

Abstract
In this study, we analyze the neuropathological and biochemical alterations involved in the pathogenesis of a neurodegenerative/movement disorder during different developmental stages in juvenile rats with a mutant Myosin5a (Myo5a). In mutant rats, a spontaneous autosomal recessive mutation characterized by the absence of Myo5a protein expression in the brain is associated with a syndrome of locomotor dysfunction, altered coat color, and neuroendocrine abnormalities. Myo5a encodes a myosin motor protein required for transport and proper distribution of subcellular organelles in somatodendritic processes in neurons. Here we report marked hyperphosphorylation of alpha-synuclein and tau, as well as region-specific buildup of the autotoxic dopamine metabolite, 3,4-dihydroxyphenyl-acetaldehyde (DOPAL), related to decreased aldehyde dehydrogenases activity and neurodegeneration in mutant rats. Alpha-synuclein accumulation in mitochondria of dopaminergic neurons is associated with impaired enzymatic respiratory complex I and IV activity. The behavioral and biochemical lesions progress after 15 days postnatal, and by 30–40 days the animals must be euthanized because of neurological impairment. Based on the obtained results, we propose a pleiotropic pathogenesis that links the Myo5a gene mutation to deficient neuronal development and progressive neurodegeneration. This potential model of a neurodevelopmental disorder with neurodegeneration and motor deficits may provide further insight into molecular motors and their associated proteins responsible for altered neurogenesis and neuronal disease pathogenesis.
Keywords: Myo5a mutation, α-synuclein/tau-P, Dying back neurodegeneration, Dopamine metabolism alteration, Mitochondria complex I-IV, Autophagy, TEM
1. Introduction
In this study, we analyze the in vivo neuropathological and biochemical alterations involved in the pathogenesis of a neurodegenerative/movement disorder within different developmental stages (15-day-old to 30-day-old juvenile rats) in a mutant Myosin5a (Myo5a) Berlin-Druckrey (BD-IV) rat model. Cells, including neurons, typically use two types of molecular motors to transport and properly distribute their organelles. Neurons use microtubule-dependent motors (kinesins and dynein) to drive long-range, bidirectional transport of organelles within axons. In contrast, short-range organelle movements in somatodendritic compartments are primarily driven by actin-based motors such as myosins. Among the approximately 18 myosin classes in vertebrates, myosin V class motors have received the most attention with regard to short-range organelle and macromolecule transport within neurons.
Mammals possess three class V myosin genes (Myo5a, Myo5b, and Myo5c), which encode the heavy chains of myosins Va, Vb, and Vc, respectively. Myosin Va is the most widely expressed isoform in the central nervous system, being present virtually throughout the brain (Bridgman, 1999, Bridgman, 2009). Myosin Va plays a major role in cytoskeleton dynamics in the nervous system. Myosin Va can also bind directly to kinesins via its interaction with neurofilaments (Huang et al., 1999), suggesting a role in mediating movements of other cytoskeletal proteins, membranous organelles, or membrane-associated proteins (Goldstein, 2003).
Mutations in Myo5a have been shown to cause pigmentation and neurological defects in humans and animals. Mutation of human Myo5a causes Griscelli syndrome type 1 (OMIM #214450), a rare autosomal recessive disorder (Griscelli and Prunieras, 1978, Pastural et al., 1997) resulting in pigmentary defects and a neurological disorder with marked delay in motor development, hypotonia, and mental retardation (Menasche et al., 2000). Mutations in horse Myo5a cause Lavender foal syndrome (OMIA 001501–9796) which is a recessive lethal disorder of Arabian foals characterized by a dilute coat color and a range of neurological signs (Brooks et al., 2010). Myo5a is also mutated in dilute mice (Mercer et al., 1991) which have suggested that myosin Va is important for melanosome transport in melanocytes and smooth endoplasmic reticulum trafficking in dendritic spines of Purkinje cells (Takagishi et al., 1996). Dilute-opisthotonus is a spontaneous Myo5a gene mutation in the rat, and phenotypes of the homozygote (dop/dop) are similar to those of the myosin Va-deficient mouse. Also, dilute-opisthotonus rats displayed the absence of smooth endoplasmic reticulum and inositol 1,4,5-triphosphate receptors in the cerebellar Purkinje cell dendritic spines(Dekker-Ohno et al., 1996, Futaki et al., 2000, Takagishi and Murata, 2006). Most ‘dilute’ alleles also produce a neurologic defect characterized by convulsions and opisthotonus, apparent at 8–10 days of age and continuing until the death of the animal at 2–3 weeks of age (Copeland et al., 1983, Jenkins et al., 1981, Mercer et al., 1991, Strobel et al., 1990).
In summary, myosin Va supports multiple functions within neurons that together encompass a diverse array of fundamental processes that continuously mediate transport of organelles, membranous cargo, secretory vesicles, mRNA, and lipids, emphasizing their roles in neuronal development, axonal transport, dendritic spine structure, and synaptic plasticity (Rudolf et al., 2011).
A spontaneous inherited autosomal recessive rat model for neurodegeneration was developed in our laboratory in a colony of Sprague-Dawley (SD) rats maintained at the Texas A&M University Lab Animal Facility, USA (Stoica et al., 2007). This phenotype has been maintained in a line of rats through sib mating, and pedigree information demonstrates clear autosomal-recessive transmission of this trait. Due to the fact that SD rats are an outbred strain, we transferred the movement disorder phenotype into a syngeneic black hooded rat, Berlin-Druckrey IV (BD-IV) by inbreeding. The affected BD-IV offspring can be identified during the first few days post-natal (dpn) by the gray color of their coat, which co-segregates with the dilute-opisthotonus phenotype. The clinical phenotype is characterized by progressive development of tremor, spasticity and rigidity, bradykinesia, and postural instability, which are obvious by 15 dpn.
In an initial report, we described the endocrine and neuropathological findings in Sprague-Dawley affected rats. The cerebellum was mostly characterized by dystrophic dendrites, atrophy, and Purkinje cell loss. The microscopic appearance of CNS lesions suggested that the neuronal change is a “dying back” type of neuronal alteration, progressing from the dendrites to the perikaryon and nucleus (Stoica et al., 2007). In the next two publications we reported a progressive increase in α-syn (Stoica et al., 2012) and deregulated microRNAs (Lungu et al., 2013) in the affected mutant BD-IV rats. These findings indicate that the gene mutated in the mutant homozygous rat model has pleiotropic systemic developmental effects, which need to be further investigated. The pigmentation defects of dilute myosin Va mutants in human and animal disease and their underlying mechanisms are well understood (Reissmann and Ludwig, 2013). However, the mechanisms responsible for the neurological deficits remain obscure; which underscores the novelty and significance of our investigation. The mutant phenotype did not change by transferring the mutation from SD to BD-IV rat strain. Both the sire and the dam are carriers for the specific gene that contributes to the disorder.
Here we present the new complex and less understood neurological findings associated with this mutant rat model. Through whole-genome sequencing, we identified a mutation in Myo5a that segregates with the pigmentation and neurologic phenotype in the “mutant” strain. The corresponding protein expression is lacking in “mutant” rat brain, indicating that the mutation behaves as a null allele. We present new data that tau protein is highly phosphorylated and discuss the interplay between α-syn and tau (α-syn/tau-P) and their potential role in neurodegeneration in this developmental disorder animal model. In addition, we document the negative consequences associated with Myo 5a mutation on dopamine (DA) metabolism and mitochondrial function, which may provide insights into the molecular mechanism of neurodegenerative disorders. This report is a continuation of our ongoing study seeking to decifer the complexity of the neuropathogenesis associated with the Myo5a mutation.
2. Results
2.1. Mutants carry a loss of function mutation in Myo5a
To determine the nature of the mutation that is associated with the neurologic phenotype in the homozygous (mutant, affected) rats, we performed whole-genome next generation sequencing on six different rats from different pedigrees (Table in Fig. 1). Of the six rats sequenced, four were affected and two were unaffected control samples. After aligning to the rat genomic reference rn4, the mapped sequence reads were determined to have a mean base pair coverage of 28.16X across all samples. Causative variant selection was first narrowed to regions of homozygosity shared among all affected samples. Within these regions only a single deleterious variant segregated appropriately. All affected samples were found to carry homozygous thymine-to-cytosine single nucleotide variants at chr8: 79,923,871 (for rn4, or c81,656,600 for rn5 (http://rgd.mcw.edu/)) (Fig. 1B). One control rat was found to be a heterozygous carrier, while the other was homozygous for the wild type allele. This mutation disrupted the splicing donor site directly adjacent to exon 4 of Myo5a.
Fig. 1.
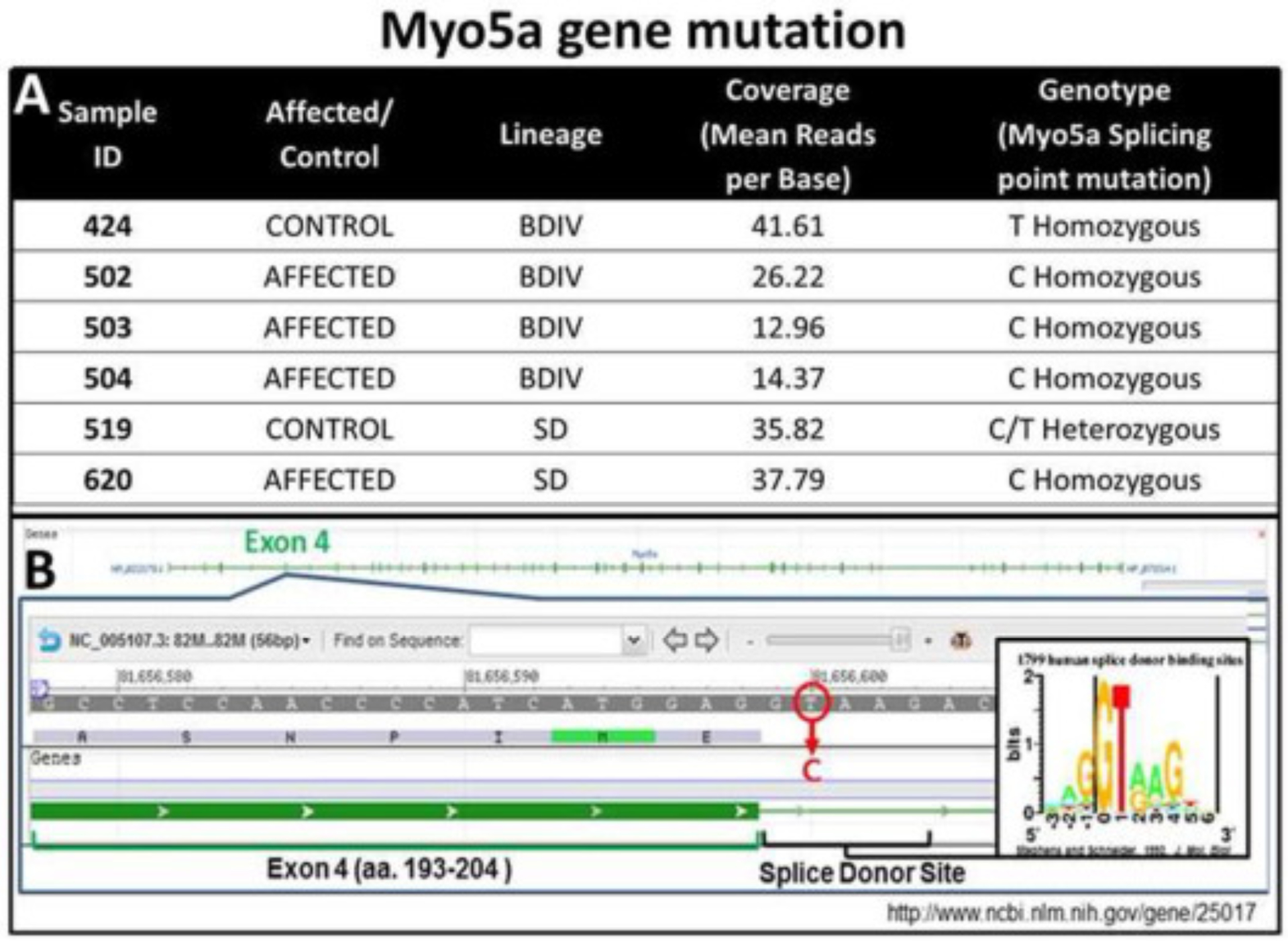
Myo5a gene mutation. (A) Myo5a gene mutation on chromosome 8, exon 4 in BD-IV mutant rats and (B) whole-genome next generation sequence results from these rats.
As such, it is highly probable that intron 4 is retained in the final mRNA product causing a frame shift that leads to early truncation of the protein or a nonsense-mediated decay of the transcript. These predictions are consistent with the following observations: First, the non-affected siblings of BD-IV rats are black hooded rats whereas the BD-IV mutants (affected-homozygous) show a gray discoloration of their coat that can be recognized at 3–4 dpn (Fig. 2A). As is typical for Myo5a mutations, the silvery-gray hair of the mutant rat shows pigment clumping due to accumulation of melanosomes in the hair shaft when compared with the normal hair shaft appearance (Fig. 2A and B) (Reissmann and Ludwig, 2013). Second, transmission electron microscopy confirmed the lack of smooth endoplasmic reticulum in Purkinje cell spines (Fig. 2C), as previously described in Myo5a mutant mice (Jones et al., 2000, Mercer et al., 1991, Takagishi et al., 1996) and rats (Dekker-Ohno et al., 1996). Finally, western blot analysis showed the complete lack of Myo5a protein, demonstrating that the splice mutation indeed leads to the loss of the Myo5a protein (Fig. 2D). This finding demonstrates the phenotypic similarity with the reported findings for the Myo5a null mouse (Jones et al., 2000) and rat dilute-opisthotonus (Takagishi and Murata, 2006) models.
Fig. 2.
