation is that the progress of this reaction can be monitored by eye, and the completion of the reaction can be judged by the disappearance of characteristic dark color of Ag2O.
Graphical Abstract

Introduction
Glycosyl chlorides, once prominent glycosyl donors, have helped shape the modern synthetic glycochemistry.[1] The story of chemical glycosylation started with exploration of glycosyl chlorides way back in the late 19th century by Michael.[2] Those first glycosylations employed per-acetylated glycosyl chloride as the glycosyl donor for reaction with phenoxide. A notable advancement of glycosylations with chlorides was made by Koenigs and Knorr who utilized simple alcohols as glycosyl acceptors instead of charged nucleophiles.[1a] Those reactions were conducted in the presence of silver salts as acid scavengers because the active role of silver salts as promoters of glycosylation was yet unknown. Only after extensive studies over the following decades, scientists began appreciating that silver salts are able to mediate glycosylation by helping dissociate the anomeric carbon-halogen bonds.[3] However, activation of glycosyl halides commonly requires stoichiometric amounts of expensive or toxic reagents such as silver(I)[1a, 4] or mercury(II) salts.[5] Some halides are cumbersome to synthesize, store, and apply due to their proclivity to hydrolyze. As a result, modern glycosyl donors, thioglycosides, trichloroacetimidates, and others, outshadowed the application of glycosyl halides in glycan synthesis.
Recently, Ye et al.[6] and Jacobsen et al.[7] largely resurrected glycosyl chlorides as glycosyl donors by demonstrating that these compounds can be activated under organocatalytic conditions with urea or thiourea-based catalysts, used along with stoichiometric additives. We have recently reported that glycosyl chlorides can be activated with catalytic ferric chloride.[8] Glycosylation with benzylated donors under these benign reaction conditions was typically completed in a couple of hours. The activation of electronically deactivated, benzoylated glycosyl chlorides could also be achieved with catalytic ferric chloride. However, these glycosylations were rather slow (16 h), and the yields remained moderate, typically within a 60–80% range. Nevertheless, these results were a significant improvement over other promoters and catalysts that previously failed to activate those unreactive substrates.
Over the course of our recent study with glycosyl bromides, we discovered that slow silver-promoted glycosylations can be dramatically accelerated in the presence of acid additives. Thus, glycosylation in the presence of 2.0 equiv of silver(I) oxide that typically requires many hours or even days to complete, became very swift (5–15 min) upon addition of 0.20 equiv of trimethylsilyl trifluoromethanesulfonate (TMSOTf).[9] An effort dedicated to studying the reaction mechanism, made it possible to reduce the amount of Ag2O to only 0.50 equiv and replace TMSOTf with TfOH.[10] Although we achieved a significantly improved outcome of the Koenigs-Knorr-like glycosylation reactions, a few limitations and uncertainties remained. First, glycosidation of bromide donors containing the nitrogen atom such as derivatives of glucosamine were very slow and provided poor yields. This limitation was presumably due to the competing protonation of the nitrogen atom with TfOH that led to partial deactivation of the donor. Second, lower reaction rates and fair product yields were also seen with thioglycoside acceptors. This phenomenon was presumably due to the competing interaction of silver oxide with the sulfur atom that led to partial deactivation of the promoter system. Additionally, while glycosidation of supposedly deactivated, benzoylated glycosyl bromides was swift and high-yielding, glycosidation of benzylated glycosyl bromides was much slower and hence much less efficient.
To improve the outcome of this new reaction, we turned our attention to investigating glycosyl chlorides as donors. Mechanistically, the activation of bromides and chlorides would be similar. As proposed in our previous study, after initial interaction of the donor with Ag2O, the resulting species A produce a strongly ionized species B due to interaction of TfOH (Scheme 1). This intermediate will rapidly dissociate producing AgX that precipitates out of the solution. Also produced at this stage is AgOH that loses water regenerating Ag2O. Water is then scavenged by molecular sieves present in all of our reaction. Depending on the protecting group at C-2, glycosyl cation C will be stabilized either via an acyloxonium (2-O-benzoyl) or oxacarbenium (2-O-benzyl) ion. The subsequent nucleophilic attack of the glycosyl acceptor (ROH) occurs with regeneration of TfOH that is available for the next catalytic cycle. Since this reaction is driven by the irreversible formation of the AgX bond, we reasoned that glycosyl chloride activation would be favored by silver chloride formation.[11] This could also minimize propensity of silver interact with other heteroatoms in the system. Reported herein is our study of glycosyl chlorides using the new acid-catalyzed Koenigs-Knorr promoter system.
Scheme 1.
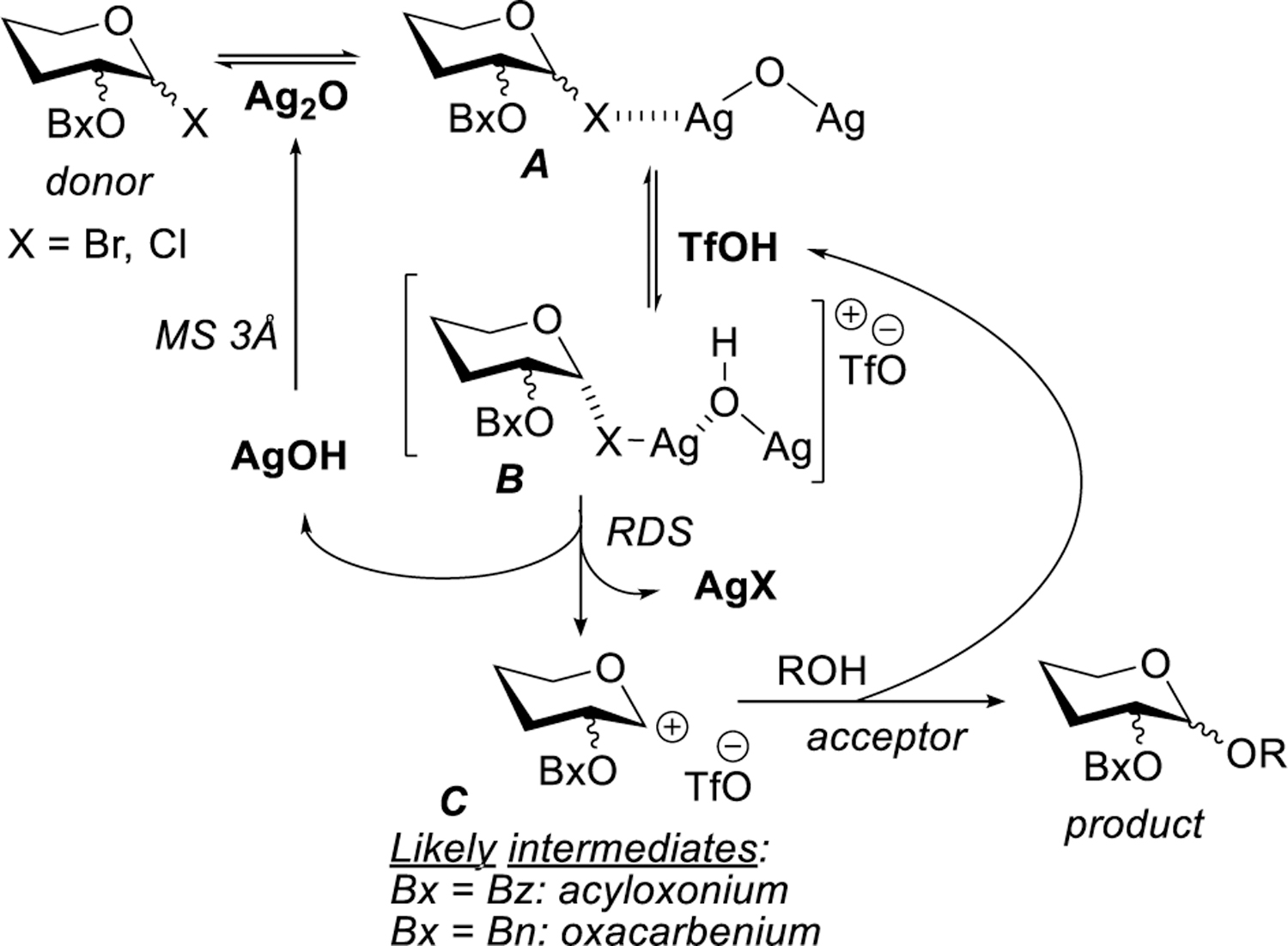
Activation of glycosyl halides in the presence of Ag2O/TfOH
Results and Discussion
We first investigated glycosidation of benzoylated glycosyl chlorides 1–3 (Table 1)[12] with a series of standard glycosyl acceptors 4–7 (Figure 1).[13] After preliminary screening of the reaction conditions we determined that the activation of glucosyl chloride 1 with 0.50 equiv of silver(I) oxide and 0.25 equiv of TfOH offers the best combination of rates and yields. As listed in Table 1, glycosylation of primary acceptor 4 gave disaccharide 12 in 98% yield in 30 min (entry 1). These optimized conditions compare very favorably with those used for the glycosyl bromide activations, 0.50 equiv of silver(I) oxide and 0.35–0.40 equiv of TfOH.[10] Glycosylation of secondary acceptors 5–7 gave similar results. Thus, glycosylation of a relatively unreactive 4OH acceptor 5 afforded disaccharide 13 in 90% yield in 30 min (entry 2). Glycosylations of acceptors 6 and 7 were equally impressive. Disaccharides 14 and 15 were obtained in 98% and 91% yield, respectively, in 30 min (entries 3 and 4). As clearly evident from our results, we have not seen any noticeable decline in reaction rates with sterically hindered glycosyl acceptors. While 0.25 equiv TfOH worked universally for all acceptors investigated, some reactions could be smoothly driven to completion using as little as 0.15 equiv of TfOH (data is not shown). All glycosidations of chloride 1 were completely 1,2-trans diastereoselective due to the assistance of the neighboring participating group. As expected, glycosidations of benzoylated glycosyl chloride 1 in the presence of Ag2O only, standard Koenigs-Knorr reaction conditions, were very sluggish, and only trace amounts of disaccharides were observed after 48 h. The reaction did not proceed at all in the presence of TfOH-only proving the cooperative catalysis nature of the activation pathway.
