

Abstract
Bacteriophage T7 and T7-like bacteriophages are valuable genetic models for lytic phage biology that have heretofore been intractable with in vivo genetic engineering methods. This manuscript describes that the presence of λ Red recombination proteins makes in vivo recombineering of T7 possible, so that single base changes and whole gene replacements on the T7 genome can be made. Red recombination functions also increase the efficiency of T7 genome DNA transfection of cells by ~100-fold. Likewise, Red function enables two other T7-like bacteriophages that do not normally propagate in E. coli to be recovered following genome transfection. These results constitute major technical advances in the speed and efficiency of bacteriophage T7 engineering and will aid in the rapid development of new phage variants for a variety of applications.
Keywords: bacteriophage engineering, phage therapy, bacteriophage genetics, recombineering
1. Introduction
Recently, interest has grown in rapid, efficient, and scalable engineering of bacteriophages, particularly virulent phages like T7 [1]. Increased interest in bacteriophage therapy has focused attention on T7 [2] due to its rapid growth, large burst size (~250 particles per infection), general resistance to restriction by the bacterial host, and rapid degradation of the host genome [3]. Phage T7 has been studied intensively for more than fifty years. The viral genome sequence and most gene functions have been defined. When T7 infects E. coli strains, rapid cell death and lysis occurs, usually within twenty minutes, with the release of progeny phage particles that spread the infection. Under optimized conditions, the time period from phage infection to host cell lysis is less than 20 min at 37 °C [4,5]. T7 is strictly lytic and is not capable of transduction, except under very special conditions [6]. Lytic or virulent phages are excellent models for developing phage therapy systems for use in combating antibiotic resistant bacteria. However, many of the attributes that make T7 a preferred system for phage therapy applications also present challenges. Standardized methods for in vivo genetic engineering, like that of recombineering, have not yet been successful using the T7 genome as a target. Here we introduce novel methods for recombineering the T7 genome and detail several intrinsic genes that are useful in the selection of recombinants.
One such gene, The bacterial trxA gene encodes thioredoxin, which is required by the T7 DNA polymerase as a processivity factor. An E. coli host with a defect in trxA blocks wild-type T7 phage growth, however, when trxA is cloned into T7, its replication and development are restored. This property allows trxA to be used as a genetic marker for positive selection of trxA recombinants into T7 genomes. This genetic approach is analogous to using antibiotic resistance genes for selecting recombination events in a bacterial genome [7]. However, even with a selectable marker, engineering virulent bacteriophages like T7 can be complicated, expensive, and time consuming. Past T7 engineering strategies have included standard in vitro molecular cloning and in vitro packaging of the genome [1], cloning and transformation of a plasmid carrying desired markers followed by infection of T7 phage to allow homologous recombination between the plasmid and the phage [8], and in vitro synthesis and assembly of full length genomes carried on yeast artificial chromosomes [8]. Here, we used the Red recombination system from phage λ as a tool to rapidly engineer genetic elements efficiently and with high-fidelity into T7 in vivo. It has already been shown that certain bacteriophages can readily be modified using BRED technology or other recombineering systems [9,10]. The λ Red recombineering system consists of a 5′-3′ double-strand DNA (dsDNA) exonuclease (Exo) and a single strand binding protein that anneals complementary DNA strands (Beta) [11]. A third function, Gam, is also commonly expressed in conjunction with λ Exo and Beta, and although not essential for recombineering, it stimulates dsDNA recombination about 10-fold [12] by disabling the potent bacterial RecBCD exonuclease and thereby helping to preserve linear dsDNA substrates electroporated into the cell [13]. Here, we have optimized the Red system specifically for modification of the bacteriophage T7 genome.
T7 genomic DNA can be introduced into cells by either infection or electroporation [14]. During phage T7 infection, the phage DNA initially enters the cell by injection of the left end of the genomic DNA, which is subsequently drawn further into the cell by coupling DNA entry to transcription by both the host and phage RNA polymerases [15]. This process establishes a temporal order of gene entry and expression from the left-most early genes to the distal late genes. The timing of DNA replication is also dependent on this mechanism of entry [16,17,18]. By contrast, when the entire genome is transferred into the cell by electroporation, the relative timing of both gene expression and DNA replication is altered. Here we find that T7 DNA, introduced by either infection or electroporation, can be genetically modified in vivo by recombineering using λ Red-mediated recombination. We also demonstrate that the efficiency of phage recovery following electroporation of T7 and other T7-like phage genomes is enhanced by λ Red recombination functions. The methods presented here provide new avenues for making synthetic phage variants that can be employed for different applications.
2. Results and Discussion
2.1. λ Red-Mediated Recombination Enhances Transfection of T7 and T7-Like Phages
T7 infection of E. coli is a multi-step process. After the phage attaches to the cell surface, its DNA is injected by a specialized event that requires transcription-mediated mobilization of the DNA from the viral particle into the cell by the host RNA polymerase [15]. In our initial experiments, we tested if introduction of purified phage genomic DNA by electroporation could produce functional phage particles. This process, also referred to as transfection, yielded very few plaques. The plaque yield could be increased several-fold by deletion of the E. coli K12-specific restriction endonuclease gene, hsdR. More importantly, we found that expression of the λ Red recombination genes increased T7 transfection efficiency by greater than 10 orders of magnitude (Figure 1A). Perhaps due to faster recombination and better preservation of the page genome.
Figure 1.