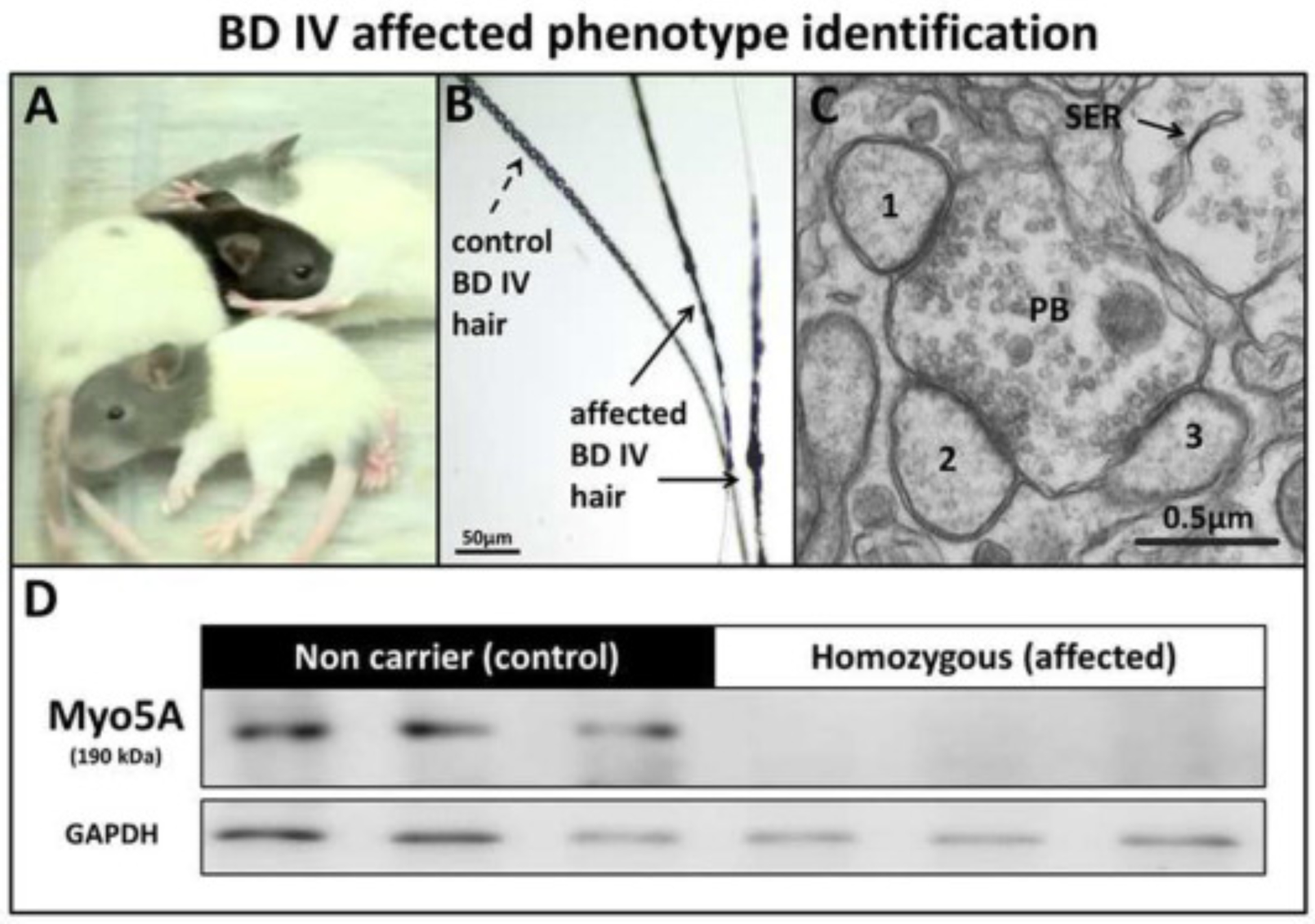
BD-IV affected phenotype identification. (A) Gray color of the head of affected rats compared with the control (non-affected littermate), (B) Accumulation of melanosomes within the hair shaft (arrows), (C) Ultrastructural cross-sectional profile of a single cerebellum large parallel fiber bouton (PB) making contact with 1, 2, 3 Purkinje cell (PC) spines from the molecular layer, showing absence of smooth endoplasmic reticulum (SER) in affected rats at 25 dpn (Bar = 0.5 μm). (D) Western blot showing loss of Myo5a protein in the brain of affected rats compared to control rats at 30 dpn.
2.2. Upregulation of α-syn/tau-P in the brain of Myo5a homozygous mutant rats
Previously, we reported upregulation of α-syn in the brain of BD-IV mutant homozygous rats (Stoica et al., 2012) without a systematic evaluation of phosphorylated α-syn/tau. The analysis of α-syn/tau-P was not previously reported in our BD-IV mutant rat or other Myo5a mutant models. Preliminary immunohistochemical examination using specific tau-P antibodies suggested an increased tau distribution and localization in the brain of mutant rats. Based on our preliminary data, reported previously (Stoica et al., 2007, Stoica et al., 2012), our aim was to investigate the expression of α-syn/tau-P in the brain of homozygous mutants compared to control littermates and their possible contribution to the neurodegeneration observed in affected rats.
Western blot (WB) analysis was performed at two time points, an early stage of disease development (15 dpn) and a later stage (30 dpn), and the levels of protein expression were compared between affected (mutant) and control (non-affected) littermates. The anatomical regions selected for WB analysis were based on previously published data, predominant lesions detected on histopathological examination, and evidence of a disrupted dopominergic system. These regions were: cerebellum (CR), brain stem (BS), mesencephalon (MS), olfactory bulb (OB), striatum (ST), hippocampus (HC), and frontal cortex (FC). The expression of α-syn-P was significantly increased in the CR, OB and HC brain regions examined in the BD-IV affected rats at 15dpn compared to 15dpn control rats (Fig. 3B), with no differences detected in tau-P (3A). The α-syn-P level was increased in CR, BS, MS, FC, ST and HC, but significantly decreased in OB, at 30 dpn in affected rats compared to the control (Fig. 3D). There was significantly increased expression of tau-P in the CR, BS, FC, OB, ST and HC but no significant elevation in the MS in homozygous rats compared with control littermates at 30 dpn (Fig. 3C). We analyzed and compared the tau/α-syn-P temporal changes between 15 and 30 dpn in control/affected mutant BD-IV in 7 brain regions (Table 1). The protein expression levels are presented as % changes between 15 and 30 dpn. In the control rats, tau-P expression decreased 65–95% in every region examined, except FC where it is increased 2-fold at 30 dpn (Table 1A). In the affected rat brain regions, there was a decrease of 23–65% in tau-P expression in all regions, except FC, which shows a 6.1-fold increase and HC, where expression is 45% elevated. In the control rat brain regions, α-syn-P expression decreased 15–86%, except in the FC, where it increased 2-fold (Table 1B). In affected rats, α-syn-P expression decreased in MS, OB, HC, and increased in CB, BS, FC and ST.
Fig. 3.
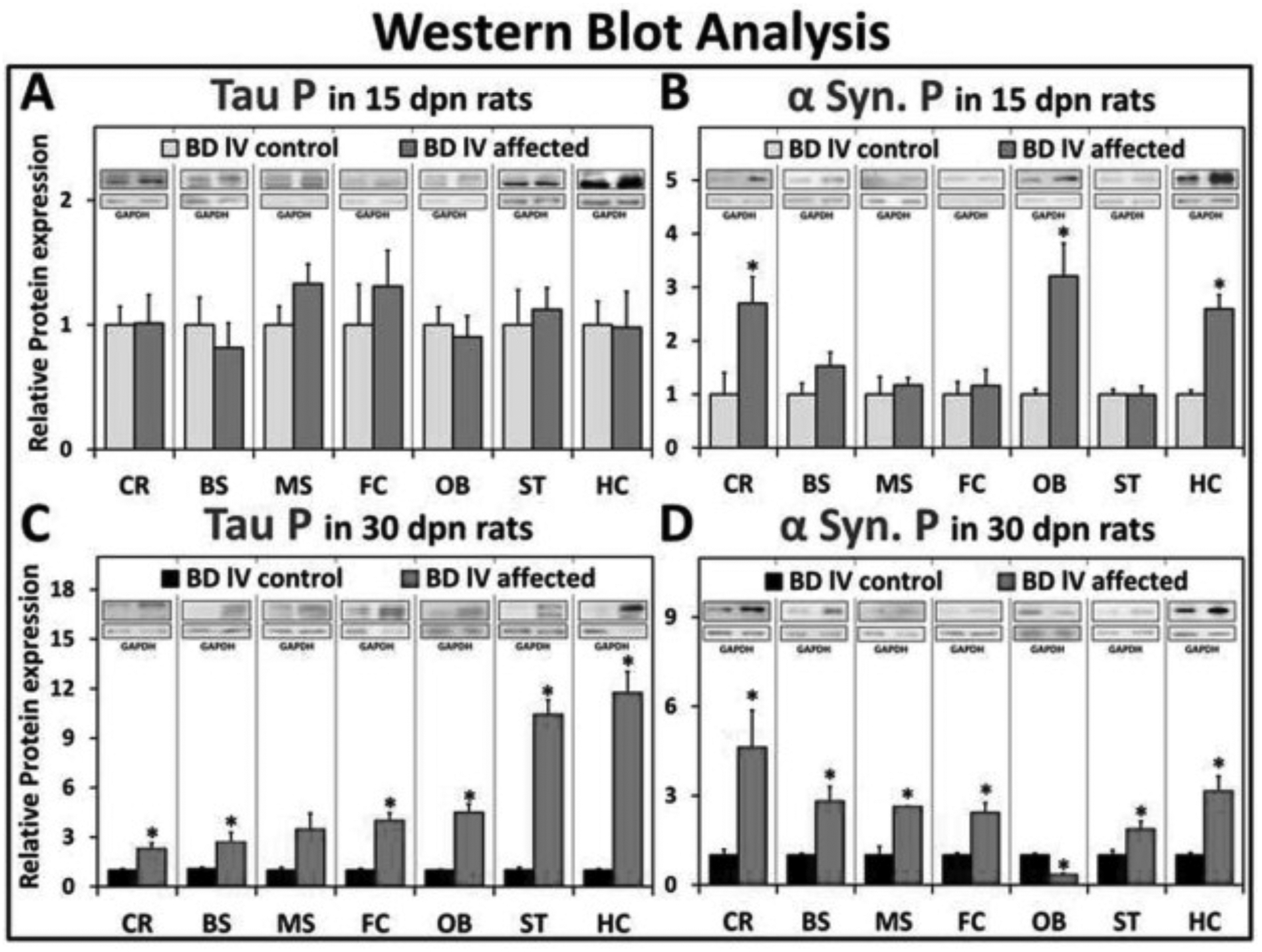
Western blot analysis. Graphic representation of tau P levels in affected and control rats at (A) 15 and (C) 30 dpn from western blot analysis. Western blot analysis graphic representation of α-syn P levels in affected and control rats at (B) 15 dpn and (D) 30 dpn. (Data is shown as mean ± S.E.M. P-values calculated using student unpaired t-test. N3–6, *p < .05). Western blots are shown as representatives (1 μg loaded for FC, HC @ 30 dpn/Tau P/affected rats (as indicated by GAPDH) and all others are loaded as 10 μg).
Table 1.
Brain α-syn/tau-P changes at 15 vs 30 dpn. The table illustrates changes in the level of tau/α-syn-P expression when comparing 15 dpn with 30 dpn in control/affected rats. (A) Shows % changes in the level of tau-P between 15 and 30 dpn in 7 brain regions of control/affected rats. In the control, tau-P expression is decreased in every region examined, except FC which is increased 2-fold. In the affected rat brain regions there is a decrease in tau-P expression, except FC showing a 6.1-fold increase and HC a 45% elevation. (B) In the control rat brain regions α-syn-P is decreased, except FC, where it is increased, 2-fold. In affected rats α-syn-P shows decreases in MS, OB, HC, and increases in CB, BS, FC (4.3-fold) and ST.
| A | Tau P (control/affected rats) | |||||
|---|---|---|---|---|---|---|
| Section | Sample | Change | P value | Sample | Change | P value |
| Cerebellum | control | ↓ 65 % | <0.05 | affected | ↓ 23 % | - |
| Brain Stem | control | ↓ 80 % | <0.02 | affected | ↓ 38 % | - |
| Mesencephalic | control | ↓ 86 % | <0.002 | affected | ↓ 65 % | <0.01 |
| Frontal Cortex | control | ↑ 2 fold | <0.05 | affected | ↑ 6.1 fold | <0.001 |
| Olfactory Bulb | control | ↓ 82 % | <0.002 | affected | ↓ 43 % | - |
| Striatum | control | ↓ 95 % | <0.001 | affected | ↓ 55 % | <0.03 |
| Hippocampus | control | ↓ 1 % | <0.001 | affected | ↑ 45 % | - |
| B | α Syn. P (control/affected rats) | |||||
| Section | Sample | Change | P value | Sample | Change | P value |
| Cerebellum | control | ↓ 25 % | <0.05 | affected | ↑ 23 % | - |
| Brain Stem | control | ↓ 15 % | - | affected | ↑ 59 % | - |
| Mesencephalic | control | ↓ 86 % | - | affected | ↓ 63 % | <0.04 |
| Frontal Cortex | control | ↑ 2 fold | <0.01 | affected | ↑ 4.3 fold | <0.003 |
| Olfactory Bulb | control | ↑ 7 % | - | affected | ↓ 89 % | <0.01 |
| Striatum | control | ↓ 40 % | <0.02 | affected | ↑ 12 % | - |
| Hippocampus | control | ↓ 53 % | <0.001 | affected | ↓ 43 % | <0.02 |
P-values calculated using student unpaired t-test. N3–6, “↑” shows increased expression and “↓” shows decreased expression between 15 and 30 dpn
Total tau/α-syn was analyzed by WB at 30 dpn (Fig. 4). The 15 dpn did not show any significant changes when control and affected groups were analyzed and compared (data not shown). WB shows that the total tau was significantly increased in BS, MS, OB, and FC but not in HC in the affected rats compared with control littermates at 30 dpn. Total α-syn at 30 dpn was significantly increased in BS, MS, OB, HC and FC in the affected rats compared with control littermates.
Fig. 4.
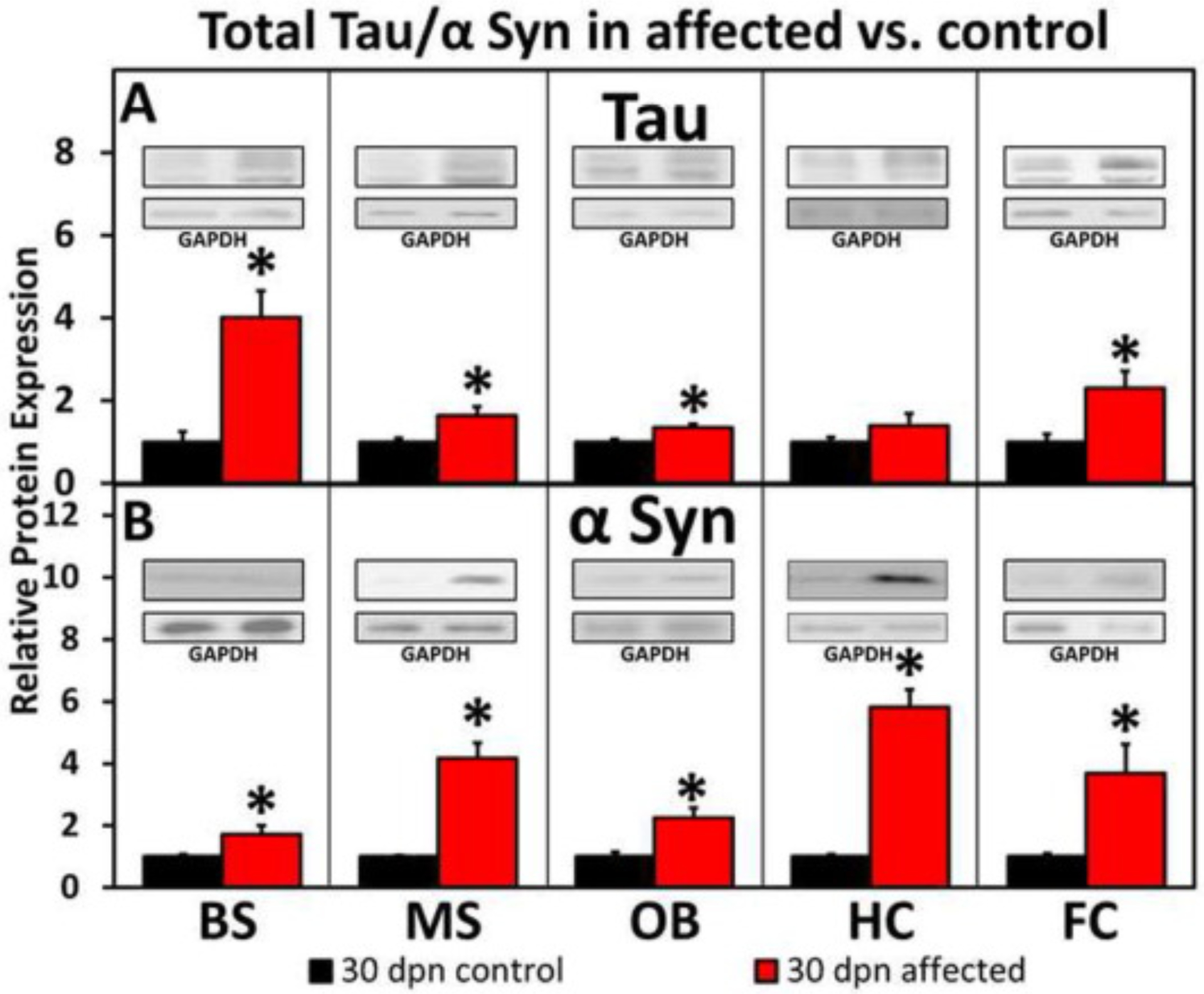
Western blot analysis of total tau/α-syn in affected versus control rats at 30 dpn. (A) Shows that the total tau was significantly increased in BS, MS, OB, and FC but not in HC in the affected rats compared with control littermates at 30 dpn. (B) Total α-syn at 30 dpn was significantly increased in BS, MS, OB, HC and FC in the affected rats compared with control littermates. (Data is shown as mean ± S.E.M. P-values calculated using student unpaired t-test. N3–6, *p < .05). Western blots are shown as representatives (1 μg loaded for HC @ 30 dpn/total Tau/affected rats (as indicated by GAPDH) and all others are loaded as 10 μg).