Table 1.
Glycosidation of benzoylated glycosyl chlorides 1–3 in the presence of Ag2O and TfOH
 | ||
|---|---|---|
| Entry | Donor + Acceptor (TfOH equiv) | Product, Yielda |
| 1 | 1 + 4 (o.25) |  |
| 12, 98% | ||
| 2 | 1 + 5 (o.25) |  |
| 13, 90% | ||
| 3 | 1 + 6 (o.25) | 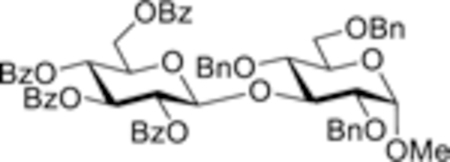 |
| 14, 98% | ||
| 4 | 1 + 7 (o.25) |  |
| 15, 91% | ||
| 5 | 1 + 8 (o.25) |  |
| 16, 98% | ||
| 6 | 2 + 4 (o.50) |  |
| 17, 99% | ||
| 7 | 2 + 5 (o.50) |  |
| 18, 99% | ||
| 8 | 2 + 6 (o.50) |  |
| 19, 99% | ||
| 12 | 2 + 7 (o.50) |  |
| 20, 99% | ||
| 9 | 2 + 9 (0.50) |  |
| 21, 73% | ||
| 10 | 2 + 10 (0.50) |  |
| 22, 75% | ||
| 11 | 2 + 11 (0.50) |  |
| 23, 60% | ||
| 13 | 3 + 4 (o.50) |  |
| 24, 98% | ||
| 14 | 3 + 5 (o.50) |  |
| 25, 98% | ||
| 15 | 3 + 6 (o.50) |  |
| 26, 98% | ||
| 16 | 3 + 7 (o.50) |  |
| 27, 99% | ||
– all yields are isolated yields after column chromatography
Figure 1.

Standard glycosyl acceptors 4–7 and thioglycoside acceptors 8–11 used in this study
Glycosylation of thioglycoside acceptor 8 also proceeded very smoothly affording disaccharide 16 in 98% yield in 30 min (entry 5). This glycosylation reaction represents an important strategic step because it encompasses selective activation of one leaving group over another.[14] This approach is commonly used in expeditious oligosaccharide synthesis[15] because disaccharide 16 can be used as the glycosyl donor for subsequent chain elongation directly. However, glycosylation of thioglycoside acceptors was somewhat inefficient in our previous studies with ferric chloride promoter[8] or with glycosyl bromides as donors.[9–10] In addition, these glycosylations could be prone to competing aglycone transfer reactions,[16] which was not observed in this case.
With notable success with glucosyl donor 1, we turned our attention to investigating benzoylated chlorides of the D-galacto and D-manno series, 2 and 3, respectively. Glycosidations of both mannosyl and galactosyl chlorides were somewhat less efficient under the established reaction conditions for glucosyl chloride 1, 0.50 equiv of silver(I) oxide and 0.25 equiv of TfOH. The reactions were slower, and remained incomplete, even in prolonged experiments (16 h). After a brief screening of the reaction conditions, we found that increasing the amount of TfOH to 0.50 equiv is optimal for driving these reactions to completion. As in case of glucosyl chloride 1, we have achieved very effective, rapid, and high-yielding reactions with both primary and secondary glycosyl acceptors. As listed in Table 1, glycosylation of acceptors 4–7 with galactosyl chloride 2, produced the respective disaccharides 17–20 in 30 min nearly quantitatively (99% yield in all experiments, entries 6–9). A large-scale glycosylation using 1.0 gram of donor 2 and acceptor 5 was also performed. During this experiment glycosyl acceptor 5 was completely consumed and disaccharide 18 was obtained in 83% yield.
To elaborate on our previous success with glycosylating thioglycoside acceptor 8 (see entry 5) we investigated other thioglycoside acceptors 9–11.[17] Glycosylations of primary thioglycoside acceptors 9 and 10 with galactosyl chloride 2 gave promising results producing the respective disaccharides 21 and 22 in 73–75% yields in 30 min. Glycosylation of a less reactive secondary acceptor 11 led to disaccharide 23 in a moderate yield of 60% in 1 h. The lower yield, in part, can be attributed to small amounts of a by-product resulting from a competing aglycone transfer reaction.[16] Nevertheless, this result favorably compares to inefficient glycosylations of thioglycoside acceptors in our previous studies with glycosyl bromides as donors.[9–10]
A very similar outcome was achieved with mannosyl donor 3. Thus, glycosylation of acceptors 4–7 afforded the corresponding disaccharides 24–27 in 30 min in 98–99% yield (entries 10–13). These glycosylations were also 1,2-trans selective, and β-galactosides and α-mannosides were all obtained with complete diastereoselectivity. As evident from the product yields, these glycosylations are spot-to-spot, and are practically free of by-products beyond trace amounts of hemiacetal resulting from hydrolysis of the donor and 1→1-linked disaccharide resulting from glycosylation of the hemiacetal. While 0.50 equiv TfOH worked universally for all acceptors investigated, some reactions with galactosyl and mannosyl chlorides could be smoothly driven to completion using as little as 0.25 equiv of TfOH.
Having obtained excellent results with all per-benzoylated chlorides, we turned our attention to investigating per-benzylated chlorides 28–30 (Table 2).[8, 12a–c, 18] It is well established that the building block reactivity and stereoselectivity can be modulated through the choice of protecting groups.[19] According to Fraser-Reid’s seminal work on the armed-disarmed strategy, benzylated (electronically activated, armed) building blocks are more reactive than their acylated (Bz, disarmed) counterparts.[20] Our recent discovery that benzylated glycosyl bromide does not follow this trend when activated in the presence of Ag+/TMSOTf or TfOH was striking.[9–10] The observed lower reactivity of benzylated glycosyl bromides resulted in reduced yields and their glycosidations required longer reaction times to complete.
Table 2.