λ Red function enhances transfection by T7 and T7-related phage genomic DNA. (A) LT976 strain contains a λ defective prophage that expresses Red recombination functions only when induced at 42 °C (see Table 1). The light-colored bars indicate cells grown at 32 °C without Red expression; the dark-colored bars indicate cells induced at 42 °C to express Red recombination functions. Standard error of the mean value is shown and indicates p < 0.005 in Student t-test, comparing induced samples to uninduced controls. Cells, either induced at 42 °C or maintained at 32 °C, were electroporated with ~50 ng of respective phage DNAs (see X-axis, ~1 phage genome per cell). The phage DNA was introduced into LT976 by electroporation as described in Materials and methods. The Y-axis indicates total number of infectious centers per electroporation reaction. The phages Pharr and Poteet were scored on their cognate host Klebsiella pneumoniae 1760b, and the phage K1-5 was scored on its cognate host E. coli K1 (see Table 1). Note that without induction of Red (32 °C no transfectants were found for phage K1-5. (B) In a separate experiment, the RecA defective strain DH5α carries the plasmid pSIM18 or pSIM28. These plasmids contain a defective prophage with genes to express Exo, Beta and Gam or just Gam, respectively. Expression is induced by a shift to 42°. Transfection of the T7 genome is as described in Methods. Bars show level of phage yields following transfection. Negligible infective centers are found in the absence of Exo and Beta.
Next, we found that the λ Red-mediated enhancement of T7 phage yields following genome electroporation extends to other T7-like bacteriophages. Bacteriophage K1-5 specifically infects E. coli K1 and E. coli K5 strains. Phage K1-5 cannot attach to and productively infect E. coli K12 [19]. In this experiment, plaque formation on E. coli K1, the natural host of the K1-5 phage, is used to determine if viable phages are generated following electroporation of the K1-5 genome into E. coli K12. No plaque-forming K1-5 phages were recovered after K1-5 genomic transfection of E. coli K12 strains. However, reasonable yields (~103) of plaque forming derivatives were found after transfection of E. coli K12 strains expressing the Red system (Figure 1A).
Transfections by two other T7-like phage genomes, Pharr and Poteet, whose cognate host is Klebsiella pneumoniae were also tested. Red-mediated transfection of DNA genomes isolated from Pharr and Poteet yielded 103 to 104 plaques in Klebsiella when λ Red function was provided, compared to <10 plaques in the absence of Red (Figure 1A).
Quantitative results demonstrated that phage yields during transfection of T7 genomic DNA increase coordinately with increasing genomic DNA transfected per culture and saturate when there is approximately one T7 genome transfected per λ Red expressing cell (Figure 2). These data suggest that the Red system has its stimulatory effect on a single T7 genome and does not require two genomes to enter the same cell. If the latter were true, we would expect a strong stimulation of yield when the number of T7 genomes transfected approached the number of cells being transfected. Instead, there appears to be a slight decrease in yield when the number of genomes transfected increases.
Figure 2.

The number of infectious centers recovered from electroporated T7 genomic DNA increases with concentration. One nanogram of T7 genomic DNA is approximately equivalent to 2.3 × 107 genomes, and each electroporation reaction contains ~1 × 108 E. coli host cells (LT976). Thus 50 ng of phage DNA = ~1.1 genomes/cell. Electroporation was carried out on cells induced for λ Red recombination functions, then incubated in LB for growth at 30°, before plating on TB agar in the presence of E. coli B plating bacteria for determining total plaque forming units at each genome concentration used. Transfection reaches saturation where one T7 genome is transfected per cell. Infectious centers generated per electroporation reaction are indicated on the Y-axis. Addition of >1 genome/cell appears to be somewhat inhibitory to recombineering and may represent competition of a second molecule for circularization.
2.2. T7 Transfection Requires the Exo and Bet Gene Functions and Is Independent of the Host RecA Function
We examined phage yields following T7 DNA transfection of recA minus strains that either expressed all the λ Red functions exo, bet and gam or were defective for exo and bet, but expressed gam. When Exo and Beta were present, high levels of transfection occurred even in the recA mutant. In the absence of Exo and Beta, transfection was greatly reduced (Figure 1B).
λ Red functions are known to be able to generate circular DNAs from linear DNA molecules having terminal homologous repeats [20]. T7 phage carries 160 bp DNA repeats at the ends of its genome and could thereby be circularized by Red recombination. Exactly how such a recombinant T7 circular DNA leads to T7 gene expression and replication remains to be determined, but it is known that the early T7 promoter is effectively transcribed by the host RNA polymerase on circular DNA [21]. This transcription leads to early gene expression and the potential for lytic development.
2.3. λ Red-Mediated Oligo Recombination Enables Directed Nucleotide Changes within the Phage T7 Genome
We next tested the ability of Red-mediated recombination to modify the T7 genome at precise locations. We first focused on g17, which encodes the tail fiber protein, a primary factor in determining the host range of T7 [7]. Starting with a T7 phage that contained a nonsense (amber) mutation in the tail fiber gene, g17am267 [5], we tested how efficiently it could be converted to wild-type g17 by a 71nt single-strand DNA (ssDNA) oligo containing the wild type sequence. We used one or the other of two complementary oligos: one (ARP292) corresponding in sequence to the leading strand, and its complement (ARP293) corresponding to the lagging strand used during T7 DNA replication (see Table 2). Initially, we focused on recombineering using transfected T7 genomic DNA. Strain ARP3771s carries an amber tRNA suppressor allele plus the λ Red recombination genes. This strain was made competent for recombineering and then electroporated with the mutant genomic T7 g17am267 DNA (50 ng) together with one of the amber mutation-correcting 70 nt oligos. The electroporated cells were allowed to recover and the resulting lysate was titered on E. coli B and E. coli B40. Since E. coli B40 contains the tRNA suppressor allele, supD, all phage produced by DNA transfection should plate on this strain, regardless of the g17 allele. Only the phages that are recombinant for the g17 amber mutant and are now wild type for the tail fiber gene g17 should grow on E. coli B. We calculated the frequency of these wild type recombinants over a range of oligo concentrations, keeping the T7 genomic DNA concentration high and constant (Figure 3). To monitor the spontaneous reversion of the amber mutation to wild type, we included a control in which only the T7 genomic DNA was added (see legend to Figure 3). No revertants were found among total transfectants indicating a reversion frequency of less than 2.3 × 10−5.
