

Abstract
Bone development occurs through a series of synchronous events that result in the formation of the body scaffold. The repair potential of bone and its surrounding microenvironment — including inflammatory, endothelial and Schwann cells — persists throughout adulthood, enabling restoration of tissue to its homeostatic functional state. The isolation of a single skeletal stem cell population through cell surface markers and the development of single-cell technologies are enabling precise elucidation of cellular activity and fate during bone repair by providing key insights into the mechanisms that maintain and regenerate bone during homeostasis and repair. Increased understanding of bone development, as well as normal and aberrant bone repair, has important therapeutic implications for the treatment of bone disease and ageing-related degeneration.
The musculoskeletal system provides the physical scaffold for the mammalian body. Bones of the human skeleton provide attachment sites for muscles, tendons and ligaments, enabling locomotion. Bones also contain the microenvironments for adult haematopoiesis to occur. The crucial role of bone in mammalian physiology is further highlighted by the body’s unique ability to repair bone through regeneration, restoring it to a fully functional, pre-injury state. Studies have shown that a regulated balance of activity between bone-forming osteoblasts and bone-resorbing osteoclasts — the two main cellular constituents of bone — is responsible for this repair capacity. Previous research on the role of osteoblasts has highlighted the importance of gradients of morphogens, such as bone morphogenetic protein (BMP) and sonic hedgehog (SHH), during bone repair. These morphogen gradients, among others, are also essential during bone development (osteogenesis).
The osteoblast lineage is of great interest in medicine owing to its implications in bone development and disease. Although a certain degree of repair capacity is maintained throughout adulthood, the ability to repair bone diminishes substantially during ageing, potentially leading to osteoporosis. Therefore, this Review examines areas of synergy and diversity in the bone developmental and repair processes. We discuss the cell types involved in osteogenesis and the molecular signalling pathways that are essential for bone formation. This Review also explores the function of critical genes and transcription factors during bone development. Additionally, the functions of different cells and signalling pathways during bone repair are described, as well as their role in bone development. Finally, we assess the dysfunctional cellular and molecular signalling that results in clinical bone disease, thus informing the current state of science and potential gaps in knowledge.
Cell types involved in osteogenesis
The skeletal lineage includes a diverse group of cells that maintain and repair bone during homeostasis and injury, respectively. This lineage of cells includes osteoblasts, osteocytes and chondrocytes1–4. These skeletal cell types are involved mainly in the formation of bone and cartilage, whereas the cells that are responsible for bone resorption, known as osteoclasts, are derived from the haematopoietic lineage. Normal bone homeostasis is maintained through a balance between osteoblast and osteoclast activity; however, during the ageing process, especially in postmenopausal women, osteoclast activity surpasses osteoblast activity, resulting in increased overall bone resorption and weaker bones5.
Osteoblasts
Osteoblasts are the main cells responsible for bone formation. These cells secrete extracellular matrix proteins such as type I collagen, osteopontin, osteocalcin and alkaline phosphatase; multiple osteoblasts interact with one another to create a unit of bone known as an osteon3. The deposition of calcium, in the form of hydroxyapatite, with type I collagen provides structural support to the skeleton3.
The specification of osteoblasts towards the skeletal lineage can be divided into three distinct stages of increasing differentiation: osteoprogenitor, preosteoblast and osteoblast1,2 (FIG. 1). Initially, expression of the transcription factor SOX9 marks the commitment to an osteoprogenitor cell. SOX9 expression also directs cell differentiation towards a chondrocyte cell fate. Chondrocytes are the only cell type found in healthy cartilage, where they produce a cartilaginous matrix consisting of collagen and proteoglycans. The subsequent expression of Runt-related transcription factor 2 (RUNX2) in the osteoprogenitor cell signifies the commitment to a preosteoblast6. During the maturation stage, WNT-β-catenin signalling acts on preosteoblasts to induce the expression of osterix (OSX; also know as SP7), which defines the cell’s differentiation to an osteoblast6. Ultimately, the expression of RUNX2 and OSX marks the commitment to a mature osteoblast.
Fig. 1 |. Bone homeostasis.
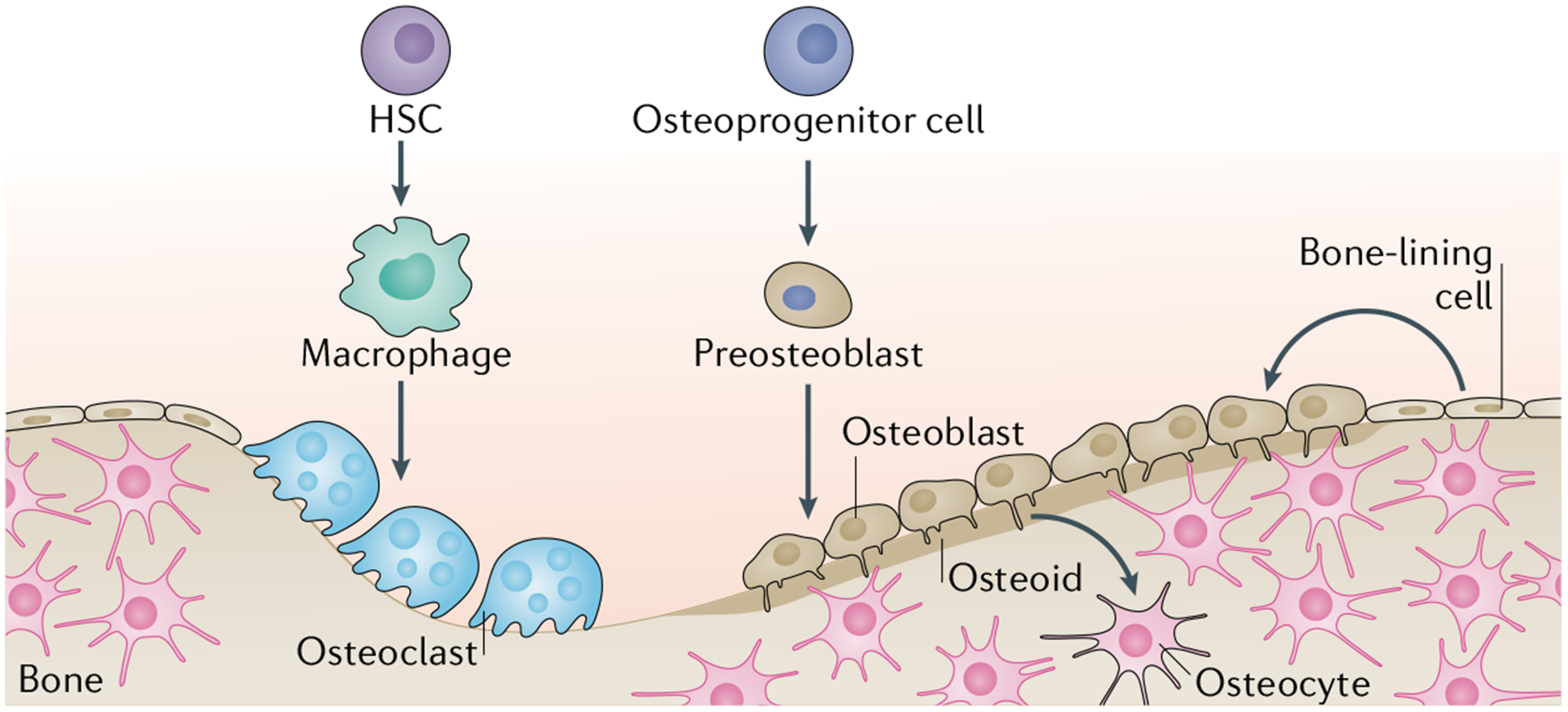
Bone homeostasis is achieved through the activity of osteoblast lineage cells and osteoclast lineage cells. Osteoblast lineage cells such as the osteoid (which is the unmineralized portion of bone matrix) secrete hydroxyapatite and calcium to promote bone mineralization and the formation of osteocytes. Osteoclast lineage cells resorb bone. The balance between the activity of the two cell lineages results in bone homeostasis. HSC, haematopoietic stem cell.