Glycosidation of benzylated glycosyl chlorides 28–30 in the presence of Ag2O and TfOH
 | ||
|---|---|---|
| Entry | Donor + Acceptor (TfOH equiv) | Product, yield,a ratio α/β |
| 1 | 28 + 4 (o.25) |  |
| 31, 97%, α/β = 1.1/1 | ||
| 2 | 28 + 5 (o.25) |  |
| 32, 99%, α/β = 1.5/1 | ||
| 3 | 28 + 6 (o.25) |  |
| 33, 99%, α/β = 1.3/1 | ||
| 4 | 28 +7 (0.25) |  |
| 34, 95%, α/β = 1.5/1 | ||
| 5 | 29+ 4 (0.50) |  |
| 35, 99%, α/β = 1.2/1 | ||
| 6 | 29 + 5 (0.50) |  |
| 36, 99%, α/β = 2.4/1 | ||
| 7 | 29 + 6 (0.50) | 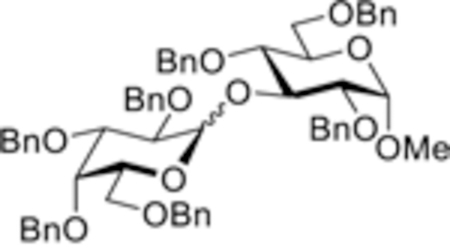 |
| 37, 99%, α/β = 2.4/1 | ||
| 8 | 29 + 7 (0.50) |  |
| 38, 99%, α/β = 4.9/1 | ||
| 9 | 30 + 4 (0.50) |  |
| 39, 98%, α/β = 1.3/1 | ||
| 10 | 30 + 5 (0.50) |  |
| 40, 98%, α only | ||
| 11 | 30 + 6 (0.50) |  |
| 41, 98%, α only | ||
| 12 | 30 + 7 (0.50) |  |
| 42, 98%, α/β =3.6/1 | ||
– all yields are isolated yields after column chromatography
Therefore, investigation of benzylated glycosyl chlorides under these reaction conditions appealed to us more than just broadening the scope of the methodology. A reaction between glycosyl donor 28 and primary glycosyl acceptor 4 in the presence of 0.50 equiv of silver(I) oxide and 0.25 equiv of TfOH afforded disaccharide 31 in 97% yield is only 30 min (α/β = 1.1/1, Entry 1, Table 2). This result was quite pleasing, particularly in the light of results previously achieved with the respective glucosyl bromide under similar reaction conditions (18 h, 46%).[9] The outcome of this reaction also indicates no particular reactivity difference between benzoylated and benzylated chlorides under these reaction conditions. No reactivity difference was verified by a direct competition experiment between donors 1 and 28. However, it is possible that reducing the equivalence of TfOH or modulating other factors could lead to reaction conditions under which the reactivity difference could be observed. Glycosylation of secondary glycosyl acceptors 5–7 with donor 28 was equally impressive. The corresponding disaccharides 32–34 were obtained in 95–99% yield in 30 min (α/β = 1.3–1.5 /1, entries 2–4).
With a notable success with glucosyl donor 28, we turned our attention to investigating benzylated chlorides of the D-galacto and D-manno series, 29 and 30, respectively. Again, a majority of glycosidations of galactosyl and mannosyl chlorides in the presence of 0.25 equiv of TfOH were somewhat slower, practically stalled after 30 min, and remained incomplete even after 16 h. Like in the case of benzoylated chlorides of these series, increasing the amount of TfOH to 0.50 equiv was found to be optimal for driving all of these reactions to completion. Very effective, rapid, and high-yielding reactions we achieved with both primary and secondary glycosyl acceptors. As listed in Table 2, glycosylation of acceptors 4–7 with galactosyl chloride 29 afforded the respective disaccharides 35–38 in 30 min nearly quantitatively (99% yield in all experiments, α/β = 1.2–4.9/1, entries 5–8). Similarly, glycosylation of acceptors 4–7 with mannosyl chloride 29 produced the corresponding disaccharides 39–42 in 98% yield in all experiments in 30 min (α/β = from 1.3/1 to α-only, entries 9–12). We note that some reactions between galactosyl and mannosyl chlorides and reactive acceptors could be smoothly driven to completion using as little as 0.25 equiv of TfOH.
Having obtained excellent results with all per-benzoylated and per-benzylated chlorides of neutral sugars, we turned our attention to investigating N-phthaloyl protected glucosamine chloride 43 (Table 3). This was of particular interest because previous attempts to glycosidate glucosamine bromides resulted in poor yields and long reaction times. As aforementioned, this was attributed to the competing protonation of the nitrogen atom that led to partial deactivation of the donor, and further attempts to glycosidate glucosamine bromides were ceased. It has become a common knowledge that 2-aminosugars may have a very different reactivity profile in comparison to their neutral sugar counterparts.[21] Glycosidation of aminosugars often requires different methods specifically designed for these substrates. We also included sialyl chlorides 44 and 45 (Table 3).[22] Being a common aminosugar in mammalian and microbial glycans, sialic acids represent a special case of glycosylation that spans beyond effects of the amino group functionality.[23] The chemical synthesis of α-sialosides is considered challenging, and mild methods enhancing the product yields and suppressing common side reactions (elimination and hydrolysis) are needed.
Table 3.
Glycosidation of 2-phthalimido chloride 43 and sialyl chlorides 44 and 45 with glycosyl acceptors 4–7 or 50 in the presence of Ag2O and TfOH
 | ||
|---|---|---|
| Entry | Donor + Acceptor (Ag2O equiv) |
Product, yield,a ratio α/β |
| 1 | 43 + 4 (1.5) |  |
| 46, 97% | ||
| 2 | 43 + 5 (1.5) |  |
| 47, 76% | ||
| 3 | 43 +6 (1.5) |  |
| 48, 72% | ||
| 4 | 43 +7 (1.5) |  |
| 49, 68% | ||
| 5b |  |
 |
| 44 + 50 (2.0) | 51, traces | |
| 6b | 45 + 49 (2.0) |  |
| 52, 97%, α/β = 1/1.7 | ||
– all yields are isolated yields after column chromatography;
- performed at –72 oC (2 h), then at rt for 22 h
First test reaction with 2-phthalimido chloride donor 43 showed that the promoter deactivation could also be the case with glycosyl chlorides. Reactions with 0.50 or even 1.0 equiv of Ag2O were incomplete and led to lower product yields. Further optimization of our reaction conditions in application to glycosidation of N-phthaloyl protected glucosamine chloride 43 showed the necessity to increase the amount of Ag2O to 1.50 equiv, whereas 0.50 equiv of TfOH was sufficient. Under these modified reaction conditions, glycosidation of glycosyl donor 43 with primary acceptor 4 gave disaccharide 46 in 97% yield in 30 min (entry 1, Table 3). Glycosylations of secondary glycosyl acceptors 5–7 with donor 43 were somewhat less efficient. The corresponding disaccharides 47–49 were obtained in commendable yields of 68–76% in 30 min (entries 2–4). These glycosylations were all completely 1,2-trans stereoselective due to the participation of the 2-phthalimido group. Prolonged experiments (beyond standard 30 min) did not help to achieve higher yields.
Lastly, we investigated glycosidation of sialyl chlorides. Unfortunately, reaction between sialyl donor 44 and galactosyl acceptor 50 only produced disaccharide 51 in trace amounts, even in the presence of 2.0 equiv sialyl donor 44 and up to 2 equiv of Ag2O (to donor, entry 5, Table 3). Having attributed this result to the deactivating nature of the acetamido moiety[24] we turned to investigating the N-acetylacetamido donor 45 because this type of protection is known to enhance the reactivity of sialyl donors.[25] Glycosylation between 45 (2.0 equiv) and 50 was conducted in the presence of 2.0 equiv Ag2O and 0.50 equiv TfOH (to donor 45). To prevent competing eliminations that hamper many types of sialylation reactions, the reaction was started at −78 °C and after 2 h the reaction was allowed to slowly warm to rt. As a result, we obtained disaccharide 52 in 97% yield in 24 h (α/β = 1/1.7, entry 6). This result is on a par or even surpasses those obtained with modern sialyl donors.[23b]
In conclusion, this study showed how glycosyl chlorides can be activated in a similar manner as glycosyl bromides using 0.50 equiv of Ag2O and 0.25–0.50 equiv of TfOH. Efficient glycosylations of benzoylated glucosyl, galactosyl, and mannosyl chlorides have all been performed with a variety of differently protected primary and secondary acceptors providing high yields and fast reaction times. Furthermore, glycosidations of benzylated glucosyl, galactosyl, and mannosyl chlorides have been performed with similar efficiency. Lastly, nitrogen containing glucosamine and sialic acid chlorides were also successfully glycosidated, but these reactions required excess silver oxide. Another convenient feature of this glycosylation is that the progress of this reaction can be monitored by eye, and the completion of the reaction can be judged by the disappearance of characteristic dark color of Ag2O. This is because since only the minimal amount of Ag2O is used to catalyze this reaction, it gets entirely converted in AgCl, which is a white crystalline solid. Attempts to improve the stereoselectivity of glycosidation of glycosyl bromides and chlorides using the silver salts and TfOH promoter system are currently underway in our laboratory.
Experimental
General.
Column chromatography was performed on silica gel 60 (70–230 mesh), reactions were monitored by TLC on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. Solvents were removed under reduced pressure at <40 °C. CH2Cl2 and ClCH2CH2Cl (1,2-DCE) were distilled from CaH2 directly prior to application. Pyridine was dried by refluxing with CaH2 and then distilled and stored over molecular sieves (3 Å). Anhydrous DMF was used as it is. Molecular sieves (3 Å or 4 Å), used for reactions, were crushed and activated in vacuo at 390 °C during 8 h in the first instance and then for 2–3 h at 390 °C directly prior to application. Optical rotations were measured using a Jasco polarimeter. 1H-NMR spectra were recorded in CDCl3 at 300 or 600 MHz, 13C-NMR spectra were recorded in CDCl3 at 75 or 151 MHz. Accurate mass spectrometry determinations were preformed using Agilent 6230 ESI TOF LCMS mass spectrometer
Synthesis of Glycosyl Donors
2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosyl chloride (1)
was obtained from 2,3,4,6-tetra-O-benzoyl-D-glucopyranose[26] as described previously,[8] and its analytical data for were the same as those reported previously.[12a, b]
2,3,4,6-Tetra-O-benzoyl-α-D-galactopyranosyl chloride (2).