Our WB data were supported by a strong α-syn-P immunolabeling of MS, HC neuron’s perikarya and neuropil in affected rats (Fig. 5B and D, respectively) when compared with control rats (Fig. 5A and C, respectively). The cerebellar nuclei (CR) showed a perikaryal granular pattern for α-syn-P in the affected rats (Fig. 5F), suggesting aggregation and co-localization with the synapses at 30 dpn. Confocal image illustrate co-localization of αsyn with synapses in affected substantia nigra (Fig. 6).
Fig. 5.
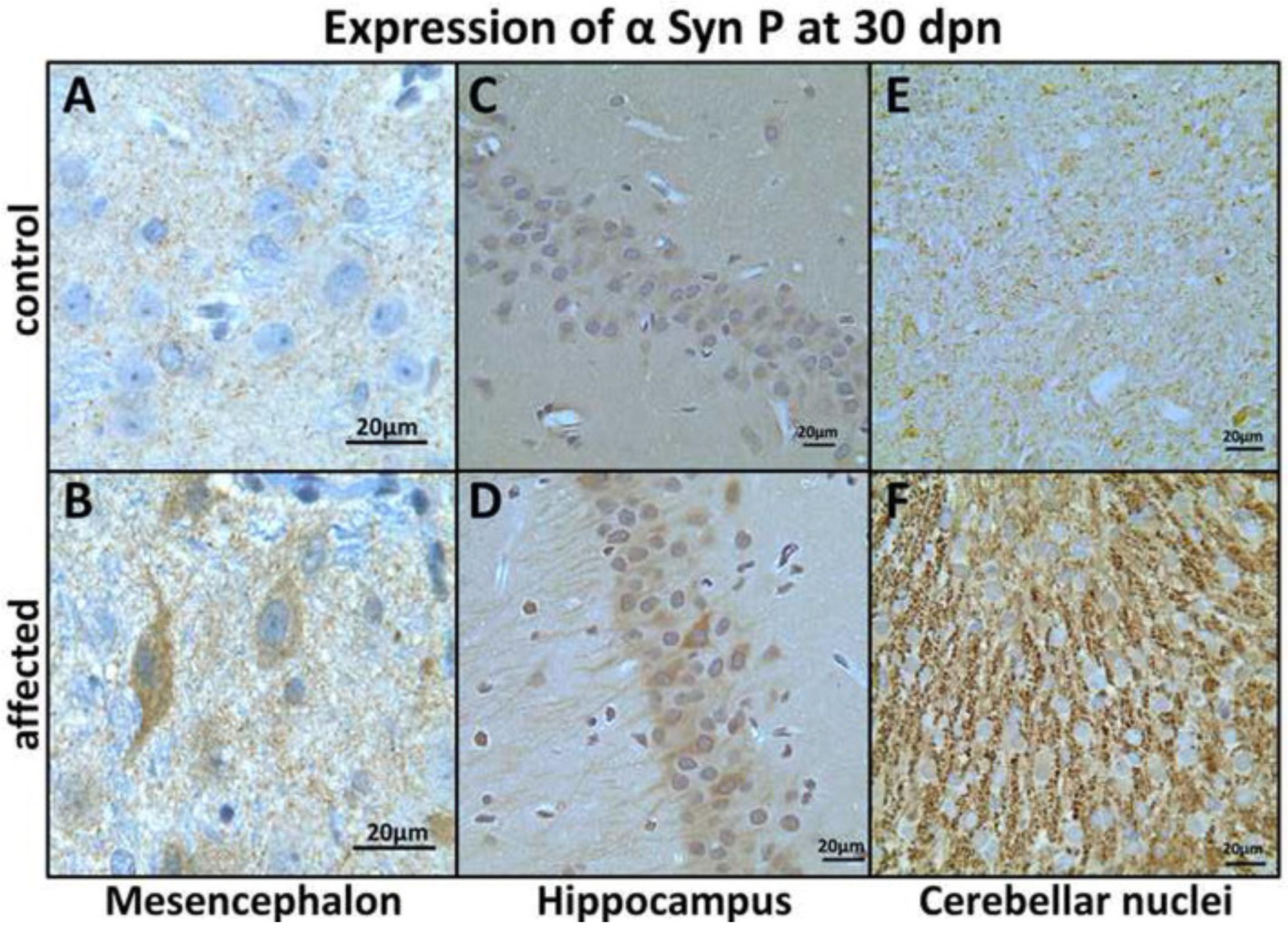
Expression of α-syn-P in mesencephalon (MS), hippocampus (HC) and cerebellar nuclei (CR) at 30 dpn. MS neuronal α-syn-P immunohistochemistry in (A) control showing punctate neuropil labeling and (B) strong neuronal perikaryal and punctate neuropil α-syn-P labeling in affected rats at 30 dpn (Bar = 20 μm). (C) HC pyramidal neurons show weak immunolabeling for α-syn-P in control rats and (D) strong perikaryal and axonal labeling of pyramidal neurons in affected brain HC. (E) CR showing weak α-syn-P labeling in control and (F) strong granular perikaryal synaptic labeling at 30 dpn in the affected rats. (Bar = 20 μm).
Fig. 6.
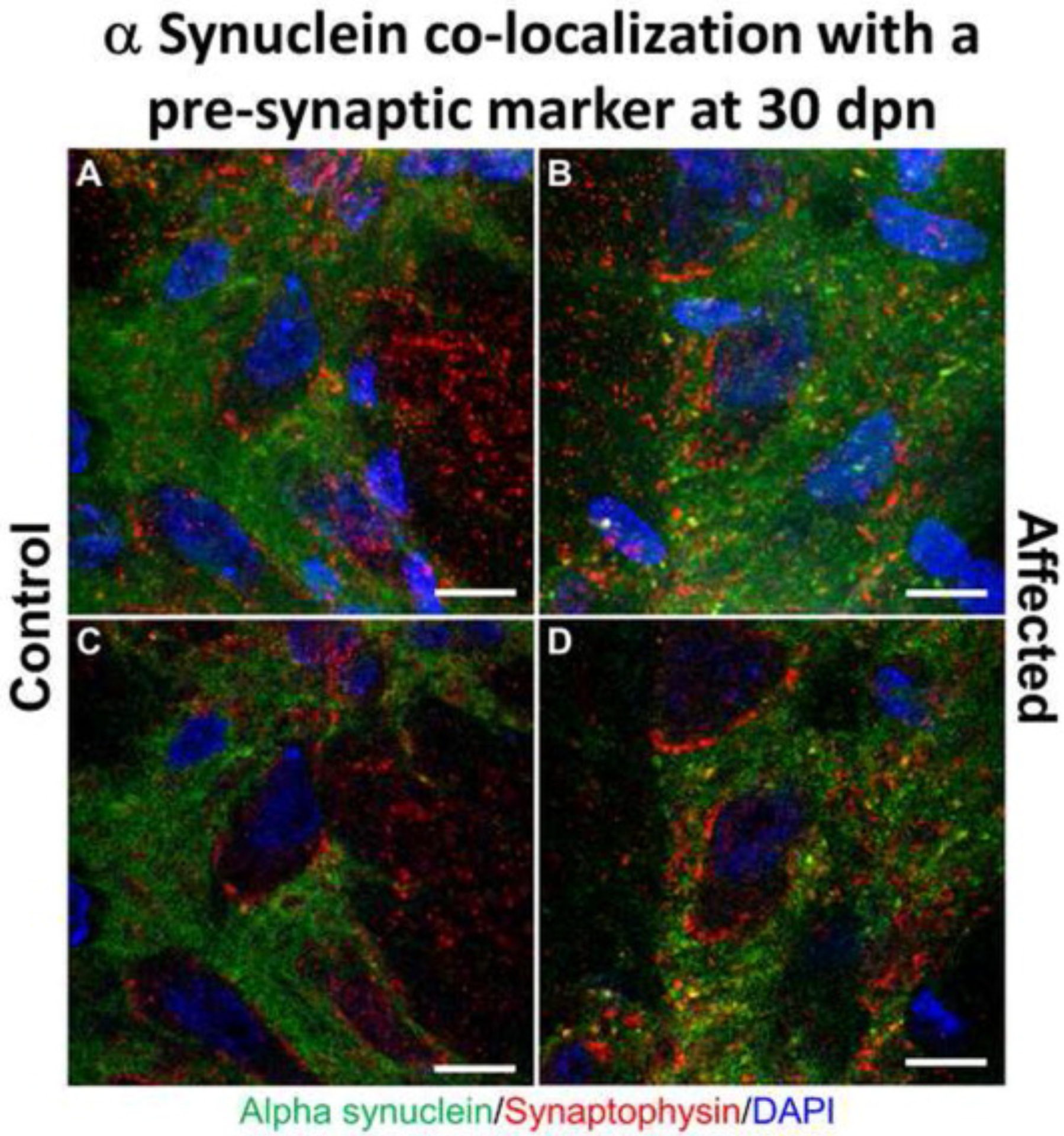
Alpha synuclein co-localization with pre-synaptic marker synaptophysin in the brain substantia nigra in Myosin5a mutants. Z-stack projection confocal immunofluorescence images of control (A) and affected (B) brains at 30 dpn staining for α-syn (green) and synaptophysin (red) demonstrating co-localization of α-syn with the synapse in the substantia nigra (Bar = 10 μm). Regions of overlap are yellow/orange. Single slice of z-stack confocal images for control (C) and affected (D) rats demonstrating colocalization of presynaptic marker synaptophysin and α-syn.
Results of the tau-P WB analysis were supported by IHC findings, which also demonstrated strong labeling in the FC, HC, ST, CR, BS and OB in the mutants compared with their control littermates. The IHC pattern was mostly axonal in the HC (Fig. 7C and E), ST (Fig. 8B), CR (Fig. 9B), and BS (Fig. 9D). In the ST (caudate-putamen), the tau-P immunolabeling was strong primarily in the nerve fiber projections in the affected compared with the control littermates. Perikaryal labeling is also present in the FC (Fig. 7B), OB (Fig. 8D) and BS (Fig. 9D). In the OB immunolabeling was strong and affected the mitral cell perikarya and internal granular layer neuropil in affected rats. In contrast, tau-P labeling of mitral cells was weak in control, non-affected rats at 30 dpn (Fig. 8C and D).
Fig. 7.
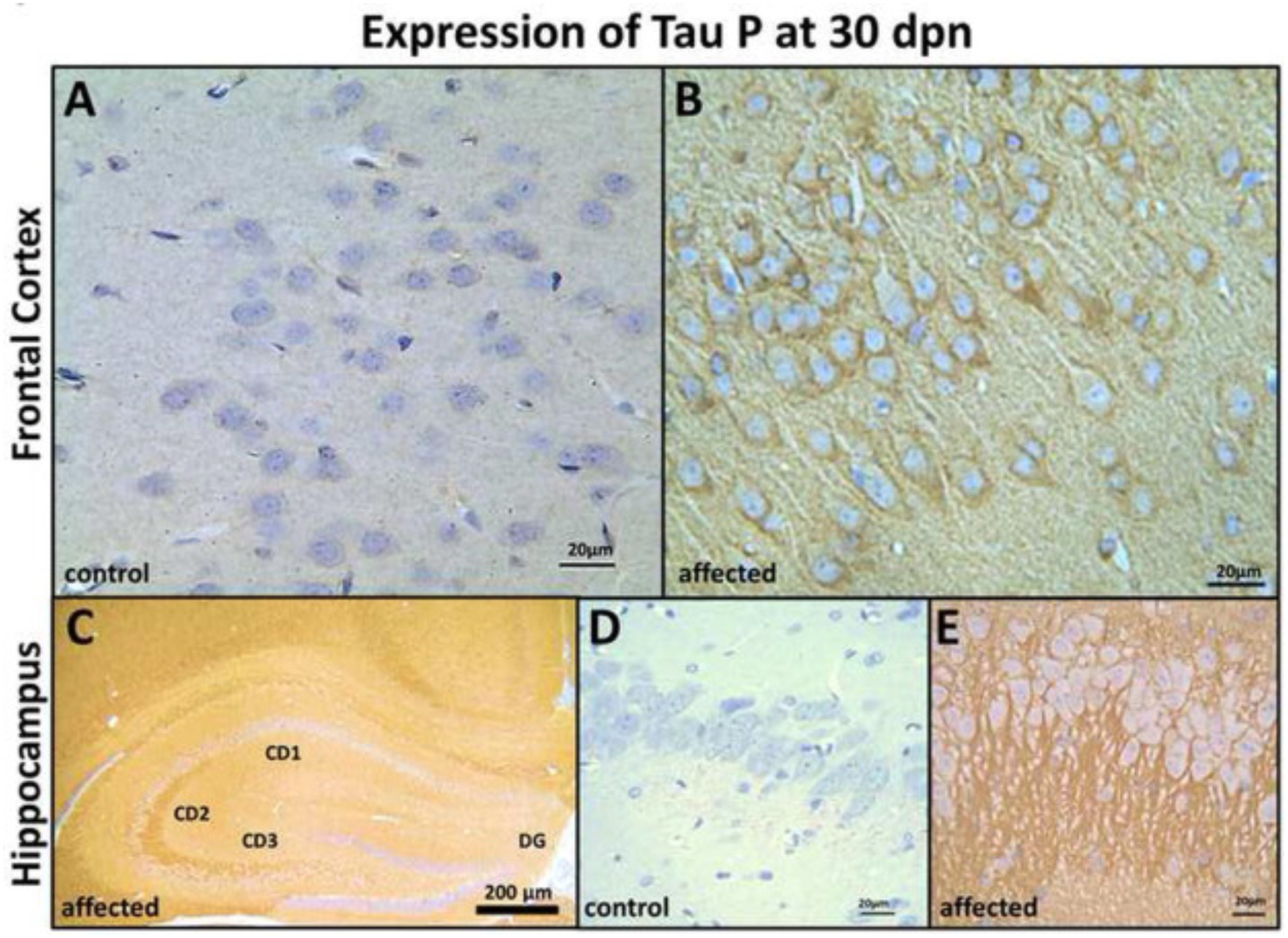
Frontal cortex (FC) and Hippocampus (HC) tau-P expression in control and affected rats at 30 dpn. FC immunohistochemistry in control rats (A) showing weak tau-P immunolabeling, and affected rats (B) showing strong labeling of axonal, perikaryal localization of tau protein (Bar = 20 μm). (C) Hippocampal formation showing strong immunolabeling of CD2 and CD3 region in the affected rats, but there is no labeling of dentate gyrus (DG) and less labeling of the CD1 region (Bar = 200 μm). Immunohistochemistry in control rats (D) showing weak tau-P immunolabeling and (E) pyramidal neurons axonal and perikaryal showing strong labeling for tau-P protein in the affected rats. (Bar = 20 μm).