Table 2.
Oligonucleotides a.
| Name | Nucleotide Sequence b | Notes |
|---|---|---|
| ARP261 | TAACGCTTCACTCGAGGCGTTTTTCGTTATGTATAAAAAGGAGCACACC/ATGGCTAACGTAATTAAAACCGTTTTGACTTACCAG | λ pL g17 forward oligo |
| ARP262 | GCTTCCCAGCCAGCGTTGCGAGTGCAGTACTCATTCGTTTTATACCTCTG/ATTACTCGTTCTCCACCATGATTGCATTAGG | λ pL g17 reverse oligo |
| ARP292 | ACCAGAACTCATGGCAAGCACGTAATGAAGCCTTA CAGTTCCGTAATGAGGCTGAGACTTTCAGAAACCAA |
T7 g17Q170UAG-CAG leading strand oligo |
| ARP293 | TTGGTTTCTGAAAGTCTCAGCCTCATTACGGAA CTGTAAGGCTTCATTACGTGCTTGCCATGAGTTCTGGT |
T7 g17Q170UAG-CAG lagging strand oligo |
| ARP301 | GGCAGTGACCCGCTTCCCGTTCGTCCGTCTGTTACTCAAACGAATCAAGGAGGTGTTCTG/ATGAGCGATAAAATTATTCACCTGACTGAC | T7 g1.7<>trxA forward |
| ARP302 | CACTCTGAGCAAGATGTGAAGTCATCAGATAGGCTGTCGGCAGGTGGGGTTGACTTGAAG/TTACGCCAGGTTAGCGTCGAGGA | T7 g1.7<>trxA reverse |
| ARP309 | ACCAACACGCCAGGCTTATTCCTGTGGAGTTATAT/TCCTAATTTTTGTTGACACTCTATC | trxA<>tetA forward |
| ARP310 | TTTTTAGCGACGGGGCACCCGAACATGAAATTCCC/ATCAAAGGGAAAACTGTCCATATGC | trxA<>tetA reverse |
| ARP321 | ACCAGAACTCATGGCAAGCACGTAATGAAGC | g17am267 70 bp dsDNA forward |
| ARP322 | CGCTTGGTTTCTGAAAGTCTCAGCCTCATTACGG | g17am267 70 bp dsDNA forward |
| ARP356 | CGAAATAATCTTCTCCCTGTAGTCTCTTAGATTTACTTTAAGGAGGTCAA/ATGAGCGATAAAATTATTCACCTGACTGAC | g17<>trxA forward |
| ARP357 | AGGTACAGTCATTGTTGTTATCTGACCCTCTACCAATGTACCAGTTATTC/TTACGCCAGGTTAGCGTCGAGGA | g17<>trxA reverse |
a Oligos were obtained desalted from Integrated DNA Technologies in Coralville, IA. b “/” indicates the border between 5′ homology to recombineering targets and 3′ homology used for primer PCR. The underlined CAG or CTG are the codon positions in the oligos ARP292 and ARP293 that correct the amber mutation in g17 used in these studies.
Figure 3.

Recombineering can correct a point mutation following transfection of the T7 genome. The T7 g17am267 mutant genomic DNA (50 ng) was co-electroporated with the substrate DNAs (pmole amounts on X-axis) into the amber suppressor strain ARP3771 expressing the λ Red functions. Single-strand oligonucleotide substrates ARP292 (circles) and ARP293 (rectangles) of 71 nucleotides in length carry the wild type coding information. Their sequences correspond, respectively, to the leading and lagging strands used in T7 DNA replication. The dsDNA substrate (triangles) is a PCR product corresponding in length and sequence to the single-stranded oligos used here. T7 g17+ wild type recombinant frequencies (Y-axis) were calculated by the following equation: (phage titer on E. coli B)/(phage titer on E. coli B supD). Values represent an average of at least three biological replicates with standard errors of the mean indicated. A control T7 genome transfection without added DNA substrate generated no wild type T7 revertants among an average of 4.4 × 104 total plaques recovered on E. coli B supD in eight experiments, indicating a reversion frequency < 2.3 × 10−5.
Both oligos generated wild type recombinants with the lagging strand oligo having a higher number of recombinants than the leading strand oligo (Figure 3). The sequence of the lagging strand oligo corresponds to that of Okazaki fragments made during the process of T7 DNA replication of gene g17 [18]. This finding is consistent with other reports that demonstrate Red-dependent oligo recombination occurs primarily at the replication fork, and recombination frequency on the lagging strand is higher than on the leading strand [11].
A dsDNA PCR product amplified from the wild-type T7 gene g17 was also tested and shown to generate wild type T7 recombinants (Figure 3). The dsDNA substrate was more efficient than ssDNA oligonucleotides at the lower DNA concentrations. We expect this result since double-strandedness protects this DNA from degradation by the multiple single strand exonucleases in E. coli. We know that ssDNA oligos are very susceptible to these single strand exonucleases, and this is most apparent at low oligo concentrations. Sawitzke et al. [22] demonstrated that high oligo concentrations were necessary to titrate the single strand exonuclease activities and thereby protect oligos so that they are available for recombination.
We have found that λ recombineering can be used to modify the T7 genome following transfection of its DNA genome and also following infection by the T7 phage particle. Cells expressing λ Red functions were infected with the tail fiber mutant phage T7 g17am267. Lysates of this phage were prepared in the host E. coli B40, which contains a tRNA suppressor for the amber mutation. The suppressor allows the amber mutation to be translated and thereby produce the essential GP17 protein.