Thionyl chloride (294 mg, 2.47 mmol) was added dropwise to a solution of 2,3,4,6-tetra-O-benzoyl-D-galactopyranose[27] (705 mg, 1.24 mmol) in 1,2-dichloroethane (10 mL) containing N,N-dimethylformamide (0.5 mL) and the resulting mixture was stirred under argon for 20 min at 0 °C. After that, the volatiles were removed under reduced pressure. The residue was dissolved in a mixture of ethyl acetate/hexane (25 mL, 1/1, v/v) and filtered through a pad of silica gel (10 g). The pad of silica gel was additionally eluted with a mixture of ethyl acetate and hexane (75 mL, 1/1, v/v) and the combined eluate was concentrated in vacuo to afford the title compound as colorless foam in 94% yield (681 mg, 1.11 mmol). Analytical data for 2 were essentially the same as reported previously.[12c]
2,3,4,6-Tetra-O-benzoyl-α-D-mannopyranosyl chloride (3).
A mixture of oxalyl chloride (820 mg, 1.48 mmol) in dichloromethane (6.0 mL) and added dropwise to a solution of 2,3,4,6-tetra-O-benzoyl-D-mannopyranose[28] (1.28 g, 2.15 mmol) in dichloromethane (20 mL) containing N,N-dimethylformamide (0.7 mL) and the resulting mixture was stirred under argon for 1.5 h at 0 °C. After that, the volatiles were removed under reduced pressure. The residue was dissolved in a mixture of ethyl acetate/ hexane (30 mL, 1/1, v/v) and filtered through a pad of silica gel (15 g). The pad of silica gel was additionally eluted with a mixture of ethyl acetate and hexane (90 mL, 1/1, v/v) and the combined eluate was concentrated in vacuo to afford the title compound as colorless foam in 94% yield (1.19 g, 1.77 mmol). Analytical data for 3 were essentially the same as that reported previously.[29]
2,3,4,6-Tetra-O-benzyl-α-D-glucopyranosyl chloride (28)
was obtained from 2,3,4,6-tetra-O-benzyl-D-glucopyranose[30] as described previously,[8] and its analytical data for were the same as those reported previously.[12b]
2,3,4,6-Tetra-O-benzyl-α-D-galactopyranosyl chloride (29)
was obtained from 2,3,4,6-tetra-O-benzyl-D-galactopyranose[31] as described previously,[8] and its analytical data were the same as those reported previously.[12c]
2,3,4,6-Tetra-O-benzyl-α-D-mannopyranosyl chloride (30)
was obtained from 2,3,4,6-tetra-O-benzyl-D-mannopyranose[32] as described previously,[8] and its analytical data were the same as those reported previously.[18b]
3,4,6-Tri-O-benzoyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl chloride (43).
Thionyl chloride (0.45 g, 3.76 mmol) was added dropwise to a solution of 3,4,5-tri-O-benzoyl-2-deoxy-2-phthalimido-D-glucopyranose[33] (1.17 g, 1.88 mmol) in dichloroethane (70 mL) containing DMF (5.0 mL) and the resulting mixture was stirred under argon for 1 h at 0 °C. The volatiles were then removed under reduced pressure. The residue was dissolved in a mixture of ethyl acetate/ hexane (50 mL, 1/1, v/v) and passed through a pad of silica gel (25 g). The pad of silica gel was additionally eluted with a mixture of ethyl acetate/ hexane (75 mL, 1/1, v/v) and the combined eluate was concentrated under reduced pressure to afford the title compound as a white foam in 87% yield (1.05 g, 1.64 mmol). Analytical data for 43: Rf = 0.70 (ethyl acetate/hexane, 1/1, v/v); [α]D21 61.3 (c 3.2, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.07 (d, J = 7.6 Hz, 2H, aromatic), 7.95 – 7.65 (m, 8H, aromatic), 7.62 – 7.39 (m, 5H, aromatic), 7.38 – 7.19 (m, 4H, aromatic), 6.41 (d, J1,2 = 9.3 Hz, 1H, H-1), 6.29 (dd, J3,4 = 9.7 Hz, 1H, H-3), 5.81 (dd, J4,5 =10.1 Hz, 1H, H-4), 4.80 (dd, J2,3 = 9.9 Hz, 1H, H-2), 4.68 (dd, J6a,6b = 12.4, 1H, H-6a), 4.53 (dd, 1H, H-6b), 4.38 (m, J5,6a = 2.5 Hz, J5,6b = 4.8 Hz, 1H, H-5) ppm; 13C NMR (75 MHz, CDCl3): δ 166.2, 165.6, 165.1, 134.5, 133.6, 133.5, 133.3, 131.2, 129.9 (x4), 129.8 (x3), 129.5, 128.5 (x7), 128.4 (x3), 128.3, 123.9 (x2), 85.8, 76.0, 71.0, 69.3, 62.8, 57.8 ppm; HRMS [M+Na]+ calcd for C35H26ClNO9Na 662.1188, found 662.1201.
Methyl (4,7,8,9-tetra-O-acetyl-5-acetamido-3,5-dideoxy-β-D-glycero-D-galacto-non-2-ulopyranosyl chloride)onate (44)
was obtained from methyl (2,4,7,8,9-penta-O-acetyl-5-acetamido-3,5-dideoxy-D-glycero-D-galacto-non-2-ulopyranos)onate[34] as described previously,[22a] and its analytical data was in accordance with that previously reported.[22b]
Methyl (4,7,8,9-tetra-O-acetyl-5-(N-acetyl)acetamido-3,5-dideoxy-β-D-glycero-D-galacto-non-2-ulopyranosyl chloride)onate (45)
was obtained from methyl (2,4,7,8,9-penta-O-acetyl-5-(N-acetyl)acetamido-3,5-dideoxy-D-glycero-D-galacto-non-2-ulopyranos)onate[35] as described previously, and its analytical data was in accordance with that previously reported.[22c]
Synthesis of Disaccharides
General procedure for glycosylations in the presence of Ag2O and TfOH.
A mixture of a glycosyl donor (0.05 mmol), glycosyl acceptor (0.035 mmol), and freshly activated molecular sieves (3 Å, 150 mg) in CH2Cl2 (1.0 mL) was stirred under argon for 1 h at rt. The mixture was cooled to 0 °C, Ag2O (0.025 mmol) was added and the resulting mixture was stirred under argon for 10 min at 0 °C. TfOH (0.25 or 0.50, see Tables) was added and the reaction mixture was stirred under argon for 30 min at 0 °C. After that, the solid was filtered off and washed sucessively with CH2Cl2. The combined filtrate (~40 mL) was washed with saturated aq. NaHCO3. (10 mL). The organic phase was separated, dried with magnesium sulfate, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate – toluene gradient elution) to afford the respective disaccharides in yields and stereoselectivites listed in Tables and below. Anomeric ratios (or anomeric purity) were determined by comparison of the integral intensities of relevant signals in 1H NMR spectra.
