

SUMMARY
Lipid droplet (LD) formation from the endoplasmic reticulum (ER) is accompanied by the targeting and accumulation of specific hydrophobic, membrane-embedded proteins on LDs. The determinants of this process are unknown. Here we study the hydrophobic membrane motifs of two Drosophila melanogaster proteins, GPAT4 and ALG14, that utilize this pathway, and we identify crucial sequence features that mediate LD accumulation. Molecular dynamics simulations and studies in cells reveal that LD targeting of these motifs requires: deeply inserted tryptophans that have lower free energy in the LD oil phase, and positively charged residues near predicted hairpin hinges that become less constrained in the LD environment. Analyzing hydrophobic motifs from similar LD-targeting proteins, it appears that the distribution of tryptophan and positively charged residues distinguishes them from non-LD targeting membrane motifs. Our studies identify specific sequence features and principles of hydrophobic membrane motifs that mediate their accumulation on LDs.
Keywords: Lipid droplets, endoplasmic reticulum, protein targeting, glycerol-3-phosphate acyltransferase 4 (GPAT4), LiveDrop, UDP-N-acetylglucosaminyltransferase subunit (ALG14)
Graphical Abstract

eTOC Blurb
Olarte et al. identify mechanistic determinants for targeting of hydrophobic membrane motifs from the endoplasmic reticulum to lipid droplets. Evidence indicates that positively charged and tryptophan residues within these motifs adopt energetically favored orientations at the lipid droplet monolayer, thus driving protein accumulation on lipid droplets.
INTRODUCTION
Lipid droplets (LDs) are unusual organelles, consisting of an organic phase of neutral lipids, such as triacylglycerols (TGs) and sterol esters, bounded by a monolayer of phospholipids (Tauchi-Sato et al., 2002; Walther et al., 2017). This oil-water interface harbors specific proteins that vary in different cell types and species (Bersuker et al., 2018; Bouchoux et al., 2011; Brasaemle et al., 2004; Currie et al., 2014; Ding et al., 2012). LD proteins number in the tens to hundreds, and many are involved in lipid metabolism. For instance, enzymes of both TG synthesis (Poppelreuther et al., 2018; Wilfling et al., 2013) and lipolysis (Zechner, 2015) localize to the surface of LDs. Additionally, enzymes for the biosynthesis of ergosterol (in yeast) and phosphatidylcholine localize to LDs (Currie et al., 2014; Krahmer et al., 2011).
Proteins target LDs from the cytosol or from the endoplasmic reticulum (ER) bilayer membrane (Bersuker and Olzmann, 2017; Kory et al., 2016; Olzmann and Carvalho, 2019). LDs appear not to possess dedicated protein targeting machinery, such as the translocon in the ER, nor are they known to utilize biochemical landmarks, such as phosphoinositides, to recruit and bind proteins. Instead, proteins target LDs by recognizing the unique physical properties of the phospholipid monolayer. Molecular dynamics simulations showed that surface phospholipid packing defects are more frequent, more persistent, and larger on LDs than on bilayer membranes (Bacle et al., 2017; Prevost et al., 2018). Cytosolic proteins with amphipathic helices recognize these phospholipid packing defects at the LD monolayer surface (Kory et al., 2016; Prevost et al., 2018).
Proteins may also access the LD monolayer surface from the ER bilayer membrane. Such targeting requires membrane continuity between the outer leaflet of the ER membrane and the monolayer surface of LDs, either when LDs bud from the ER or at later steps in LD biogenesis via ER-LD bridges (Wilfling et al., 2014; Wilfling et al., 2013). Glycerol-3-phosphate acyl transferase 4 (GPAT4) catalyzes the first step in glycerolipid biosynthesis (Chen et al., 2008; Nagle et al., 2008) on the cytosolic leaflet of the ER membrane (Coleman and Bell, 1978), and is an important substrate for this targeting pathway. In Drosophila S2 cells, during LD induction, GPAT4 relocalizes from the ER and accumulates on a subset of LDs, where it helps mediate their expansion (Wilfling et al., 2013). Although this targeting reaction typically happens hours after LD formation is initiated, a 56-amino acid, hydrophobic, membrane-embedded motif of GPAT4 (known as LiveDrop) is sufficient to localize to LD surfaces during their formation (Wang et al., 2016; Wilfling et al., 2013). Further, LiveDrop not only targets LDs but rapidly accumulates on their surface, making it an excellent model substrate to investigate the molecular mechanisms that drive the accumulation of hydrophobic, membrane-embedded motifs on the surface of LDs.
Here, we utilized LiveDrop and the membrane-embedded portion from another D. melanogaster LD protein, UDP-N-acetylglucosaminyltransferase subunit (ALG14), as model substrates to dissect the ER-to-LD targeting mechanism. We identify specific sequence requirements and define principles for the accumulation of these hydrophobic, membrane-embedded motifs on LDs. Analysis of motifs from other LD proteins utilizing this pathway suggests that these are general sequence determinants.
RESULTS
The Hydrophobic, Membrane-Embedded Motif LiveDrop Accumulates on Nascent LDs
We sought to identify a minimal sequence motif that is sufficient to mediate GPAT4 accumulation on LDs. Consistent with previous reports (Wang et al., 2016; Wilfling et al., 2013), a central hydrophobic sequence of D. melanogaster GPAT4 (LiveDrop, amino acids 160–215), which is predicted to be membrane-embedded (Fig. 1A, S1A), targeted to and accumulated on LDs induced by oleate incubation (Fig. 1B, 1C). Similar to full-length GPAT4, LiveDrop exhibited little depletion of signal from LDs during repeated bleaching of the ER, indicating that the protein has a slow off-rate once it is bound to LDs (Fig. 1D). Also, LiveDrop targeted to LDs faster than full-length GPAT4; all nascent LDs contained LiveDrop as early as 30 min after LD induction, whereas full-length GPAT4 accumulated on LDs after hours of incubation with oleate (Fig. 1B, 1C) (Wilfling et al., 2013). For this and similar experiments, we measured expression levels of the tested constructs by quantifying the fluorescence signal of each protein variant per cell. For instance, LiveDrop was expressed in cells at levels similar to or higher than full-length GPAT4 (Fig. S1B). The different timing of LD targeting for full-length GPAT4 and LiveDrop suggests that there is a mechanism that prevents full-length GPAT4 from targeting to newly forming LDs.
Figure 1. The Hydrophobic, Membrane-Embedded Motif LiveDrop Accumulates on Nascent LDs.
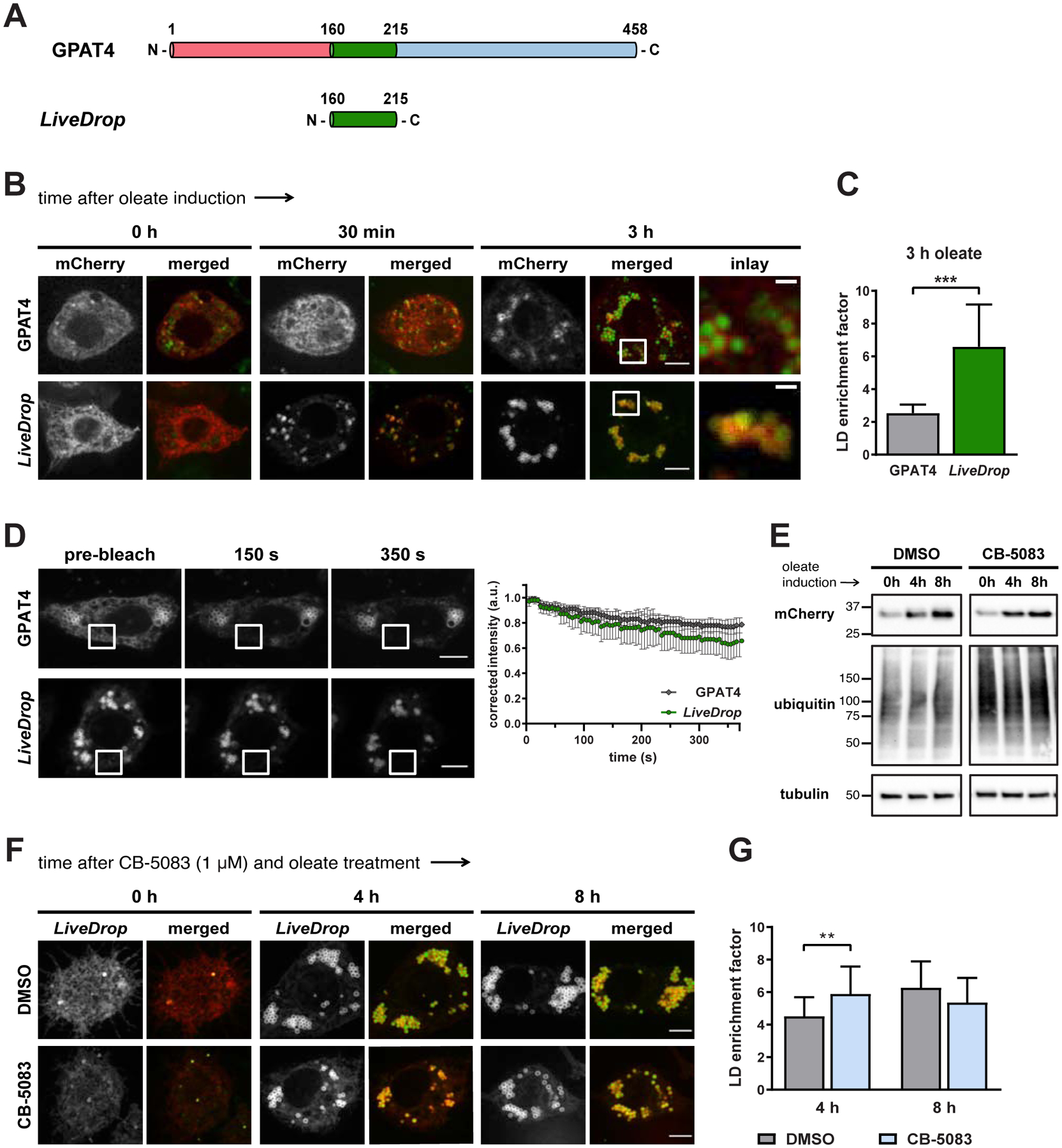
(A) Schematic representation of D. melanogaster GPAT4 (1–458 aa), including the LiveDrop motif (160–215 aa, green).
(B) LiveDrop, but not full-length GPAT4, targets LDs during their biogenesis. Drosophila S2 cells transfected with mCherry-tagged GPAT4 or mCherry-tagged LiveDrop (red) were imaged after incubation with oleate. LDs were stained with BODIPY (green). Scale bar, 5 μm (merged), 1 μm (inlay).
(C) Mean values + SD (n > 15) of the protein signal on LDs after 3 h of oleate treatment. ***, p < 0.001.
(D) (left) LiveDrop and full-length GPAT4 accumulate on LDs but do not move back to the ER. Repeatedly bleaching the mCherry signal in the indicated regions did not reduce the LD signal over time. Scale bar, 5 μm. (right) Mean values ± SD of the normalized fluorescence intensity on LDs for both mCherry-GPAT4 (n = 7) and mCherry-LiveDrop (n = 9) after 5 and 3 h of oleate treatment, respectively. a.u., arbitrary units.
(E) Inhibiting VCP/p97 with 1 μM CB-5083 leads to accumulation of ubiquitylated proteins in S2 cells expressing mCherry-LiveDrop. The samples shown were ran on the same SDS-PAGE gel.
(F) Inhibiting VCP/p97 with CB-5083 does not significantly change the enrichment of LiveDrop (red) on LDs. LDs were stained with BODIPY (green). Scale bar, 5 μm.
(G) Mean values + SD (n > 23) of the protein signal on LDs after 4 and 8 h of oleate treatment. **, p < 0.01.
To test whether the earlier targeting of LiveDrop to LDs may be due to its smaller size compared with full-length GPAT4, we increased its size by fusing fluorophores to both its N- and C-termini. This modification caused a modest reduction in LD targeting at 3 h, but the LiveDrop variant was still highly enriched on LDs (Fig. S1C, S1D, S1E), suggesting that features other than size prevent the targeting of full-length GPAT4 during initial LD formation. Indeed, a C-terminal segment of full-length GPAT4 (amino acids 216–458) was necessary and sufficient for delaying LD targeting (Fig. S1F, S1G, S1H). How this C-terminal region of the protein exerts that is currently unknown.
Testing Several Models for LiveDrop Accumulation on LDs
In one model for ER-to-LD protein targeting and accumulation, proteins equilibrate between the ER and LDs, but the ER pool is selectively degraded, leaving only the LD pool (Ruggiano et al., 2016). To specifically test for a role of ER-associated protein degradation (ERAD) in LD targeting, we inhibited VCP/p97, an ATPase important for the extraction of proteins from the ER membrane (Ye et al., 2017). Inhibiting VCP/p97 led to the accumulation of ubiquitylated proteins (Fig. 1E) but did not increase the visible ER pool or notably change the LD enrichment of LiveDrop (Fig. 1F, 1G, S1I). Thus, selective degradation of the ER protein pool is unlikely to explain the LD accumulation of LiveDrop.
In an alternative model, GPAT4 could bind to a protein on LDs, thereby causing its accumulation on LDs. However, this seems unlikely because overexpressing LiveDrop does not saturate LD targeting (Fig. S1J, S1K). It is also improbable because D. melanogaster LiveDrop targets LDs in evolutionarily diverse species, such as yeast (Ruggiano et al., 2016) and human (Chung et al., 2019; Wang et al., 2016), in which protein-protein interactions are generally not conserved.