Fig. 8.
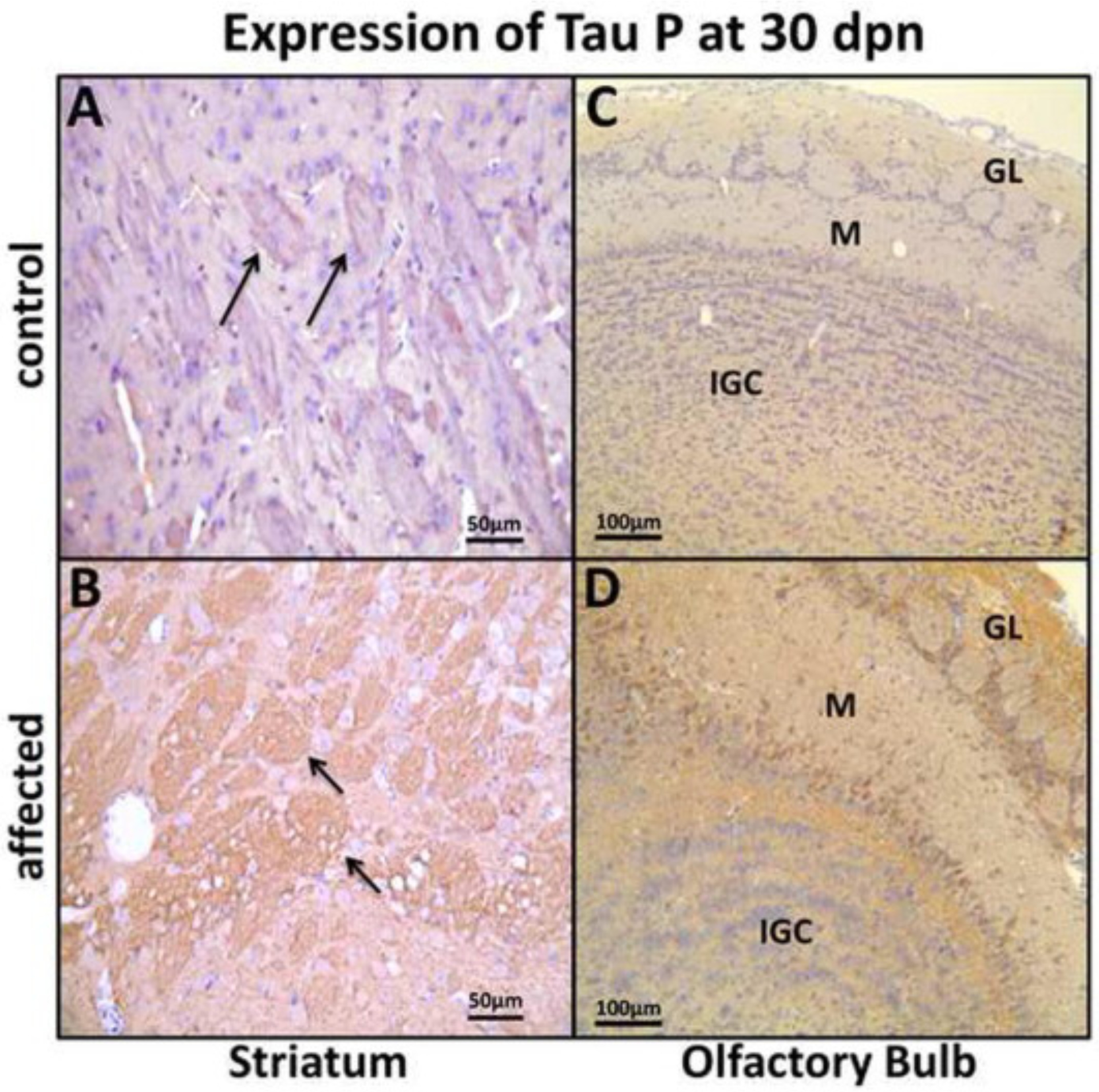
Expression of tau P in striatum (ST) and olfactory bulb (OB) at 30 dpn. (A) ST from a control rat showing weak tau-P labeling of nerve fiber projections (arrows) and (B) affected rats showing strong labeling of fiber projections (arrows) at 30 dpn (Bar = 50 μm). (C) OB showing weak immunolabeling in control rats and (D) strong perikarya mitral cells labeling (M), internal granular cells layer (IGC) and, granular layer (GL) neuropil in affected rats. (Bar = 100 μm).
Fig. 9.
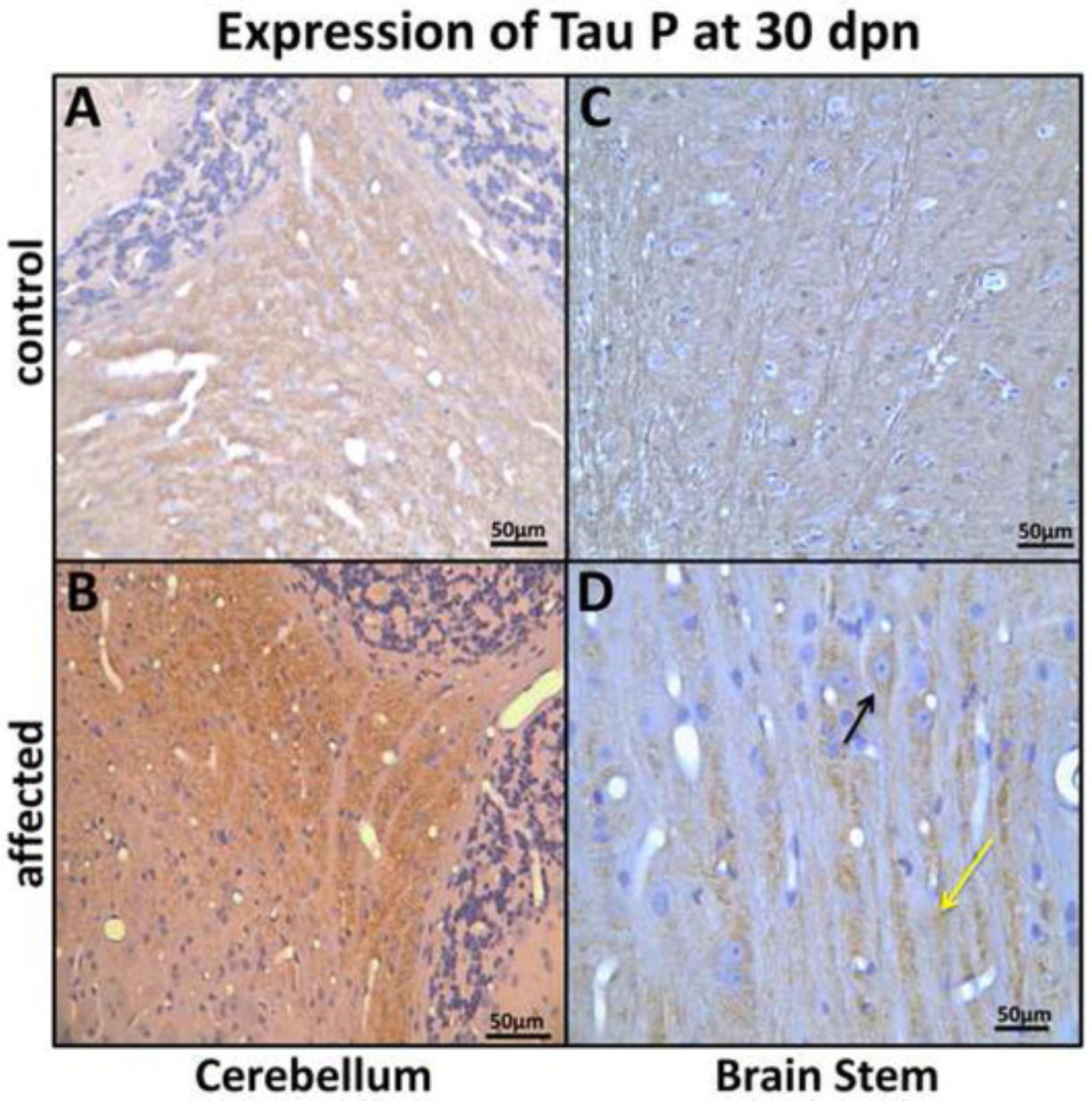
Expression of tau P in cerebellum nuclei (CR), and brain stem (BS) at 30 dpn. (A) CR nuclei from a control rat showing weak tau-P labeling and (B) an affected rat showing strong tau-P labeling in the CR nuclei (Bar = 50 μm). (C) BS pontocerebellar fibers showing weak immunolabeling in control and (D) showing strong tau-P labeling, perikaryal (black arrow) and axonal (yellow arrow) in affected rats. (Bar = 50 μm).
Taken together, WB and immunohistochemical data suggest that α-syn/tau-P was significantly up-regulated in specific anatomical brain areas at 30 dpn in BD-IV mutant rats compared to the control rats. Over-expression of α-syn-P preceded the tau-P increase, being significantly elevated at 15 dpn in CR, OB and HC in the mutants compared with their control littermates.
2.3. Ultrastructural pathological changes in Myo5a expression in the brain of homozygous rats
To determine the ultrastructural changes that occur in the mutant brain, we performed TEM on the CB, ST, OB and BS sections from affected and non-affected rats sacrificed at 25 dpn. The ultrastructural cross-section profile of CB parallel large fibers making contact with Purkinje cell spines from the molecular layer showed absence of smooth endoplasmic reticulum in affected homozygous rats expressing movement disorder (Fig. 2C). OB projections (Fig. 10B and E) and BS showed axonal and perikaryal autophagosome formation. The ultrastructure image is representative of n = 4. This phenotype was also, biochemically supported by elevation of LC3, an autophagy biomarker (Fig. 10C and D).
Fig. 10.
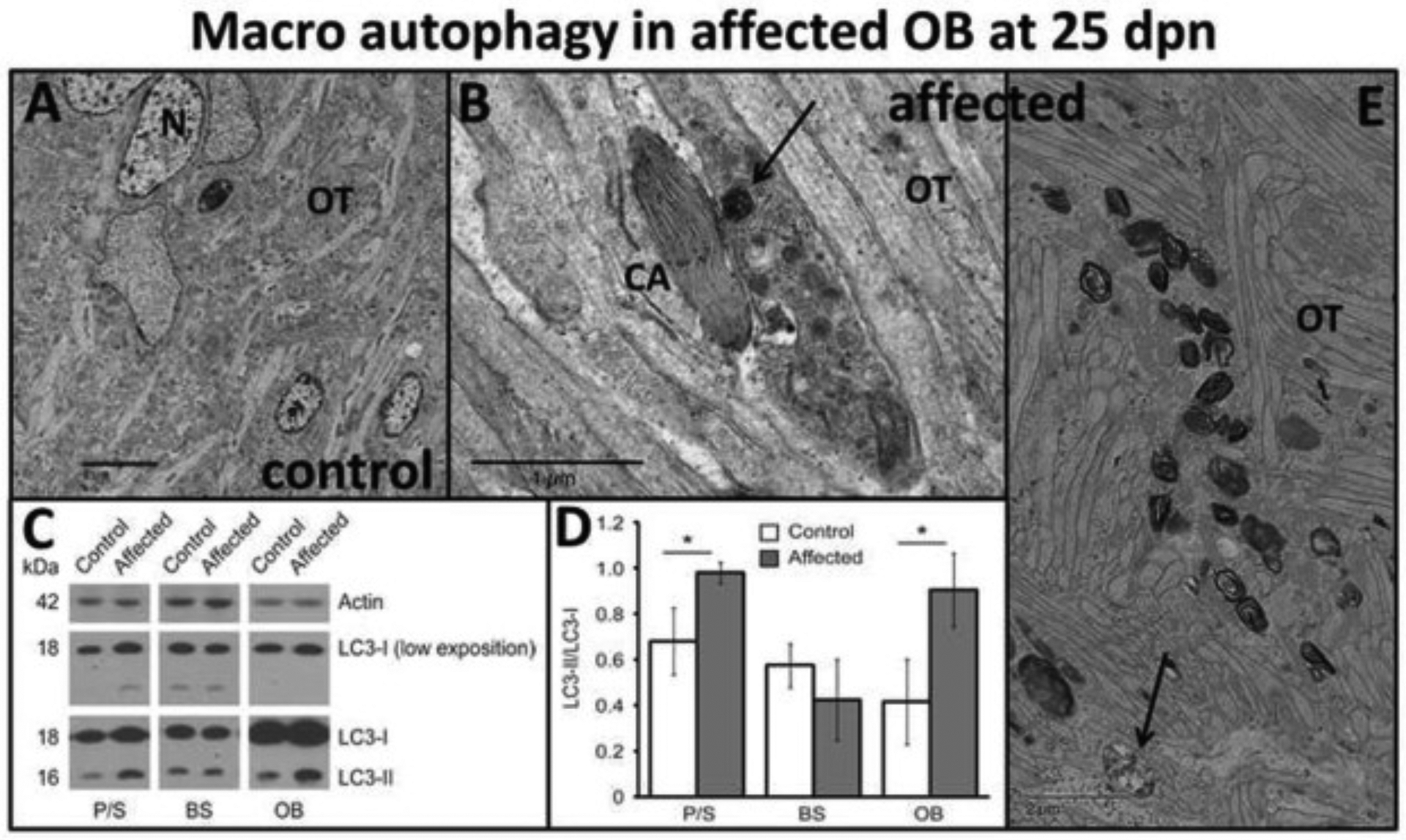
Olfactory tract (OT) pathological alterations. (A) Normal ultrastructural sagittal section through olfactory tract (OT) projections and neurons (N) in control rats at 25 dpn and (B, E) ultrastructural sagittal section showing axonal accumulation of macro autophagosomes (B, black arrow) in different degrees of development in the OT of affected rats at 25 dpn. (B) Also, adjacent there is an axon containing smooth endoplasmic reticulum in a crystalline-like configuration (CA). Left lower corner in E represents a typical early autophagosome consisting of accumulation of organelles and lysosomes surrounded by a capsule (arrow). [Bar = 5 μm (A), Bar = 1 μm (B), Bar = 2 μm (E)]. Autophagosome biomarkers: (C) LC3I to LC3II conversion. Representative immunoblots of protein extracted from putamen/striatum (P/S), brain stem (BS) and olfactory bulb (OB) of control and affected rats (25 dpn), probed with antibodies (lower exposure of LC3-I were used for quantification). (D) Graph shows the densitometric analysis expressed as the LC3-II/LC3-I ratio. Data is shown as mean ± S.E.M. *p < .05 (n = 3 animals for each experimental condition).