A fraction of these osteoblasts will undergo apoptosis, whereas a subset of osteoblasts secrete extracellular matrix components and embed into the matrix of the bone, forming osteocytes7. Osteocytes account for most of the cells found in mature mineralized bone and coordinate bone maintenance through interactions between osteoblasts and osteoclasts7–9 (FIG. 1). The process of bone maintenance is sensitive to mechanical forces; during mechanical unloading, osteocytes express receptor activator of nuclear factor-κB ligand (RANKL), which promotes bone resorption through the activation of osteoclasts10. Conversely, in response to mechanical loading, osteocytes decrease the expression of Dickkopf-related protein 1 (DKK1) and sclerostin, leading to increased bone formation through activation of WNT-β-catenin signalling in osteoblasts9. Osteocytes respond to hormonal and mechanical signals to tightly control bone remodelling through signalling pathways that are discussed later.
Osteoclasts
Osteoclasts are large multinucleated cells whose primary function is bone resorption. These cells originate from the haematopoietic lineage and differentiate to mature osteoclasts through the interaction of macrophage colony-stimulating factor (M-CSF) and RANKL11 (FIG. 1). M-CSF promotes proliferation of osteoclast precursors, whereas RANKL promotes differentiation of osteoclast precursors to mature osteoclasts11. The expression of RANKL is necessary for osteoclast function as mice lacking RANKL are unable to resorb bone12,13. During bone remodelling, osteoclastogenesis begins with recruitment of osteoclast precursors by osteocytes that express RANKL. The expression of RANKL and M-CSF within the bone marrow compartment initiates the differentiation of osteoclast precursors to osteoclasts, leading to the start of bone remodelling. Under homeostasis, the ratio of bone formation and bone resorption follows a tightly controlled programme to ensure consistency in bone mass. For instance, the release of active transforming growth factor-β (TGFβ) and insulin-like growth factor 1 (IGF1) after resorption of the bone matrix triggers osteoblast differentiation to replace the resorbed matrix with new bone matrix14. An imbalance of this control leads to osteoporosis, the most common disease associated with upregulation of osteoclast activity, which occurs when bone resorption exceeds bone formation (discussed in detail later)14. Conversely, pathologically increased osteoblast activity leads to heterotopic ossification, which is signified by bone formation at an extraskeletal site (discussed in detail later)15.
Stem and progenitor cells
Osteoblast differentiation requires a multitude of steps from a stem cell differentiating into a mature osteoblast. Mesenchymal stem cells (MSCs) and skeletal stem cells (SSCs) have both been shown, separately, to give rise to osteoblasts; however, the relationship between these two stem cell populations has not been precisely determined. In this Review, both populations are considered separately to discuss their role in osteoblast differentiation. Furthermore, throughout osteoblast differentiation, specific genes are expressed to mark their maturation at each stage during development and repair. Therefore, to study the function of each intermediary cell type, transgenic mice are used to understand the lineage specification process. The Cre recombinase (Cre)-loxP system enables conditional gene inactivation by which Cre excises the ‘target’ DNA sequence, corresponding to the gene of interest, that has been flanked by two 34-bp DNA sequences termed the ‘loxP sites’16. TABLE 1 outlines the numerous transgenic mouse models that can be used to study specific cell types during osteoblast differentiation, along with the advantages and disadvantages of each model.
Table 1 |.
Cre models applied for osteoblast lineage studies
| Cre model | Targeted cells | Advantages | Disadvantages | Refs |
|---|---|---|---|---|
| Sox9 | SSCs, osteochondroprogenitor cells | Overcomes embryonic lethality of Sox9 heterozygous mutant mice; expressed throughout the lifetime of mice from the neonatal stage to old age | Potential for off-target Cre-mediated recombination in intestines and pancreas | 203 |
| Grem1 | Osteochondroprogenitor cells, bone stromal cells | Expression of cells concentrated in metaphysis; distinct from mesenchymal stem cells | Potential for off-target Cre-mediated recombination in intestines; low level of fibroblast CFUs (~1%) in in vitro studies forself-renewal | 36 |
| Osx | SSCs, osteoblast progenitor cells | Expression is specific to osteoblasts; expression is associated with invading blood vessels during development | Unable to study precursor cells that have not committed to the osteoblast lineage fate | 204 |
| Prrxl | SSCs | Expressed throughout the lifetime of mice from the neonatal stage to old age; presence of self-renewal properties for bulk cell culture in vitro | Potential off-target effects around the eye | 205 |
| Sost | Mature osteocytes | Used a Sost gene within a BAC clone for better expression; no off-target effects present in osteoblasts or myocytes | Potential off-target effects in haematopoietic cells and osteoclasts | 206 |
| Pthrp (also known as Pthih) | SSCs, osteochondroprogenitor cells, bone stromal cells | Stromal contribution; expressed throughout the lifetime of mice from the neonatal stage to old age | Involved in long-term maintenance of skeletal integrity; predilection for chondrogenesis over osteogenesis | 42 |
| Runx2 | Mature osteoblasts | Can be used to determine gene function in mature osteoblasts; robust expression and recombination | Reporter leakage into cartilage cells | 204 |
| Ctsk | SSCs in the periosteum | Can be used to determine the function of SSCs found in the periosteum | Potential deletion of gene in germline cells | 43 |
| Hoxa11 | SSCs in the periosteum | Labelling of cells from development until adulthood allows cell lineage tracing | Hox11+ SSCs only mark a subset of the entire SSC population | 26,28,207 |
BAC, bacterial artificial chromosome; CFU, colony-forming unit; Cre, Cre recombinase; SSC, skeletal stem cell.
Mesenchymal stem cells
MSCs, first discovered in the bone marrow, are a multipotent, non-haematopoietic stem cell population. MSCs have the capacity to differentiate into mature cell types of mesenchymal tissues such as bone, cartilage and fat17,18. Specifically, seminal work established the osteogenic potential of MSCs through heterotopic transplantation of bone marrow cells19–21. Through the addition of exogenous factors, MSCs have the ability to be directed towards an osteogenic cell differentiation fate17,18. For example, the addition of dexamethasone, β-glycerol phosphate and ascorbate to cultured human MSCs results in osteogenic cell differentiation. The cells form nodules with increased expression of alkaline phosphatase17. Studying the differentiation path of MSCs towards osteoblasts provides insight into key lineage specification moments during the differentiation process.
Transcription factors enable the initiation and promotion of MSC differentiation towards an osteogenic fate. Specifically, RUNX2 and OSX are the main transcription factors whose activation commits the cells to the osteogenic lineage. The expression of RUNX2 is preceded by the upregulation of GLUT1, which results in feedforward regulation22. GLUT1 is a glucose transporter, and its upregulation facilitates increased glucose uptake by cells. This sequence of events indicates that osteoblast differentiation requires a high energy demand, which is met by increased glucose transport and uptake through upregulation of GLUT1. This feedforward regulation explains impaired bone healing in patients with diabetes, as their cells become insensitive to glucose uptake22,23.