We also considered whether global, structural features of LiveDrop account for its LD accumulation. Specifically, the LiveDrop sequence contains two hydrophobic, likely α-helical stretches of amino acids that are separated near the midpoint by a methionine (M184) and a proline (P185) (Fig. 2A, S1A). With 21 amino acids on each side of these residues, the two hydrophobic stretches are relatively long for transmembrane domains. Thus, we considered whether this results in a mismatch between the transmembrane domain lengths and the ER membrane thickness, potentially putting LiveDrop under a strain that could be relieved by targeting to LDs, where the hydrophobic phase is essentially unlimited in thickness. This hypothesis has analogies to protein sorting along the secretory pathway, where the protein transmembrane domain length generally matches the bilayer thickness of the targeted organelle (Sharpe et al., 2010). However, shortening each of the hydrophobic α-helical segments of LiveDrop by four amino acids (roughly one α-helical turn) (Fig. S2A) did not affect its LD targeting (Fig. S2B, S2C, S2D), suggesting that hydrophobic mismatch is not a major contributing mechanism. Moreover, M184 and P185 of the LiveDrop sequence were not required for LD targeting (Fig. S2E, S2F, S2G), further suggesting a rigid hairpin is not strictly needed for LD targeting.
Figure 2. LD Accumulation of LiveDrop Requires Specific Sequence Features.
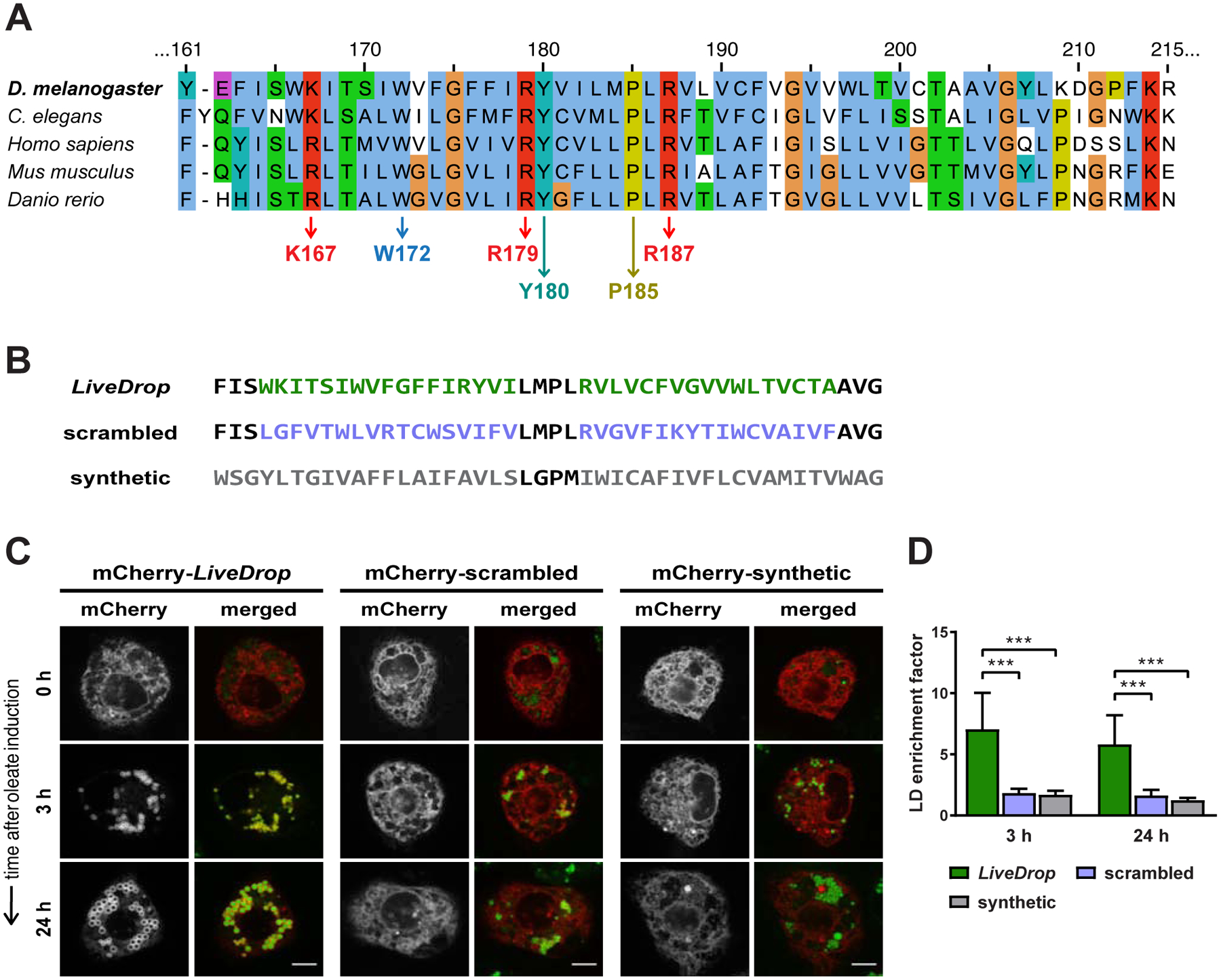
(A) Sequence alignment of the D. melanogaster LiveDrop motif (160–215 aa) and other GPAT4 ortholog sequences from representative species. Conserved residues are highlighted, including P185 (dark yellow), positively charged residues (K167, R179, R187, red), and large hydrophobic residues (W172, Y180, sky blue or teal).
(B) Amino acid sequences of LiveDrop (green), a scrambled LiveDrop variant (violet), and a synthetic hydrophobic α-helical motif (gray).
(C) The scrambled LiveDrop variant and the synthetic hydrophobic motif do not target LDs. S2 cells transfected with mCherry-tagged versions of the protein motifs in (B) (red) were incubated with oleate throughout the indicated time points and imaged by confocal microscopy. LDs were stained with BODIPY (green). Scale bar, 5 μm.
(D) Mean values + SD (n > 12) of the protein signal on LDs after 3 and 24 h of oleate treatment. ***, p < 0.001.
LD Accumulation of LiveDrop Requires Specific Sequence Features
Since the overall hydrophobic character and many specific residues of the LiveDrop membrane-embedded sequence are evolutionarily conserved (Fig. 2A, S3A), we generated a scrambled version of LiveDrop with the same length and hydrophobicity but different amino acid order (Fig. 2B). The scrambled variant inserted into the ER membrane with the same topology as wild-type LiveDrop (N- and C-termini in the cytosol) (Fig. S2H) but failed to accumulate on LDs (Fig. 2C, 2D, S2I). Similarly, a synthetic hydrophobic motif, with a sequence of amino acids designed according to their propensity to form an α-helix in bilayer membranes (Fig. 2B) (Hedin, 2010), localized to the ER and had the same topology as wild-type LiveDrop (Fig. S2H) but failed to accumulate on LDs (Fig. 2C, 2D, S2I). These results indicate that both the specific amino acid order and composition of the LiveDrop sequence are crucial for LD targeting.
Simulations of LiveDrop Suggest Conformational Changes and Energetic Contributions of Specific Sequence Features to LD Targeting
To gain insights into how specific sequence features of LiveDrop determine its LD accumulation we performed molecular dynamics simulations. Since the structure of LiveDrop has not been resolved experimentally, we predicted it using the Rosetta ab initio structure generation tool (Raman et al., 2009; Rohl et al., 2004), and validated it with MEMSAT-SVM (Jones, 2007; Jones et al., 1994; Nugent and Jones, 2009), TOPCONS (Tsirigos et al., 2015), and RosettaMP (Alford et al., 2015) as described in Methods. The ten best-scoring structures were similar, showing two α-helices closely connected by a hinge at P185 resulting in a hairpin. We next computationally inserted and relaxed LiveDrop in a bilayer membrane and LD monolayer, each with a composition resembling the mammalian ER and LDs (Bartz et al., 2007; Chitraju et al., 2012). In the LD system, the neutral lipid layer was composed of 1:1 TG and cholesteryl oleate.
During equilibration there was a shift in the helix-helix separation of LiveDrop in the monolayer versus bilayer simulations. To verify this structural change was sufficiently sampled, we used umbrella sampling (Torrie and Valleau, 1977) to calculate the free energy profile (potential of mean force (PMF)) of helix-helix separation (Fig. S4A). This revealed that the most energetically stable conformation of LiveDrop is more open in the bilayer (~1.7-nm separation) than in the monolayer (~0.7-nm separation) (Fig. 3A, S4A). As a result, a number of residues changed orientation. W172 relocalized from the bilayer midplane to just below the phospholipid glycerol plane in the monolayer (Fig. 3A). The positively charged R179 and R187, originally found at the luminal interface of the bilayer, relocalized to the monolayer surface and the organic phase, surrounded by water molecules, respectively (Fig. 3A). A similar solvation effect was observed for P185, which transitioned from the luminal interface of the bilayer to the organic phase in the monolayer (Fig. 3A). These results were further validated in 1–2-μs molecular dynamics simulations lacking the influence of sterol esters in the oil phase of the LD system (Fig. S4B) (see Methods). In both of these monolayer and bilayer simulations, the depth of the central P185 was stabilized (Fig. 3B).
Figure 3. Simulations of LiveDrop Suggest Conformational Changes and Energetic Contributions of Specific Sequence Features to LD Targeting.

(A) Equilibrated simulation structures of LiveDrop in a bilayer (left) and monolayer (right) highlighting key residues: R179 and R187 (pink); W166, W172, W197, and P185 (red); and A203 (gray). The phospholipid headgroups and tails are shown in light and medium blue, respectively, the neutral lipid phase in green, and water molecules in dark blue.
(B) P185 depth with respect to the upper phosphate plane of the bilayer (blue) and monolayer (orange). The solid lines show the running averages and the filled areas show the standard deviations from four and two simulations of the bilayer and monolayer, respectively. Extending the simulations in the monolayer to 2 μs (not shown) did not reveal notable changes in the depth or conformation during the last 1 μs.
(C) (upper panel) Free energy of insertion of LiveDrop hydrophobic residues into the bilayer (blue) and monolayer (orange). The defined hairpin regions were residues position (α-helix top (H1 top, H2 top), middle (H1 mid, H2 mid), or hinge) are shown. (lower panel) Free energy differences between the monolayer and bilayer (ΔFM-B). Bars are colored yellow or violet if the residue is more stable in the monolayer (FM < FB) or in the bilayer (FM > FB), respectively.
(D) Free energy differences between the monolayer and bilayer (ΔFM-B) for the phenylalanine (left panel, black) and tryptophan (right panel, black) residues, and for their corresponding valine mutants (blue).
Since it is not computationally feasible to calculate the free energy of membrane insertion for a protein of this size, we estimated the driving force for LD accumulation by calculating the free energy profiles (PMFs) of individual residues inserting into a bilayer membrane or LD monolayer (see Methods for justification). Permeation PMFs for seven amino acids of varying hydrophobicity were calculated using transition-tempered metadynamics (Dama et al., 2014). The PMFs for phenylalanine, tryptophan, and tyrosine demonstrated greater stability at all depths in the monolayer than in the bilayer (Fig. S4C). In the phospholipid tail region (~2-nm membrane depth) particularly, the normally high energy barrier for bilayer permeation (Norman and Nymeyer, 2006; Yau et al., 1998) is significantly reduced in the monolayer (Fig. S4C) due to the formation of hydrogen bonds between the residues’ side chains and the polar ester groups of phospholipids and core TG molecules. This stabilization effect continues into the neutral lipid layer where each of the reported aromatic residues has slightly lower free energy than in bulk water (Fig. S4C). Specifically, the imino group of tryptophan and the hydroxyl group of tyrosine are stabilized by forming hydrogen bonds with the glycerol moieties of TG and associated water molecules, suggesting these residues have a preference for the LD environment.
Besides this general trend, there were specific changes in free energy due to the conformational changes of LiveDrop. Based on the depth of each residue in the bilayer or monolayer minimum free energy structures (Fig. 3A), we calculated their respective insertion free energies from the single amino acid PMFs (Fig. S4C) (see Methods). The difference between those for the bilayer and monolayer estimates the change in free energy for LiveDrop to accumulate on LDs (Fig. 3C). Notably, the hydrophobic residues positioned in the middle of the α-helices (Table 1) and, therefore, located near the energetically unfavorable bilayer midplane (Fig. S4C), have a large preference for the LD monolayer (Fig. 3C), where they redistribute to more favorable depths. Two large hydrophobic tryptophan residues (W172 and W197) in particular gained the largest free energy difference by relocating to the monolayer (Fig. 3C). This is consistent with amino acid distribution studies, which show that tryptophan and tyrosine residues preferentially occur at the interface regions of membrane proteins (Landolt-Marticorena et al., 1993). Thus, the position of tryptophan (and potentially tyrosine) residues within LiveDrop contributes significantly to its accumulation on LDs.
Table 1.
Stretch of residues assigned to the defined regions of each predicted hairpin motif
| Predicted hairpins | N-terminus | H1 top | H1 mid | Hinge | H2 mid | H2 top | C-terminus |
|---|---|---|---|---|---|---|---|
| LiveDrop | HYE | FISWKITSI | WVFGFFIRY | VILMPLRVL | VCFVGVVWL | TVCTAAVG | YLKDGPFKR |
| ALG14 hp | QS | WLSSIFTSL | WALLWSCYLV | WRDRPQLILCN | GPGTCVPFCYAA | YLWRLLGRL | PS |
| Spastin hp | GYSSSVHKQN | LYVVSF | PIIFLF | NVLRSLI | YQLFCI | FRYLYG | ASTKVIYRPH |
| LDAH hp | MMESPNG | WVFTKVA | MPLYSVFGYIF | FSFFNFLPVWLR | LMLIQIYFLIF | SIPRQFL | GTALKYS |
| AGPAT3 hp | RLCS | LVNFVCW | AVFSLSCIFY | YVITSLLAANW | TAFITALSVL | GLFYWLM | GQAINK |
| DGAT2 hp | RLQ | ILVTAFF | TSMLLILLSVS | FLLVAGSLIYGG | LLVRSLMVTY | LAYVFVH | HKKTQS |
| FATP hp | QRRRRQR | FLVIFRFFC | ATVAFGLAIACV | IYTLHTMGW | IFAVLVALVALL | LTKPGWRWF | YIAGATA |
| SelT hp | DPPGLNYYLS | KMIFALKII | IIVSVVSAVSPF | TFLGLNTPSWW | SHMQANKIYACM | MIFFLGNML | EAQLISSGA |
The amino acid sequence of each of the predicted hairpin motifs was divided into five regions. In each case, the predicted α-helices (H1 and H2) were divided into top (H1 top, H2 top) and middle (H1 mid, H2 mid) regions. The residues predicted to connect the α-helices were defined as the hinge region. Short sequence overhangs at the N- and C-termini were included when expressing these motifs in cells. The tryptophan and positively charged residues are underlined.