Methyl 6-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (12)
was obtained from donor 1 and acceptor 4[13a] under the general glycosylation method as a colorless foam in 98% yield. Analytical data for 12 was in accordance with that previously reported.[36]
Methyl 4-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-2,3,6-tri-O-benzyl-α-D-glucopyranoside (13)
was obtained from donor 1 and acceptor 5[13a] under the general glycosylation method as a colorless foam in 90% yield. Analytical data for 13 was in accordance with that previously reported.[36]
Methyl 3-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-2,4,6-tri-O-benzyl-α-D-glucopyranoside (14)
was obtained from donor 1 and acceptor 6[13a] under the general glycosylation method as a colorless foam in 98% yield. Analytical data for 14 was in accordance with that previously reported.[13a]
Methyl 2-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-3,4,6-tri-O-benzyl-α-D-glucopyranoside (15)
was obtained from donor 1 and acceptor 7[13a] under the general glycosylation method as a colorless foam in 91% yield. Analytical data for 15 was in accordance with that previously reported.[37]
Ethyl 6‐O‐(2,3,4,6‐tetra‐O‐benzoyl‐β‐D‐glucopyranosyl)‐2,3,4‐tri‐O‐benzoyl‐1‐thio‐β‐D‐glucopyranoside (16)
was obtained from donor 1 and acceptor 8[13b] under the general glycosylation method as a colorless foam in 98% yield. Analytical data for 16 was in accordance with that previously reported.[38]
Methyl 6-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (17)
was obtained from donor 2 and acceptor 4 under the general glycosylation method as a colorless foam in 99% yield. Analytical data for 17 was in accordance with that previously reported.[39]
Methyl 4-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-2,3,6-tri-O-benzyl-α-D-glucopyranoside (18)
was obtained from donor 2 and acceptor 5 under the general glycosylation method as a colorless foam in 99% yield. Analytical data for 18 was in accordance with that previously reported.[37]
Methyl 3-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-2,4,6-tri-O-benzyl-α-D-glucopyranoside (19)
was obtained from donor 2 and acceptor 6 under the general glycosylation method as a colorless foam in 99% yield. Analytical data for 19 was in accordance with that previously reported.[40]
Methyl 2-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-3,4,6-tri-O-benzyl-α-D-glucopyranoside (20)
was obtained from donor 2 and acceptor 7 under the general glycosylation method as a colorless foam in 99% yield. Analytical data for 20 was in accordance with that previously reported.[40]
Tolyl 6-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-2,3,6-tri-O-benzoyl-1-thio-β-D-glucopyranoside (21)
was obtained from donor 2 and acceptor 9[17a] under the general glycosylation method as a colorless foam in 73% yield. Analytical data for 21: Rf = 0.72 (ethyl acetate/toluene, 1/4, v/v); [α]D23 63.6 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.06 (m, 4H, aromatic), 7.94 (m, 4H, aromatic), 7.87 – 7.69 (m, 6H, aromatic), 7.67 – 7.04 (m, 26H, aromatic), 6.00 (dd, J4’,5’ = 3.1 Hz, 1H, H-4’), 5.81 (dd, J2’,3’ = 8.0 Hz, 1H, H-2’), 5.74 (dd, J4,5 = 9.5 Hz, 1H, H-4), 5.58 (dd, J3’,4’ = 10.4 Hz, 1H, H-3’), 5.30 (dd, J2,3 = 9.7 Hz, 1H, H-2), 5.26 (dd, J3,4 = 9.4 Hz, 1H, H-3), 5.03 (d, J1’,2’ = 8.0 Hz, 1H, H-1’), 4.80 (d, J1,2 = 9.9 Hz, 1H, H-1), 4.60 (dd, 1H, H-6b’), 4.39 (dd, J6a’,6b’ = 11.2 Hz, 1H, H-6a’), 4.28 (dd, J5’,6a’ = 6.6, J5’,6b’ = 6.4Hz, 1H, H-5’), 4.12 – 3.91 (m, 3H, H-5, 6a, 6b), 2.30 (s, 3H, CH3) ppm; 13C NMR (75 MHz, CDCl3): δ 166.1, 165.7, 165.6, 165.4 (x2), 165.0, 138.9, 134.0 (x2), 133.6 (x2), 133.4, 133.3 (x3), 130.1 (x3), 129.9 (x6), 129.8 (x3), 129.4 (x2), 129.3, 129.0, 128.8 (x2), 128.7 (x2), 128.6 (x5), 128.5 (x4), 128.4 (x6), 128.3 (x4), 127.5, 101.6, 85.9, 78.7, 74.1, 71.8, 71.4, 70.4, 69.7, 69.4, 68.3, 68.1, 61.9, 21.2 ppm; HRMS [M+Na]+ calcd for C68H56O17SNa 1199.3130, found 1199.3150.
Phenyl 6-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-2,3,4-tri-O-benzyl-1-thio-β-D-glucopyranoside (22)
was obtained from donor 2 and acceptor 10[17b] under the general glycosylation method as a colorless foam in 75% yield. Analytical data for 22: Rf = 0.73 (ethyl acetate/toluene, 1/4, v/v); [α]D23 55.6 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.07 (m, 4H, aromatic), 7.82 (m, 4H, aromatic), 7.69 – 7.02 (m, 32H, aromatic), 5.97 (dd, J4’,5’ = 3.0 Hz, 1H, H-4’), 5.85 (dd, J2’,3’ = 10.3 Hz, 1H, H-2’), 5.53 (dd, J3’,4’ = 3.3 Hz, 1H, H-3’), 4.88 (d, J1’,2’ = 7.8 Hz, 1H, H-1’), 4.78 (dd, 2J = 10.6 Hz, 2H, CH2Ph), 4.71 (dd, 2J = 10.6 Hz, 2H, CH2Ph) 4.65 (dd, 1H, H-6b’) 4.63 (dd, 2J = 11.0 Hz, 2H, CH2Ph) 4.62 (dd, J2,3 = 9.6 Hz, 1H, H-2), 4.44 (d, J1,2 = 10.1 Hz, 1H, H-1), 4.41 (dd, J6a’,6b’ = 7.0 Hz, 1H, H-6a’), 4.22 (dd, 1H, H-6b), 4.20 (dd, 1H, H-5’), 3.89 (dd, J6a,6b = 11.4, 1H, H-6a), 3.61 (dd, J3,4 = 8.7 Hz, 1H, H-3), 3.49 (m, J5,6a = J5,6b = 4.6 Hz, 1H, H-5), 3.43 (dd, J4,5 = 8.2 Hz, 1H, H-4), 3.40 (dd, J2,3 = 9.1 Hz, 1H, H-2) ppm; 13C NMR (75 MHz, CDCl3): δ 166.1, 165.6, 165.2, 138.3, 138.0, 137.8, 133.6, 133.4 (x2), 133.2 (x2), 130.1, 129.9 (x4), 129.8, 129.5, 129.3, 129.2 (x2), 129.1 (x3), 128.8, 128.7 (x2), 128.6 (x2), 128.5 (x6), 128.4 (x4), 128.3 (x5), 128.0, 127.9 (x2), 127.8 (x2), 125.4, 101.3, 87.2, 86.6, 80.4, 78.9 (x2), 77.4, 75.8, 75.5, 75.0, 71.8, 71.3, 69.7, 68.1, 67.5, 61.9, 21.5 ppm; HRMS [M+Na]+ calcd for C67H60O14SNa 1143.3596, found 1043.3608.
Ethyl 4-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-2,3,6-tri-O-benzyl-1-thio-β-D-glucopyranoside (23)
was obtained from donor 2 and acceptor 11[17c] under the general glycosylation method as a colorless foam in 60% yield. Analytical data for 23: Rf = 0.70 (ethyl acetate/toluene, 1/4, v/v); [α]D23 3.2 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.10–7.7o (m, 8H, aromatic), 7.65 – 7.04 (m, 27H, aromatic), 5.86 (br. d, 1H, H-4’), 5.72 (dd, J2’,3’ = 8.5 Hz, 1H, H-2’), 5.39 (dd, J3’,4’ = 3.2 Hz, 1H, H3’), 5.07 (dd, 2J = 11.1 Hz, 2H, CH2Ph), 4.97 (d, J1’,2’ = 7.9 Hz, 1H, H-1’), 4.77 (dd, 2J = 10.3 Hz, 2H, CH2Ph), 4.55 (dd, 2J = 10.0 Hz, 2H, CH2Ph), 4.40 (dd, 1H, H-6b’), 4.38 (d, J1,2 = 11.6 Hz, 1H, H-1), 4.19 (dd, J6a,6b = 9.4 Hz, 1H, H-6a’) 4.14 (dd, J4,5 = 9.4 Hz, H-4), 3.94 (m, J5’,6a = J5’,6b = 6.6 Hz, 1H, H-5’), 3.75–3.54 (m, 3H, H-3, 6a, 6b), 3.41 (dd, J2,3 = 9.2 Hz, 1H, H-2), 3.24 (dd, 1H, H-5), 2.78 – 2.56 (m, 2H, SCH2CH3), 1.28 (t, J = 7.0 Hz, 3H, SCH2CH3) ppm; 13C NMR (151 MHz, CDCl3): δ 165.8, 165.4 (x2), 164.9, 139.0, 138.1, 138.0, 133.4 (x2), 133.3, 133.2, 129.8 (x5), 129.7 (x2), 129.6 (x2), 129.5, 129.1, 129.0, 128.8, 128.6 (x2), 128.5 (x3), 128.4 (x2), 128.3 (x2), 128.2 (x3), 128.1 (x2), 128.0 (x2), 127.7, 127.3 (x2), 100.3, 85.1, 84.4, 81.0, 78.6, 76.5, 75.5, 75.3, 73.5, 71.8, 71.1, 70.4, 67.9 (x2), 61.4, 24.8, 15.1 ppm; HRMS [M+Na]+ calcd for C63H60O14SNa 1095.3596, found 1095.3609.