MSCs have been shown to maintain expression of genes characteristic of embryonic development, such as the Hox genes. Hox genes encode evolutionarily conserved transcription factors that control skeletal patterning24,25. Hox gene expression is regionally restricted and regulates the morphology of specific vertebral and long bone elements24,25. For example, Hox6 expression is present in the ribs, and Hox11 (also known as Tlx1) expression is present in the zeugopod (radius, ulna, tibia and fibula) of mice postnatally to adulthood25. Furthermore, Hoxa11+ cells from postnatal development of the zeugopod to adulthood display characteristics of osteoblast progenitors26–28. When the function of the Hoxa11 alleles in ulnar fracture healing in adult mice was studied, Hoxa11 mutant mice exhibited perturbed fracture repair28. Specifically, cells of the Hoxa11+ mesenchymal population had decreased osteoblast differentiation potential25,29. Taken together, these studies indicate that mesenchymal cells regionally restricted to the skeleton maintain expression of developmental genes, whose regulation has a role in bone repair.
Skeletal stem cells
The isolation of SSCs in mice and humans on the basis of distinctive immunophenotypic cell surface markers is a relatively new concept30–32 (FIG. 2). Thus, we focus here on the ability to isolate mouse and human skeletal progenitors and their role during repair1,4,33,34. Of note, the term ‘skeletal stem cells’ has evolved over time and continues to be refined.
Fig. 2 |. Skeletal stem cell hierarchy.
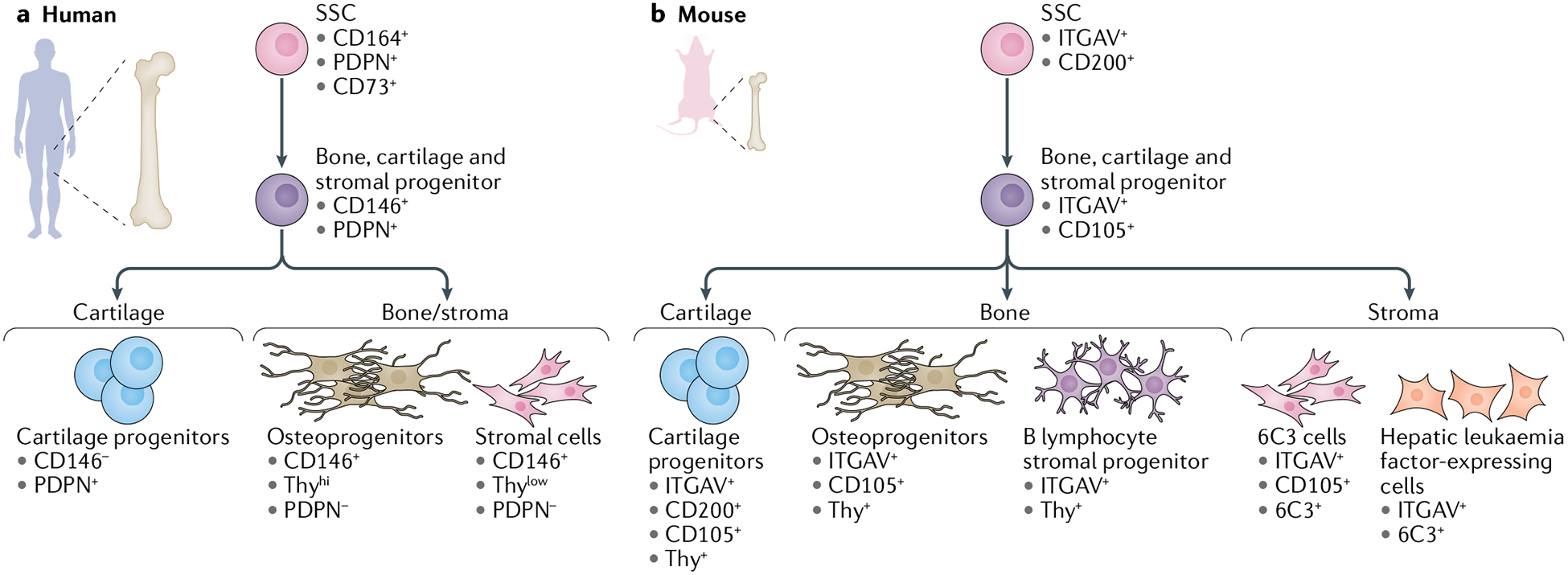
Skeletal stem cells (SSCs) (and their progenitors) in mice and humans can be isolated on the basis of distinctive immunophenotypic cell surface markers. a | Human SSC hierarchy immunophenotypic profile, beginning with a human SSC at the apex, differentiating into a human bone, cartilage and stromal progenitor and into its committed progenitors: human cartilage progenitors, human osteoprogenitors and human stromal cells. b | Mouse SSC immunophenotypic profile, beginning with a mouse SSC at the apex, differentiating into a mouse bone, cartilage and stromal progenitor and into its committed progenitors: mouse cartilage progenitors, mouse osteoprogenitors, mouse B lymphocyte stromal progenitors, mouse 6C3 cells and mouse hepatic leukaemia factor-expressing cells.
A subpopulation of cells isolated from the fetal mouse long bone growth plate has the ability to differentiate into bone, cartilage and bone stroma30. This cell population was identified with use of a ubiquitous actin rainbow reporter system that randomly marks dividing cells and their progeny, allowing fluorescent tracing of ‘clonal’ cell clusters arising from a single parent cell30,35. Clonal regions were observed in the growth plate of long bones, indicating the presence of a restricted progenitor cell. Additionally, isolation of cells from the growth plate was achieved by fluorescence-activated cell sorting. In vitro and in vivo studies showed that CD45−TER119−TIE2−ITGAV+CD200+ single sorted cells have the ability to generate serial colony-forming units, differentiate into bone, cartilage and stromal cells, and support haematopoiesis30.
The fetal human growth plate is of particular interest owing to clonal observations made in the fetal mouse growth plate. With use of a single-cell RNA sequencing approach, analysis revealed a set of 76 gene candidates expressed in mouse SSCs, mouse bone, cartilage and stromal progenitor cells (BCSPs) and their respective human orthologues31. By focusing on the cell surface markers that were specifically enriched in the growth plate and not in the diaphysis, the study authors selected a list of potential markers. Flow analysis and immunofluorescent staining of the growth plate revealed that CD45−CD235a−TIE2−CD31−PDPN+CD73+CD164+ human cells had the capability to generate colony-forming units from single cells in vitro and have the ability to differentiate into bone, cartilage and stroma in the sub-renal space of a mouse. In addition, co-transplantation of haematopoietic cells with CD45−CD235a−TIE2−CD31−PDPN+CD73+CD164+ human cells in irradiated immunodeficient NSG mice supported haematopoiesis31. The role of SSCs has been implicated in development, but to date no studies have successfully demonstrated how SSCs contribute to the developing skeleton36. A transgenic mouse model specifically marking SSCs would enable these studies; however, such a model is not yet available.