We also addressed whether residue identity is important for the energetic stabilization of LiveDrop on LDs by calculating the free energy difference associated with changing F174, F176, F177, and F192, as well as W166, W172, and W197, individually to valine (Fig. 3D). Changing phenylalanines to valines in the LiveDrop sequence did not considerably alter the free energy differences between monolayer and bilayer (Fig. 3D). However, free energy calculations for mutating tryptophans to valines predicted a decrease in LD targeting for W172V and W197V, and an increase in LD targeting for W166V (Fig. 3D).
Positively Charged and Tryptophan Residues Are Required for Efficient LD Targeting of LiveDrop in Cells
To experimentally address the predictions derived from our simulations, we tested the LD targeting role of the positively charged and large hydrophobic residues in LiveDrop. Positively charged residues are unusual for a membrane-embedded protein segment, but can localize at the membrane-water interface by interacting with the negatively charged phospholipid headgroups, in a phenomenon known as “snorkeling” (Segrest et al., 1990). We individually mutated each of these residues (i.e., K167, R179, and R187) to alanine and assessed the LD targeting capacity of the mutant variants. R179A resulted in a targeting defect, reducing LD accumulation by ~44% at 3 h and ~47% at 24 h of oleate treatment. In contrast, K167A or R187A had no apparent effect in LD targeting (Fig. 4A, 4B, S5A). Combining R179A with K167A and R187A did not affect ER targeting nor membrane protein topology (Fig. S5B), but it reduced LD accumulation to a similar extent as R179A alone (by ~40% at 3 h and ~47% at 24 h) (Fig. 4C, 4D, S5C). Thus, R179 appears to be the most relevant positively charged residue in LiveDrop for mediating LD accumulation.
Figure 4. Positively Charged and Tryptophan Residues Are Required for Efficient LD Targeting of LiveDrop in Cells.
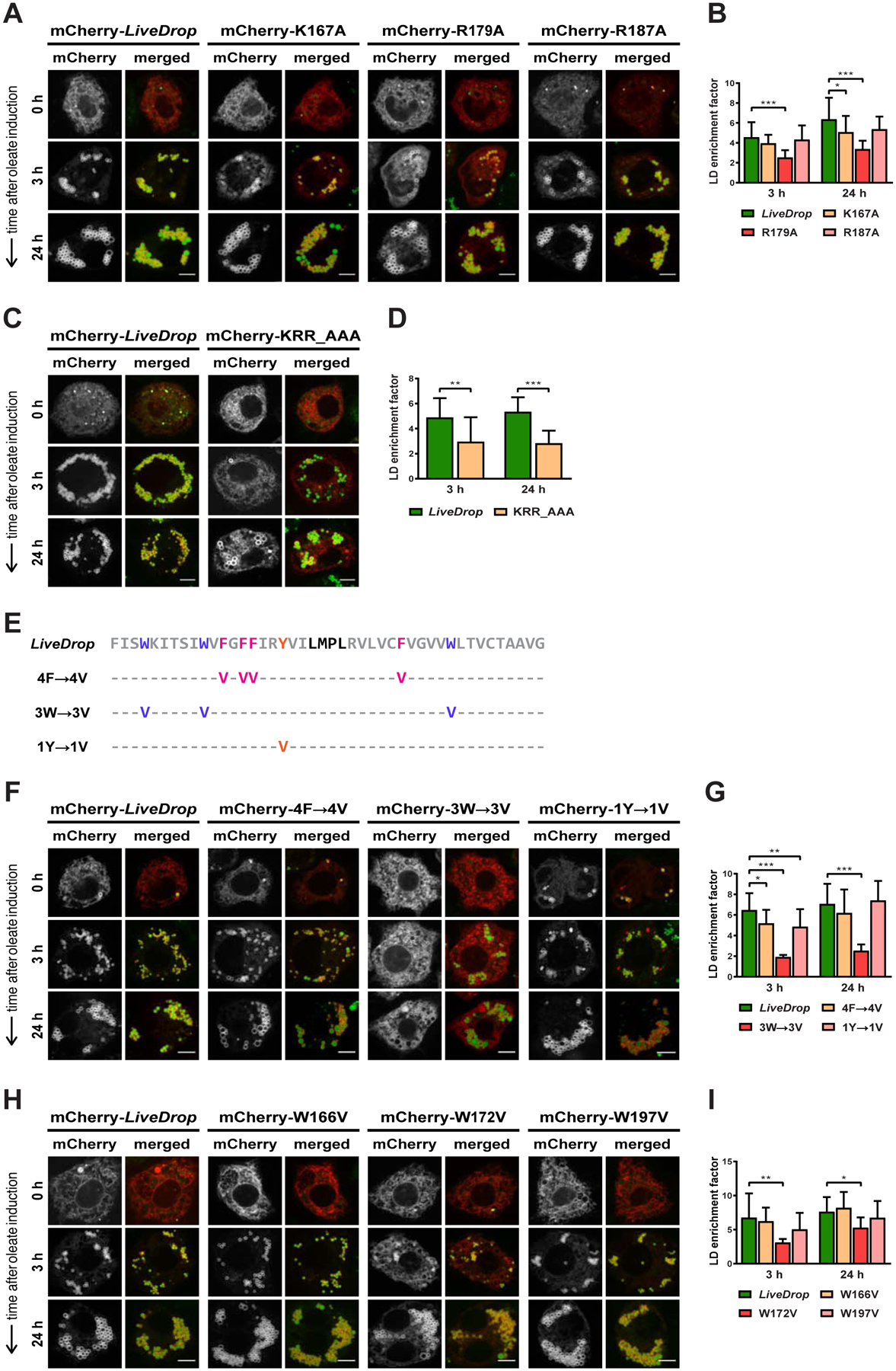
(A) Of the three LiveDrop variants with single positive charge mutations (K167A, R179A, R187A), only R179A causes a reduction in LD accumulation.
(B) Mean values + SD (n > 18) of the protein signal on LDs after 3 and 24 h of oleate treatment. *, p < 0.05; ***, p < 0.001.
(C) A LiveDrop variant with the three positively charged residues (K167, R179, R187) exchanged for alanines (KRR_AAA) is compromised in LD accumulation to a similar extent as the single R179A mutation.
(D) Mean values + SD (n > 10) of the protein signal on LDs after 3 and 24 h of oleate treatment. **, p < 0.01; ***, p < 0.001.
(E) Amino acid sequence of LiveDrop variants in which the phenylalanines (4F→4V, magenta), tryptophans (3W→3V, purple), and tyrosine (1Y→1V, orange) are individually mutated to valines. The predicted hinge of the LiveDrop sequence (gray) is shown in black. For each LiveDrop variant, the amino acid positions indicated with a hyphen (−) remain the same as in the original sequence.
(F) The LiveDrop variant with mutated tryptophans (3W→3V) does not accumulate on LDs, but the variants with mutated phenylalanines (4F→4V) and tyrosine (1Y→1V) target LDs in a manner comparable to the wild-type sequence.
(G) Mean values + SD (n > 10) of the protein signal on LDs after 3 and 24 h of oleate treatment. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
(H) LiveDrop variants with single tryptophan mutations (W166V, W187V) target LDs similar to the wild-type sequence, except for W172V, which shows reduced LD accumulation.
(I) Mean values + SD (n > 10) of the protein signal on LDs after 3 and 24 h of oleate treatment. *, p < 0.05; **, p < 0.01.
For (A), (C), (F), and (H), S2 cells were transfected with mCherry-tagged versions of the LiveDrop variants (red), incubated with oleate throughout the indicated time points, and imaged by confocal microscopy. LDs were stained with BODIPY (green). Scale bar, 5 μm.
Our simulations also predicted that large hydrophobic residues are important for LiveDrop accumulation on LDs. Consistently, when we mutated all large hydrophobic residues to small hydrophobic valine residues (Fig. S5D), the resulting protein variant appeared to insert into the ER membrane but failed to accumulate on LDs (Fig. S5E, S5F, S5G). To determine which specific type of the large hydrophobic residues was most crucial, we individually mutated the phenylalanines, tryptophans, or the tyrosine to valines (Fig. 4E). Mutating the three tryptophans (W166, W172, and W197) in the LiveDrop sequence abolished LD accumulation (Fig. 4F, 4G, S5H) without changing the ER membrane topology (Fig. S5I). In contrast, mutating either the phenylalanines or the tyrosine did not considerably alter LD targeting (Fig. 4F, 4G, S5H). Mutating each of the LiveDrop tryptophan residues individually revealed that W172, which is the conserved tryptophan with a calculated high free energy contribution (see Fig. 3C, 3D), was not essential for LD accumulation but caused the greatest reduction at early time points of LD induction (~54% at the 3 h time point) (Fig. 4H, 4I, S5J).
Our simulation and experimental results are consistent with the hypothesis that the bilayer anchoring and subsequent reorientation in the monolayer of positively charged residues, R179 and R187, and the differences in the free energies of tryptophans mediate LD accumulation of LiveDrop. To test whether these features are sufficient to mediate LD accumulation, we added either type of feature alone or in combination at their original positions to the scrambled-LiveDrop sequence (see Fig. 2B). The scrambled LiveDrop variant harboring both the positively charged and tryptophan residues showed modestly enriched signal on LDs over the ER protein pool when compared with the original scrambled LiveDrop variant (~39% increase at the 24 h time point) (Fig. S6A, S6B, S6C). Thus, repositioning the positively charged and tryptophan residues enables some LD accumulation, yet not to the extent found for wild-type LiveDrop, which likely harbors additional determinants mediating LD accumulation.
We were curious to examine the contribution of the hairpin sequence motif to full-length GPAT4 targeting to LDs. Full-length GPAT4 lacking the LiveDrop motif failed to insert into the ER membrane but was able to target LDs from the cytosol at both the 3 h and 24 h time points (Fig. S6D, S6E, S6F), indicating sequences outside the hairpin motif can contribute to targeting. Mutating the three LiveDrop tryptophan residues in the context of full-length GPAT4 reduced but did not abolish LD accumulation (Fig. S6D, S6E, S6F). Together, these experiments indicate that targeting of full-length GPAT4 to LDs involves more determinants than those we uncovered for the LiveDrop motif alone.
The Predicted ALG14 Hairpin Motif Is Sufficient to Target LDs and It Relies on Similar LD Targeting Features and Principles as LiveDrop
To identify other proteins with motifs that are sufficient to target LDs from the ER during early time points of LD biogenesis, we examined the D. melanogaster LD proteins (Krahmer et al., 2013) with a predicted hydrophobic hairpin motif that are known to localize to the ER. Among these (Table 2), we selected ALG14, which is an ER protein of the lipid-linked oligosaccharide synthesis pathway (Gao et al., 2005) and has been reported to be a LD protein in yeast (Currie et al., 2014) and humans (Saka et al., 2015). ALG14 is predicted to contain a hydrophobic hairpin motif (amino acids 83–137), consisting of two α-helical segments separated by a proline residue in position 108 (P108) (Fig. S3B, S7A).
Table 2.