Cells being prepared for recombineering were mixed with the T7 g17am267 mutant phage to allow adsorption during the time that they were on ice prior to recombineering. Unabsorbed phages were eliminated by washing steps. The electroporation conditions and the substrate oligos used for recombineering were the same as those used for the transfection and recombineering of phage genomic DNA. The products of the recombineering reactions were assayed on both E. coli B and B40 to determine the frequency of wild type recombinants (see Methods, Figure 4). Spontaneous reversion of the amber mutant to wild-type was observed at a frequency of approximately one in 106 viable phage (see Figure 4 and its legend), which is consistent with previous reports documenting the reversion frequency of T7 amber mutants [5,23,24]. Providing the oligo without λ Red functions present revealed that phage T7′s own recombination and replication functions have little if any ability to mediate oligo recombination, whereas λ Red functions greatly enhance targeting T7 genome recombination by as much as 10,000-fold at high oligo concentration (Figure 4).
Figure 4.

Recombineering corrects a point mutation after infection by a T7 phage particle. The lagging strand single-strand oligonucleotide (ARP293) of 71 nucleotides in length was used as a substrate during recombineering to correct the phage mutation T7 g17am267. The amber suppressor strain ARP3771 was infected with the T7 g17am267 mutant phage at a multiplicity of infection of ~5 phage per bacterium. Cells were either induced to express Red function and then electroporated with the leading and lagging strand oligonucleotides ARP292 and ARP293 at several oligo concentrations (see x-axis). Electroporation reactions were outgrown at 37 °C in SOC media until lysis. Frequency of T7 g17+ recombinants (y-axis) was calculated by the following equation: (recombinant phage titer on E. coli B)/(total phage titer on E. coli B40 supD). Values represent an average of at least three biological replicates. Lysates, in which no lagging strand oligo DNA was added, revealed a reversion frequency of ~1.1 × 10−6 (±4.1 × 10−7).
2.4. Gene Replacement by λ Red-Mediated Recombineering
Thioredoxin is the product of the bacterial trxA gene. TrxA is a processivity factor for the T7 DNA polymerase and is therefore essential for T7 [25]. Thus, wild-type T7 phage cannot form plaques on E. coli if the trxA gene is deleted from the bacterial chromosome. However, if the trxA gene is incorporated into and expressed from the T7 genome itself, phage replication and plaque forming ability can be restored even if the E. coli strain is deleted for trxA.
We have replaced the non-essential T7 nucleotide kinase gene g1.7 [7] with the trxA gene by recombineering. In this experiment, the entire T7 genome was introduced into cells by electroporation along with a double-stranded PCR product of trxA to target replacement of T7 gene g1.7. Recombineering to insert and replace g1.7 with the trxA DNA was very efficient (0.6%) (Figure 5).
Figure 5.
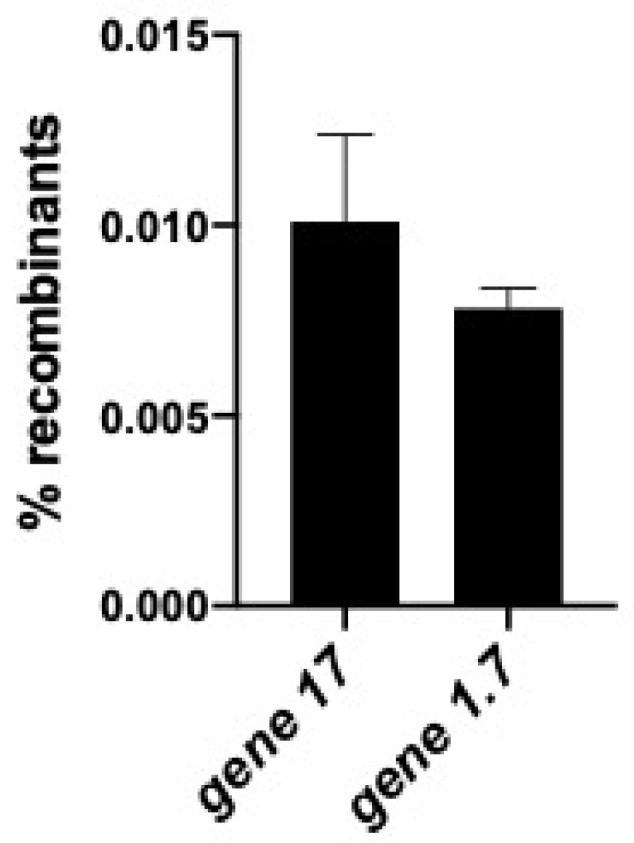
Replacement of T7 genes g1.7 and g17 with the bacterial trxA gene. Recombineering was used to replace two different T7 genes with the bacterial trxA gene. One gene, g1.7, is non-essential; the other gene, g17, is essential and required for plaque formation. The trxA dsDNA PCR fragments, g1.7<>trxA and g17<>trxA were amplified using the bacterial trxA gene as a template to generate the trxA gene flanked by T7 DNA homology that targets recombination to g1.7 or g17, respectively. Oligos ARP301 and ARP302 used for PCR are designed with trxA having T7 flanking homologies to replace g1.7 with trxA. Oligos ARP356 and ARP357 used for PCR are designed with trxA having T7 flanking homologies to replace g17 with trxA (Table 2). LT976 cells were induced for Red-mediated recombination and then co-electroporated with genomic T7 DNA and either the g1.7 or g17 targeted trxA PCR products. The electroporation reactions were out-grown in SOC medium at 37 °C. Gene 1.7 trxA replacements were detected on E. coli B 3783, a host that lacks the trxA gene in its genome. The g17 replacements were detected on ARP3750 at 42 °C. Non-recombinant T7 cannot grow on these two bacteria that lack trxA. T7 recombinant frequencies in the lysates were calculated by the following equations: [(T7 g1.7<>trxA recombinant phage titer on E. coli B3783 ΔtrxA/(total phage titer on E. coli B)] × 100; [(T7 g17<>trxA recombinant phage titer on ARP3750)/(total phage titer on ARP3607)] × 100. Values represent an average of at least three biological replicates with the standard error of the mean calculated. The identity and accuracy of the gene substitutions on the recombinant phages were confirmed by sequence.