The identification of mouse and human SSCs was based on the combination of transgenic mouse models, flow cytometry and single-cell sequencing30,31,37. Although these advancements enabled the discovery of SSCs, it is important to highlight the limitations of the known SSC hierarchy. One of the most important technical and conceptual limitations of mouse and human SSCs is the loss of spatial information on cell isolation, which limits the ability to precisely locate and understand their role in the native bone niche. In addition, the SSCs were identified in the long bones of mice and humans, bones that form through endochondral ossification. However, different skeletal compartments develop through distinct processes; for example, bones of the skull use intramembranous ossification in development and repair38. Whether the identity of the SSC is shared across two different skeleton compartments (cranial versus long bones) is yet to be defined. In addition, the cell surface marker profiles across mice and humans are known to be different. Indeed, mouse and human SSCs might be different cell types that merely happen to behave similarly in the in vitro and in vivo systems studied. Further work is required to help explain the discrepancy between the immunophenotypic cell surface profile across the two species so as to evaluate the relevance of these SSCs to the broader biological context of bone development and repair.
In the context of injury, postnatal SSCs have an important role in enacting a bone repair paradigm to promote healing4. For instance, in response to fracture injury, mouse BCSPs express CD49f and are activated to contribute to bone repair39. By contrast, these fracture-elicited mouse BCSPs are absent in the uninjured bone. The injury-induced activation of SSCs is also captured in humans. With use of a novel human xenograft model, a unicortical injury on human fetal phalanges displayed an elevated frequency of human SSCs and exhibited increased osteogenic potential in vitro31. In diabetic mice, SSCs have decreased expansion and differentiation abilities, resulting in poor fracture healing through the repression of Indian hedgehog (IHH) signalling40. Downregulation of IHH signalling is also recapitulated in femoral and knee specimens of human patients with diabetes40. However, this effect can be rescued in mice with the exogenous delivery of IHH or SHH, which successfully improves bone healing by inducing SSC expansion40.
A unique SSC-driven repair process has also been observed in the craniofacial region, a skeletal component that heals through intramembranous ossification. During mandibular distraction osteogenesis in mice, focal adhesion kinase (FAK)-responsive SSCs acquire an embryonic neural crest identity and promote regeneration41. With inhibition of FAK, SSC-driven bone regrowth is severely impaired, resulting in a fibrous intermediate in place of bone41. These studies highlight the importance of SSCs in postnatal bone repair. However, no studies to date have successfully demonstrated how SSCs contribute to the developing skeleton36. Further studies are required to elucidate their specific roles during embryonic development.
Many groups have made significant progress in understanding the role of skeletal progenitors in bone remodelling and maintenance by combining two approaches: the Cre-loxP system (TABLE 1), to enable lineage tracing, and fluorescence-activated cell sorting, to select for the SSC population on the basis of its cell surface marker profile. These studies have identified compartment-specific skeletal progenitors and highlight the ability to lineage trace these skeletal progenitors through development42,43. Studies building on the capacity to lineage-trace skeletal progenitors will help elucidate their role during bone development. In addition, future advancements should address the rigor of methods for isolating cells from freshly harvested tissue, which can be subject to the loss of fragile cell types. Improved isolation methods, preventing the deterioration of sparse cell populations and surface antigens that might be necessary for understanding development, might reveal additional subpopulations. Coupled with emerging novel single-cell transcriptional analyses and assay for transposase-accessible chromatin sequencing methods, the relationships of these cell populations to existing skeletal progenitors can be established44,45.
Regulation of cell lineage specification
Skeletal formation occurs through orchestrated, temporal events enabling the bone to form and ossify over the period of development. The expression of transcription factors at certain stages in bone development guides the progenitor cells to differentiate towards an osteoblast fate. Along with the genetic changes, signalling pathways enable the cells to communicate with their environment. The external cues given by the signalling pathways synchronize cellular differentiation. The transcription factors and signalling pathways necessary for bone development become activated as bone regrows after an injury. This section discusses the essential transcription factors and signalling pathways for osteoblast differentiation in the context of bone development and injury.
Embryonic origins of bones
During skeletal development, osteoblasts arise from multiple embryonic germ layers. Osteoblasts of the craniofacial region originate from neural crest cells, which are derived from the neural ectoderm38,46. The neural ectoderm develops into the tissues of the craniofacial region, including the skull, dentin of teeth and bones of the face. By contrast, the long bones of the skeleton originate from paraxial mesoderm (somites) (which gives rise to the axial skeleton) and lateral plate mesoderm (which gives rise to the appendicular skeleton)38.
Bone formation during embryogenesis occurs in two distinct processes: intramembranous ossification or endochondral ossification. Intramembranous ossification begins with the condensation of mesenchymal populations that directly differentiate into bone38. The flat bones of the body, including the skull, mandible, maxilla and clavicle, originate from this process. By contrast, endochondral ossification is an intricate process signified by the development of bone through a cartilage intermediate38,47. During endochondral ossification, the cells in the middle of the mesenchymal condensations develop into chondrocytes, which begin to secrete cartilage matrix. The cells surrounding the newly differentiated chondrocytes form the perichondrium, which defines the border of the developing skeleton38. The cells defining the border withdraw from the cell cycle and undergo hypertrophy, prompting differentiation of osteoblasts from the perichondrium. Chondrocyte hypertrophy is essential to activate osteoblast differentiation. Blood vessels begin to infiltrate the hypertrophic cartilage, which triggers osteoblast differentiation and eventual formation of the bone marrow cavity. The infiltration of blood vessels serves multiple functions: it facilitates recruitment of chondro-resorptive cells (which degrade existing cartilage) and osteoprogenitors (which promote osteoblastogenesis) and enables perichondrial cells to enter the developing bone marrow cavity47,48. Blood vessel infiltration into the hypertrophic cartilage is signified by the development of a primary ossification centre.
The primary ossification centre continues to expand during embryonic development and is eventually succeeded by the formation of secondary ossification centres. The secondary ossification centres develop in the growing ends of the bones (epiphyses)49. The compartments of the growing bones can be distinguished by their locations within the bone. Between the epiphysis and the diaphysis (midshaft) is the metaphysis, which contains the epiphyseal growth plate and can be further categorized into different zones of development38,49 (FIG. 3). The reserve zone contains quiescent chondrocytes. The proliferative zone is a site of rapid replication of chondrocytes; as these cells divide, they orient themselves parallel to the growing bone, thus becoming arranged in a columnar fashion38,47. As the chondrocytes migrate further away from the epiphysis, they halt proliferation and begin to enlarge (hypertrophy) to contribute to skeletal growth. Most of these hypertrophic chondrocytes undergo apoptosis, followed by calcification of the cartilaginous matrix, while the remaining chondrocytes become osteoblasts and contribute to the growing skeleton47,48. The invasion of vessels into the bone cavity recruits osteoclasts to resorb the calcified cartilage matrix and osteoblasts to lay down new mineralized bone tissue, promoting the ossification of the newly formed bone38. Postnatal bone growth continues into adolescence until bone remodelling is complete and maturity is achieved, marked by the closure of the growth plate-containing metaphysis and fusion of the epiphysis and diaphysis.
Fig. 3 |. Long bone anatomy.