List of protein candidates for the ER-to-LD targeting pathway
| # | Uniprot code | Organism | Annotation symbol | Protein name | Predicted hairpin | Hairpin sequence | Source | Localization category |
|---|---|---|---|---|---|---|---|---|
| 1 | Q9VXU7 | D. melanogaster | CG6308 | ALG14 | 83–137 aa | QSWLSSIFTSLWALLWSCYLVWRDRPQLILCNGPGTCVPFCYAAYLWRLLGRLPS | LD Proteome (Krahmer et al., 2013) | ER-LD |
| 2 | Q0KHU5 | D. melanogaster | CG32699 | LPCAT | 58–102 aa | EIDSHIEVAKIYVLTVLLLPIRVVGCVLSLISAWMFACIGLYGMT | LD Proteome (Krahmer et al., 2013) | ER-LD; C-LD |
| 3 | Q9VMV6 | D. melanogaster | CG3887 | SelT-like protein | 80–151 aa | DPPGLNYYLSKMIFALKIIIIVSVVSAVSPFTFLGLNTPSWWSHMQANKIYACMMIFFLGNMLEAQLISSGA | LD Proteome (Krahmer et al., 2013) | ER |
| 4 | Q961F9 | D. melanogaster | CG4729 | AGPAT3 | 315–369 aa | RLCSLVNFVCWAVFSLSCIFYYVITSLLAANWTAFITALSVLGLFYWLMGQAINK | LD Proteome (Krahmer et al., 2013) | ER |
| 5 | Q9Y140 | D. melanogaster | CG7601 | SDR reductase | 10–84 aa | APSSDWNVLYWVLGTVLMPVALPLAIINIWQRFQAQKFRNQLPGKVVLITGASSGLGESLAHVFYRAGCRVILAA | LD Proteome (Krahmer et al., 2013) | A |
| 6 | B7Z0W3 | D. melanogaster | CG1021 | Dementin | 555–638 aa | GIDNSNARALVVKLINVVLTILQVVLLLVATAAGIIMPFLKTRVRVLTTFLSICFVIFVIRQWPDVQDIGSGLVRHLKQSLVVK | LD Proteome (Krahmer et al., 2013) | C |
| 7 | Q7JW12 | D. melanogaster | CG11007 | Thioredoxin TM protein | 67–132 aa | IRSRKTGSVTMINYLASSFLYTKVANAILWAYADFRYGLGFLLLCVLVGMVLPEPSYRGPEHITYF | LD Proteome (Krahmer et al., 2013) | ER-LD |
| 8 | A1ZBD6 | D. melanogaster | CG15078 | MCTP | 785–847 aa | GYLASLGESTINTFNFSVPELTWLAVVLLLGAILVLHFVPLRWLLLFWGLMKFSRRLLRPNTI | (Joshi et al., 2018; Krahmer et al., 2013) | C |
| 9 | Q7K1C2 | D. melanogaster | CG1942 | DGAT2 | 15–70 aa | RLQILVTAFFTSMLLILLSVSFLLVAGSLIYGGLLVRSLMVTYLAYVFVHHKKTQS | (Wilfling et al., 2013) | ER |
| 10 | E1JI03 | D. melanogaster | CG5295 | ATGL/Brummer | 312–377 aa | FKHRGIKLLSLPATVPMDFLLATISKISALTPKLTKQARVMVEDLILQLNHAMRIRLLNKFTLYDQ | (Murugesan et al., 2013) | C-LD |
| 11 | A0A0B4K FE4 | D. melanogaster | CG8732 | ACSL | 1–67 aa | MDSFWVQSAIGAIKAIAFVYDIITLPVYLVLQKPWKRRQDSRRVKAKPINQKMLVDESKYAPDDIEA | (Poppelreuther et al., 2012) | A |
| 12 | Q9VJ58 | D. melanogaster | CG10372 | UBXD8 | 74–136 aa | FLQQVFSANMPGGRTVSRVPSGPVPRSFTGIIGYVINFVFQYFYSTLTSIVSAFVNLGGGNEA | (Suzuki et al., 2012) | ER-LD |
| 13 | Q8I0P1 | D. melanogaster | CG5977 | Spastin | 105–155 aa | GYSSSVHKQNLYVVSFPIIFLFNVLRSLIYQLFCIFRYLYGASTKVIYRPH | (Chang et al., 2019) | ER-LD |
| 14 | E1JHE4 | D. melanogaster | CG46149 | FATP | 87–151 aa | QRRRRQRFLVIFRFFCATVAFGLAIACVIYTLHTMGWIFAVLVALVALLLTKPGWRWFYIAGATA | (Kory et al., 2015) | ER |
| 15 | Q9W0H3 | D. melanogaster | CG9186 | LDAH | 151–212 aa | MMESPNGWVFTKVAMPLYSVFGYIFFSFFNFLPVWLRLMLIQIYFLIFSIPRQFLGTALKYS | (Kory et al., 2017) | ER-LD |
| 16 | Q6UX53 | H. sapiens | --- | ALDI | 1–40 aa | MDILVPLLQLLVLLLTLPLHLMALLGCWQPLCKSYFPYLM | (Zehmer et al., 2008) | A |
| 17 | Q9H8H3 | H. sapiens | --- | AAM-B | 1–40 aa | MELTIFILRLAIYILTFPLYLLNFLGLWSWICKKWFPYFL | (Zehmer et al., 2008) | ER-LD |
| 18 | Q9Y679–3 | H. sapiens | --- | AUP1 | 12–75 aa | LFDSHRLPGDCFLLLVLLLYAPVGFCLLVLRLFLGIHVFLVSCALPDSVLRRFVVRTMCAVLGL | (Stevanovic and Thiele, 2013) | ER-LD |
| 19 | P56539 | H. sapiens | --- | Caveolin 3 (Cav3) | 50–111 aa | GTYSFDGVWKVSYTTFTVSKYWCYRLLSTLLGVPLALLWGFLFACISFCHIWAVVP CIKSYL | (Stevanovic and Thiele, 2013) | ER-LD |
| 20 | Q15738 | H. sapiens | --- | NSDHL | 290–338 aa | APKYHIPYWVAYYLALLLSLLVMVISPVIQLQPTFTPMRVALAGTFHYY | (Stevanovic and Thiele, 2013) | ER-LD |
The selected LD proteins and their respective predicted hairpin motifs were identified based on the secondary-structure analysis of the D. melanogaster LD proteome and examining the literature (‘Source’ column). Only in the case of proteins lacking a D. melanogaster homologue we selected the corresponding human sequence. Evaluating the subcellular localization of the predicted hairpin motifs allowed us to categorize them as: only cytosolic (C), only ER-localized (ER), cytosolic and targets LDs (C-LD), ER-localized and targets LDs (ER-LD), and aberrant (A) (‘Localization category’ column).
We predicted the structure of the ALG14 hairpin with the RosettaMP ab initio tool (Alford et al., 2015). The two best-scoring structures were inserted into a bilayer membrane and LD monolayer for molecular dynamics simulations (see Methods). As shown in representative snapshots (Fig. 5A), two positively charged residues (R105 and R107) and one negatively charged residue (D106) localized to the luminal interface of the bilayer membrane and became solvated by water molecules in the organic phase of the LD system. This hydration stabilizes the charged side chains, since they do not reach the phosphate level of the monolayer. One of the charged residues in particular, R105, dramatically reoriented toward the phospholipids in the monolayer (Fig. 5A), resembling the changes observed for R179 in LiveDrop (see Fig. 3A).
Figure 5. The ALG14 Hairpin Is Sufficient to Target LDs and It Relies on Similar LD Targeting Features as LiveDrop.
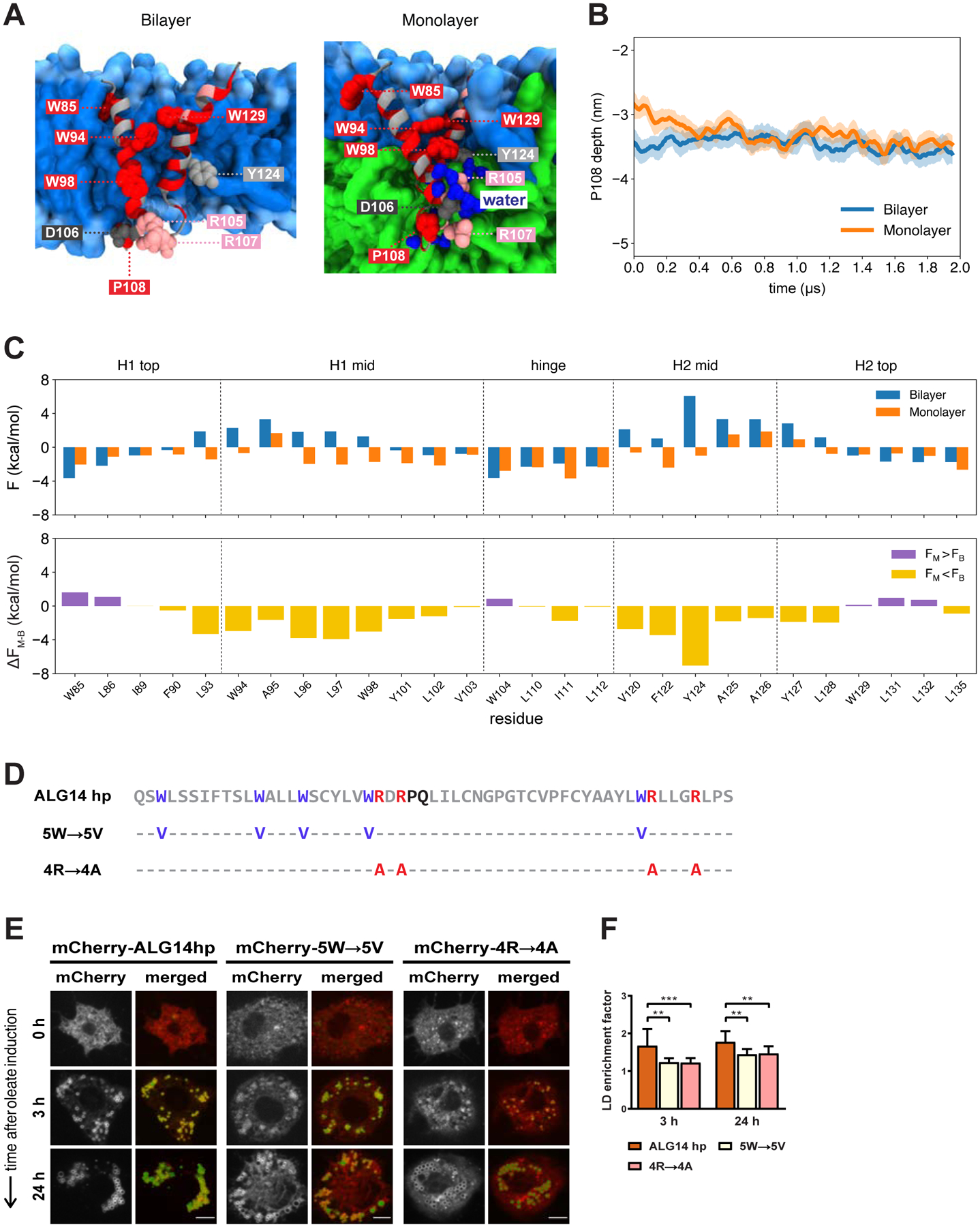
(A) Representative structures of the ALG14 hairpin in a bilayer (left) and monolayer (right) highlighting key residues: R105 and R107 (pink); D106 (dark gray); W85, W94, W98, W129, and P108 (red); and Y124 (light gray). The phospholipid headgroups and tails are shown in light and medium blue, respectively, the neutral lipid phase in green, and water molecules in dark blue.
(B) P108 depth with respect to the upper phosphate plane of the bilayer (blue) and monolayer (orange). The solid lines show the running averages and the filled areas show the standard deviations from four simulations of both the bilayer and monolayer, respectively.
(C) Same as Figure 3C for the ALG14 hairpin.
(D) Amino acid sequence of the ALG14 hairpin (ALG14 hp, gray) and its sequence variants in which the tryptophan (5W→5V, purple) and positively charged (4R→4A, red) residues are individually mutated to valines and alanines, respectively. The predicted hinge of the ALG14 hairpin sequence is shown in black. Amino acid positions indicated with a hyphen (−) remain the same as in the original sequence.
(E) Both of the ALG14 hairpin sequence variants with mutated tryptophans (5W→5V) or positively charged arginines (4R→4A) are compromised in LD accumulation. S2 cells transfected with mCherry-tagged versions of the ALG14 hairpin and its variants (red) were incubated with oleate throughout the indicated time points and imaged by confocal microscopy. LDs were stained with BODIPY (green). Scale bar, 5 μm.
(F) Mean values + SD (n > 13) of the protein signal on LDs after 3 and 24 h of oleate treatment. **, p < 0.01; ***, p < 0.001.
Same as for LiveDrop, we estimated which residues contribute to the LD accumulation of the predicted ALG14 hairpin by calculating the free energy changes associated with the different membrane depths of specific hydrophobic residues in the bilayer and monolayer environments. We confirmed that the individual residues had a consistent depth throughout the simulations, including the central P108 residue (Fig. 5B). Consistent with LiveDrop (see Fig. 3C), most of the energy stabilization leading to LD accumulation comes from the hydrophobic residues located in the middle section of each α-helix (Fig. 5C, Table 1). Specifically, a phenylalanine (F122), tryptophans (W94, W98), and tyrosines (Y101, Y124) are predicted to prefer the monolayer environment (Fig. 5C). However, similar residues (i.e., W85, F90, W104, and W129) found at different regions of the predicted ALG14 hairpin, such as the top ends and hinge region, do not contribute to LD accumulation (Fig. 5C), indicating that both residue identity and position within a hairpin motif are important for LD accumulation. Also similar to LiveDrop, a number of the hydrophobic residues showing an energetic preference for LDs (Fig. 5C) are conserved across species (Fig. S3B).
To test experimentally whether the large hydrophobic and positively charged residues are responsible for the LD accumulation of the predicted ALG14 hairpin, we first generated a sequence variant where the large hydrophobic tryptophans were exchanged for valines (Fig. 5D). This reduced LD accumulation by ~26% at 3 h and ~18% at 24 h of oleate treatment (Fig. 5E, 5F, S7B). Likewise, mutating the positively charged residues also reduced LD targeting (~27% at 3 h and ~17% at 24 h) (Fig. 5D, 5E, 5F, S7B).
The Distribution of Tryptophan and Positively Charged Residues Differentiates LD Targeting from Non-LD Targeting Hairpin Motifs
To examine whether the features critical for LD accumulation of LiveDrop and the predicted ALG14 hairpin operate more broadly, we generated a list of candidates for ER to LD localization based on the D. melanogaster LD proteome and examining the literature (Table 2). We expressed the predicted hairpins of each protein candidate and classified them based on their subcellular localization (Table 2). We excluded predicted hairpin motifs that failed to insert into the ER (e.g., MCTP, dementin) or appeared to target LDs from the cytosol (e.g., ATGL/Brummer). Regarding the motifs that localized to the ER and targeted LDs (Table 2), most showed low to moderate LD accumulation (e.g., UBXD8, AUP1, AAM-B, NSDHL). However, the predicted hairpin motifs of spastin and LD-associated hydrolase (LDAH) accumulated on LDs efficiently during early time points of LD biogenesis, similar to LiveDrop, constituting LD targeting motifs (Fig. 6A, 6B, S7C). Conversely, the predicted hairpin motifs of AGPAT3, DGAT2, FATP, and SelT-like protein appeared to insert into the ER but were not sufficient to target LDs (Fig. 6C, 6D, S7D), defining a set of non-LD targeting motifs.
Figure 6. The Distribution of Tryptophan and Positively Charged Residues Differentiates LD Targeting from Non-LD Targeting Hairpin Motifs.

(A) The predicted hairpin motifs of the LD proteins spastin (spastin hp) and LDAH (LDAH hp) target the ER and efficiently accumulate on LDs.