We have shown above that a non-essential T7 gene (g1.7) can be replaced very efficiently with trxA using recombineering. We now wanted to replace the essential tail fiber gene, g17, with trxA and used recombineering to generate recombinant T7 g17<>trxA phages. Because g17 makes the tail fibers used for phage attachment to the cell surface, T7 recombinants lacking g17 will not be able to attach and infect E. coli. Therefore, to detect these recombinants, we constructed a special strain of E. coli, ARP3750, which expresses Gp17 tail fiber protein constitutively (see methods). Wild-type T7 cannot grow on this trxA mutant strain since the bacterium and wild type T7 lack trxA. A T7g17<>trxA recombinant phage carrying trxA would be able to grow because the essential Gp17 tail fibers are expressed from the bacterial chromosome. Figure 5 shows that in this strain, trxA recombinants replace the essential gene g17 at a frequency of ~1.0%.
We have observed a ~1.0% frequency of gene replacement by trxA at the essential g17 tail fiber locus, and a similar 0.6% frequency of replacement by trxA at the nonessential g1.7 locus (Figure 5). The generation of tail fiber recombinants in which the g17 tail fiber gene of phage T7 is replaced by other bacteriophage type tail fiber genes can now be detected by screening a few thousand plaques generated after transfection. Recombineering methods for exchanging tail fiber genes opens the potential for extending T7′s host range and thereby weaponizing T7-type phages against drug resistant bacteria [26]. Indeed, Yehl et al. [27] recently demonstrated that they could extend the host range of bacteriophage T3 by mutagenizing the tail fiber gene. These works are complementary, as engineering the mutants would be greatly facilitated by using recombineering methods described herein.
Lytic or virulent phages are excellent models for developing anti-bacterial systems to combat antibiotic resistant bacteria. Here we have engineered the T7 genome with the goal of generating a general platform for modification of T7 that can be easily manipulated and has potential for biomedical applications.
3. Materials and Methods
3.1. Media
For standard cultivation of bacteria, cells were grown in liquid L Broth (LB) containing 0.5% NaCl, 1.0% (w/v) tryptone, and 0.5% (w/v) yeast extract. LB Agar plates contained in addition 1.5% agar. For sucrose counter-selection, NaCl was omitted from LB agar and 6% (wt/vol) sucrose was added. To select for antibiotic resistant genetic markers, the following antibiotic concentrations were used: 12.5 μg/mL tetracycline, 30 μg/mL ampicillin, and 200 μg/mL hygromycin.
SOC medium was used for recovery of cells following electroporation, and contained 2% (w/v) tryptone, 0.5% (w/v) yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 20 mM glucose. Phages were diluted in TMG, containing 10 mM Tris-HCl, pH 7.5, 10 mM MgSO4, and 0.1% gelatin. Plaque assays were carried out on Tryptone Broth (TB) agar plates containing 0.5% NaCl, 1.0% (w/v) tryptone, and 1.0% (w/v) agar.
3.2. Bacterial Strains
Bacterial strains used in this study are listed in Table 1. Strains specific to this paper were generated by recombineering and/or P1 transduction. The method for strain construction is provided in Table 1. All oligos used in strain construction were purchased from Integrated DNA Technologies (IDT, Coralville, IA, USA) and are listed in Table 2. Final constructions were verified by PCR analyses and sequencing. Details of individual strain constructions will be provided upon request.
Table 1.
Bacterial strains, Bacteriophages, and Plasmids used in this study.
| Bacterial Strains | Relevant Genotype | Source, Reference |
|---|---|---|
| E. coli strains | ||
| E. coli B | Wild-type | Ian Molineux collection |
| E. coli B40 | E. coli B supD | Ian Molineux collection |
| E. coli B3783 | E. coli B ΔtrxA<>*tetA | This work; recombineering pSIM18; gene replacement hsdR<>bla |
| E. coli K1 | Wild-type | Sankar Adhya collection |
| E. coli K12 strains: | ||
| W3110 | rph-1 inv(rrnD-rrnE) | Laboratory stock strain |
| DY330 | W3110 [λ cI857 gam+bet+exo+ Δ(cro-bioA)] | {Thomason et al., 2014} |
| T-SACK | W3110 araD<>tetA-sacB-amp fliC<>cat argG:kan | {Li et al., 2013} |
| LT976 | W3110 hsdR<>bla λ cI857 gam+bet+exo+ Δ(cro-bioA) |
Lynn Thomason collection; recombineering; DY330, gene replacement hsdR<>bla |
| ARP3607 | W3110 hsdR<>bla λ cI857 pL g17 Δ(cro-bioA) |
This work; recombineering; gene replacement LT976, (cIII-galK)<>gp17 |
| ARP3750 | W3110 hsdR<>bla ΔtrxA<>tetA λcI857pL g17 Δ(cro-bioA) |
This work |
| ARP3620 | LT976 ΔtrxA<>tetA | This work |
| ARP3771 | LT976 supF:Tn10 | This work; transduction LT976 X (P1) |
| ARP3816 | W3110 hsdR<>bla | This work; transduction W3110 X (P1) LT976 |
| DH5α | fhuA2 lac(del)U169 phoA glnV44 Φ80’ lacZ(del)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | Laboratory stock strain |
| Klebsiella pneumoniae Kpn1760 | Cured of resistance plasmid pKpQIL at 42 °C | Karen Frank collection |
| Bacteriophages and Plasmids | ||
| T7 | Wild-type | Sankar Adhya collection |
| T7 (amber267) | T7 g17Q170UAG | Ian Molineux collection |
| K1-5 | Wild-type | Sankar Adhya collection |
| Pharr | Isolated from sewage | Jason Gill collection |
| Poteet | Isolated from sewage | Jason Gill collection |
| pSIM18 HygroR | pSC101 PLgam+bet+exo+ | {Thomason et al., 2014} |
| pSIM28 HygroR | pSC101, PL-gam+ | (http://redrecombineering.ncifcrf.gov/) |
* “<>” represents a convention used by some bacterial geneticists to denote “[gene] replaced by [gene]” (e.g., ΔtrxA<>tetA).