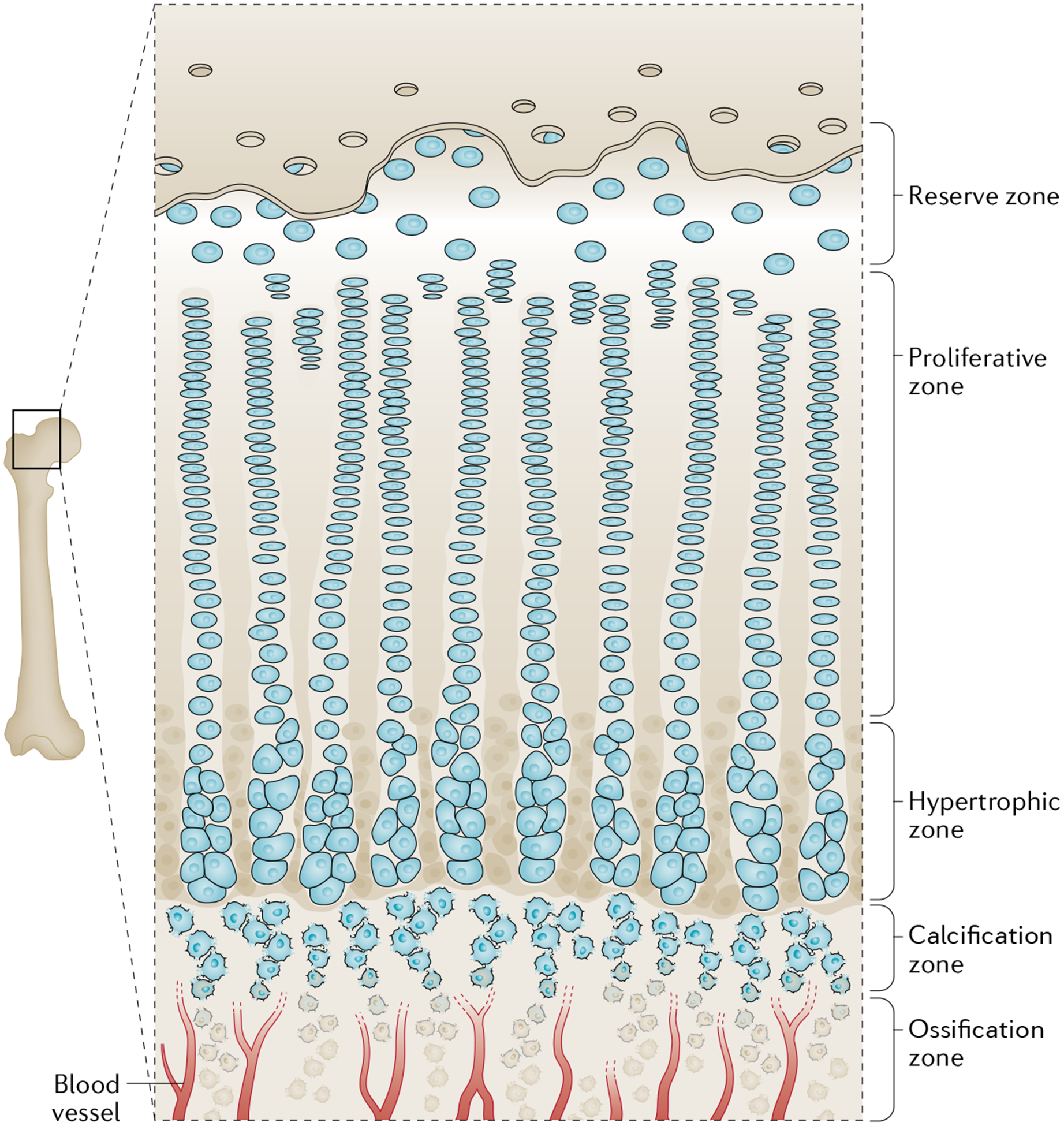
The metaphysis contains the epiphyseal growth plate, the site of new longitudinal bone growth. This area can be categorized into five zones: the reserve, proliferative, hypertrophy, calcification and ossification zones. The reserve zone contains quiescent chondrocytes found towards the epiphyseal end of the bone. The proliferative zone contains chondrocytes that undergo rapid proliferation. The hypertrophy zone contains chondrocytes that stop proliferating and begin to undergo rapid growth. The calcified zone contains cells that begin to undergo apoptosis and their matrix begins to calcify. The ossification zone contains mature and terminally committed osteoblasts that help lay down mineralized bone.
Transcriptional regulation
The activation of transcription factors at specific moments during differentiation provides the necessary cues to specify the functions of the osteoprogenitor as the cell commits to an osteoblast. This section discusses the essential transcription factors for osteoblast differentiation such as SOX9, RUNX2, OSX and activated transcription factor 4 (ATF4). Furthermore, TABLE 2 highlights the transcription factors and protein-coding genes involved throughout the differentiation process. These specific transcription factors and protein-coding genes were chosen because they interact with the essential transcription factors discussed in the following sections to ensure proper osteoblast differentiation.
Table 2 |.
Transcription factors and protein-coding genes involved in osteoblast differentiation
| Transcription factor or gene | Effect | Mode of action | Refs |
|---|---|---|---|
| Transcription factor | |||
| AP-1 | Regulates osteoblast homeostasis | Enhances RUNX2 expression | 76 |
| FIAT | Reduces osteoblast activity | Antagonizes ATF4 function | 208 |
| FOXO | Increases osteoblast numbers | Interacts with ATF4 | 209 |
| GLI1 | Stimulates osteoblast differentiation | Enhances RUNX2 activity | 92 |
| GLI2 | Stimulates osteoblast differentiation | Enhances RUNX2 activity | 210 |
| GLI3 | Suppresses osteoblast differentiation | Inhibits DNA binding of RUNX2 | 211 |
| HAND2 | Inhibits intramembranous osteoblast differentiation in the mandible | Inhibits RUNX2 | 212 |
| HOX11 (also known as TLX1) | Globally patterns the appendicular skeleton | Enhances RUNX2 expression | 28 |
| HOXA2 | Suppresses osteoblast differentiation | Downregulates RUNX2 expression | 213 |
| OSX | Stimulates osteoblast differentiation | Interacts with MSX2 | 65 |
| RUNX2 | Stimulates osteoblast differentiation | Acts as a scaffold regulatory factor involved in skeletal gene expression | 214 |
| TAZ | Stimulates osteoblast differentiation | RUNX2 co-activator | 215 |
| TWIST | Inhibits osteoblast differentiation | Inhibits RUNX2 | 216 |
| Protein-coding gene | |||
| BGLAP | Regulates bone remodelling | Binds to apatite and calcium | 217 |
| COL1A1 | Stimulates osteoblast differentiation | Binds to calcium ion | 218 |
| DKK1 | Negative regulator of bone growth | Inhibits LRP5 or LRP6 interaction | 219 |
| PTK2 | Stimulates osteoblast differentiation | Integrin signal transduction | 41,220 |
| SOST | Negative regulator of bone growth | Inhibition of canonical WNT signalling | 221 |
AP-1, activator protein 1; ATF4, activated transcription factor 4; FIAT, factor inhibiting ATF4-mediated transcription; FOXO, forkhead box protein O; LRP, low-density lipoprotein receptor-related protein; OSX, osterix; RUNX2, Runt-related transcription factor 2.
SOX9.
SOX9, a member of the SRY-related high mobility group box-containing (SOX) transcription factor family, controls skeletal development during endochondral Ossification50. Studies in mice have emphasized the importance of SOX9 as the integral transcription factor for chondrogenic commitment49, which is essential for establishing the intermediate primordium for eventual ossification by osteoblasts. SOX9 also drives cartilage differentiation by activating chondrogenic genes such as Col2a1 (REFS50,51). Mouse embryo chimaeras consisting of Sox9−/− and Sox9+/+ pluripotent cells were revealed to contain only Sox9+/+ cells in the cartilage condensation52. Furthermore, loss of Sox9 perturbs cartilage differentiation and inactivates chondrogenic marker expression, eventually causing cell death53,54.