(B) Mean values + SD (n > 19) of the protein signal on LDs after 3 and 24 h of oleate treatment. **, p < 0.01; ***, p < 0.001.
(C) The predicted hairpin motifs of the LD proteins AGPAT3 (AGPAT3 hp), DGAT2 (DGAT2 hp), FATP (FATP hp), and SelT-like protein (SelT hp) target the ER, but they are not sufficient to target LDs.
(D) Mean values + SD (n > 10) of the protein signal on LDs after 3 and 24 h of oleate treatment. For (A) and (C), S2 cells were transfected with mCherry-tagged versions of the predicted hairpin motifs (red), incubated with oleate throughout the indicated time points, and imaged by confocal microscopy. LDs were stained with BODIPY (green). Scale bar, 5 μm.
(E) Scores plot from the principal component analysis (PCA) of the parameters derived from the predicted hairpin sequences previously classified as LD targeting and non-LD targeting motifs. The first two principal components (PC1 and PC2) are plotted against each other. PC1 accounts for 29.29% of the variance, and PC2 accounts for 21.16%.
(F) and (G) Distribution of the tryptophan (left, green scale) and positively charged residues (right, red scale) throughout defined regions of the indicated hairpin motifs (see Table 1 for details).
Analyzing the sequences of the LD targeting and non-LD targeting motifs (Fig. S7E, S7F), we found no simple correlation between LD accumulation and the number of positively charged, large hydrophobic, or tryptophan residues (Fig. S7G, Table S1). We, thus, determined a range of properties for these sequences, including molecular weight, hydrophobicity, net charge, number of charged residues, number of large hydrophobic residues, and residue distribution variables (Table S2). Principal component analysis of these parameters (Table S2) with respect to LD accumulation yielded two major principal components that explain 30% and 21% of the overall variance between the eight motifs (Fig. 6E). Strikingly, the motifs cluster in groups corresponding to the experimentally determined LD targeting and non-LD targeting groups (Fig. 6E). Although no single variable can differentiate the two groups, the overall number of positively charged residues and specific residue distribution variables, such the number of positively charged residues at the hairpin hinge region and the number of tryptophan residues in the middle section of the first hairpin α-helix, make the strongest contributions to the two major principal components (Fig. S7H, Table S2). Accordingly, tryptophan residues are found in different hairpin regions of the LD targeting motifs, possibly enriched in the first α-helix of these predicted hairpins (Fig. 6F), whereas in the non-LD targeting ones, they are only found at the top ends and hinge region (Fig. 6G). Similarly, positively charged residues were consistently found at the hinge region of the LD targeting motifs (Fig. 6F), but they were absent from that region in all the non-LD targeting motifs (Fig. 6G).
DISCUSSION
A number of proteins are originally inserted, via membrane-embedded motifs, in the ER and can subsequently relocalize to the surfaces of LDs. This can occur during LD formation for some proteins, or at later time points for others, possibly via ER-LD membrane bridges (Wilfling et al., 2013). What drives proteins from the ER bilayer membrane to target to and accumulate at the LD monolayer is unknown.
Based on our data for the hydrophobic, membrane-embedded motifs of GPAT4 and ALG14, a model emerges. The pathway commences with the initial insertion of these motifs into the ER membrane. How LiveDrop or the hydrophobic motif of ALG14 insert into the ER membrane is unknown, but candidate pathways for this include the canonical transloconmediated pathway (Gorlich et al., 1992) and the Pex19/Pex3 pathway posited for other membrane-embedded LD proteins (Schrul and Kopito, 2016). The latter is apparently used for the ER insertion of UBXD8, a protein that targets to LDs from the ER, similar to GPAT4 (Schrul and Kopito, 2016). In the ER, these hydrophobic sequences likely adopt a hairpin conformation in which positively charged residues in the hinge region (such as LiveDrop’s R179) interact with the phospholipid headgroups in the luminal side of the ER membrane. This anchors the protein there and, thus, forces large hydrophobic residues into a less favorable environment within the phospholipid tail region of the membrane.
As LDs begin to form in the ER, these hydrophobic hairpins migrate onto the monolayer surface of LDs that are continuous with the ER bilayer membrane. Here, bilayer membrane anchoring is lost, and key residues are able to move to more energetically favorable positions in the environment of the monolayer-bound LD. In particular, large hydrophobic residues, such as tryptophans and sometimes tyrosines, achieve a lower free energy state in the oil phase of the LD than in the midplane of the bilayer membrane. Our simulations suggest that this is due to hydrogen bonding between specific hydrophilic groups in these amino acids (e.g., the imino group of tryptophans or the hydroxyl group of tyrosines) and the headgroups of phospholipids, the glycerol moieties of TG, or coordinating water molecules. Since mutating individual tryptophans of LiveDrop had little effect on LD targeting compared with the mutation of all three, the sum of free energies gained from multiple residues on LDs appears to be crucial for the preference of protein motifs for the LD surface. However, since the rescue of LD targeting by reintroducing the positively charged and hydrophobic tryptophan residues was modest, additional sequence features must contribute to LiveDrop accumulation on LDs. These could be, for instance, the sequence context around each key residue, necessary to position them in specific geometries, or the sum of minor free energy differences between the bilayer and monolayer for other residues.
Statistical analysis of biophysical and sequence-derived properties allowed us to classify the predicted hairpins of other LD proteins correctly. The main feature correlating with LD accumulation was the presence of positively charged residues around the hairpins’ hinge region. In our LiveDrop simulations, the positively charged residues (R179 and R187) relocalized from the luminal leaflet of the ER bilayer to the LD monolayer surface and to the hydrated glycerol moieties of the neutral lipids in the LD, respectively. Similarly, the positively charged residue R105 of the predicted ALG14 hairpin relocalized from the luminal leaflet of the bilayer towards the monolayer surface, suggesting that this may be a general feature of the membrane-embedded motifs that move from the ER to the LD surface.
Among the hydrophobic ER-to-LD targeting motifs we analyzed, we found that spastin is an exception, since it does not contain tryptophans in its predicted membrane-embedded domain. However, this sequence is overall quite different from either LiveDrop or the predicted ALG14 hairpin, inasmuch as it is shorter and it does not contain a central proline (see Fig. S7E). Thus, its conformation may differ from a hairpin and other LD targeting determinants may apply.
From our studies, a mechanism emerges for how ER membrane proteins target and accumulate on the monolayer surface of LDs. Specifically, membrane-embedded motifs of these proteins appear to recognize and conformationally adapt to the unique properties of the LD oil-water phase boundary. Future work identifying more sequences that are sufficient to mediate ER-to-LD targeting and accumulation will uncover additional targeting determinants and enable the refinement of this model.
STAR METHODS
RESOURCE AVAILABILITY Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tobias C. Walther (twalther@hsph.harvard.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Cell Culture
The Drosophila cells used in this study belong to the S2R+ cell line (sex: male) and were provided by Prof. Norbert Perrimon (Harvard Medical School). Cells were cultured at 26°C in Schneider’s Drosophila Medium (Life Technologies) containing 10% fetal bovine serum (FBS) (Gibco), 50 units/ml penicillin, and 50 mg/ml streptomycin.
METHOD DETAILS
Protein Sequence Design
The sequence of the scrambled LiveDrop variant was designed by moving around the residues that form the predicted α-helices of LiveDrop in an arbitrary way, both within their respective α-helix and across α-helices. The ends as well as the predicted hinge of the LiveDrop sequence remained untouched. For the synthetic hydrophobic motif, we designed two theoretical α-helical sequences separated by a short kink in the middle, following the statistical distribution of amino acids along a transmembrane α-helix (Hedin, 2010). In both cases, the overall length and hydrophobicity of the designed sequences were not changed with respect to the original LiveDrop sequence. TOPCONS (Tsirigos et al., 2015) and PSIPRED (Jones, 1999) were used to predict the secondary structure of the designed sequences and to check whether these were consistent with a hairpin conformation (i.e., two α-helices separated by a short loop).
Plasmid DNA Construction
Synthetic double-stranded DNA (gBlocks® Gene Fragments) encoding different protein variants (indicated in Table S3) were purchased from Integrated DNA Technologies and subcloned into the pENTR™/SD/D-TOPO™ vector (Thermo Fisher Scientific) according to manufacturer’s instructions. For the construct mCherry-LiveDrop-mCherry, the insert that was subcloned into the pENTR™/SD/D-TOPO™ vector (Thermo Fisher Scientific) was generated by PCR amplification. In all cases, the resulting entry clones were subsequently in vitro recombined with the destination vector pA-CherryW (Guo et al., 2008) for Drosophila expression, using the Gateway™ LR Clonase™ II Enzyme mix (Thermo Fisher Scientific). Specific protein variants harboring single-point mutations (indicated in Table S3) were generated using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) according to manufacturer’s instructions. The primers used for site-directed mutagenesis are listed in Table S4. The plasmids that were not originally made for this study are also included in Table S3 and, in those cases, the corresponding source is mentioned in place of the insert generation strategy.
Transfection
Plasmid transfection into Drosophila S2 cells was performed with Effectene® Transfection Reagent (Qiagen) according to manufacturer’s instructions. For live-cell imaging experiments, cells were cultured and transfected in 24-well plates. In the case of experiments destined to be analyzed by western blot, we cultured and transfected cells in 6-well plates or 100-mm dishes depending on the amount of starting material needed. For all experiments, plasmids were expressed for 24 hours post-transfection.
LDs Induction
In all applicable cases, cells were treated, as described (Wang et al., 2016), with 1 mM oleic acid (Sigma) complexed with 0.5% bovine serum albumin (BSA) (Sigma) for the indicated times of LD induction.
Microscopy
For all live-cell imaging experiments, cells previously cultured and transfected in 24-well plates were transferred to glass-bottom dishes (MatTek Corporation) coated with Concanavalin A (Sigma), diluted in a 1:1 ratio with culture medium, and allowed to settle down for 30–45 min. Cells were subsequently treated with 0.5 μg/ml BODIPY™ 493/503 (Thermo Fisher Scientific) to stain neutral lipids, and shortly after imaged in the absence of oleate. After imaging the steady state condition, cells were incubated with oleate to induce LD formation and further imaged throughout specific time points.
Spinning-disk confocal microscopy was performed using a Nikon Eclipse Ti inverted microscope equipped with a spinning disk confocal head (Yokogawa CSU-X1), which was operated through NIS-Elements (Nikon). In all cases, images were acquired with a 100x ApoTIRF 1.4 NA objective (Nikon). Fluorophores were excited with 488- or 561-nm laser lines and fluorescence was detected by an iXon Ultra 897 electron-multiplying charge-coupled device (EMCCD) camera (Andor). Multicolor images were acquired sequentially.
Fluorescence Loss in Photobleaching
Fluorescence loss in photobleaching (FLIP) experiments were performed on cell samples previously cultured, transfected, and treated as for regular live-cell imaging experiments. Imaging was done as described above and the bleaching pulses were performed using the Galvo Miniscanner (Nikon). Fields containing at least two cells expressing the construct of interest were selected; one cell was used for FLIP analysis and the other one as an internal control for photobleaching due to imaging. For FLIP analysis of cells expressing mCherry-tagged proteins, a region in the ER was defined and repeatedly bleached (with 30% 561-nm laser power for 300 μs) every 30 seconds, resulting in a 370-second movie comprising twelve bleaching pulses. At the same time, single-section images were acquired (with the 561-nm laser line) every 5 seconds before the first bleaching step and throughout the rest of the movie.
VCP/p97 Inhibition
To inhibit VCP/p97, cells were set up for live-cell imaging as described above and treated with culture medium containing either 1 μM of the compound CB-5083 (Selleck Chemicals), a potent inhibitor of VCP/p97 (Le Moigne et al., 2017; Zhou et al., 2015), or an equivalent volume of dimethyl sulfoxide (DMSO) (Sigma) as negative/vehicle control. After 45–60 min of incubation, cells were imaged (steady state), treated with oleate, and further imaged throughout specific time points of LD induction. To confirm that VCP/p97 was being efficiently inhibited, parallel cell samples (cultured in 6-well plates) were incubated with similar amounts of CB-5083, treated with oleate throughout the same time points of LD induction, harvested, and processed for western blot (see section below) to probe for the endogenous amounts of ubiquitylated proteins.
Protein Extraction and Western Blot
Cells were harvested by resuspension and centrifugation at 1,800 rpm for 5 min. The resulting pellets were washed once with ice-cold PBS, resuspended in lysis buffer (150 mM NaCl, 20 mM Tris-HCl [pH 8.0], 1% Triton™ X-100 (Sigma), and cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail (Roche)), and rocked end-over-end at 4°C for 30 min. The cell lysates were then cleared by centrifugation at 7,000 × g for 5 min at 4°C. After measuring the protein concentration using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific), 20–30 μg of protein from each lysate were mixed with 4x Laemmli sample buffer (containing 10% 2-mercaptoethanol) and heated up to 60°C for 15 min. Protein samples were thereafter separated by SDS-PAGE using 4–15% Mini-PROTEAN® TGX™ Precast Protein Gels (Bio-Rad).
For western blot analysis, gels were transferred to 0.2 μm pore-size nitrocellulose membranes (iBlot™ 2 Transfer Stacks, Thermo Fisher Scientific) using the iBlot™ 2 Gel Transfer Device (Thermo Fisher Scientific). Membranes were incubated with blocking buffer (TBS-T supplemented with 5% non-fat dry milk) at room temperature for 60 min, treated with primary antibodies (1:1000 dilution in blocking buffer), and further incubated at 4°C with gentle shaking overnight. Membranes were subsequently washed three times in TBS-T for 10 min, incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10000 dilution in blocking buffer) at room temperature for 60 min, and washed again there times in TBS-T for 10 min. Protein signal was detected by treating the membranes with SuperSignal™ West Pico, Dura, or Femto chemiluminescent substrates (Thermo Fisher Scientific).