Striatal, myelinated and non-myelinated projections appear to be degenerating, accompanied by severely disrupted axonal microtubule structures and vacuolar changes. Similar degenerative changes also characterized the neuronal cell body (Fig. 11B).
Fig. 11.
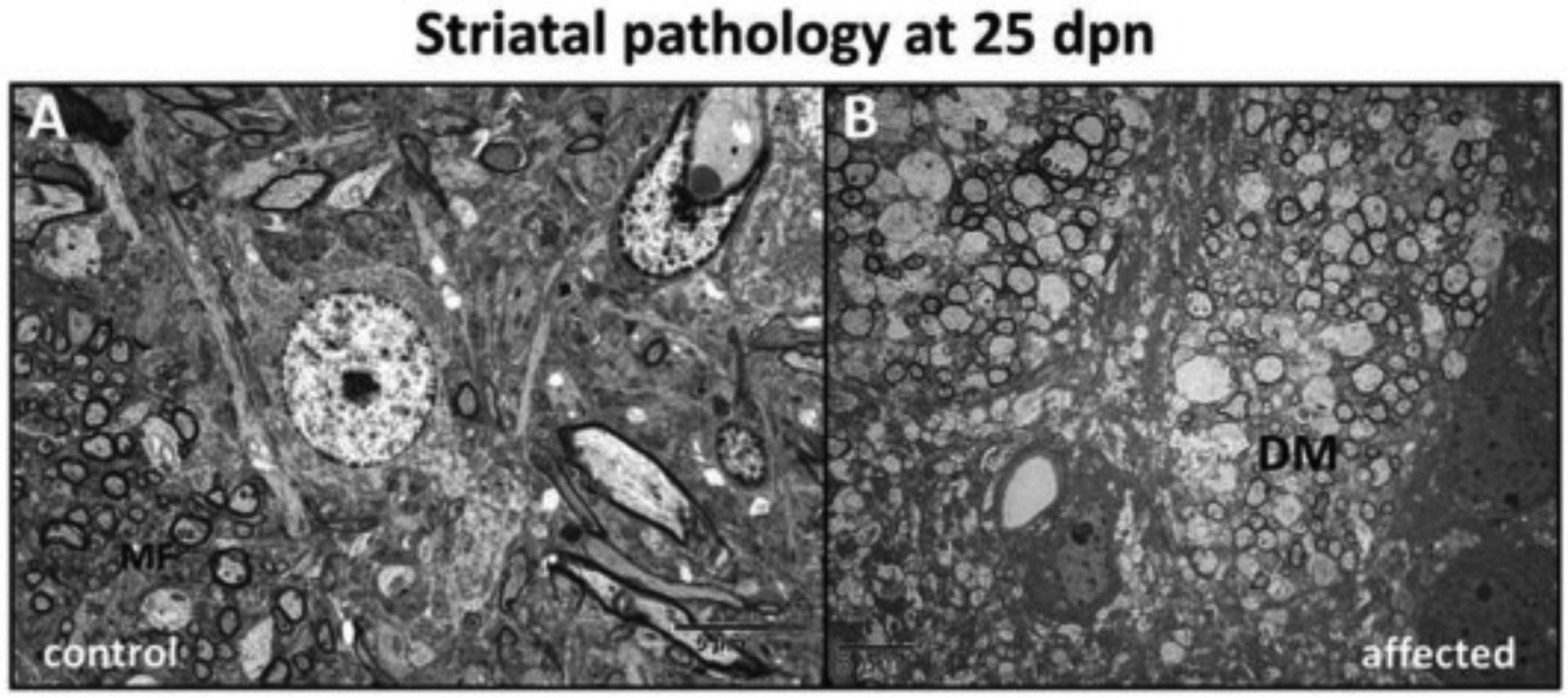
Striatum (ST) pathological alterations. (A) Striatal section from a control rat showing myelinated fibers (MF) (B) The axons showed demyelination and swelling characteristic of degeneration (DM) in affected rat dorsal striatum at 25 dpn. (Bar = 5 μm).
2.4. Increased mitochondrial accumulation of α-syn and complex I-IV alterations in affected mutant rats
α-Syn is thought to mediate different pathogenic effects depending on its localization (Boassa et al., 2013). α-Syn localized in the synapse is thought to play a role in presynaptic DA recruitment (Yavich et al., 2004). Since we observe an increase in the total amount of α-syn (Stoica et al., 2012), (Fig. 4), as well as α-syn-P (Fig. 3, Table 1), we wished to determine the subcellular localization of the increase in α-syn in mutant brains. Confocal immunofluorescence images of mutant and control brains at 30 dpn, following staining for α-syn and COX IV, demonstrated partial co-localization of α-syn with the perikaryal mitochondria in the substantia nigra pars compacta (SNpc) in affected but not control rats (Fig. 12A–E).
Fig. 12.
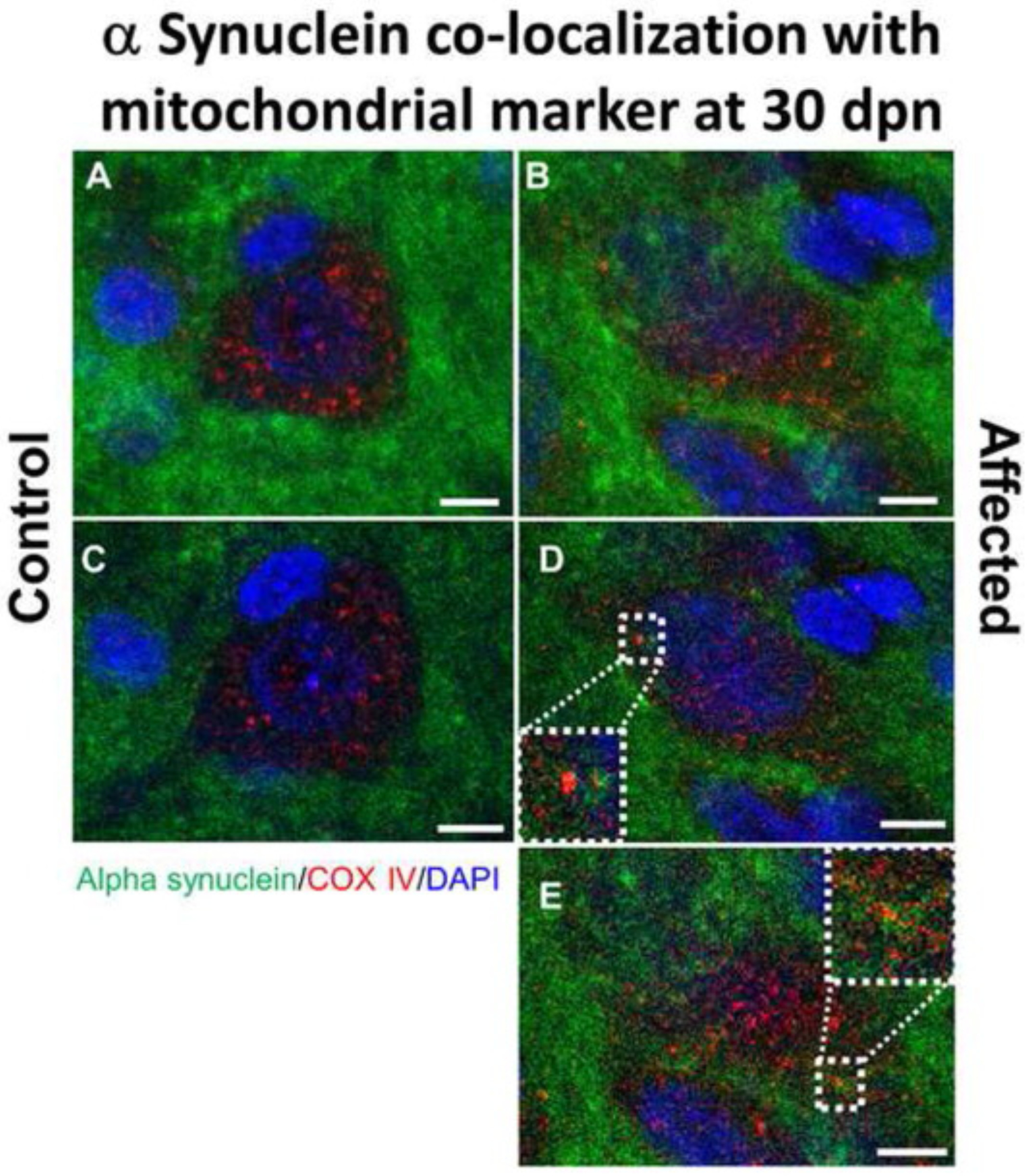
Alpha synuclein co-localization with mitochondrial marker COX IV in the brain substantia nigra in Myosin5a mutants. Z-stack projection confocal immunofluorescence images of control (A) and affected (B) brains at 30 dpn staining for α-syn (green) and COX IV (red). Following staining for α-syn and COX IV, demonstrated partial co-localization of α-syn with the perikaryal mitochondria in the substantia nigra pars compacta (SNpc) in affected but not control rats (Bar = 5 μm). Regions of overlap are yellow/orange. Single slice of z-stack confocal images for control (C) and affected (D, E) rats demonstrating colocalization of mitochondrial marker COX IV and α-syn. Dotted white line indicates zoomed in area to show colocalized pixels.
The analysis of electron transport chain (ETC) activity was performed on rat brain samples from three affected and three control animals; FC, MS, and OB. All three regions from the mutant (affected) animals exhibit partial (25–40%), but statistically significant, reductions in complex I (NADH dehydrogenase) and complex IV (cytochrome c oxidase) activities (Fig. 13A–C). The complex III (cytochrome c oxidoreductase) activity was significantly increased and citrate synthase (CS) activity was mildly (15–40%) elevated. The presented ETC enzyme activities were first normalized to the corresponding CS activity and then expressed relative to the control activities so that the relative changes in activities shown reflect changes in specific activities and are not solely due to differences in cellular mitochondrial content. These observations suggest that the mutant rat brains were exhibiting mild to moderate dysfunction of mitochondrial energy metabolism/oxidative phosphorylation.
Fig. 13.
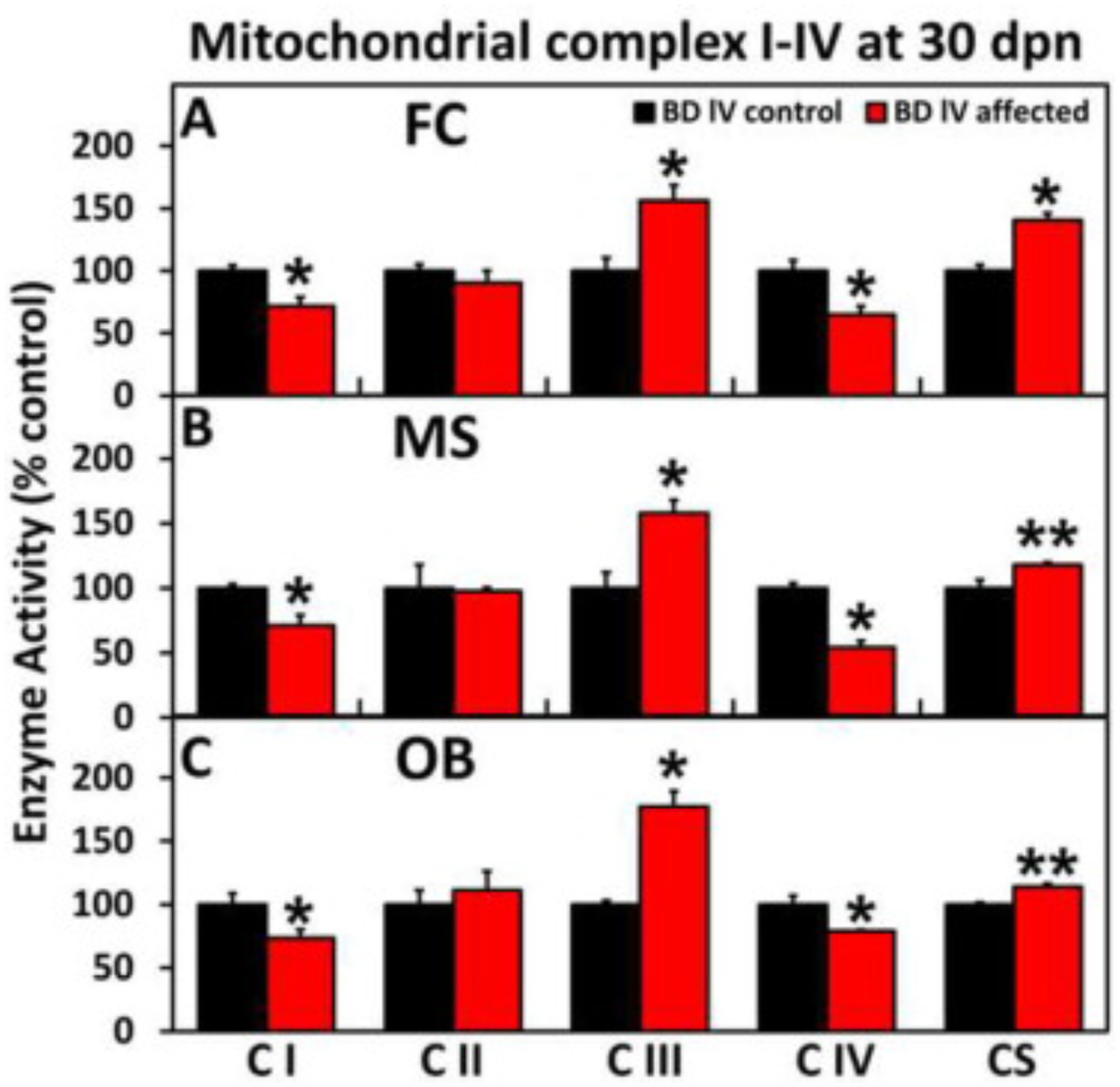
ETC Complex I-IV enzyme activity in the (A) frontal cortex, (B) mesencephalon and (C) olfactory bulb of affected and control BD-IV rats at 27 dpn. All three regions from the mutant (affected) animals show a significant reduction in complex 1 (NADH dehydrogenase) and IV (cytochrome c oxidase) activities. The complex III and citrate synthase (CS) activities are increased.