In humans, heterozygous mutations within the SOX9 locus lead to the development of an early lethal skeletal syndrome named ‘campomelic dysplasia’55. Individuals with campomelic dysplasia are born with skeletal defects such as micrognathia, dwarfism and other skeletal malformations56. Normally, the expression of SOX9 throughout adulthood enables continual maintenance of developing cartilage and bone growth51,52.
RUNX2.
RUNX2 is a transcription factor that drives skeletal development (TABLE 2). RUNX2 binds to core-binding factor subunit-β to form a heterodimer that regulates the expression of osteoblast genes such as those encoding osteopontin (Spp1), bone sialoprotein 2 (Ibsp) and osteocalcin 2 (Bglap2)57–59. In mice, Runx2 is strongly expressed in immature osteoblasts but expression decreases in mature osteoblasts57. Runx2 deletion in mice results in complete osteoblast absence and loss of Spp1, Ibsp and Bglap2 expression60–62. Therefore, RUNX2 activity during early skeletal development is essential for osteoblast differentiation.
By contrast, the role of RUNX2 in mature osteoblasts is not well understood and requires further investigation. Conditional Runx2-knockout studies under the mature osteoblast promoter Col1a1 led to conflicting results. Deletion of the Runt domain in exon 4 led to no phenotypic changes62, whereas truncation of Runx2 through exon 8 deletion resulted in reduced bone formation63. These findings suggest that Runx2 maintains osteoblasts in the immature state, thereby arresting osteoblast maturation and the completion of bone formation63. Furthermore, these findings are consistent with the observed reduction of Runx2 expression in mature osteoblasts64.
OSX.
OSX, an osteoblast transcription factor, is required for normal skeletal development (TABLE 2). OSX triggers differentiation of immature osteoblasts to mature osteoblasts and eventually into osteocytes65–67. In mice, as Osx expression increases during osteoblast maturation, Runx2 expression decreases simultaneously68. The deletion of Osx in mouse embryos leads to failure of bone formation and lack of expression of osteoblast genes such as Sparc (which encodes osteonectin) and Spp1 (REF.68). Postnatal inactivation of Osx leads to perturbed bone formation with the absence of trabeculae and more porous cortical bone, abnormal cartilage accumulation, and osteocyte dysfunction along with a decrease in osteocyte gene expression such as expression of Sost and Dkk1 (REFS69,70) (TABLE 2).
In bones undergoing endochondral ossification, the heterogeneous population of immature osteoblasts, osteoclasts and blood vessels is able to invade the cartilage primordial matrix; however, without the presence of OSX, ossification does not occur68. Furthermore, the cells in the condensed mesenchyme of the membranous skeletal elements are unable to differentiate into osteoblasts without the expression of Osx68. Runx2 expression is still present in skeleton-forming cells even in the absence of Osx expression. Therefore, OSX acts downstream of RUNX2, indicating that during the lineage specification of osteoblasts, RUNX2 promotes immature osteoblast differentiation, whereas OSX is required for mature osteoblast differentiation.
ATF4.
ATF4 is a leucine zipper-containing transcription factor and a component of the cAMP response element-binding protein (CREB) family71,72. Originally, ATF4 was identified as a nuclear binding protein enriched in osteoblasts that was required for the activation of BGLAP2 and terminal differentiation of osteoblasts. Mutations in ATF4 result in skeletal abnormalities, such as decreased bone mass associated with Coffin-Lowry syndrome73. Furthermore, an Atf4-knockout mouse model exhibited decreased bone mass and bone strength resulting from delayed formation of bone trabeculae71,74. This knockout model has provided insights into the role of ATF4 in osteoblast differentiation and bone homeostasis71,74.
The regulatory effects of ATF4 on bone homeostasis occur through two mechanisms. First, ATF4 regulates osteoblast differentiation through post-transcriptional modification, enabling the synthesis of type I collagen, an essential role of the osteoblast74. Second, ATF4 interacts cooperatively with RUNX2 by forming a complex with SATB2, enhancing the activity of both transcription factors and promoting osteogenesis75. Furthermore, ATF4 activity is suppressed by factor inhibiting ATF4-mediated transcription (FIAT). ATF4 and FIAT (also known as TXLNG) are expressed in osteoblasts72. The negative feedback between ATF4 and FIAT regulates activation of BGLAP2: ATF4 binds to osteocalcin-specific element 1, leading to BGLAP2 expression and further extending the role of ATF4 in bone homeostasis (TABLE 2).
AP-1.
The activator protein 1 (AP-1) transcription factor complex has been implicated as an important regulator in bone development, particularly for osteoblast homeostasis (TABLE 2). AP-1 activity can be induced through TGFβ, parathyroid hormone and 1,25-dihydroxyvitamin D76. The AP-1 complex is composed of a variety of members from the FOS, JUN and ATF families76. Mutations of the genes encoding these transcription factors result in skeletal defects. Specifically, mutations in Junb result in osteopenia owing to osteoblast defects; however, mutations in Jund result in increased bone mass from increased bone formation76,77. Furthermore, the AP-1 complex preferentially binds to an osteoblast-specific enhancer, Ce1, in the Runx2 promoter78. For instance, Fosb in the AP-1 complex binds to Ce1, and ΔFosb, the naturally occurring short isoform of Fosb, causes the development of osteosclerosis owing to increased osteoblast differentiation78. Overexpression of FOS-related antigen 1 (FRA1), a FOS-related protein encoded by the FOS target gene Fosl1, also results in osteosclerosis79,80. The increase in FRA1 expression accelerated the differentiation of osteoprogenitors into osteoblasts, resulting in an increased number of active osteoblasts79. Therefore, the regulation of the JUN, FOS and ATF factors within the AP-1 complex functions to modulate the activity of osteoblasts in bone formation and homeostasis.
Signalling pathways in bone development
Differentiation of the osteoblast lineage (discussed earlier) is regulated by various signalling pathways (FIG. 4). Although the signalling pathways are discussed separately in this section, the pathways function in a coordinated manner to ensure appropriate bone development and repair.
Fig. 4 |. Developmental signalling pathways regulating osteoblast differentiation.