Protease Protection Assays
Cells previously cultured in 100-mm dishes and transfected with the double-tagged constructs of interest were harvested as described above. The cell suspensions from at least three 100-mm dishes expressing the same construct were combined during the harvesting step. The resulting pellets were kept in ice (no freezing) and directly processed for microsome isolation. To this end, each pellet, corresponding to a different construct, was resuspended in 1–2 ml of homogenization buffer (250 mM sucrose, 20 mM Tris-HCl [pH 7.4], 10% glycerol, 1mM EDTA, and cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail (Roche)), transferred to a pre-chilled glass homogenizer, and lysed by applying 20–30 gentle strokes using a loose-fitting pestle. The cell lysates were centrifuged (600 × g for 5 min at 4°C) to remove remaining intact cells and the nuclear fractions, and the resulting supernatants were subjected to a second centrifugation (8,000 × g for 10 min at 4°C) to remove the mitochondrial fractions. A third centrifugation step (100,000 × g for 1 h at 4°C) was performed to yield the pellets corresponding to the microsomal fractions. These were resuspended in 300–500 μl of homogenization buffer lacking protease inhibitor, and their protein concentration was measured using the NanoDrop™ 2000 Spectrophotometer (Thermo Fisher Scientific).
For the protease protection assays, we set up individual reactions containing 100 μg of protein from the corresponding fresh microsome sample in a final volume of 100 μl of homogenization buffer lacking protease inhibitor (i.e., 1 μg/μl protein concentration). For each construct of interest, individual reactions were subjected to three different treatments: non-treated (negative control), trypsin (12.5 μg/ml final concentration) (Sigma), and both trypsin and Triton™ X-100 (1% final concentration) (Sigma). After adding the respective treatments, all reactions were incubated at 30°C for 30 min and then put back in ice. To stop trypsin digestion, soybean trypsin inhibitor (0.05 μg/μl final concentration) (Sigma) was immediately added and reactions were incubated at room temperature for 5 min. We then treated all reactions with 4x Laemmli sample buffer (containing 10% 2-mercaptoethanol) and boiled them up to 95°C for 5 min. In all cases, a reaction volume equivalent to at least 30 μg of protein was further used for SDS-PAGE and western blot analysis, which were performed as described above.
Antibodies
Western blot analysis was done using the Living Colors® mCherry monoclonal antibody from Takara (Cat# 632543), the ubiquitin (P4D1) monoclonal antibody from Cell Signaling Technology (Cat# 3936S), the anti-α-tubulin monoclonal antibody from Sigma (Cat# T5168), the ANTI-FLAG® M2 monoclonal antibody from Sigma (Cat# F1804), and the anti-GRP78/BiP (ET-21) polyclonal antibody from Sigma (Cat# G9043). Secondary antibodies conjugated to horseradish peroxidase (HRP) against mouse (m-IgGκ-HRP, Cat# sc-516102) and against rabbit (mouse anti-rabbit IgG-HRP, Cat# sc-2357) were acquired from Santa Cruz Biotechnology.
Sequence Alignments and Analyses
For Figure 2A, the sequences of LiveDrop and a set of GPAT4 ortholog proteins from representative species were aligned in Clustal Omega (Sievers et al., 2011) using default settings. The resulting alignment file was further formatted using Jalview (Waterhouse et al., 2009) to generate the final display version of the sequence alignment.
The conservation profiles of the residues in the LiveDrop and predicted ALG14 hairpin sequences, shown in Figure S3, were generated by performing high throughput sequence alignments of each protein motif using EVcouplings (Marks et al., 2011). The “image view” option of the alignment viewer tool was utilized to display the sequence alignments in the format of color-coded conservation maps.
HELIQUEST (Gautier et al., 2008) was used to calculate the physicochemical and sequence-derived parameters of LiveDrop and the LD targeting and non-LD targeting motifs identified in Figure 6. These calculated parameters were included as variables in the matrix utilized for principal component analysis (see Table S2).
Simulation Methods
Structure Generation
The LiveDrop structure was generated using the Rosetta ab initio structure generation tool (Bradley et al., 2005; Raman et al., 2009; Rohl et al., 2004). Fragment files were created using the Robetta server (Kim et al., 2004; Song et al., 2013). Fifty thousand structures were generated, and the ten structures with the best scores were chosen for analysis. These ten structures contained the same main structural elements (i.e., a kink at P185 and two transmembrane α-helices on either side of P185). The top-scoring structure was used for all simulations. We also predicted the structure with MEMSAT-SVM (Jones, 2007; Jones et al., 1994; Nugent and Jones, 2009), TOPCONS (Tsirigos et al., 2015), and the RosettaMP ab initio structure generation tool (Alford et al., 2015; Barth et al., 2007; Barth et al., 2009; Koehler Leman et al., 2017; Koehler Leman et al., 2015; Yarov-Yarovoy et al., 2006), which gave structures with similar helicity and features as those from Rosetta. The best Rosetta generated structure was then solvated in a cubic water box and relaxed with 1 ns of NVT equilibration and 20 ns of NPT equilibration.
The ALG14 hairpin structure was generated using the RosettaMP ab initio structure generation tool. Fragment files were created using the Robetta server, and the membrane topology was predicted using OCTOPUS (Viklund and Elofsson, 2008). More than ten thousand structures were generated and the two structures with the best scores were used. A low-resolution structure was refined to atomic level detail using high-resolution refinement as described (Alford et al., 2015). The three principal axes of the ALG14 hairpin structure were aligned with the x, y, and z directions before inserting into either a bilayer or monolayer system.
Molecular Dynamics Simulations
Molecular dynamics simulations of LiveDrop (1 μs in the bilayer and 2 μs in the monolayer) and the predicted ALG14 hairpin (2 μs), in both a bilayer and monolayer system, were conducted. In the case of the LiveDrop simulations, the initial protein structure for the final production run was taken from the corresponding umbrella sampling minimum representation (see below). For the ALG14 hairpin simulations, the initial protein structures were predicted using RosettaMP as described above. The CHARMM-GUI membrane builder (Jo et al., 2008) was used to insert the protein motifs into a bilayer membrane. The bilayer system was composed of 133–137 POPC, 55 DOPE, and 16 SAPI molecules per leaflet in TIP3P water (Jorgensen et al., 1983) and 0.15 M NaCl solution. A detailed description of the lipid composition used in each simulation is shown in Table S5. The monolayer system was generated by including an 8-nm-thick TG layer between the two leaflets of the original bilayer, so that both systems have the same lipid composition. To build a monolayer system, three segments were prepared separately and later combined using VMD (Humphrey et al., 1996). For the first segment, a bilayer system, including a given protein motif, was built using the CHARMM-GUI builder. The protein, the upper membrane leaflet, and the water and ions above the bilayer midplane were selected and used as the upper component in a final monolayer system. For the second segment, a separate bilayer membrane was built using the CHARMM-GUI builder. The corresponding lower leaflet and water and ions below the bilayer midplane were selected and used as the lower component in a final monolayer system. The third segment, corresponding to the neutral lipid layer, was built using Packmol (Martinez et al., 2009). All three segments were further constructed such that they had the same X and Y dimensions. The three segments were then combined with extra 1-nm spacing along the Z axis between the TG segment and the other membrane segments. Any TG molecules that had bad contacts with the membrane components were removed from the system. Additional ions were added, when needed, to make the system neutral.
All-atom simulations were performed using the GROMACS (version 2016 and 2018) simulation engine (Abraham et al., 2015) with the CHARMM36 lipid and protein force fields (Best et al., 2012; Klauda et al., 2010; MacKerell et al., 1998). Simulations were integrated with a 2-fs timestep. The Particle Mesh Ewald algorithm (Essmann et al., 1995) was used to evaluate long-range electrostatic interactions with a real space cutoff of 1.0 nm. Lennard-Jones interactions were cut-off at 1.0 nm with the potential-shift-Verlet method, and the neighbor list was updated every 100 steps. Long-range dispersion was corrected for energy and pressure. The pressure was maintained semi-isotropically using the Parrinello-Rahman barostat (Parrinello and Rahman, 1981) at a pressure of 1.0 bar, a compressibility of 4.5×10−5/bar, and a coupling time constant of 2.0 ps. Bonds to hydrogen were constrained using the LINCS algorithm (Hess, 2008). The temperature was maintained at 310 K using the stochastic velocity rescaling thermostat (Bussi et al., 2007) with a coupling time constant of 0.1 ps. Biased simulations were also conducted in GROMACS, with the addition of the PLUMED2 plugin (Tribello et al., 2014). For biased simulations, non-bonded van der Waals interactions were cut-off at a distance of 1.2 nm between atoms and were switched to zero in the case of distances between 1.0 and 1.2 nm. The Particle Mesh Ewald algorithm was used with a real space cutoff of 1.2 nm. The pressure was maintained semi-isotropically with a coupling time constant of 5.0 ps.
LiveDrop Umbrella Sampling Simulations
To confirm that the conformational changes in the LiveDrop α-helices converged, umbrella sampling simulations (Torrie and Valleau, 1977) were run on LiveDrop. The protein was first inserted into the bilayer and monolayer systems using the procedure described by Javanainen (Javanainen and Martinez-Seara, 2016). Briefly, the protein was placed next to the membrane patch, with the water and ions removed from the system. Restraints were placed on the protein backbone, as well as on the phosphorous atoms of the lipids. A lateral pressure of 1000 bar was applied to the system, pushing the protein into the membrane. Three lipids were removed from the cytosolic membrane leaflet to maintain the area of both leaflets equal. The systems were then relaxed for 2 ns of NPT equilibration. TIP3P water and 0.15 M NaCl were added to both systems. Finally, the membrane-protein systems were equilibrated according to the CHARMM-GUI membrane equilibration procedure (Jo et al., 2007; Jo et al., 2008; Lee et al., 2016; Wu et al., 2014). The bilayer membrane comprised 258 POPC, 107 DOPE, and 32 SAPI molecules (see Table S5). The monolayer system comprised 218 POPC, 91 DOPE, 26 SAPI, 91 TG, and 92 cholesteryl oleate molecules in total (see Table S5).
Umbrella sampling simulations were run biasing the distance between the center of geometry of the LiveDrop residues W166 and A203. Harmonic restraints with a force constant of 104 kJ/mol/nm2 were placed on each of the ~80 umbrella sampling windows, which were spaced every 0.05 nm over a distance range of 0.5–4.5 nm. Each window was run for 50 ns in the bilayer and for 40 ns in the monolayer. The first 10 ns were considered equilibration and they were not used for calculating the potential of mean force (PMF). The starting structures for each window were generated by conducting metadynamics (Laio and Parrinello, 2002) on the same collective variable as the umbrella sampling, for both the bilayer and monolayer. Hills were deposited with a height of 0.003 kJ/mol/nm every 1 ps. The simulation length of each of the four metadynamics runs was at least 1 μs. Each of the starting structures for the umbrella sampling was then chosen from the metadynamics runs, where the distance between residues W166 and A203 was within 0.02 nm of the equilibrium window distance. The PMF was then calculated using the Weighted Histogram Analysis Method (WHAM) package (Grossfield; Kumar et al., 1995; Roux, 1995) with a bin spacing of 0.01 nm. The error in the simulations was estimated by dividing the equilibrated trajectories into four blocks, calculating the PMF for each block, and then determining the standard deviation of the block PMFs.
Single Amino Acid Permeation
The PMFs for single amino acid permeating through a bilayer and monolayer were conducted using Transition-Tempered Metadynamics (Dama et al., 2014; Sun et al., 2016), biasing the z-component of the position vector connecting the center of mass of the membrane and the center of mass of the amino acid. The bilayer system comprised 16 POPC, 7 DOPE, and 2 SAPI molecules per leaflet in TIP3P water and 0.15 M NaCl solution. The monolayer system included 35 TG molecules between the two bilayer leaflets. Acetylated N-terminus (ACE) and methylated C-terminus (CT1) patches were applied to the termini of all amino acids evaluated. For each amino acid, four independent simulation runs, each 2 μs long, were conducted in each membrane system. The final PMF was obtained by averaging the PMFs obtained from the four simulations. The Gaussian function was deposited every 2 ps with a height of 0.001 kJ/mol and a scaled width of 0.025 for the bilayer and 0.018 for the monolayer. The bias factor was set to 10. The two transition basins were located at −0.2 and 0.2 for the bilayer and at −0.3 and 0.3 for the monolayer. The latter distance values are based on a scaled distance for biasing simulations, where the z-dimension of a system was considered equal to 1 and centered at 0. Simulations were conducted in the canonical ensemble (NVT). Single amino acid permeation calculations were run for most of the amino acids bearing hydrophobic side chains, including alanine, valine, isoleucine, leucine, phenylalanine, tyrosine, and tryptophan.
Free Energy Calculations
The thermodynamic force driving protein accumulation on LDs was approximated based on the changes in free energy for amino acids transitioning from a bilayer to a monolayer. These changes in free energy are based on the bilayer and monolayer structures combined with the previously described permeation free energy profiles for single hydrophobic amino acids. To estimate these changes in free energy, the following process was used: (i) The depth of each residue was calculated as the z component of the position vector connecting the center of mass of the upper phospholipid phosphorus and the center of mass of a given residue. For LiveDrop, the residue depths were calculated using the minimum energy structures obtained from the umbrella sampling simulations, and for the predicted ALG14 hairpin, residue depths were calculated by time-averaging all the corresponding simulations from 500 ns to 2 μs. (ii) The permeation PMF for a given residue was shifted so that the free energy is set to zero in bulk water, and the depth of the upper phosphorus atoms are set to zero to correspond with the structural depth. (iii) Based on the permeation PMF profiles, the corresponding free energy of each hydrophobic residue in the protein motifs was estimated at its calculated depth. Comparing the free energy of each residue in the bilayer and monolayer estimates its change in free energy when transitioning from the ER to LDs. Given the large changes in stability as a function of bilayer or monolayer depth, the changes in position are expected to dominate the change in free energy for a full protein to relocate from the ER bilayer to the LD monolayer. This is further justified by the thermodynamic cycle shown in Figure S8, which depicts the minimal change in association energies for amino acids in a bilayer versus a monolayer environment, compared to the large changes based on position and increased stability in the monolayer relative to the bilayer midplane (shown in Figure S4C).