The deletion trxA<>tetA was constructed on the bacterial chromosome of LT976 by recombineering to replace trxA with tetA [28] to generate ARP3620. The tetA cassette was amplified from T-SACK [29] using primers ARP309 and ARP310, which add 50 bp of homology from the regions flanking the trxA gene to the flanks of the tetA cassette. The tetA insertion in ARP3620 was moved by P1 transduction [30] to generate the trxA deleted strains E. coli B3783 and ARP3750.
To cultivate T7 phages that lack g17, which encodes the essential tail fiber protein, we constructed strain ARP3607 to express g17 in trans. The g17 coding region, flanked by λ homologies, was generated by PCR using oligos ARP261 and ARP262. This linear fragment was inserted into a defective λ prophage by recombineering in strain LT976 to allow expression of Gp17 from the pL promoter of λ [12].
3.3. Bacteriophages
Bacteriophages used in this study are listed in Table 1. T7 phage was obtained from the collection of Ian Molineux, and Klebsiella phages Pharr and Poteet were isolated in collaboration with the laboratory of Jason Gill and Ry Young at Texas A&M. Phage K1-5 was a laboratory stock of S. Adhya. Pharr and Poteet genomic DNAs were a generous gift from Jason Gill.
All phage lysates were grown in TB by diluting an appropriate overnight bacterial culture 1:5 into fresh TB and adding a single phage plaque picked up by Pasteur pipet. Cultures were grown until lysis occurred, then chloroform was added, and cell debris was cleared by centrifugation.
3.4. Preparation of Genomic DNA
Genomic DNAs of ~40 kb in length from the phages T7, Pharr, Poteet, or K1-5 were extracted from lysates, having titers of ~1.0 × 1010 PFU/mL, using the Qiagen lambda Midi Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s recommendations. Phage genomic DNA was diluted to a working concentration of 100 ng/µL in ddH2O for all subsequent manipulations.
3.5. Recombineering Using Transfected Bacteriophage Genomic DNA
Red-recombination proficient cells (ARP3771) were made competent for Red recombination and electroporation as described in [28]. Briefly, log phase cells were shifted from a 32 °C to a 42 °C shaking water bath for 15 min to induce λ Red functions and subsequently cooled in an ice bath to prepare the cells for electroporation. Electroporation reactions were prepared [31,32] by mixing 50 μL of cells with 50 ng genomic DNA); when required, ssDNA or dsDNA substrates containing the desired genetic changes were added. The cells and DNA were transferred to a pre-chilled 0.1 cm electroporation cuvette (Bio Rad, Hercules, CA, USA) and electroporated at 1.3 kV in a Bio Rad Micropulser (Bio Rad, Hercules, CA, USA); the voltage used for electroporation was decreased to 1.3 kV, (below the standard 1.8 kV used for E. coli) to accommodate the large size of T7 genomic DNA [33]. The electroporation mix was allowed to recover in 10 mL SOC medium and processed for either infective centers or total phage yield as described below.
To assess the number of infectious centers following transfection, cells were diluted from the electroporation into 10 mL SOC and shaken for 15 min in baffled flasks in a 32 °C water bath, before pelleting at 15,000× g for 1 min. Cell pellets were suspended in 1 mL TB and then serially diluted in TMG. These dilutions were mixed with plating bacteria and layered on TB agar plates to determine the number of transfected cells that generate an infectious phage resulting in a plaque.
When monitoring phage yield after complete lysis, electroporation reactions were mixed with 10 mL SOC and incubated in a 32 °C shaking water bath for 60 min, or until lysis, and at which time lysis was completed by addition of chloroform before centrifugation to remove cell debris. Resulting lysates were diluted and assayed on selective and non-selective hosts (see below), respectively, to calculate recombination frequencies, relative to total phage yield.
3.6. Recombineering Following Infection by Bacteriophage T7
Red-recombination proficient cells (ARP3771) were prepared as described above to express the Red proteins from the defective λ prophage, and 100 μL of a T7 phage lysate containing 2.4 × 1010 PFU/mL were added to the 15-mL recombination competent cultures, equivalent to an average multiplicity of infection of 5. After 15 min, cells were washed as described above to prepare them for electroporation and to remove free phage. Electroporation reactions were prepared by mixing 50 μL of electrocompetent cells, 0.5 μL of single-strand or double-strand DNA containing the desired genetic modification. The cells and DNA were transferred to a pre-chilled 0.1 cm electroporation cuvette and electroporated at 1.8 kV. Cells recovered in 10 mL SOC medium by incubation at 32 °C for 1 h at which time each culture received chloroform and was centrifuged to remove cell debris. The resulting cleared lysates were assayed on non-selective and selective strains of E. coli to determine total and recombinant phage titers (details below).