Methyl 6-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (24)
was obtained from donor 3 and acceptor 4 under the general glycosylation method as a colorless foam in 98% yield. Analytical data for 24 was in accordance with that previously reported.[37]
Methyl 4-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-2,3,6-tri-O-benzyl-α-D-glucopyranoside (25)
was obtained from donor 3 and acceptor 5 under the general glycosylation method as a colorless foam in 98% yield. Analytical data for 25 was in accordance with that previously reported.[36]
Methyl 3-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-2,4,6-tri-O-benzyl-α-D-glucopyranoside (26)
was obtained from donor 3 and acceptor 6 under the general glycosylation method as a colorless foam in 98% yield. Analytical data for 26 was in accordance with that previously reported.[41]
Methyl 2-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-3,4,6-tri-O-benzyl-α-D-glucopyranoside (27)
was obtained from donor 3 and acceptor 7 under the general glycosylation method as a colorless foam in 98% yield. Analytical data for 27 was in accordance with that previously reported.[41]
Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranosyl)-α-D-glucopyranoside (31)
was obtained from donor 28 and acceptor 4 under the general glycosylation method as a colorless foam in 97% yield (α/β = 1.1/1). Analytical data for 31 was in accordance with that previously reported.[42]
Methyl 2,3,6-tri-O-benzyl-4-O-(2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranosyl)-α-D-glucopyranoside (32)
was obtained from donor 28 and acceptor 5 under the general glycosylation method as a colorless foam in 99% yield (α/β = 1.5/1). Analytical data for 32 was in accordance with that previously reported.[43]
Methyl 2,4,6-tri-O-benzyl-3-O-(2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranosyl)-α-D-glucopyranoside (33)
was obtained from donor 28 and acceptor 6 under the general glycosylation method as a colorless foam in 99% yield (α/β = 1.3/1). Analytical data for 33 was in accordance with that previously reported.[44]
Methyl 3,4,6-tri-O-benzyl-2-O-(2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranosyl)-α-D-glucopyranoside (34)
was obtained from donor 28 and acceptor 7 under the general glycosylation method as a colorless foam in 95% yield (α/β = 1.5/1). Analytical data for 34 was in accordance with that previously reported.[45]
Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (35)
was obtained from donor 29 and acceptor 4 under the general glycosylation method as a colorless foam in 99% yield (α/β = 1.2/1). Analytical data for 35 was in accordance with that previously reported.[46]
Methyl 2,3,6-tri-O-benzyl-4-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (36)
was obtained from donor 29 and acceptor 5 under the general glycosylation method as a colorless foam in 99% yield (α/β = 2.4/1). Analytical data for 36 was in accordance with that previously reported.[47]
Methyl 2,4,6-tri-O-benzyl-3-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (37)
was obtained from donor 29 and acceptor 6 under the general glycosylation method as a colorless foam in 99% yield (α/β = 2.4/1). Analytical data for 37 was in accordance with that previously reported.[48]
Methyl 3,4,6-tri-O-benzyl-2-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (38)
was obtained from donor 29 and acceptor 7 under the general glycosylation method as a colorless foam in 99% yield (α/β = 4.9/1). Analytical data for 38 was in accordance with that previously reported.[49]
Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-mannopyranosyl)-α-D-glucopyranoside (39)
was obtained from donor 30 and acceptor 4 under the general glycosylation method as a colorless foam in 98% yield (α/β = 1.3/1). Analytical data for 39 was in accordance with that previously reported.[50]
Methyl 2,3,6-tri-O-benzyl-4-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-α-D-glucopyranoside (40)
was obtained from donor 30 and acceptor 5 under the general glycosylation method as a colorless foam in 98% yield. Analytical data for 40 was in accordance with that previously reported.[51]
Methyl 2,4,6-tri-O-benzyl-3-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-α-D-glucopyranoside (41)
was obtained from donor 30 and acceptor 6 under the general glycosylation method as a colorless foam in 98% yield. Analytical data for 41 was in accordance with that previously reported.[52]
Methyl 3,4,6-tri-O-benzyl-2-O-(2,3,4,6-tetra-O-benzyl-α/β-D-mannopyranosyl)-α-D-glucopyranoside (42)
was obtained from donor 30 and acceptor 7 under the general glycosylation method as a colorless foam in 98% yield (α/β = 3.6/1). Analytical data for 42 was in accordance with that previously reported.[45]
Methyl 6-O-(3,4,6-tri-O-benzoyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (46)
was obtained from donor 42 and acceptor 4 under the general glycosylation method using 1.0 equiv of Ag2O and 0.5 equiv of TfOH as a colorless foam in 97% yield. Analytical data for 46 was in accordance with that previously reported.[53]
Methyl 4-O-(3,4,6-tri-O-benzoyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl)-2,3,6-tri-O-benzyl-α-D-glucopyranoside (47)
was obtained from donor 42 and acceptor 5 under the general glycosylation method using Ag2O (1.50 equiv) and TfOH (0.50 equiv) as a colorless foam in 76% yield. Analytical data for 47: Rf = 0.60 (ethyl acetate/toluene, 1/4, v/v); [α]D21 31.1 (c 2.05, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.04 – 7.93 (m, 2H, aromatic), 7.88 – 7.78 (m, 2H, aromatic), 7.75 – 7.69 (m, 2H, aromatic), 7.69 – 7.59 (m, 2H, aromatic), 7.55 – 7.12 (m, 26H, aromatic), 6.16 (dd, J3’,4’ = 9.3 Hz, 1H, H-3’), 5.77 (d, J1’,2’ = 8.4 Hz, 1H, H-1’), 5.60 (dd, J4’,5’ = 9.7 Hz, 1H, H-4’), 4.98 (dd, 2J = 11.8 Hz, 2H, CH2Ph), 4.60 (dd, 2J = 12.2 Hz, 2H, CH2Ph), 4.51 (dd, J2’,3’ = 10.7 Hz, 1H, H-2’) 4.50 (d, J1,2 = 3.0 Hz, 1H, H-1), 4.44 (dd, 2J = 2.7 Hz, 2H, CH2Ph), 4.34 (dd, J6a’,6b’ = 12.2, 1H, H-6a’), 4.14 (dd, 1H, H-6b’), 4.08 (dd, J4,5 = 9.2 Hz, 1H, H-4), 3.90 (dd, J3,4 = 9.2 Hz, 1H, H-3), 3.70 – 3.53 (m, J5’,6b’ = 3.3 Hz, 2H, H-5, 5’), 3.53 – 3.47 (m, 2H, H-6a, 6b), 3.43 (dd, J2,3 = 9.5 Hz, 1H, H-2), 3.26 (s, 3H, OCH3) ppm; 13C NMR (151 MHz, CDCl3): δ 166.2, 165.9, 165.2, 139.6, 138.5, 138.4, 133.5, 133.5, 133.1, 130.0 (x6), 129.9 (x4), 129.1, 128.7, 128.5 (x12), 128.4 (x3), 128.2 (x3), 127.9, 127.6 (x3), 127.3, 127.2 (x3), 98.4, 97.6, 80.3, 79.5, 75.7, 75.0, 73.7, 73.0, 71.9, 71.4, 70.4, 69.5, 68.5, 63.2, 55.8, 55.5 ppm; HRMS [M+Na]+ calcd for C63H57NO15Na 1090.3620, found 1090.3627.
Methyl 3-O-(3,4,6-tri-O-benzoyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl)-2,4,6-tri-O-benzyl-α-D-glucopyranoside (48)
was obtained from donor 43 and acceptor 6 under the general glycosylation method using Ag2O (1.50 equiv) and TfOH (0.50 equiv) as a colorless foam in 72% yield. Analytical data for 48: Rf = 0.60 (ethyl acetate/toluene, 1/4, v/v); [α]D21 6.4 (c 2.34, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.97 (m, 2H, aromatic), 7.91–7.76 (m, 6H, aromatic), 7.75 – 7.64 (m, 2H, aromatic), 7.53 – 7.37 (m, 3H, aromatic,), 7.37 – 7.20 (m, 15H, aromatic), 7.20 – 7.10 (m, 6H, aromatic), 6.43 (dd, J3’,4’ = 9.6 Hz, 1H, H-3’), 6.01 (d, J1’,2’ = 8.3 Hz, 1H, H-1’), 5.70 (dd, 1H, H-4’), 5.06 (d, 2J = 11.2 Hz, 1H, CHPh), 4.86 (dd, 2J = 12.6 Hz, 2H, CH2Ph), 4.61 (dd, J2’,3’ = 10.7 Hz, 1H, H-2’), 4.51 (dd, J6a’,6b’ = 12.0 Hz, 1H, H-6a’), 4.44 (dd, 2J = 9.0 Hz, 2H, CH2Ph), 4.40 (dd, 1H, H-6b’), 4.38 (dd, J3’,4’ = 12.2 Hz, 1H, H-3), 4.37 (d, 1H, CHPh), 4.17 (dd, J5’,6a’ = 3.1 Hz, 1H, H-5’), 4.13 (d, J1,2 = 6.0 Hz, 1H, H-1), 3.84 (d, 2J = 12.6 Hz, 1H, CHPh), 3.67–3.43 (m, 4H, H-4, 5, 6a, 6b), 3.23 (dd, J2,3 = 9.6 Hz, 1H, H-2), 3.08 (s, 3H, OCH3) ppm; 13C NMR (151 MHz, CDCl3): δ 166.3, 165.8, 165.4, 138.8, 138.5, 138.1, 133.5, 133.4, 133.0 (x8), 129.9, 129.1, 128.9, 128.6 (x3), 128.52 (x10), 128.4 (x3), 128.4 (x3), 128.3 (x2), 128.2, 128.1 (x4), 127.8, 127.6, 98.5, 97.7, 81.0, 78.7, 76.0, 74.9, 74.0, 73.6, 71.7, 71.1, 70.8, 69.6, 68.7, 63.5, 56.0, 55.0 ppm; HRMS [M+Na]+ calcd for C63H57NO15Na 1090.3620 found 1090.3617.