2.5. Dopamine (DA) metabolism in mutant rats
Historically, the majority of studies on DA metabolism within the CNS has been carried out on the striatal level, which is considered the final target of the nigrostriatal DA pathway that contains the highest DA concentration. Investigation of the striatal pathology and dopaminergic system alterations previously described in our BD-IV rat model (Guatteo et al., 2016, Stoica et al., 2012), required examination of the DA metabolic alteration in this mutant rat model. Impaired detoxification of biogenic aldehydes is important in the pathophysiology of neurodegeneration. Accumulation of potentially toxic aldehydes in the striatum raised our interest in investigating the possibility of aldehyde detoxification system involvement in DA metabolism in the mutant rats.
In the ST, mutant affected BD-IV rats showed a non-significant increase of DA, a significant increase of neurotoxic DOPAL, as well as significant DOPAL:DA and DOPAL:DOPAC elevations when compared with control littermates (Fig. 14A–D).
Fig. 14.
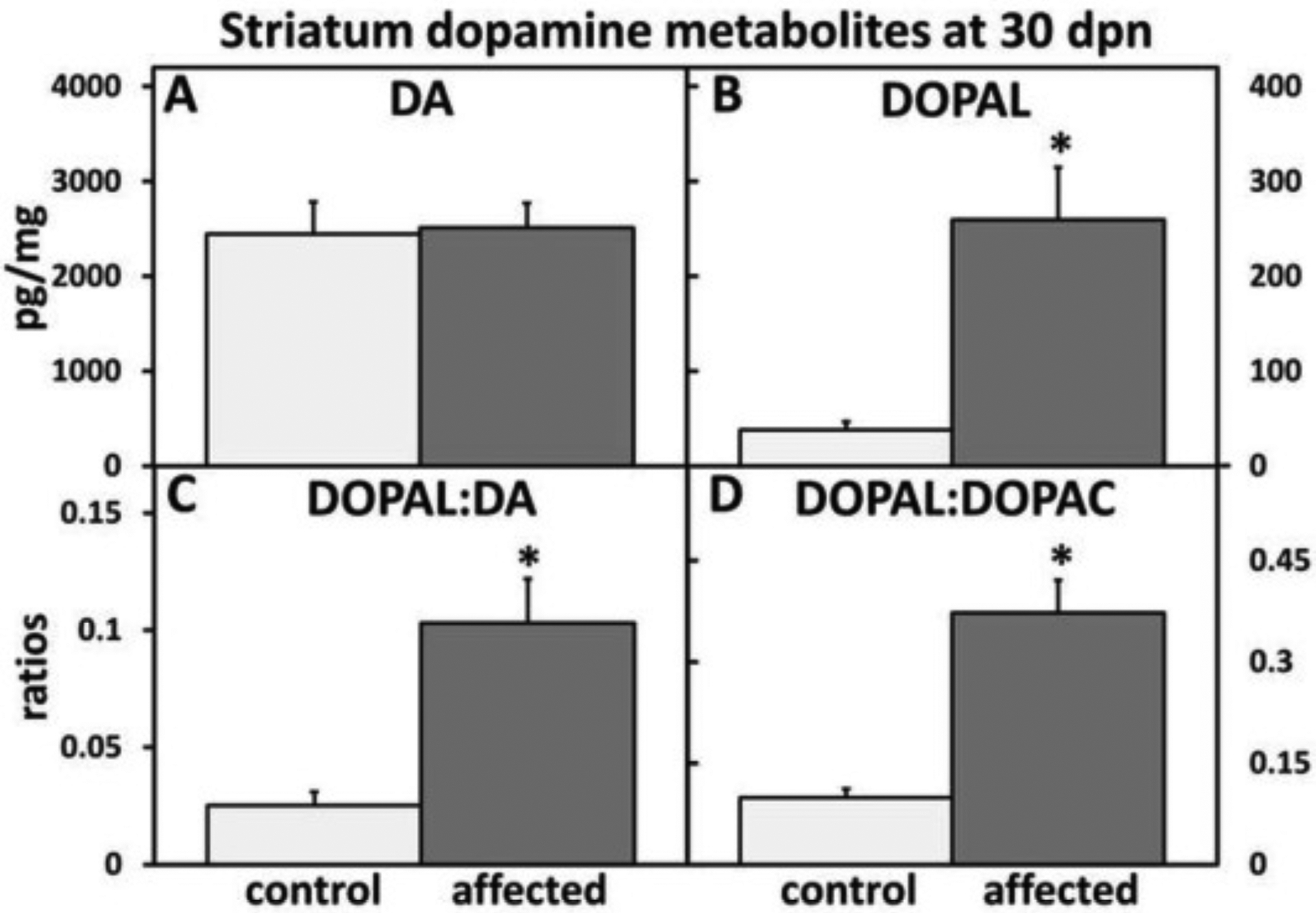
Striatum (ST) dopamine (DA) metabolism alteration in affected versus control rats at 30 dpn. (A) DA was slightly elevated in affected rat striatum. (B) There was a significant increase in neurotoxic DOPAL in the ST of affected rats. There was a significant increased in (C) DOPAL:DA and (D) DOPAL:DOPAC ratios in affected ST. (n = 4–6 animals per experimental condition).
WB analysis showed a significant decrease in mitochondrial ALDH1A2 and ALDH2 (Fig. 15A–D) protein levels in the ST of affected rats at 28 dpn when compared with control littermates.
Fig. 15.
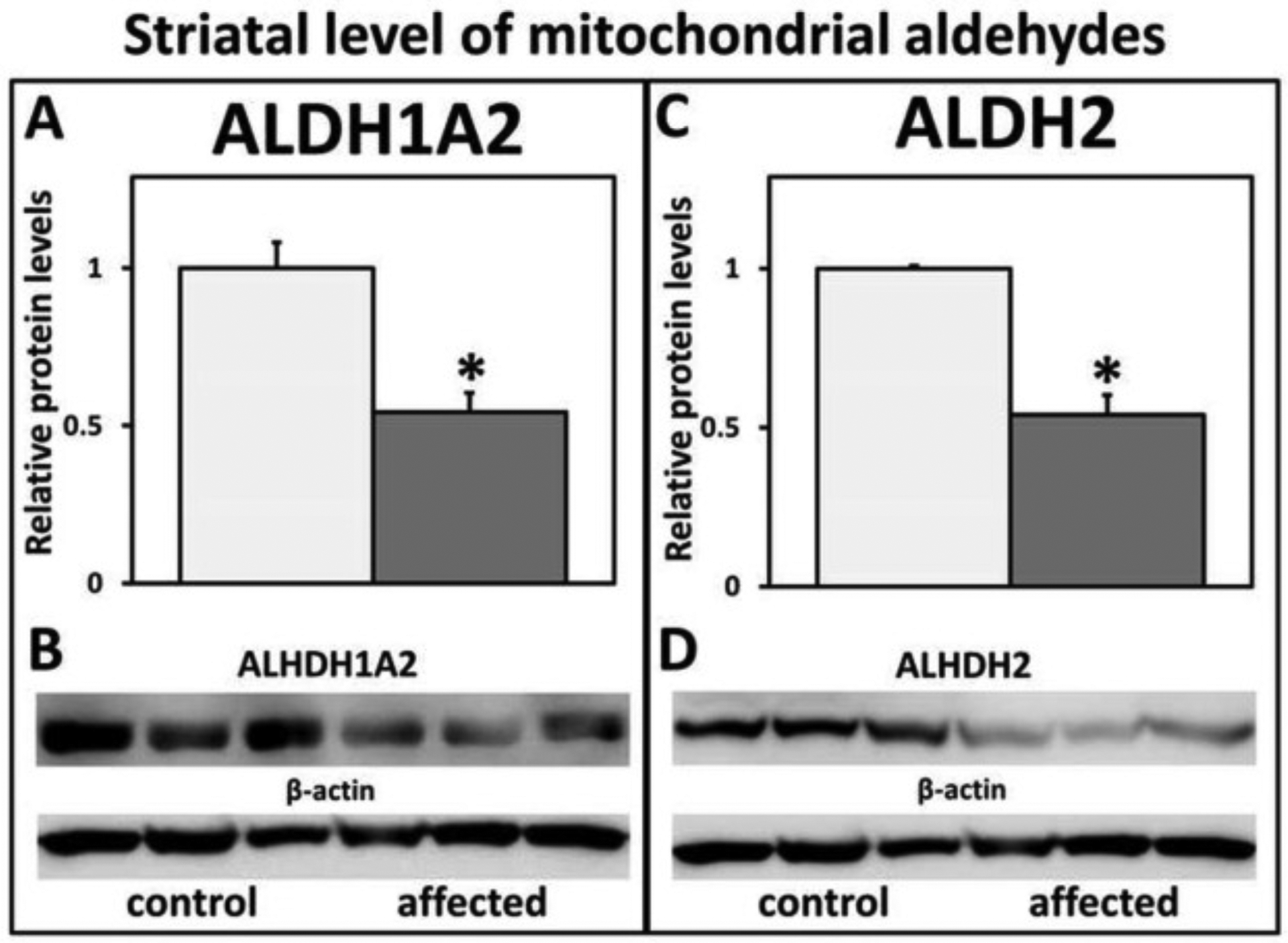
Western blot analysis and relative protein levels of striatum ALDH1A2 and ALDH2 in control versus affected rats. There was a significant decrease of (A, B) ALDH1A2 and (C, D) ALDH2 mitochondrial protein levels in the striatum (ST) of affected rats compared with control littermates at 28 dpn.
3. Discussion
3.1. Mapping of the rat mutation to the Myo5a gene
This study was performed on newborn, juvenile mutant BD-IV rats in which the mutated molecular motor Myo5a protein plays an important role in neurogenesis and was also associated with neuronal disease pathogenesis. The process of neural progenitor cell replication, differentiation and migration is regulated by molecular motors and their associated proteins as essential steps in neurogenesis. Their roles are diverse and poorly understood or unknown. The kinesin, dynein, and myosin superfamily molecular motors have fundamental roles in neuronal function, plasticity, morphogenesis, and survival by transporting cargos such as synaptic vesicle precursors, neurotransmitter and neurotrophic factor receptors, and mRNAs within axons, dendrites, and synapses. Molecular genetics has revealed additional roles for molecular motors in brain wiring, neuronal survival, neuronal plasticity, higher brain function, and control of central nervous system and peripheral nervous system development (Hirokawa et al., 2010).
Here we show the genomic mapping data of a Myo5a mutation, that spontaneously arose in a rat strain, associated with a severe neurological phenotype, and further describe the histological, ultrastructural, biochemical and molecular findings obtained through detailed characterization of this animal model. Through generations of backcrossing the mutant, which we refer to as “mutant”, to a syngeneic background and performing WGS of six rat genomes at ~30X coverage, we identified a mutation in Myo5a that uniquely segregated with the “dilute” phenotype (Strobel et al., 1990, Takagishi et al., 1996, Takagishi and Murata, 2006). The gene encoding Myo5a is an actin-based motor molecule, which is part of a large group of unconventional myosins. This protein is the predominant neuronal type and is highly expressed in the brain, and also in the skin. Myo5a protein is not expressed in the brain of mutant homozygous BD-IV rats (Fig. 2D), suggesting a null mutation for Myo5a. Myo5a has been suggested to play diverse roles in axonal and dendritic transport.
Several interesting morphological and biochemical findings are supportive of our “delayed neuro developmental and neurodegeneration associated with Myos5a mutation” hypothesis which were reported by our lab in the first publication on this rat model (Stoica et al., 2007), when we were unaware of the Myo5a mutation. This supportive evidence includes: persistence of cerebellar external granular layer, undifferentiated neurons in the adrenal medulla, disrupted hypothalamic-pituitary-thyroid axis, demyelination with a decrease in basic myelin protein, and significant reduction in body weight and bone development in affected compared to control littermates. In addition, ultrastructural examination of brain tissues from ST, SN, OB and BS showed extensive post-synaptic degeneration, perikaryal, axonal, mitochondrial swelling and cristae fragmentation, dendrite and spine alterative changes previously reported by our lab (Stoica et al., 2012). The present data are also supportive of our hypothesis showing a developmental stage-specific change in α-syn/tau-P protein levels in the anatomical regions examined. The level of expression differs with anatomical region examined, and was directly related to the stage of neonatal brain development. Toward 30 dpn, there was a general decrease in the expression of phosphorylated proteins in both affected and control littermates except FC, where expression remains significantly elevated, primarily in affected rats. Elevation of α-syn/tau-P indicated that FC brain region is not completely developed in both affected and control rats at 30 dpn. An unexpected significant decrease in α-syn-P in the OB in mutant brain might be related to the accumulation of macro-autophagosomes in the OB projections, which might block the axonal traffic of α-syn and neurotrasmitters to the OB. There was a shift from phosphorylated to non-phosphorylated proteins in both affected and control rats at 30 dpn. The decrease in the protein levels was delayed in the affected rats compared with controls.
Absence of Myo 5a protein expression in the brain of affected rats, due to their known or less understood protein/protein interactions, creates a pleiotropic domino–like effect that negatively dysregulates the normal homeostatic environment, resulting in excessive accumulation of α-syn/tau-P proteins, malfunction, neuronal degeneration and death in the affected rats. The neural basis of the motor coordination deficit is not completely understood but may be related to the absence of myosin Va during development of the nervous system in either cerebellar or non cerebellar structures, as it was suggested to occur in the viable myosin Va mutant dilute-neurological (d-n) mice (Miyata et al., 2011). The movement disorder observed in our rat model, we believe, relates to cerebellar, striatal lesions and a delay in myelination during development of the nervous system that might also contribute to motor incoordination (Lungu et al., 2013, Stoica et al., 2007, Stoica et al., 2012).
3.2. Potential role of α-syn/tau-P in neurodegeneration associated Myo5a mutation in BD-IV rats
Excessive accumulation of α-syn/tau-P proteins in the brain of affected rats raises the question of whether this accumulation might be responsible for the observed neurodegeneration associated with Myos5a mutation. In this study, we show that there is an increase in α-syn/tau-P, which are both post-translationally modified forms of proteins that are linked to neurodegenerative diseases in humans and animal models. In the homozygous mutant rat brain, biochemical and pathological changes were characterized by over-expression of α-syn/tau-P, a slight increase in the DA level with aberration of its metabolic pathway, mitochondrial alterations, and pathological changes characterized by dying back neuronal degeneration, all of which are associated with the lack of Myo5a protein expression in the brain of mutant BD-IV rats. Mutated α-syn has been implicated in the pathogenesis of several neurodegenerative disorders called α-synucleinopathies (Goldstein, 2003, Jellinger 2015, Spillantini et al., 1997, Yasuda et al., 2013). Also, excessive accumulation of wild type α-syn was reported to be responsible for neurodegeneration in the absence of its mutation (Byers et al., 2011). Interestingly, we find that there was an increase in the α-syn that cofractionates with the mitochondria. Alpha-syn accumulation in synapses and mitochondria of dopaminergic neurons was associated with decreased mitochondria complex I and IV activity (Fig. 13). The citrate synthase (CS) activity was slightly (15–40%) elevated in the mutants. Complex III (cytochrome c oxidoreductase) activity was also increased, which is a phenomenon observed in other models of secondary mitochondrial complex I deficiency and suggestive of a compensatory, adaptive response (Havlickova Karbanova et al., 2012, Wu et al., 2015). The elevation suggests that the cell is responding to reductions in ETC activities with a compensatory proliferation of mitochondria. These findings are suggestive of a dysfunction in mitochondrial energy metabolism/oxidative phosphorylation resulting in deregulation of normal neuronal homeostasis. All of these findings could be incriminated in the described neurodegeneration related to over-expression of α-syn-P in affected rats.