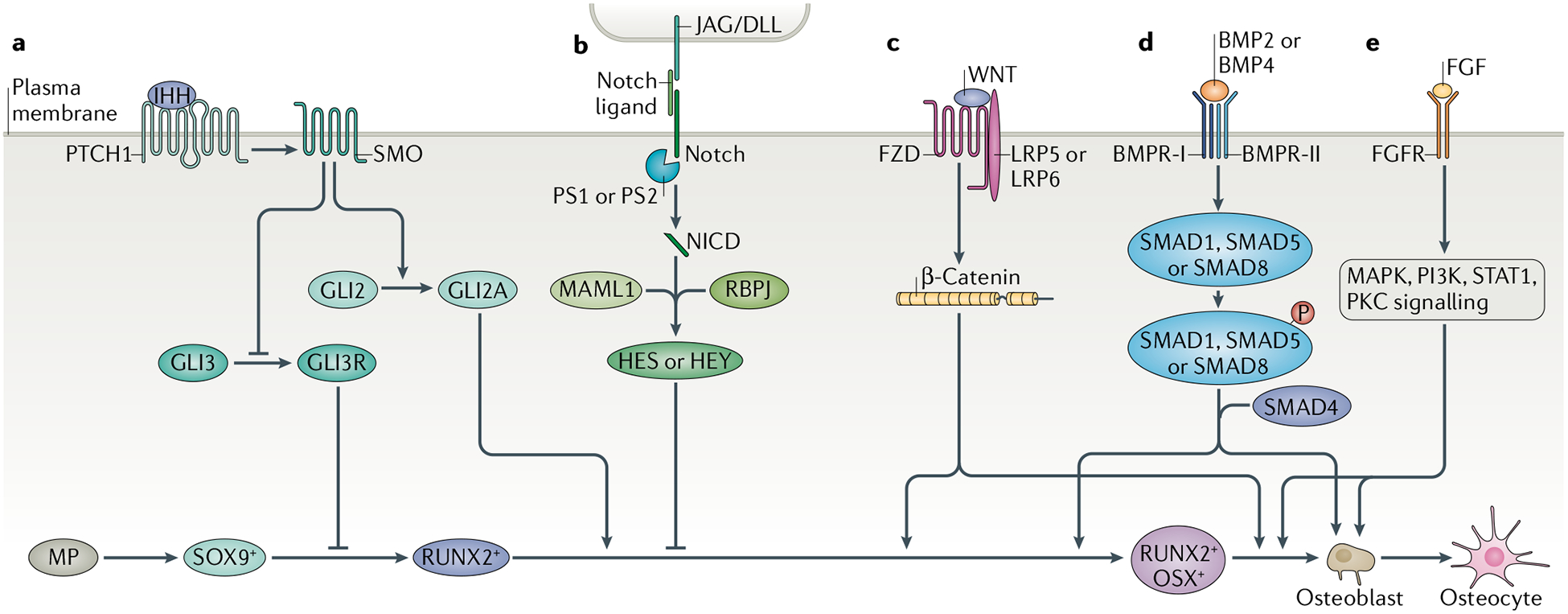
Various signalling pathways function in a coordinated manner to ensure appropriate bone development and repair. a | Mesenchymal progenitors (MP) are initially marked by SOX9+ expression committing them to the osteochondroprogenitor lineage. Indian hedgehog (IHH) binds to Smoothened homologue (SMO) to prevent the cleavage of GLI3 to GLI3 repressor (GLI3R) and to activate GLI2 activator (GLI2A), thus leading to expression of SOX9 and Runt-related transcription factor 2 (RUNX2) in osteochondroprogenitor cells. b | Notch signalling is a negative regulator of osteoblast differentiation. Notch binding to Jagged (JAG) or Delta-like protein (DLL) causes proteolytic cleavage of Notch, allowing Notch intracellular domain (NCID) to interact with RBPJ and Mastermind-like protein 1 (MAML1) to affect the downstream targets Hairy and Enhancer of Split (HES) and HES-related with YRPW motif (HEY), leading to the inhibition of osteoblast differentiation. c | Canonical WNT signalling acts as a positive regulator of osteoblast differentiation. WNT ligand binding to low-density lipoprotein receptor-related protein (LRP5) or LRP6 and Frizzled (FZD) leads to the accumulation of β-catenin, thus allowing its translocation to the nucleus to affect gene expression, including increasing the expression of RUNX2 and osterix (OSX), which marks the commitment to mature osteoblasts. d | Bone morphogenetic protein (BMP) signalling induces osteoblast differentiation. Binding of BMP2 or BMP4 leads to the phosphorylation of SMAD1, SMAD5 or SMAD8. They form a complex with SMAD4 and then enter the nucleus to control gene expression, allowing the transition of RUNX2+OSX+ cells to mature osteoblasts. e | Fibroblast growth factor (FGF) binds to cell surface tyrosine kinase FGF receptors (FGFR1-FGFR4), leading to a cascade of intracellular signalling events. FGF signalling controls preosteoblast proliferation, osteoblast differentiation and the function of mature osteoblasts. BMPR-I, bone morphogenetic protein receptor type 1; BMPR-II, bone morphogenetic protein receptor type 2; PKC, protein kinase C; PS, presenilin; PTCH1, Patched homologue 1.
Hedgehog signalling.
In 1980, Hedgehog (HH) was identified as an important gene for creating the differences between the development of the anterior and posterior sections of individual body segments in Drosophila81. The control of the anterior-posterior body axis by HH has been shown to be conserved throughout multiple species, including mammals82,83.
HH is a secreted protein that functions as a morphogen through a paracrine signalling mechanism. The pathway consists of three homologues — SHH, IHH and desert hedgehog (DHH) — with the SHH pathway being the best studied84. Patched homologue 1 (PTCH1) and Smoothened homologue (SMO) are transmembrane proteins that are the receptors for the HH signalling pathway. In the absence of HH, PTCH inhibits SMO85,86. When HH binds to PTCH, its inhibition of SMO is removed, activating downstream transcriptional gene targets such as the GLI family of transcription factors84–86 (FIG. 4a).
In limb development, SHH functions as an important morphogen in anteroposterior patterning of the developing limb bud. In addition to SHH itself, GLI2 and GLI3, the transcription factor targets of SHH signalling, are required for normal skeletal development87,88. Absence of either Gli2 or Gli3 results in abnormal skeletal development and embryonic lethality in mice; GLI1 is not necessary for skeletal development89–91 but it does, however, act synergistically with GLI2 and GLI3 in osteogenesis. In an in vitro system, overexpression of Gli1 upregulated the transcription of early osteogenesis-related genes such as Runx2 and Osx92.
Along with its importance in development, the HH pathway also has a vital role in bone formation during endochondral ossification. Osteoblast differentiation requires the regulation of Gli1 or Gli2 through HH signalling93. Furthermore, osteoblast differentiation is lost in the absence of IHH signalling, resulting in incomplete endochondral ossification and incomplete subsequent bone formation94,95. Specifically, downregulation of IHH signalling decreases the osteoblast differentiation potential of mouse SSCs, which further results in poor fracture healing40. Additionally, microarray analysis indicates the upregulation of Ptch1 in mouse BCSPs during the initial 3 days of femoral fracture healing39. Therefore, HH signalling is important for osteoblast differentiation of mouse SSCs enabling the cells to undergo endochondral ossification and repair bone. Postnatally, osteoblasts express IHH, with the transcriptional target GLI2 having a key role in activating osteoblast-specific genes. HH signalling targets the activation of the transcription factor GLI2, which in turn activates RUNX2, the master regulator of osteoblast differentiation93.
Furthermore, HH signalling interacts with the WNT and BMP pathways to regulate bone formation. The WNT pathway is required downstream of HH signalling to regulate osteoblast differentiation; additionally, the presence of β-catenin is required for HH signalling96,97. Finally, HH signalling interacts with the BMP pathway during endochondral ossification. The BMP pathway acts downstream of HH signalling to regulate osteoblast differentiation from perichondrial cells98.
Notch signalling.
Notch signalling requires direct cell-to-cell contact. Notch receptors (NOTCH1, NOTCH2, NOTCH3 and NOTCH4) expressed on cells interact with the Jagged (JAG1 and JAG2) and Delta-like (DLL1, DLL3 and DLL4) families to initiate intracellular signalling99. This binding leads to proteolytic cleavage of the γ-secretase complex with the help of presenilin 1 (PS1) or PS2, which releases Notch intracellular domain (NCID)100. This domain translocates to the nucleus to interact Hairless LAG-2 family transcription factor RBPJ to initiate the Mastermind-like protein 1 (MAML1) activator complex99,100. The activator complex induces expression of Notch target genes, including the genes encoding Hairy and Enhancer of Split (HES) and HES-related with YRPW motif (HEY)99–101 (FIG. 4b).