Methyl 2-O-(3,4,6-tri-O-benzoyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl)-3,4,6-tri-O-benzyl-α-D-glucopyranoside (49)
was obtained from donor 43 and acceptor 7 under the general glycosylation method using Ag2O (1.50 equiv) and TfOH (0.50 equiv) as a colorless foam in 68% yield. Analytical data for 49: Rf = 0.65 (ethyl acetate/toluene, 1/4, v/v); [α]D21 72.1 (c 2.35, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.12 – 8.02 (m, 2H, aromatic), 7.94 – 7.88 (m, 2H, aromatic), 7.76 – 7.65 (m, 2H, aromatic), 7.60 – 7.08 (m, 21H, aromatic), 7.08 – 6.95 (m, 3H, aromatic), 6.84 (m, 4H, aromatic), 6.23 (dd, J3’,4’ = 9.7 Hz, 1H, H-3’), 5.84 (d, J1’,2’ = 8.4 Hz, 1H, H-1’), 5.72 (dd, J4’,5’ = 9.7 Hz, 1H, H-4’), 5.10 (d, J1,2 = 3.3 Hz, 1H, H-1), 4.77 (dd, J2’,3’ = 10.6 Hz, 1H, H-2’), 4.72 (dd, J6a’,6b’ = 11.6 Hz, 1H, H-6a’), 4.53 (dd, 2J = 12.1 Hz, 2H, CH2Ph), 4.44 (dd, 1H, H-6b’), 4.40 (dd, 2J = 12.1 Hz, 2H, CH2Ph), 4.39 (s, 2H, CH2Ph), 4.28 (ddd, J5’,6a’ = 2.7 Hz, J5’,6b’ = 5.0 Hz, 1H, H-5’), 3.85 (dd, J3,4 = 8.7 Hz, 1H, H-3), 3.73 (dd, J2,3 = 9.8 Hz, 1H, H-3), 3.72 – 3.54 (m, 3H, H-5, 6a, 6b), 3.57 (dd, J4,5 = 10.7 Hz, 1H, H-4), 3.32 (s, 3H, OCH3) ppm; 13C NMR (151 MHz, CDCl3): δ 166.0, 165.6, 165.1, 138.5, 138.0 (x2), 133.8, 133.4, 133.2 (x2), 129.8 (x5), 129.7 (x3), 129.4, 128.8, 128.5, 128.4 (x4), 128.3 (x3), 128.2 (x2), 128.12 (x3), 127.9 (x5), 127.8 (x4), 127.6, 127.5, 126.5, 126.0 (x2), 100.1, 99.2, 82.9, 80.3, 77.7, 74.9, 74.6, 73.5, 72.2, 71.2, 69.9, 69.8, 68.5, 63.0, 55.2, 54.8; HRMS [M+Na]+ calcd for C63H57NO15Na 1090.3620 found 1090.3615.
1,2:3,4-Di-O-isopropylidene-6-O-[methyl (4,7,8,9-tetra-O-acetyl-5-(N-acetyl)acetamido-3,5-dideoxy-D-glycero-α-D-galacto-non-2-ulo-pyranosyl)onate]-α-D-galactopyranose (52)
A mixture of a glycosyl donor 45[22c] (29.5 mg, 0.053 mmol), glycosyl acceptor 50 (6.8 mg, 0.026 mmol), and freshly activated molecular sieves (3 Å, 150 mg) in CH2Cl2 (1.0 mL) was stirred under argon for 1 h. The mixture was cooled to −78 °C, Ag2O (24.8 mg, 0.11 mmol) was added, and the resulting mixture was stirred for 10 min. TfOH (4.0 mg, 0.027 mmol) was then added, and the resulting mixture was stirred under argon for 2 h at −78 °C. After that, the reaction mixture was allowed to warm to rt over the course of 6 h and left stirring for additional 16 h at rt. The solid was filtered off and washed sucessively with CH2Cl2. The combined filtrate (~40 mL) was washed with saturated NaHCO3 (10 mL). The organic phase was separated, dried with magnesium sulfate, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (acetone – hexane gradient elution) to give the title compound as a clear syrup in 97% yield (19.6 mg, 0.025 mmol). Analytical data for 52 was in accordance with that previously reported.[54]
Large-scale glycosylation.
A mixture of donor 2 (1049 mg, 1.72 mmol), acceptor 5 (533.0 mg, 1.15 mmol), and freshly activated molecular sieves (3.0 g) in CH2Cl2 (50 mL) was stirred under argon for 2 h at rt. The mixture was cooled to 0 °C, Ag2O (199 mg, 0.86 mmol) was added, and the resulting mixture was stirred under argon for 10 min. TfOH (129 mg, 0.086 mmol) was then added, and the resulting mixture was stirred under argon for 30 min at o °C. After that, the solid was filtered off and washed sucessively with CH2Cl2. The combined filtrate (~150 mL) was washed with saturated aq. NaHCO3 (30 mL). The organic phase was separated, dried with magnesium sulfate, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate – toluene gradient elution) to afford 18 (996 mg, 0.95 mmol) in 83% yield
A competition experiment.
A mixture of benzoylated donor 1 (30.8 mg, 0.050 mmol), benzylated donor 28 (33.0 mg, 0.050 mmol), glycosyl acceptor 4 (16.3 mg, 0.035 mmol), and freshly activated molecular sieves (3 Å, 150 mg) in CH2Cl2 (1.5 mL) was stirred under argon for 1 h at rt. The mixture was cooled to 0 °C, Ag2O (5.8 mg, 0.025 mmol) was added, and the resulting mixture was stirred under argon for 10 min. TfOH (1.9 mg, 0.013 mmol) was then added and the resulting mixture was stirred under argon for 30 min at o °C. After that, the solid was filtered off and washed sucessively with CH2Cl2. The combined filtrate (~40 mL) was washed with saturated aq. NaHCO3 (10 mL). The organic phase was separated, dried with magnesium sulfate, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate – hexane gradient elution) to afford a mixture of disaccharides 12 and 31 in approximately equal amounts, judged by NMR.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NSF (CHE-1800350) and the National Institute of General Medical Sciences (GM111835).
Footnotes
ASSOCIATED CONTENT
Supporting Information
NMR spectra for all new and selected known compounds. This material is available free of charge via the Internet at
The authors declare no competing financial interests.