Tau is an axonal protein. The best-established function of tau is the stabilization of microtubules in mature neurons (Zempel and Mandelkow, 2014), but in disease conditions called tauopathies it redistributes to the somatodendritic compartments (Goedert, 2004). Tau phosphorylation can regulate its association with motor machinery, suggesting that signaling deregulation can lead to mislocalization (Cuchillo-Ibanez et al., 2008). It is well accepted that soluble tau is a ligand for α-syn, which can modulate the phosphorylation of tau. This stimulatory, synergistic effect of α-syn on tau phosphorylation demonstrated that α-syn has the ability to affect diverse intracellular signaling pathways (Jensen et al., 1999). Hence, in the absence of Myo5a, α-syn accumulation that occurs early, at 15 dpn in the “mutant” rat, may trigger the increase of tau-P at later stages of the neurodegenerative progression. There was a progressive phosphorylated α-syn/tau upregulation in several brain anatomical regions in the BD-IV affected rats (Fig. 3), which are integral parts of basal ganglia and dopaminergic systems. The increase in α-syn-P levels precedes increases in tau-P levels at 15 dpn. There were no significant differences in tau-P expression levels between mutant and non-affected BD-IV rats in the anatomical regions analyzed at 15 dpn. However, there was a significant elevation of tau-P expression levels in all anatomical brain regions analyzed in mutants when compared with controls littermates at 30 dpn. In addition, IHC examination shows that there was tau-P perikaryal retention in BS, HC and ST neurons in mutant rats at 30 dpn. Hyperphosphorylation of tau is interpreted as a hallmark of pathology, but can also occur physiologically (Arendt et al., 2003, Planel et al., 2007). It is accepted that the elevation of tau causes inhibition of motor-based transport (Kapitein et al., 2011, Li et al., 2011), with subsequent mitochondrial dysfunction and spine loss, as previously reported by us in this mutant model (Stoica et al., 2012). In the mutant brains, we hypothesize that there was a delay in redistribution of tau-P from the somatodentritic compartment to the axonal compartment rather than mislocalization, as reported in typical neurodegenerative diseases (Zempel and Mandelkow, 2014) where tau-P mislocalizes to the somatodendritic compartment. In the affected rats, tau-P should be axonally localized only, as it is in control littermates at 30 dpn. This argument is supported by the fact that the tau-P retention pattern is not affecting all neurons in the regions examined, and somatodentritic retention is observed in some neurons in BS, OB and CB, only, as exemplified in Fig. 7, Fig. 8, Fig. 9.
Over-expression and perikaryal retention of these two proteins was interpreted as excessive and a potential contributory factor in the pathogenesis of some of the neurodegeneration in our model. Accumulation of α-syn/tau-P, perikaryal and axonal, and alteration of the dopaminergic system in the absence of Myo5a motor movement protein might be responsible, in addition to a systemic alteration of the entire body’s homeostatic environment, for the early death in the BD-IV mutant rats.
Autophagy is a lysosomal degradation pathway that is critical for maintaining the normal cellular homeostatic environment (Nixon and Yang, 2012). Autophagy is implicated in the pathogenesis of many neurodegenerative disorders. Despite the major advances in this field, the mechanism and the complexity of autophagy’s involvement in human disease remain elusive (Webb et al., 2003). In our BD-IV mutant rat model for neurodegeneration, the defects in autophagic flux in specific brain anatomical locations was observed by TEM examination and specific biomarkers Fig. 10A–E). Autophagosome formation was analyzed at the late period of survival of affected rats, between 25 and 30 dpn. The macro-autophagosomes in various stage of development were in the olfactory tract and BS nerve fiber projections. Little early stage autophagosome formation was present in the neuronal perikarya in the BS, ST and SN. In addition, ultrastructural changes characterized by degeneration of myelinated and non-myelinated nerve fibers, perikaryal microtubule and mitochondria aggregation were observed in dorsal striatum of affected rats at 25–30 dpn (Fig. 11B), which supports the dying back neuronal degeneration phenotype in this rat model. Accumulation of autophagosomes is most likely in response to α-syn/tau-P accumulation in specific brain anatomical areas. The mechanism(s) of autophagosomes in this spontaneous mutant suggests a multifactorial origin, which adds to the complexity of brain pathology associated with Myo5a mutation. However, further study is necessary to elucidate the mechanism and role of autophagy in this spontaneous model of abnormal development and neurodegeneration.
3.3. Pathogenesis of striatal dopamine metabolism alteration in BD-IV mutant rats
The decreased levels of aldehyde dehydrogenases were associated with neurodegenerative changes affecting the dopaminergic system and the basal ganglia, supporting the hypothesis that impaired detoxification of biogenic aldehydes might be an additional contributory factor in the pathophysiology of neurodegeneration in our rat model.
In the BD-IV mutant rat model, loss of detoxifying ALDH enzymes was associated with a marked decline in motor function, loss of tyrosine hydroxylase immunoreactive neurons, altered dopamine metabolism, and increased accumulation of DOPAL, a neurotoxic metabolite of DA in the striatum. DOPAL induces α-syn aggregation in a cell-free system of dopaminergic neurons and in vivo neuronal loss with stereotactic injections into the SN of Sprague-Dawley rats (Burke et al., 2008). Most importantly, in the affected animals, striatal DOPAL is increased, and DOPAC is decreased. These findings indicate a buildup of DOPAL, due to a shift from decreased vesicular uptake to oxidative deamination of cytosolic DA and to decreased ALDH1A2 activity (Wey et al., 2012). This pattern fits well with the catecholadehyde hypothesis (Goldstein et al., 2011, Goldstein and Sims-O’Neil, 2016). Striatal DA was only slightly increased, perhaps the depletion takes longer to develop. We hypothesize that increased buildup of catecolaldehydes, such as DOPAL, comes first. These compounds are neurotoxic and induce degeneration of the DA terminals in the ST and MS. DOPAL buildup also promotes α-syn oligomerization (Burke et al., 2008, Goldstein et al., 2016).
The mitochondrial-associated ALDH2 is of particular interest in our BD-IV rat model for neurodegeneration because it metabolizes toxic aldehydes in brain tissue, including DOPAL. Another interesting finding in our rat model is the association of the α-syn increase, accumulation of DOPAL, decreased ALDHs, and alteration of mitochondrial complex I-IV activity. We hypothesize that DA is metabolized in the mitochondria to DOPAL and hydrogen peroxide. The hydroxyl radical can enter mitochondria via the voltage-dependent anion channels and inhibit complex I activity, affecting NAD levels important for aldehyde dehydrogenase conversion of DOPAL to DOPAC (Galvin, 2006).
Mitochondrial and synaptic accumulations of α–syn associated with a disrupted DA metabolic pathway and tau over-expression are novel findings associated with Myo5a mutation, in the BD-IV mutant rat model. We propose that a reciprocal interplay exists between hyperphosphorylated tau, a ligand for α-syn, which can modulate the phosphorylation of tau. In our rat model, there is a widespread accumulation of α-syn/tau-P in the specific brain areas. Tau and α-syn are presynaptic proteins involved in the normal development of nervous system but their excessive accumulation could have a pathological significance. The role of the actin-based motor Myo5a in co-operation with other molecular motor proteins may be to transport these proteins from postsynaptic/dendritic sites into axons/presynaptic sites to exert their normal physiological functions. Absence of brain Myo5a protein expression resulted in excessive axonal and synaptic accumulation of these proteins, a phenomenon that could be responsible for dying back neuronal degeneration. In addition, synaptic blockade contributed to loss of trafficking for neurotrasmitters and growth factors necessary for neuronal survival. However, further investigation of this mutant rat will likely reveal the relationship between Myo5a and pathological findings reported, and help us to better understand how different pathological events come together to ultimately result in the devastating neurological phenotype and death of the mutant animals.
3.4. Conclusions
We have identified in vivo neuropathological and biochemical alterative changes involved in the pathogenesis of a spontaneous autosomal recessive movement disorder in a mutant Myo5a juvenile rat model. The over-expression of α-syn/tau-P, striatal ultrastructural pathological changes, and an altered dopaminergic system were not previously reported to be associated with Myo5a mutations in human diseases and animal models, based on our best knowledge. In the current study we demonstrate that absence of Myo5a protein in the brain tissue of mutant homozygous BD-IV rats is associated with upregulation of α-syn/tau-P in specific brain anatomical locations. Excessive axonal terminal accumulation of these proteins precedes neuronal loss and is believed to be responsible for the dying back neuronal degeneration described in this model. This Myo5a mutation appears to alter the DA homeostasis in favor of the accumulation of the neurotoxic DOPAL metabolite in our model. This mutant juvenile rat model is a developmental disorder showing pleiotropic diverse effects associated with the Myo5a mutation.
4. Experimental procedure
4.1. Whole genome next generation sequencing (NGS)
Paired-end libraries were generated for each animal sample for whole genome sequencing using Illumina protocols. Finished DNA libraries were then sequenced on an Illumina HiSeq 2000 following standard operating procedure.
4.2. Bioinformatics analysis
Sequenced reads were mapped to the rn4 rat reference (http://rgd.mcw.edu/) using the BWA alignment tool (Li and Durbin, 2009). To ensure more accurate mutation calling, aligned read data was processed using the recalibration and local realignment functions of the Genomic Analysis Tool Kit (GATK) (McKenna et al., 2010). Variant determination was performed on a sample by sample basis using Atlas-SNP2 and Atlas-Indel to detect single nucleotide variations (SNVs) and small insertions or deletions (indels), respectively (Challis et al., 2012). Homozygosity mapping and determination of shared homozygous regions among samples were performed using custom Perl scripts.
4.3. Animals
Brains from Berlin-Druckrey IV (BD-IV) mutant (affected) and age-matched control (BD-IV non-carrier littermate) juvenile rats were used. The background of this rat colony has been previously described (Stoica et al., 2012). All animal procedures were approved by the Texas A&M University Institutional Animal Care and Use Committee.
4.4. Tissue processing and collection
For western blot analysis, rats were sacrificed by decapitation under anesthesia and different brain areas were immediately removed, placed on dry ice and stored at −80 °C until use. Brains from BD-IV affected rats (n = 3–5) and from control BD-IV rats (n = 3–4) at 15 and 30 dpn were used for western blot analysis. For immunohistochemistry (IHC), BD-IV affected (n = 3) and control (n = 3) rats at 25 dpn were perfused under anesthesia (ketamine hydrochloride, 1.5 mL/kg, 75 mg/kg [Phoenix Scientific, Inc., St. Joseph, MO, USA] and Xylazine, 0.5 mL/kg, 10 mg/kg [Bayer AG, Pittsburgh, PA, USA]) via the left ventricle with 4% paraformaldehyde (PFA) solution (Electron Microscopy Science, Hatsfield, PA, USA) in phosphate-buffered saline (PBS, pH 7.2 at 4 °C) using a peristaltic pump (Stoica et al., 2012).
4.5. Immunohistochemistry (IHC)
IHC was carried out as previously described (Stoica et al., 2012). Anti α-synuclein and Tau-P (Anti-Tau phospho S396) antibody were purchased from Abcam (Cambridge, MA, USA), anti COX IV antibody from Cell Singaling Technology (Danvers, MA, USA), anti α-synuclein phos.129 from GeneTex (Irvine, CA, USA). Following the primary antibody reaction, sections were washed and incubated with anti-mouse or anti-rabbit IgG HRP (Vector Laboratories, Burlingame, CA, USA). Sections that were not incubated with primary antibody served as negative controls. Paraffin-embedded tissue sections were pre-treated using heat mediated antigen retrieval with citrate buffer. The sections were incubated with antibody at 1:1000 dilution for 2 h at 21 °C and a biotin-conjugated goat-anti-rabbit antibody was used as a secondary at 1:250.
4.6. Immunofluorescence confocal microscopy
Rat brains were collected after 4% paraformaldehyde perfusion. The brains were then fixed overnight at 4 °C in 4% paraformaldehyde in PBS. The brains were then dehydrated in 30% sucrose in PBS. Thirty-five micrometer coronal sections were cut with a microtome and stored in cryoprotectant at −20 °C. Prior to staining, sections were washed in PBS and blocked in PBS containing 0.4% Triton X-100 and 2% donkey serum for one hour at room temperature. The sections were then incubated with primary antibody (mouse anti-clone 42/alpha synuclein, BD Transduction Laboratories; rabbit anti-COX IV #4850-from-Cell Singaling Technology), diluted 1:500 in blocking solution overnight at 4 °C. After washing in PBS, sections were incubated with secondary antibody [Alexafluor 488- and Alexafluor 555-conjugated antibodies (Invitrogen)] for 2 h at room temperature and mounted in a DAPI solution after final washing. Images were taken on a confocal laser microscope (Leica SPE).
4.7. Western blotting
Frozen brain tissues were homogenized in lysis buffer containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM CaCl2, 1mM MgCl2, 0.1% Triton X-100 and a mixture of protease inhibitors at 10 μg/ml (Sigma-Aldrich, St. Louis, MO, USA). The protein content was determined using the Bradford Assay (Bio-Rad, Hercules, CA. USA), with bovine serum albumin as the standard. Proteins (10–30 μg) were separated by 9–15% SDS polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (PALL Life Sciences, Ann Arbor, MI, USA). Membranes were incubated 1 h in blocking buffer (20 mM Tris–HCl buffered saline containing 0.1% Tween 20) at room temperature and then probed with appropriate antibodies in blocking buffer overnight. Blots were incubated with anti-rabbit Myo5A (ab11094), rabbit TAU phos.396 (ab109390), rabbit ALD1A2 (ab75674), rabbit ALDH2 (ab108306) and mouse anti GAPDH (ab9484), all purchased from Abcam. Anti-rabbit α-synuclein (SC-7011R) from Santa Cruz (Dallas, TX, USA), rabbit a-synuclein phos.129 (GTX50222) from GeneTex (Irvine, CA, USA), rabbit total TAU (A0024) from Dako (Carpinteria, CA, USA) and mouse β-actin from Sigma-Aldrich were also used. Peroxidase-labeled anti-rabbit and anti-mouse secondary antibodies were purchased from BioRad. After additional washes, the blots were incubated with chemiluminescent substrate, according to directions in the kit (Super-Signal West Pico, Pierce, Rockford, IL, USA). Mitochondrial lysates were prepared from different brain regions of affected and control rats using a mitochondrial isolation kit (Bio Chain Institute, Newark, CA, USA).