3.7. Quantitation of Phage in Lysates
Phage titers were determined by plaque assays and were carried out by infecting 200 μL of host bacteria grown overnight in LB with 100 μL of phage lysates diluted appropriately in TMG buffer. After 15 min for adsorption, the infected bacteria were mixed with 3 mL of pre-warmed (50 °C) tryptone broth agar (0.7% agar) and layered over TB-agar (1% agar) plates and incubated at room temperature overnight. E. coli and its derivatives were used as host for T7 phage; E. coli K1 was used as host for phage K1-5 phage; and Klebsiella pneumoniae strain 1760b was used as host for phages Pharr and Poteet. T7 recombinants containing an insertion of the trxA gene were titered on E. coli B3783, and T7 containing amber mutations were titered on E. coli B40.
4. Conclusions
T7 and other T7-like bacteriophages are valuable for their innate virulence and bacteria-killing properties. T7 is used as a model for lytic phage biology, and its genetics have been studied for over 50 years. Historically, however, virulent phages have been difficult to modify by in vivo genetic engineering methods. We have developed λ Red recombineering for T7 and used the Red functions to engineer changes in essential and non-essential genes of the phage. Additionally, we found that the Red recombination functions stimulate T7 phage production following transfection of T7 genomes into E. coli. This latter feature is important since the stimulation applies not only to phage T7 that uses E. coli as its natural host, but also to T7-like phages Pharr and Poteete that normally infect the pathogenic host Klebsiella pneumoniae where Red-mediated recombineering is inefficient [34]. By enabling their transfection and development in E. coli, research has been extended to these phages in a non-pathogenic environment. The methods presented here expand the tools available for engineering T7-like bacteriophages and enable facile construction of novel bacteriophages for basic research purposes and new therapeutic applications.
Acknowledgments
We thank members of the Court laboratory past and present for helpful ideas and discussions. Lynn Thomason and Nina Costantino provided advice and many of the E. coli strains. Carolyn Court provided strong support during the writing and editing of the manuscript. Robert Danner’s guidance during the inception of this project was invaluable. We are grateful for Karen Frank’s generous provision of Klebsiella pneumoniae hosts. T7 amber mutants and E. coli B hosts were from Ian Molineux. Phage and DNA of the bacteriophages Pharr and Poteete were provided by Jason Gill. Ry Young offered helpful advice during this study.
Author Contributions
Conceptualization, A.R.P., S.A. and D.L.C.; Data curation, J.D.J.; Formal analysis, J.D.J. and A.R.P.; Funding acquisition, D.L.C.; Investigation, A.R.P., J.D.J.; Methodology, J.D.J., A.R.P. and S.A.; Project administration, D.L.C.; Resources, D.L.C.; Supervision, A.R.P., S.A. and D.L.C.; Validation, J.D.J. and A.R.P.; Visualization, J.D.J., A.R.P. and A.J.R.; Writing—original draft, J.D.J. and A.R.P.; Writing—review and editing, A.J.R. and D.L.C. All authors have read and agreed to the published version of the manuscript.
Funding
Funding was provided by the Intramural Research Program of the National Institutes of Health at National Cancer Institute’s Center for Cancer Research; National Cancer Institute, National Institutes of Health [HHSN261200800001E], and partial support by NIH Director’s Challenge Innovation Award NRC-13045.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Chan L.Y., Kosuri S., Endy D. Refactoring bacteriophage T7. Mol. Syst. Biol. 2005;1:1–10. doi: 10.1038/msb4100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu T.K., Koeris M.S. The next generation of bacteriophage therapy. Curr. Opin. Microbiol. 2011;14:524–531. doi: 10.1016/j.mib.2011.07.028. [DOI] [PubMed] [Google Scholar]
- 3.Studier F.W. Bacteriophage T7. Science. 1972;176:367–376. doi: 10.1126/science.176.4033.367. [DOI] [PubMed] [Google Scholar]
- 4.Krüger D.H., Schroeder C. Bacteriophage T3 and bacteriophage T7 virus-host cell interactions. Microbiol. Rev. 1981;45:9–51. doi: 10.1128/MMBR.45.1.9-51.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Studier F. The genetics and physiology of bacteriophage T7. Virology. 1969;39:562–574. doi: 10.1016/0042-6822(69)90104-4. [DOI] [PubMed] [Google Scholar]
- 6.Chung Y.B., Hinkle D.C. Bacteriophage T7 DNA packaging. I. Plasmids containing a T7 replication origin and the T7 concatemer junction are packaged into transducing particles during phage infection. J. Mol. Biol. 1990;216:911–926. doi: 10.1016/S0022-2836(99)80010-2. [DOI] [PubMed] [Google Scholar]
- 7.Qimron U., Marintcheva B., Tabor S., Richardson C.C. Genomewide screens for Escherichia coli genes affecting growth of T7 bacteriophage. Proc. Natl. Acad. Sci. USA. 2006;103:19039–19044. doi: 10.1073/pnas.0609428103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiro R., Shitrit D., Qimron U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014;11:42–44. doi: 10.4161/rna.27766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marinelli L.J., Piuri M., Swigoňová Z., Balachandran A., Oldfield L.M., Van Kessel J.C., Hatfull G.F. BRED: A Simple and Powerful Tool for Constructing Mutant and Recombinant Bacteriophage Genomes. PLoS ONE. 2008;3:e3957. doi: 10.1371/journal.pone.0003957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oppenheim A.B., Rattray A.J., Bubunenko M., Thomason L.C., Court D.L. In vivo recombineering of bacteriophage lambda by PCR fragments and single-strand oligonucleotides. Virology. 2004;319:185–189. doi: 10.1016/j.virol.