REFERENCES
- [1].a) Koenigs W and Knorr E, Ber. Dtsch. Chem. Ges 1901, 34, 957–981; [Google Scholar]; b) Kulkarni SS and Gervay-Hague J in Handbook of Chemical Glycosylation, Vol. (Ed. Demchenko AV), Wiley-VCH, Weinheim, Germany, 2008, pp. 59–93. [Google Scholar]
- [2].Michael A, Am. Chem. J 1879, 1, 305–312. [Google Scholar]
- [3].Helferich B and Bredereck H, Liebigs Ann. Chem 1928, 465, 166–184. [Google Scholar]
- [4].Igarashi K, Honma T and Irisawa J, Carbohydr. Res 1970, 15, 329–337. [Google Scholar]
- [5].Helferich B and Wedemeyer KF, Ann 1949, 563, 139–145. [Google Scholar]
- [6].Sun L, Wu X, Xiong DC and Ye XS, Angew. Chem. Int. Ed 2016, 55, 8041–8044. [DOI] [PubMed] [Google Scholar]
- [7].Park Y, Harper KC, Kuhl N, Kwan EE, Liu RY and Jacobsen EN, Science 2017, 355, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Geringer SA and Demchenko AV, Org. Biomol. Chem 2018, 16, 9133–9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Singh Y and Demchenko AV, Chem. Eur. J 2019, 25, 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Singh Y and Demchenko AV, Chem. Eur. J 2020, 26, 1042–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Darwent B. d. in Bond dissociation energies in simple molecules, Vol. NIST Commerce Department, 1970, p. Publication number 31. [Google Scholar]
- [12].a) Tatina MB, Khong DT and Judeh ZMA, Eur. J. Org. Chem 2018, 2018, 2208–2213; [Google Scholar]; b) Encinas L and Chiara JL, J. Comb. Chem 2008, 10, 361–363; [DOI] [PubMed] [Google Scholar]; c) Chang C-W, Chang S-S, Chao C-S and Mong K-KT, Tetrahedron Lett 2009, 50, 4536–4540; [Google Scholar]; d) Pozsgay V, Tetrahedron Lett 1993, 34, 7175–7178. [Google Scholar]
- [13].a) Ranade SC, Kaeothip S and Demchenko AV, Org. Lett 2010, 12, 5628–5631; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pornsuriyasak P, Jia XG, Kaeothip S and Demchenko AV, Org. Lett 2016, 18, 2316–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kaeothip S and Demchenko AV, Carbohydr. Res 2011, 346, 1371–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Smoot JT and Demchenko AV, Adv. Carbohydr. Chem. Biochem 2009, 62, 161–250. [DOI] [PubMed] [Google Scholar]
- [16].Li Z and Gildersleeve J, J. Am. Chem. Soc 2006, 128, 11612–11619. [DOI] [PubMed] [Google Scholar]
- [17].a) Huang X, Huang L, Wang H and Ye XS, Angew. Chem. Int. Ed 2004, 43, 5221–5224; [DOI] [PubMed] [Google Scholar]; b) Pfaeffli PJ, Hixson SH and Anderson L, Carbohydr. Res 1972, 23, 195–206; [Google Scholar]; c) Van Steijn AMP, Kamerling JP and Vliegenthart JFG, Carbohydr. Res 1992, 225, 229–245. [DOI] [PubMed] [Google Scholar]
- [18].a) Grob VD, Squires TG and Vercellotti JR, Carbohydr. Res 1969, 10, 595–597; [Google Scholar]; b) Gómez AM, Pedregosa A, Casillas M, Uriel C and López JC, Eur. J. Org. Chem 2009, 2009, 3579–3588. [Google Scholar]
- [19].Bandara MD, Yasomanee JP and Demchenko AV in Selective Glycosylations: Synthetic Methods and Catalysts, Vol. (Ed. Bennett CS), Wiley, 2017, pp. 29–58. [Google Scholar]
- [20].a) Mootoo DR, Konradsson P, Udodong U and Fraser-Reid B, J. Am. Chem. Soc 1988, 110, 5583–5584; [Google Scholar]; b) Fraser-Reid B, Udodong UE, Wu ZF, Ottosson H, Merritt JR, Rao CS, Roberts C and Madsen R, Synlett 1992, 927–942 and references therein. [Google Scholar]
- [21].Bongat AFG and Demchenko AV, Carbohydr. Res 2007, 342, 374–406. [DOI] [PubMed] [Google Scholar]
- [22].a) Shpirt AM, Kononov LO, Torgov VI and Shibaev VN, Russ. Chem. Bull 2004, 53, 717–719; [Google Scholar]; b) Kononov LO and Magnusson G, Acta Chem. Scand 1998, 52, 141–144; [Google Scholar]; c) Orlova AV, Shpirt AM, Kulikova NY and Kononov LO, Carbohydr. Res 2010, 345, 721–730. [DOI] [PubMed] [Google Scholar]
- [23].a) Boons GJ and Demchenko AV, Chem. Rev 2000, 100, 4539–4565; [DOI] [PubMed] [Google Scholar]; b) De Meo C and Jones BT, Adv. Carbohydr. Chem. Biochem 2018, 75, 215–316. [DOI] [PubMed] [Google Scholar]
- [24].De Meo C and Priyadarshani U, Carbohydr. Res 2008, 343, 1540–1552. [DOI] [PubMed] [Google Scholar]
- [25].Demchenko AV and Boons GJ, Tetrahedron Lett 1998, 39, 3065–3068. [Google Scholar]
- [26].Pilgrim W and Murphy PV, J. Org. Chem 2010, 75, 6747–6755. [DOI] [PubMed] [Google Scholar]
- [27].Mikamo M, Carbohydr. Res 1989, 191, 150–153. [Google Scholar]
- [28].Saksena R, Zhang J and Kovac P, J. Carbohydr. Chem 2002, 21, 453–470. [Google Scholar]
- [29].Pozsgay V, Coxon B and Yeh H, Bioorg. Med. Chem 1993, 1, 237–257. [DOI] [PubMed] [Google Scholar]
- [30].Damager I, Erik Olsen C, Lindberg Møller B and Saddik Motawia M, Carbohydr. Res 1999, 320, 19–30. [DOI] [PubMed] [Google Scholar]
- [31].Barua PMB, Sahu PR, Mondal E, Bose G and Khan AT, Synlett 2002, 2002, 0081–0084. [Google Scholar]
- [32].Zeng J, Vedachalam S, Xiang S and Liu X-W, Org. Lett 2011, 13, 42–45. [DOI] [PubMed] [Google Scholar]
- [33].Auzanneau F-I, Forooghian F and Pinto BM, Carbohydr. Res 1996, 291, 21–41. [PubMed] [Google Scholar]
- [34].Byramova NE, Tuzikov AB and Bovin NV, Carbohydr. Res 1992, 237, 161–175. [Google Scholar]
- [35].Demchenko AV and Boons GJ, Chem. Eur. J 1999, 5, 1278–1283. [Google Scholar]
- [36].Garcia BA and Gin DY, J. Am. Chem. Soc 2000, 122, 4269–4279. [Google Scholar]
- [37].Pornsuriyasak P and Demchenko AV, Chem. Eur. J 2006, 12, 6630–6646. [DOI] [PubMed] [Google Scholar]
- [38].Pornsuriyasak P and Demchenko AV, Tetrahedron: Asymmetry 2005, 16, 433–439. [Google Scholar]
- [39].Codee JDC, Van den Bos LJ, Litjens REJN, Overkleeft HS, Van Boeckel CAA, Van Boom JH and Van der Marel GA, Tetrahedron 2004, 60, 1057–1064. [Google Scholar]
- [40].Hasty SJ, Kleine MA and Demchenko AV, Angew. Chem. Int. Ed 2011, 50, 4197–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Singh Y, Wang T, Geringer SA, Stine KJ and Demchenko AV, J. Org. Chem 2018, 83, 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nigudkar SS, Parameswar AR, Pornsuriyasak P, Stine KJ and Demchenko AV, Org. Biomol. Chem 2013, 11, 4068–4076.23674052 [Google Scholar]
- [43].Pougny JR, Nassr MAM, Naulet N and Sinay P, Nouveau J. Chem 1978, 2, 389–395. [Google Scholar]
- [44].Chiba H, Funasaka S and Mukaiyama T, Bull. Chem. Soc. Jpn 2003, 76, 1629–1644. [Google Scholar]
- [45].Ito Y, Ogawa T, Numata M and Sugimoto M, Carbohydr. Res 1990, 202, 165–175. [DOI] [PubMed] [Google Scholar]
- [46].Vankar YD, Vankar PS, Behrendt M and Schmidt RR, Tetrahedron 1991, 47, 9985–9992. [Google Scholar]
- [47].Wegmann B and Schmidt RR, J. Carbohydr. Chem 1987, 6, 357–375. [Google Scholar]
- [48].Kobashi Y and Mukaiyama T, Bull. Chem. Soc. Jpn 2005, 78, 910–916. [Google Scholar]
- [49].Premathilake HD and Demchenko AV, Beilstein J. Org. Chem 2012, 8, 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hotha S and Kashyap S, J. Am. Chem. Soc 2006, 128, 9620–9621. [DOI] [PubMed] [Google Scholar]
- [51].Nguyen HM, Chen YN, Duron SG and Gin DY, J. Am. Chem. Soc 2001, 123, 8766–8772. [DOI] [PubMed] [Google Scholar]
- [52].Jayakanthan K and Vankar YD, Carbohydr. Res 2005, 340, 2688–2692. [DOI] [PubMed] [Google Scholar]
- [53].Kamkhachorn T, Parameswar AR and Demchenko AV, Org. Lett 2010, 12 3078–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Crich D and Li W, Org. Lett 2006, 8, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.