4.8. Electron transport chain (ETC) complexes I-IV assays
An ETC activity assay was performed as previously described (Wu et al., 2015). For enzymatic assays of respiratory chain complexes I-IV, sufficient amounts of tissue or cells were collected. Potassium phosphate buffer (25 mM, pH 7.5) was added to a final volume of 300 mL and the samples were sonicated (5 s pulse x4, 60% power) using a Microson XL2000 Ultrasonic Cell Disruptor (Misonix, Farmingdale, NY). For experiments using isolated mitochondria, the mitochondria were also disrupted by sonication as described above. The spectrophotometric kinetic assays were performed at 30 °C in a volume of 175 mL using a monochromator microplate reader (Tecan M200). Complex I activity (NADH:ubiquinone oxidoreductase) was determined by measuring oxidation of NADH at 340 nm (using ferricyanide as the electron acceptor) in a reaction mixture of 25 mM potassium phosphate (pH 7.5), 0.2 mM NADH, and 1.7 mM potassium ferricyanide. Complex II activity (succinate sehydrogenase) was determined by measuring the reduction of the artificial electron acceptor 2,6-dichlorophenol-indophenol (DCIP) at 600 nm in a reaction mixture of 25 mM potassium phosphate (pH 7.5), 20 mM succinate, 0.5 mM DCIP, 10 mM rotenone, 2 mg/mL antimycin A, and 2 mM potassium cyanide. Complex III activity (ubiquinol:cytochrome c oxidoreductase) was determined by measuring the reduction of cytochrome c at 550 nm in a reaction mixture of 25 mM potassium phosphate (pH 7.5), 35 mM reduced decylubiquinone, 15 mM cytochrome c, 10 mM rotenone, and 2 mM potassium cyanide. Complex IV activity (cytochrome C oxidase) was determined by measuring the oxidation of cytochrome C at 550 nm in a reaction mixture of 10 mM potassium phosphate (pH 7.5) and 0.1 mM reduced cytochrome C. Citrate synthase activity was determined by measuring the reduction of 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) at 412 nm, which is coupled to the reduction of acetyl-CoA by citrate synthase in the presence of oxaloacetate. The reaction mixture consists of 10 mM potassium phosphate (pH 7.5), 100 mM DTNB, 50 mM acetyl-CoA, and 250 mM oxaloacetate. All activities were calculated as nmoles/min/mg protein, and expressed as a percentage of control activity. Experiments were performed on 6 independent samples for each genotype.
4.9. HPLC analysis
Tissue contents of DA, 3,4-dihydroxyphenylacetaldehyde (DOPAL), and 3,4-dihydroxyphenylacetic acid (DOPAC) were assayed in the laboratory of the Clinical Neurocardiology Section of the National Institute of Neurological Disorders and Stroke at the National Institutes of Health, as described previously (Casida et al., 2014, Goldstein et al., 2011). Briefly, after tissue homogenization, the supernatant was subjected to batch alumina extraction to partially purify the catechols, and the alumina eluate was injected into a liquid chromatographic system with post-column series electrochemical detection.
4.10. Transmission electron microscopy (TEM) studies
CNS samples from five affected and five control littermates were processed for TEM as previously described (Stoica et al., 2007). Briefly, rats were deeply anesthetized with pentobarbital (10 mg/kg intraperitoneally) and then perfused through the aorta via the left ventricle with PBS, pH 7.4, followed by fresh 4 °C Karnovsky’s type fixative (2% glutaraldehyde and 2% paraformaldehyde). The perfused brain tissues were cut into 1-mm cubes and left overnight at 4 °C in the same fixative. The tissues were then washed twice in 0.1 M cacodylate buffer, and post fixed in 1.3% osmium tetroxide in 2,4,6-collidine, pH 7.4. Thin sections for ultrastructural evaluation were cut on an LKB ultramicrotome with a diamond knife, stained with uranyl acetate and lead citrate, and examined with a Zeiss 10 electron microscope (Carl Zeiss NTS, LLC, Thornwood, NY, USA).
4.11. Statistical analysis
Densitometry analysis of western blot bands was performed using the public domain NIH Image-J program (http://rsb.info.nih.gov/nih-image). Western blots were analyzed using the Odyssey program and band densities were normalized to the density of the loading controls. Information shown in Table 1 was calculated as a% changes between 15 dpn and 30 dpn in control rats and between 15 dpn and 30 dpn in affected rats. All data are presented as means ± standard error of the mean (S.E.M.). P-values were calculated using Student’s unpaired t-test. Statistical significance was taken at P < .05.
Highlights.
Loss of MYO5A results in structural and functional alterations in the rat brain.
α-Syn/Tau-P is significantly elevated in mutant brain regions.
Myo5a mutant rats exhibit alterations in dopamine metabolism in the striatum.
α-Syn accumulation is associated with alteration of mitochondrial enzyme activity.
Neuropathological changes are characteristic of dying back degeneration.
Delayed neurodevelopment and neurodegeneration are associated with a Myo5a mutation.
Acknowledgements
We thank Dr. Gina Lungu, former Research Associate in Dr. G. Stoica’s Laboratory for her past research contribution on this rat model and for technical assistance to Jean Kovar for supervision of the rat colony; Dr. Ambrus Andy, for help with immunohistochemistry, Ross Payne, for help with electron microscopy, and Dr. Cindy Meininger and Dr. Louise Abbott for critical review of this manuscript. This research was funded in part by the MJ Fox Foundation for Parkinson’s Disease (Rapid Response Innovation Awards, 2010, 2013).
Abbreviations
- α-Syn-P
α-synuclein phosphorylated
- Tau-P
tau phosphorylated
- mutant
homozygous mutant BD-IV rats
- BD-IV
Berlin Druckrey
- SD
Sprague Dawley
- Myo5a
Myosin5a gene
- DA
dopamine
- DOPAL
3,4-dyhydroxyphenyl-acetaldehyde
- DOPAC
dihydroxyphenylacetic acid
- ALDH
aldehyde dehydrogenases
- miRNA
microRNAs
- ETC
electron transport chain
- TEM
transmission electron microscopy
- NGS
whole genome next generation sequencing
- IHC
immunohistochemistry
- BS
brain stem
- ST
striatum
- FC
frontal cortex
- OB
olfactory bulb
- CR
cerebellum
- MS
mesencephalon
- HC
hippocampus
- SNpc
substantia nigra pars compacta
- PBS
phosphate-buffered saline
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- WB
western blot
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest to report.
Ethical approval
All procedures performed in these studies involving animals were in accordance with published ethical standards. All animal procedures were approved by the Texas A&M University Institutional Animal Care and Use Committee.
References
- Arendt T, et al. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals J. Neurosci, 23 (2003), pp. 6972–6981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boassa D, et al. Mapping the subcellular distribution of alpha-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: implications for Parkinson’s disease pathogenesis J. Neurosci, 33 (2013), pp. 2605–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgman PC Myosin Va movements in normal and dilute-lethal axons provide support for a dual filament motor complex J. Cell Biol, 146 (1999), pp. 1045–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgman PC Myosin motor proteins in the cell biology of axons and other neuronal compartments Results Probl. Cell Differ, 48 (2009), pp. 91–105 [DOI] [PubMed] [Google Scholar]
- Brooks SA, et al. Whole-genome SNP association in the horse: identification of a deletion in myosin Va responsible for Lavender Foal Syndrome PLoS Genet, 6 (2010), p. e1000909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke WJ, et al. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine Acta Neuropathol, 115 (2008), pp. 193–203 [DOI] [PubMed] [Google Scholar]
- Byers B, et al. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress PLoS One, 6 (2011), p. e26159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casida JE, et al. Benomyl, aldehyde dehydrogenase, DOPAL, and the catecholaldehyde hypothesis for the pathogenesis of Parkinson’s disease Chem. Res. Toxicol, 27 (2014), pp. 1359–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis D, et al. An integrative variant analysis suite for whole exome next-generation sequencing data BMC Bioinf, 13 (2012), p. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland NG, Jenkins NA, Lee BK Association of the lethal yellow (Ay) coat color mutation with an ecotropic murine leukemia virus genome Proc. Natl. Acad. Sci. U.S.A, 80 (1983), pp. 247–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchillo-Ibanez I, et al. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin FASEB J, 22 (2008), pp. 3186–3195 [DOI] [PubMed] [Google Scholar]
- Dekker-Ohno K, et al. Endoplasmic reticulum is missing in dendritic spines of Purkinje cells of the ataxic mutant rat Brain Res, 714 (1996), pp. 226–230 [DOI] [PubMed] [Google Scholar]
- Futaki S, et al. Identification of a novel myosin-Va mutation in an ataxic mutant rat, dilute-opisthotonus Mamm. Genome, 11 (2000), pp. 649–655 [DOI] [PubMed] [Google Scholar]
- Galvin JE Interaction of alpha-synuclein and dopamine metabolites in the pathogenesis of Parkinson’s disease: a case for the selective vulnerability of the substantia nigra Acta Neuropathol, 112 (2006), pp. 115–126 [DOI] [PubMed] [Google Scholar]
- Goedert M Tau protein and neurodegeneration Semin. Cell Dev. Biol, 15 (2004), pp. 45–49 [DOI] [PubMed] [Google Scholar]
- Goldstein DS, et al. Catechols in post-mortem brain of patients with Parkinson disease Eur. J. Neurol, 18 (2011), pp. 703–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, et al. Comparison of monoamine oxidase inhibitors in decreasing production of the autotoxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde in PC12 cells J. Pharmacol. Exp. Ther, 356 (2016), pp. 484–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Sims-O’Neil C Systemic hemodynamics during orthostasis in multiple system atrophy Parkinsonism Relat. Disord, 25 (2016), pp. 106–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LS Do disorders of movement cause movement disorders and dementia? Neuron, 40 (2003), pp. 415–425 [DOI] [PubMed] [Google Scholar]
- Griscelli C, Prunieras M Pigment dilution and immunodeficiency: a new syndrome Int. J. Dermatol, 17 (1978), pp. 788–791 [DOI] [PubMed] [Google Scholar]
- Guatteo E, et al. Functional alterations of the dopaminergic and glutamatergic systems in spontaneous alpha-synuclein overexpressing rats Exp. Neurol, 287 (2016), pp. 21–33 [DOI] [PubMed] [Google Scholar]
- Havlickova Karbanova V, et al. Compensatory upregulation of respiratory chain complexes III and IV in isolated deficiency of ATP synthase due to TMEM70 mutation Biochim. Biophys. Acta, 1817 (2012), pp. 1037–1043 [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease Neuron, 68 (2010), pp. 610–638 [DOI] [PubMed] [Google Scholar]
- Huang JD, et al. Direct interaction of microtubule- and actin-based transport motors Nature, 397 (1999), pp. 267–270 [DOI] [PubMed] [Google Scholar]
- Jellinger KA How close are we to revealing the etiology of Parkinson’s disease? Expert Rev. Neurother, 15 (2015), pp. 1105–1170 [DOI] [PubMed] [Google Scholar]
- Jenkins NA, et al. Dilute (d) coat colour mutation of DBA/2J mice is associated with the site of integration of an ecotropic MuLV genome Nature, 293 (1981), pp. 370–374 [DOI] [PubMed] [Google Scholar]
- Jensen PH, et al. alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356 J. Biol. Chem, 274 (1999), pp. 25481–25489 [DOI] [PubMed] [Google Scholar]
- Jones JM, et al. The mouse neurological mutant flailer expresses a novel hybrid gene derived by exon shuffling between Gnb5 and Myo5a Hum. Mol. Genet, 9 (2000), pp. 821–828 [DOI] [PubMed] [Google Scholar]
- Kapitein LC, et al. NMDA receptor activation suppresses microtubule growth and spine entry J. Neurosci, 31 (2011), pp. 8194–8209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform Bioinformatics, 25 (2009), pp. 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, et al. Novel diffusion barrier for axonal retention of Tau in neurons and its failure in neurodegeneration EMBO J, 30 (2011), pp. 4825–4837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lungu G, Stoica G, Ambrus A MicroRNA profiling and the role of microRNA-132 in neurodegeneration using a rat model Neurosci. Lett, 553 (2013), pp. 153–158 [DOI] [PubMed] [Google Scholar]
- McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data Genome Res, 20 (2010), pp. 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menasche G, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome Nat. Genet, 25 (2000), pp. 173–176 [DOI] [PubMed] [Google Scholar]
- Mercer JA, et al. Novel myosin heavy chain encoded by murine dilute coat colour locus Nature, 349 (1991), pp. 709–713 [DOI] [PubMed] [Google Scholar]
- Miyata M, et al. A role for myosin Va in cerebellar plasticity and motor learning: a possible mechanism underlying neurological disorder in myosin Va disease J. Neurosci, 31 (2011), pp. 6067–6078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Yang DS Autophagy and neuronal cell death in neurological disorders Cold Spring Harbor Perspect. Biol, 4 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastural E, et al. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene Nat. Genet, 16 (1997), pp. 289–292 [DOI] [PubMed] [Google Scholar]
- Planel E, et al. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia J. Neurosci, 27 (2007), pp. 3090–3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissmann M, Ludwig A Pleiotropic effects of coat colour-associated mutations in humans, mice and other mammals Semin. Cell Dev. Biol, 24 (2013), pp. 576–586 [DOI] [PubMed] [Google Scholar]
- Rudolf R, Bittins CM, Gerdes HH The role of myosin V in exocytosis and synaptic plasticity J. Neurochem, 116 (2011), pp. 177–191 [DOI] [PubMed] [Google Scholar]
- Spillantini MG, et al. Alpha-synuclein in Lewy bodies Nature, 388 (1997), pp. 839–840 [DOI] [PubMed] [Google Scholar]
- Stoica G, et al. Inherited tertiary hypothyroidism in Sprague-Dawley rats Brain Res, 1148 (2007), pp. 205–216 [DOI] [PubMed] [Google Scholar]
- Stoica G, et al. Potential role of alpha-synuclein in neurodegeneration: studies in a rat animal model J. Neurochem, 122 (2012), pp. 812–822 [DOI] [PubMed] [Google Scholar]
- Strobel MC, et al. Molecular analysis of two mouse dilute locus deletion mutations: spontaneous dilute lethal20J and radiation-induced dilute prenatal lethal Aa2 alleles Mol. Cell Biol, 10 (1990), pp. 501–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagishi Y, et al. The dilute-lethal (dl) gene attacks a Ca2+ store in the dendritic spine of Purkinje cells in mice Neurosci. Lett, 215 (1996), pp. 169–172 [DOI] [PubMed] [Google Scholar]
- Takagishi Y, Murata Y Myosin Va mutation in rats is an animal model for the human hereditary neurological disease, Griscelli syndrome type 1 Ann. N.Y. Acad. Sci, 1086 (2006), pp. 66–80 [DOI] [PubMed] [Google Scholar]
- Webb JL, et al. Alpha-Synuclein is degraded by both autophagy and the proteasome J. Biol. Chem, 278 (2003), pp. 25009–25013 [DOI] [PubMed] [Google Scholar]
- Wey MC, et al. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson’s disease PLoS One, 7 (2012), p. e31522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SP, et al. Increased COUP-TFII expression in adult hearts induces mitochondrial dysfunction resulting in heart failure Nat. Commun, 6 (2015), p. 8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Nakata Y, Mochizuki H alpha-Synuclein and neuronal cell death Mol. Neurobiol, 47 (2013), pp. 466–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavich L, et al. Role of alpha-synuclein in presynaptic dopamine recruitment J. Neurosci, 24 (2004), pp. 11165–11170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Mandelkow E Lost after translation: missorting of Tau protein and consequences for Alzheimer disease Trends Neurosci, 37 (2014), pp. 721–732 [DOI] [PubMed] [Google Scholar]