QUANTIFICATION AND STATISTICAL ANALYSIS
Fluorescence Image Quantification
Acquired images and movies were processed and quantified using Fiji (Schindelin et al., 2012). For all constructs in each time course experiment of LD induction, raw multi-channel images were first split into individual channels using a custom-made script. An automatic threshold was then applied to the BODIPY channel, corresponding to the LD staining, and used as a mask to identify the LD-occupied region in each image. By subtracting this from the mCherry channel, corresponding to the protein signal, we measured the mean intensity and area of the mCherry signal in the whole cell excluding LDs and in the LD-occupied region only. For each of the quantified cells, the mean intensity of the mCherry signal on LDs was divided by the mean intensity of the mCherry signal in the whole cell excluding LDs. This ratio is referred to as the “LD enrichment factor”, and it represents the extent to which each mCherry-tagged protein variant accumulates on LDs over the rest of the cell. The average of the individual LD enrichment factors measured for each construct, at each time point of LD induction, was calculated for all time course experiments.
Additionally, we measured the mean intensity of the mCherry signal in the whole cell and used it as a read out for the total amount of mCherry-tagged protein being expressed in each one of the quantified cells. The average of this value was calculated for each construct and further normalized to the average whole-cell intensity of the corresponding mCherry-LiveDrop sample included in each experiment and at each time point of LD induction. The resulting value is referred to as “normalized whole-cell intensity (WCI)” and is expressed in arbitrary units (a.u.).
In the case of FLIP experiments, the mean mCherry signal on LDs was measured for each frame throughout each movie using a custom-made script. This was done separately for both the bleached cell and non-bleached control cell included in every field of all acquired movies. For each frame, the mean mCherry signal on LDs measured from the bleached cell was divided by the corresponding value from the control cell. This resulted in a ratio which was further normalized by dividing it by the maximum ratio obtained within each movie. The values obtained are referred to as the “corrected intensity” for each frame and are expressed in arbitrary units (a.u.). For each construct tested, the average corrected intensity per movie frame was calculated and presented as a function of time in seconds.
Statistical Analyses
For all time course experiments of LD induction, the average LD enrichment factor and average normalized WCI are presented in bar graphs as mean + standard deviation (SD). For the FLIP experiment, the average corrected intensity is presented as mean ± SD for each movie frame. In all cases, the n value indicated for each experiment represents the number of cells quantified. Additional statistical details can be found in the corresponding figure legends and below.
All statistical tests were performed using GraphPad Prism version 8.1.1 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com. To evaluate the statistical difference between the mean values of the LD enrichment factors calculated for each protein variant, we treated the values giving place to each mean as independent datasets. In all cases, we first calculated whether the dataset followed a normal distribution or not by performing a Shapiro-Wilk test. When evaluating the statistical difference between the mean values of two datasets, if both of them followed a normal distribution, we used an unpaired t-test with Welch’s correction (which assumes unequal SDs), and if they did not, we used a Mann-Whitney test. When evaluating more than two datasets, we performed one-way ANOVA if all of them followed a normal distribution, followed by Dunnett’s multiple comparison test. Otherwise, we performed a Kruskal-Wallis test, followed by Dunn’s multiple comparison test.
Moreover, the statistical correlation between the LD enrichment factors measured for LiveDrop across different experiments and their respective normalized WCI values was tested. Because the values of both variables did not followed a normal distribution, we performed a non-parametric Spearman correlation test (two-tailed). Further statistical details, including the values of the correlation coefficients (r) obtained, can be found in the respective figure legend.
For all the tests performed, p-values smaller than 0.05 were considered statistically significant and denoted as * for p-values < 0.05, ** for p-values < 0.01, and *** for p-values < 0.001.
Principal Component Analysis
Principal component analysis (PCA) was conducted using R (version 3.6.0) (R_Core_Team, 2018) and the corresponding R package for PCA, FactoMineR (Lê et al., 2008). The matrix of variables analyzed is shown in Table S2.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-mCherry | Takara | Cat# 632543; RRID: AB_2307319 |
| Mouse monoclonal anti-ubiquitin | Cell Signaling Technology | Cat# 3936S; RRID: AB_331292 |
| Mouse monoclonal anti-α-tubulin | Sigma-Aldrich | Cat# T5168; RRID: AB_477579 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Polyclonal rabbit anti-GRP78/BiP | Sigma-Aldrich | Cat# G9043; RRID: AB_2279879 |
| Mouse anti-IgG kappa binding protein-HRP | Santa Cruz Biotechnology | Cat# sc-516102; RRID: AB_2687626 |
| Mouse monoclonal anti-rabbit IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2357; RRID: AB_628497 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Effectene® Transfection Reagent | Qiagen | Cat# 301425 |
| Oleic acid | Sigma-Aldrich | Cat# O1383 |
| Concanavalin A | Sigma-Aldrich | Cat# C5275 |
| BODIPY™ 493/503 | Thermo Fisher Scientific | Cat# D3922 |
| CB-5083, VCP/p97 inhibitor | Selleck Chemicals | Cat# S8101 |
| Triton™ X-100 | Sigma-Aldrich | Cat# T8787 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11836170001 |
| Trypsin from bovine pancreas | Sigma-Aldrich | Cat# T9201 |
| Trypsin inhibitor from Glycine max (soybean) | Sigma-Aldrich | Cat# T9003 |
| Critical Commercial Assays | ||
| pENTR™/SD/D-TOPO™ Cloning Kit | Thermo Fisher Scientific | Cat# K242020 |
| Gateway™ LR Clonase™ II Enzyme mix | Thermo Fisher Scientific | Cat# 11791020 |
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 200521 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| SuperSignal™ West Pico | Thermo Fisher Scientific | Cat# 34580 |
| SuperSignal™ West Dura | Thermo Fisher Scientific | Cat# 34076 |
| SuperSignal™ West Femto | Thermo Fisher Scientific | Cat# 34095 |
| Experimental Models: Cell Lines | ||
| D. melanogaster: Cell line S2: S2R+ | Drosophila RNAi Screening Center, Laboratory of Norbert Perrimon | FlyBase ID: FBtc0000150; RRID: CVCL_Z831 |
| Oligonucleotides | ||
| Primers for site-directed mutagenesis, see Table S4 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pA-CherryW | Guo et al., 2008 | N/A |
| Synthetic double-stranded DNA: gBlocks® Gene Fragments | Integrated DNA Technologies | N/A |
| Software and Algorithms | ||
| TOPCONS | Tsirigos et al., 2015 | http://topcons.cbr.su.se |
| PSIPRED | Jones, 1999 | http://bioinf.cs.ucl.ac.uk/psipred/ |
| NIS-Elements | Nikon | N/A |
| Clustal Omega | Sievers et al., 2011 | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| Jalview | Waterhouse et al., 2009 | http://www.jalview.org/ |
| EVcouplings | Marks et al., 2011 | https://evcouplings.org/ |
| HELIQUEST | Gautier et al., 2008 | http://heliquest.ipmc.cnrs.fr/ |
| Rosetta ab initio structure generation tool | Rohl et al., 2004 | https://rosettacommons.org/ |
| Robetta server | Kim et al., 2004 | https://robetta.bakerlab.org/ |
| RosettaMP | Alford et al., 2015 | https://rosettacommons.org/ |
| OCTOPUS | Viklund and Elofsson, 2008 | http://octopus.cbr.su.se/ |
| CHARMM-GUI | Jo et al., 2008 | http://www.charmm-gui.org/ |
| VMD | Humphrey et al., 1996 | https://www.ks.uiuc.edu/Research/vmd/ |
| Packmol | Martinez et al., 2009 | http://m3g.iqm.unicamp.br/packmol/home.shtml |
| GROMACS | Abraham et al., 2015 | http://manual.gromacs.org/ |
| CHARMM36m force field | Klauda et al., 2010 | http://mackerell.umaryland.edu/charmm_ff.shtml |
| Plumed2 | Tribello et al., 2014 | https://www.plumed.org/ |
| Weighted Histogram Analysis Method (WHAM) package | Laboratory of Alan Grossfield | http://membrane.urmc.rochester.edu/?page_id=126 |
| Fiji | Schindelin et al., 2012 | https://fiji.sc/ |
| GraphPad Prism 8 | GraphPad | http://www.graphpad.com/ |
| R (version 3.6.0) | R Core Team | https://www.r-project.org/ |
Highlights.
Specific membrane-embedded ER proteins target and accumulate on LDs
Minimal targeting motifs use Trp and positive-charged residues for LD accumulation
Distribution of these residues within motifs correlates with LD accumulation
ACKNOWLEDGMENTS
We thank Scot Stone (U. of Saskatchewan) for advice on the protease protection assays; Coline Prévost for help in image and movie quantification; Xihao Li (Harvard Chan School of Public Health) and Wenting Lyu for help with statistical analyses; Chenghan Li, Jesper Madsen, and Zack Jarin (Voth group) for help with simulations; Julia Koehler Leman (Flatiron Institute, Simons Foundation) for help with predicting membrane protein structures; members of the Farese & Walther laboratory, Stephen White (UC Irvine), and Gregory Voth (U. of Chicago) for useful discussions; and Gary Howard for editorial assistance. This research was supported by the National Institute of General Medical Sciences of the NIH through 5R01GM097194 (to TCW) and 5R01GM124348 (to RVF). TCW is an investigator of the Howard Hughes Medical Institute. The simulations used the Extreme Science and Engineering Discovery Environment (XSEDE), supported by NSF grant ACI-1548562. Specifically, it used the Stampede2 system at the Texas Advanced Computing Center (TACC) and the Bridges system at the Pittsburgh Supercomputing Center (PSC) through allocation number TG-MCA94P017. This work was also completed in part with resources provided by the U. of Chicago Research Computing Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindahl E (2015). Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. [Google Scholar]
- Alford RF, Koehler Leman J, Weitzner BD, Duran AM, Tilley DC, Elazar A, and Gray JJ (2015). An integrated framework advancing membrane protein modeling and design. PLoS Comput Biol 11, e1004398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacle A, Gautier R, Jackson CL, Fuchs PFJ, and Vanni S (2017). Interdigitation between triglycerides and lipids modulates surface properties of lipid droplets. Biophys J 112, 1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth P, Schonbrun J, and Baker D (2007). Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci U S A 104, 15682–15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth P, Wallner B, and Baker D (2009). Prediction of membrane protein structures with complex topologies using limited constraints. Proc Natl Acad Sci U S A 106, 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartz R, Li WH, Venables B, Zehmer JK, Roth MR, Welti R, Anderson RG, Liu P, and Chapman KD (2007). Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J Lipid Res 48, 837–847. [DOI] [PubMed] [Google Scholar]
- Bersuker K, and Olzmann JA (2017). Establishing the lipid droplet proteome: Mechanisms of lipid droplet protein targeting and degradation. Biochim Biophys Acta Mol Cell Biol Lipids 1862, 1166–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Peterson CWH, To M, Sahl SJ, Savikhin V, Grossman EA, Nomura DK, and Olzmann JA (2018). A proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Dev Cell 44, 97–112 e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, and Mackerell AD Jr. (2012). Optimization of the additive charmm all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J Chem Theory Comput 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchoux J, Beilstein F, Pauquai T, Guerrera IC, Chateau D, Ly N, Alqub M, Klein C, Chambaz J, Rousset M, et al. (2011). The proteome of cytosolic lipid droplets isolated from differentiated caco-2/tc7 enterocytes reveals cell-specific characteristics. Biol Cell 103, 499–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley P, Misura KM, and Baker D (2005). Toward high-resolution de novo structure prediction for small proteins. Science 309, 1868–1871. [DOI] [PubMed] [Google Scholar]
- Brasaemle DL, Dolios G, Shapiro L, and Wang R (2004). Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3t3-l1 adipocytes. J Biol Chem 279, 46835–46842. [DOI] [PubMed] [Google Scholar]
- Bussi G, Donadio D, and Parrinello M (2007). Canonical sampling through velocity rescaling. J Chem Phys 126, 014101. [DOI] [PubMed] [Google Scholar]
- Chang CL, Weigel AV, Ioannou MS, Pasolli HA, Xu CS, Peale DR, Shtengel G, Freeman M, Hess HF, Blackstone C, et al. (2019). Spastin tethers lipid droplets to peroxisomes and directs fatty acid trafficking through escrt-iii. J Cell Biol 218, 2583–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YQ, Kuo MS, Li S, Bui HH, Peake DA, Sanders PE, Thibodeaux SJ, Chu S, Qian YW, Zhao Y, et al. (2008). Agpat6 is a novel microsomal glycerol-3-phosphate acyltransferase. J Biol Chem 283, 10048–10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitraju C, Trotzmuller M, Hartler J, Wolinski H, Thallinger GG, Lass A, Zechner R, Zimmermann R, Kofeler HC, and Spener F (2012). Lipidomic analysis of lipid droplets from murine hepatocytes reveals distinct signatures for nutritional stress. J Lipid Res 53, 2141–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Wu X, Lambert TJ, Lai ZW, Walther TC, and Farese RV Jr. (2019). Ldaf1 and seipin form a lipid droplet assembly complex. Dev Cell 51, 551–563 e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman R, and Bell RM (1978). Evidence that biosynthesis of phosphatidylethanolamine, phosphatidylcholine, and triacylglycerol occurs on the cytoplasmic side of microsomal vesicles. J Cell Biol 76, 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie E, Guo X, Christiano R, Chitraju C, Kory N, Harrison K, Haas J, Walther TC, and Farese RV Jr. (2014). High confidence proteomic analysis of yeast lds identifies additional droplet proteins and reveals connections to dolichol synthesis and sterol acetylation. J Lipid Res 55, 1465–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dama JF, Rotskoff G, Parrinello M, and Voth GA (2014). Transition-tempered metadynamics: Robust, convergent metadynamics via on-the-fly transition barrier estimation. J Chem Theory Comput 10, 3626–3633. [DOI] [PubMed] [Google Scholar]
- Ding Y, Yang L, Zhang S, Wang Y, Du Y, Pu J, Peng G, Chen Y, Zhang H, Yu J, et al. (2012). Identification of the major functional proteins of prokaryotic lipid droplets. J Lipid Res 53, 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, and Pedersen LG (1995). A smooth particle mesh ewald method. The Journal of Chemical Physics 103, 8577–8593. [Google Scholar]
- Gao XD, Tachikawa H, Sato T, Jigami Y, and Dean N (2005). Alg14 recruits alg13 to the cytoplasmic face of the endoplasmic reticulum to form a novel bipartite udp-n-acetylglucosamine transferase required for the second step of n-linked glycosylation. J Biol Chem 280, 36254–36262. [DOI] [PubMed] [Google Scholar]
- Gautier R, Douguet D, Antonny B, and Drin G (2008). Heliquest: A web server to screen sequences with specific alpha-helical properties. Bioinformatics 24, 2101–2102. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Prehn S, Hartmann E, Kalies KU, and Rapoport TA (1992). A mammalian homolog of sec61p and secyp is associated with ribosomes and nascent polypeptides during translocation. Cell 71, 489–503. [DOI] [PubMed] [Google Scholar]
- Grossfield A Wham: An implementation of the weighted histogram analysis method.