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 11.Ellis H.M., Yu D., Ditizio T., Court D.L. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. USA. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Datta S., Costantino N., Zhou X., Court D.L. Identification and analysis of recombineering functions from Gram-negative and Gram-positive bacteria and their phages. Proc. Natl. Acad. Sci. USA. 2008;105:1626–1631. doi: 10.1073/pnas.0709089105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy K.C. Lambda Gam protein inhibits the helicase and chi-stimulated recombination activities of Escherichia coli RecBCD enzyme. J. Bacteriol. 1991;173:5808–5821. doi: 10.1128/JB.173.18.5808-5821.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ando H., Lemire S., Pires D.P., Lu T.K. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015;1:187–196. doi: 10.1016/j.cels.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molineux I.J. No syringes please, ejection of phage T7 DNA from the virion is enzyme driven. Mol. Microbiol. 2001;40:1–8. doi: 10.1046/j.1365-2958.2001.02357.x. [DOI] [PubMed] [Google Scholar]
- 16.Molineux I.J. The T7 group. In: Calendar R., editor. The Bacteriophages. 2nd ed. Oxford University Press; Oxford, UK: 2006. pp. 277–301. [Google Scholar]
- 17.Richardson C.C. Bacteriophage T7: Minimal requirements for the replication of a duplex DNA molecule. Cell. 1983;33:315–317. doi: 10.1016/0092-8674(83)90411-7. [DOI] [PubMed] [Google Scholar]
- 18.Fuller C.W., Richardson C.C. Initiation of DNA replication at the primary origin of bacteriophage T7 by purified proteins. Initiation of bidirectional synthesis. J. Biol. Chem. 1985;260:3197–3206. [PubMed] [Google Scholar]
- 19.Scholl D., Kieleczawa J., Kemp P., Rush J., Richardson C., Merril C., Adhya S., Molineux I. Genomic Analysis of Bacteriophages SP6 and K1-5, an Estranged Subgroup of the T7 Supergroup. J. Mol. Biol. 2004;335:1151–1171. doi: 10.1016/j.jmb.2003.11.035. [DOI] [PubMed] [Google Scholar]
- 20.Thomason L.C., Costantino N., Court D.L. Examining a DNA Replication Requirement for Bacteriophage λ Red- and Rac Prophage RecET-Promoted Recombination in Escherichia coli. MBio. 2016;7 doi: 10.1128/mBio.01443-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heumann H., Lederer H., Kammerer W., Palm P., Metzger W., Baer G. Large-scale preparation of a DNA fragment containing the strong promoter A1 of the phage T7. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 1987;909:126–132. doi: 10.1016/0167-4781(87)90034-0. [DOI] [PubMed] [Google Scholar]
- 22.Sawitzke J.A., Costantino N., Li X.-T., Thomason L.C., Bubunenko M., Court C., Court D.L. Probing Cellular Processes with Oligo-Mediated Recombination and Using the Knowledge Gained to Optimize Recombineering. J. Mol. Biol. 2011;407:45–59. doi: 10.1016/j.jmb.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Springman R., Keller T., Molineux I.J., Bull J. Evolution at a High Imposed Mutation Rate: Adaptation Obscures the Load in Phage T7. Genetics. 2009;184:221–232. doi: 10.1534/genetics.109.108803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drake J.W., Charlesworth B., Charlesworth D., Crow J.F. Rates of spontaneous mutation. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reutimann H., Sjoeberg B.-M., Holmgren A. Bacteriophage T7 DNA polymerase: Cloning and high-level expression. Proc. Natl. Acad. Sci. USA. 1985;82:6783–6787. doi: 10.1073/pnas.82.20.6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yosef I., Manor M., Kiro R., Qimron U. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. USA. 2015;112:7267–7272. doi: 10.1073/pnas.1500107112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yehl K., Lemire S., Yang A.C., Ando H., Mimee M., Torres M.D.T., De La Fuente-Nunez C., Lu T.K. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell. 2019;179:459–469.e9. doi: 10.1016/j.cell.2019.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomason L.C., Sawitzke J.A., Li X., Costantino N., Court D.L. Recombineering: Genetic Engineering in Bacteria Using Homologous Recombination. Curr. Protoc. Mol. Biol. 2014;106:1–16. doi: 10.1002/0471142727.mb0116s106. [DOI] [PubMed] [Google Scholar]
- 29.Li X.-T., Thomason L.C., Sawitzke J.A., Costantino N., Court D.L. Positive and negative selection using the tetA-sacB cassette: Recombineering and P1 transduction in Escherichia coli. Nucleic Acids Res. 2013;41:e204. doi: 10.1093/nar/gkt1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomason L.C., Costantino N., Court D.L. E. coli Genome Manipulation by P1 Transduction. Curr. Protoc. Mol. Biol. 2007;79:1–17. doi: 10.1002/0471142727.mb0117s79. [DOI] [PubMed] [Google Scholar]
- 31.Thomason L.C., Oppenheim A.B., Court D.L. Modifying Bacteriophage λ with Recombineering. Adv. Struct. Saf. Stud. 2009;501:239–251. doi: 10.1007/978-1-60327-164-6_21. [DOI] [PubMed] [Google Scholar]
- 32.Da Silva J.L., Piuri M., Broussard G., Marinelli L.J., Bastos G.M., Hirata R.D., Hatfull G.F., Hirata M. Application of BRED technology to construct recombinant D29 reporter phage expressing EGFP. FEMS Microbiol. Lett. 2013;344:166–172. doi: 10.1111/1574-6968.12171. [DOI] [PubMed] [Google Scholar]
- 33.Sheng Y., Mancino V., Birren B. Transformation of Escherichia coli with large DNA molecules by electroporation. Nucleic Acids Res. 1995;23:1990–1996. doi: 10.1093/nar/23.11.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei D., Sun J., Shi J., Liu P., Hao J. New strategy to improve efficiency for gene replacement in Klebsiella pneumoniae. J. Ind. Microbiol. Biotechnol. 2013;40:523–527. doi: 10.1007/s10295-013-1250-1. [DOI] [PubMed] [Google Scholar]