- Guo Y, Walther TC, Rao M, Stuurman N, Goshima G, Terayama K, Wong JS, Vale RD, Walter P, and Farese RV (2008). Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature 453, 657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedin LE (2010). Intra- and intermolecular interactions in proteins: Studies of marginally hydrophobic transmembrane alpha-helices and protein-protein interactions In Department of Biochemistry and Biophysics (Sweden, Stockholm University; ). [Google Scholar]
- Hess B (2008). P-lincs: A parallel linear constraint solver for molecular simulation. J Chem Theory Comput 4, 116–122. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). Vmd: Visual molecular dynamics. Journal of Molecular Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Javanainen M, and Martinez-Seara H (2016). Efficient preparation and analysis of membrane and membrane protein systems. Biochim Biophys Acta 1858, 2468–2482. [DOI] [PubMed] [Google Scholar]
- Jo S, Kim T, and Im W (2007). Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One 2, e880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, and Im W (2008). Charmm-gui: A web-based graphical user interface for charmm. J Comput Chem 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- Jones DT (1999). Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292, 195–202. [DOI] [PubMed] [Google Scholar]
- Jones DT (2007). Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23, 538–544. [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, and Thornton JM (1994). A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 33, 3038–3049. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983). Comparison of simple potential functions for simulating liquid water. The Journal of chemical physics 79, 926–935. [Google Scholar]
- Joshi AS, Nebenfuehr B, Choudhary V, Satpute-Krishnan P, Levine TP, Golden A, and Prinz WA (2018). Lipid droplet and peroxisome biogenesis occur at the same er subdomains. Nat Commun 9, 2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DE, Chivian D, and Baker D (2004). Protein structure prediction and analysis using the robetta server. Nucleic Acids Res 32, W526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr., and Pastor RW (2010). Update of the charmm all-atom additive force field for lipids: Validation on six lipid types. J Phys Chem B 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler Leman J, Mueller BK, and Gray JJ (2017). Expanding the toolkit for membrane protein modeling in rosetta. Bioinformatics 33, 754–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler Leman J, Ulmschneider MB, and Gray JJ (2015). Computational modeling of membrane proteins. Proteins 83, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kory N, Farese RV Jr., and Walther TC (2016). Targeting fat: Mechanisms of protein localization to lipid droplets. Trends Cell Biol 26, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kory N, Grond S, Kamat SS, Li Z, Krahmer N, Chitraju C, Zhou P, Frohlich F, Semova I, Ejsing C, et al. (2017). Mice lacking lipid droplet-associated hydrolase, a gene linked to human prostate cancer, have normal cholesterol ester metabolism. J Lipid Res 58, 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kory N, Thiam AR, Farese RV Jr., and Walther TC (2015). Protein crowding is a determinant of lipid droplet protein composition. Dev Cell 34, 351–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahmer N, Guo Y, Wilfling F, Hilger M, Lingrell S, Heger K, Newman HW, Schmidt-Supprian M, Vance DE, Mann M, et al. (2011). Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of ctp:Phosphocholine cytidylyltransferase. Cell Metab 14, 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahmer N, Hilger M, Kory N, Wilfling F, Stoehr G, Mann M, Farese RV Jr., and Walther TC (2013). Protein correlation profiles identify lipid droplet proteins with high confidence. Mol Cell Proteomics 12, 1115–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, and Kollman PA (1995). Multidimensional free-energy calculations using the weighted histogram analysis method. Journal of Computational Chemistry 16, 1339–1350. [Google Scholar]
- Laio A, and Parrinello M (2002). Escaping free-energy minima. Proc Natl Acad Sci U S A 99, 12562–12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landolt-Marticorena C, Williams KA, Deber CM, and Reithmeier RA (1993). Non-random distribution of amino acids in the transmembrane segments of human type i single span membrane proteins. J Mol Biol 229, 602–608. [DOI] [PubMed] [Google Scholar]
- Le Moigne R, Aftab BT, Djakovic S, Dhimolea E, Valle E, Murnane M, King EM, Soriano F, Menon MK, Wu ZY, et al. (2017). The p97 inhibitor cb-5083 is a unique disrupter of protein homeostasis in models of multiple myeloma. Mol Cancer Ther 16, 2375–2386. [DOI] [PubMed] [Google Scholar]
- Lê S, Josse J, and Husson F (2008). Factominer: An r package for multivariate analysis. Journal of Statistical Software 25, 1–18. [Google Scholar]
- Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, et al. (2016). Charmm-gui input generator for namd, gromacs, amber, openmm, and charmm/openmm simulations using the charmm36 additive force field. J Chem Theory Comput 12, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. (1998). All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Marks DS, Colwell LJ, Sheridan R, Hopf TA, Pagnani A, Zecchina R, and Sander C (2011). Protein 3d structure computed from evolutionary sequence variation. PLoS One 6, e28766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez L, Andrade R, Birgin EG, and Martinez JM (2009). Packmol: A package for building initial configurations for molecular dynamics simulations. J Comput Chem 30, 2157–2164. [DOI] [PubMed] [Google Scholar]
- Murugesan S, Goldberg EB, Dou E, and Brown WJ (2013). Identification of diverse lipid droplet targeting motifs in the pnpla family of triglyceride lipases. PLoS One 8, e64950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle CA, Vergnes L, Dejong H, Wang S, Lewin TM, Reue K, and Coleman RA (2008). Identification of a novel sn-glycerol-3-phosphate acyltransferase isoform, gpat4, as the enzyme deficient in agpat6−/− mice. J Lipid Res 49, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman KE, and Nymeyer H (2006). Indole localization in lipid membranes revealed by molecular simulation. Biophys J 91, 2046–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent T, and Jones DT (2009). Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics 10, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, and Carvalho P (2019). Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol 20, 137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello M, and Rahman A (1981). Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics 52, 7182–7190. [Google Scholar]
- Poppelreuther M, Rudolph B, Du C, Grossmann R, Becker M, Thiele C, Ehehalt R, and Fullekrug J (2012). The n-terminal region of acyl-coa synthetase 3 is essential for both the localization on lipid droplets and the function in fatty acid uptake. J Lipid Res 53, 888–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppelreuther M, Sander S, Minden F, Dietz MS, Exner T, Du C, Zhang I, Ehehalt F, Knuppel L, Domschke S, et al. (2018). The metabolic capacity of lipid droplet localized acyl-coa synthetase 3 is not sufficient to support local triglyceride synthesis independent of the endoplasmic reticulum in a431 cells. Biochim Biophys Acta Mol Cell Biol Lipids 1863, 614–624. [DOI] [PubMed] [Google Scholar]
- Prevost C, Sharp ME, Kory N, Lin Q, Voth GA, Farese RV Jr., and Walther TC (2018). Mechanism and determinants of amphipathic helix-containing protein targeting to lipid droplets. Dev Cell 44, 73–86 e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R_Core_Team (2018). R: A language and environment for statistical computing. (R Foundation for Statistical Computing, Vienna, Austria.). [Google Scholar]
- Raman S, Vernon R, Thompson J, Tyka M, Sadreyev R, Pei J, Kim D, Kellogg E, DiMaio F, Lange O, et al. (2009). Structure prediction for casp8 with all-atom refinement using rosetta. Proteins 77 Suppl 9, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohl CA, Strauss CE, Misura KM, and Baker D (2004). Protein structure prediction using rosetta. Methods Enzymol 383, 66–93. [DOI] [PubMed] [Google Scholar]
- Roux B (1995). The calculation of the potential of mean force using computer simulations. Computer Physics Communications 91, 275–282. [Google Scholar]
- Ruggiano A, Mora G, Buxo L, and Carvalho P (2016). Spatial control of lipid droplet proteins by the erad ubiquitin ligase doa10. EMBO J 35, 1644–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka HA, Thompson JW, Chen YS, Dubois LG, Haas JT, Moseley A, and Valdivia RH (2015). Chlamydia trachomatis infection leads to defined alterations to the lipid droplet proteome in epithelial cells. PLoS One 10, e0124630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrul B, and Kopito RR (2016). Peroxin-dependent targeting of a lipid-droplet-destined membrane protein to er subdomains. Nat Cell Biol 18, 740–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segrest JP, De Loof H, Dohlman JG, Brouillette CG, and Anantharamaiah GM (1990). Amphipathic helix motif: Classes and properties. Proteins 8, 103–117. [DOI] [PubMed] [Google Scholar]
- Sharpe HJ, Stevens TJ, and Munro S (2010). A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 142, 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, Thompson J, and Baker D (2013). High-resolution comparative modeling with rosettacm. Structure 21, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanovic A, and Thiele C (2013). Monotopic topology is required for lipid droplet targeting of ancient ubiquitous protein 1. J Lipid Res 54, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, Dama JF, Tan JS, Rose JP, and Voth GA (2016). Transition-tempered metadynamics is a promising tool for studying the permeation of drug-like molecules through membranes. J Chem Theory Comput 12, 5157–5169. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Otsuka T, Ohsaki Y, Cheng J, Taniguchi T, Hashimoto H, Taniguchi H, and Fujimoto T (2012). Derlin-1 and ubxd8 are engaged in dislocation and degradation of lipidated apob-100 at lipid droplets. Mol Biol Cell 23, 800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauchi-Sato K, Ozeki S, Houjou T, Taguchi R, and Fujimoto T (2002). The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J Biol Chem 277, 44507–44512. [DOI] [PubMed] [Google Scholar]
- Torrie GM, and Valleau JP (1977). Nonphysical sampling distributions in monte carlo free-energy estimation: Umbrella sampling. Journal of Computational Physics 23, 187–199. [Google Scholar]
- Tribello GA, Bonomi M, Branduardi D, Camilloni C, and Bussi G (2014). Plumed 2: New feathers for an old bird. Computer Physics Communications 185, 604–613. [Google Scholar]
- Tsirigos KD, Peters C, Shu N, Kall L, and Elofsson A (2015). The topcons web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res 43, W401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viklund H, and Elofsson A (2008). Octopus: Improving topology prediction by two-track ann-based preference scores and an extended topological grammar. Bioinformatics 24, 1662–1668. [DOI] [PubMed] [Google Scholar]
- Walther TC, Chung J, and Farese RV Jr. (2017). Lipid droplet biogenesis. Annu Rev Cell Dev Biol 33, 491–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Becuwe M, Housden BE, Chitraju C, Porras AJ, Graham MM, Liu XN, Thiam AR, Savage DB, Agarwal AK, et al. (2016). Seipin is required for converting nascent to mature lipid droplets. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, and Barton GJ (2009). Jalview version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Thiam AR, Olarte MJ, Wang J, Beck R, Gould TJ, Allgeyer ES, Pincet F, Bewersdorf J, Farese RV Jr., et al. (2014). Arf1/copi machinery acts directly on lipid droplets and enables their connection to the er for protein targeting. Elife 3, e01607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, Uchida A, Cheng JX, Graham M, Christiano R, Frohlich F, et al. (2013). Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the er to lipid droplets. Dev Cell 24, 384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, et al. (2014). Charmm-gui membrane builder toward realistic biological membrane simulations. J Comput Chem 35, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Schonbrun J, and Baker D (2006). Multipass membrane protein structure prediction using rosetta. Proteins 62, 1010–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau WM, Wimley WC, Gawrisch K, and White SH (1998). The preference of tryptophan for membrane interfaces. Biochemistry 37, 14713–14718. [DOI] [PubMed] [Google Scholar]
- Ye Y, Tang WK, Zhang T, and Xia D (2017). A mighty “protein extractor” of the cell: Structure and function of the p97/cdc48 atpase. Front Mol Biosci 4, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R (2015). Fat flux: Enzymes, regulators, and pathophysiology of intracellular lipolysis. EMBO Mol Med 7, 359–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehmer JK, Bartz R, Liu P, and Anderson RG (2008). Identification of a novel n-terminal hydrophobic sequence that targets proteins to lipid droplets. J Cell Sci 121, 1852–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HJ, Wang J, Yao B, Wong S, Djakovic S, Kumar B, Rice J, Valle E, Soriano F, Menon MK, et al. (2015). Discovery of a first-in-class, potent, selective, and orally bioavailable inhibitor of the p97 aaa atpase (cb-5083). J Med Chem 58, 9480–9497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.