Genetic studies in mice have revealed the importance of Notch signalling during skeletal development. In Prrx1Cre mice, inactivation of PS1, NOTCH1 and NOTCH2 in the developing limb bud resulted in an increase in the abundance of hypertrophic chondrocytes in the growth plate, leading to increased mass of the trabecular bone101,102. Notch enhances cell replication of the osteoblast lineage and suppresses osteoblast differentiation101,102. A possible explanation for the mechanism of action is an effect secondary to the inhibition of RUNX2 function100,103. NCID can directly interact with Runx2 and inhibit the transactivation of Bglap2100,103. HES and HEY proteins also inhibit RUNX2 function100,103. Increased expression of Notch1 under an osteoblast-specific promoter (Osx) hindered osteoblast lineage cells from differentiating to specialized osteoblasts, leading to osteopenia in mice104. Conversely, inactivation of Notch1 and Notch2 under the control of the Osx promoter led to an increased number of mature osteoblasts and increased cancellous bone volume105. These studies suggest the importance of Notch signalling in osteoblast lineage differentiation.
Finally, the Notch signalling pathway has been shown to be upregulated in mouse SSCs after mandibular distraction41. One hypothesis, based on previous studies of Notch signalling and osteoblast differentiation, posits that the upregulation of Notch signalling on injury might allow mouse SSCs to replicate and ensure the appropriate number of osteoblast progenitors are present for bone repair before osteoblast differentiation. However, additional research is needed to determine the relationship between Notch signalling and SSCs.
Canonical WNT signalling.
WNT ligands communicate by attaching to a cell surface receptor, eliciting an intracellular signalling cascade106. The canonical WNT pathway is characterized by the stabilization of β-catenin on activation by WNT ligands through their binding to the receptor Frizzled and low-density lipoprotein receptor-related protein 5 (LRP5) or LRP6 (REFS107–109). Binding of WNT ligands leads to accumulation of β-catenin in the cytoplasm, thus enabling it to translocate to the nucleus. In the nucleus, β-catenin associates with T cell factor (TCF)/lymphoid enhancing factor (LEF) to stimulate transcriptional activity (FIG. 4c).
Mutations causing loss of function of LRP5 lead to low bone mass and the development of osteoporosis-pseudoglioma syndrome, an autosomal recessive disorder characterized by osteoporosis and eye abnormalities110. By contrast, mutations that increase the function of LRP5 have been linked to increased bone mass in humans111,112. Although the mechanism by which LRP5 (or LRP6) and WNT ligands regulate bone mass has not been elucidated fully, both components are required to stimulate osteoblast proliferation and osteoblast maturation113. When liposomal vesicles of purified WNT3A were delivered to the site of a fracture in mice, rapid healing occurred owing to increased proliferation and earlier differentiation of osteoblast lineage cells114.
WNT antagonists have been shown to be upregulated in mouse SSCs and downstream progenitors after injury, probably to ensure the correct timing for osteoblast differentiation in the bone repair process. For example, microarray analysis of mouse BCSPs showed the upregulation of DKK1, a WNT antagonist, 3 days after a femoral fracture39. However, DKK1 was downregulated 7 days after the injury, indicating the temporality of WNT signalling for osteoblast differentiation of mouse SSCs39. Furthermore, under normal conditions, SOST, which encodes sclerostin and is upregulated in human SSCs, binds to LRPs, suppressing receptor and WNT activity8,115. Therefore, SOST acts as a negative regulator of bone formation by opposing the effects of LRP activation to ensure the bone does not overgrow. Missense mutations in the Lrp5 gene cause the receptor to have low binding affinity for sclerostin and DKK1, thus leading to accumulation of bone mass in mice exhibiting phenotypes similar to the human disease known as autosomal dominant high bone mass116. Knockout of the Lrp5 gene in mice led to decreased bone mass reminiscent of the human osteoporotic phenotype111,117. Additionally, haploinsufficiency of Lrp6 in Lrp5−/− mice results in reduced bone mass118. Whereas the lower bone mass in Lrp5-knockout mice is attributed to reduced bone formation, the lower bone mass observed in Lrp6 hypomorphic mice is due to increased bone resorption119.
Finally, the canonical WNT signalling pathway has an important role in interacting with other signalling pathways. Activation of the Notch pathway inhibits the canonical WNT pathway, causing an arrest in osteoblast differentiation120. As outlined earlier, Notch pathway activation increases osteoprogenitor proliferation while suppressing differentiation into mature osteoblasts. However, the canonical WNT pathway enables osteoblast lineage cells to progress to the final, committed osteoblast. Therefore, the balance between Notch signalling and canonical WNT signalling determines the commitment of lineage cells to osteoblasts. Additionally, BMP signalling complements WNT-induced osteogenic differentiation as both signalling pathways share common targets, such as connective tissue growth factor121–124. For example, osteogenic effects are enhanced in the presence of WNT3A and BMP9; furthermore, the induction of ectopic bone formation by BMP2 is antagonized through Dkk1 overexpression and the conditional knockout of Ctnnb1 (which encodes β-catenin)125,126. Furthermore, BMP2 might promote osteogenic differentiation through increased expression of Lrp5 and stabilization of β-catenin127.
BMP signalling.
The TGFß family consists of a large group of structurally similar proteins. TGFß is a multi-functional peptide that regulates cellular processes such as proliferation and differentiation. These proteins form TGFß complexes, which interact with cell surface serine/threonine-specific protein kinase receptors128. Subsequent intracellular signals are propagated by SMAD proteins, ultimately resulting in changes in target gene expression128. The TGFß superfamily contains the TGFß subfamily, the activin subfamily and BMPs. Specifically, BMP2 and BMP4 have been shown to be essential in osteoblast differentiation (FIG. 4d).
BMP binds to BMP receptor (BMPR) located on the cell surface. BMPR is a transmembrane protein belonging to the serine/threonine kinase family, and the protein consists of a type 1 receptor and a type 2 receptor129,130. The interaction between BMP and BMPR stabilizes the complex, resulting in the type 2 receptor phosphorylating the type 1 receptor129,131,132. The phosphorylated type 1 receptors result in propagation of the intracellular signal through the phosphorylation of the receptor-regulated SMAD family of proteins (SMAD1, SMAD5 and SMAD8)129,130. The phosphorylated SMAD protein interacts with SMAD4, which contains a nuclear import sequence, enabling the protein to be localized to the nucleus and regulate gene expression129,131,132.
Genetic studies have determined the role of Bmp2 in promoting osteoblast differentiation by targeting Runx2 downstream. Conditional knockout of Bmp2 resulted in developmental deficiencies affecting prenatal and postnatal bone formation. Specifically, bone mass and trabeculation were significantly decreased in Bmp2-knockout mice133. Studies have shown that a loss of Bmp2 alone does not stop limb osteoblast differentiation; however, a combinational knockout of Bmp2 and Bmp4 or Bmp2 and Bmp7 resulted in abnormal osteoblast differentiation133. Although Bmp2 might be dispensable for osteoblast formation during development, it is required for fracture repair. During the healing period, Bmp2-deficient mice expressed lower expression levels of Runx2, Osx, Bglap2 and Col1a1, genes which are normally upregulated during bone formation and repair134. Both Bmp2 and Smad3 were upregulated in mouse SSCs after mandibular fracture41. The gene regulation changes support the necessity of BMP signalling and SMAD protein signal transduction for osteoblast differentiation and subsequent bone repair. Furthermore, analysis of the transcriptional expression of Bmp2 in mouse SSCs and their downstream progenitors, without any prior injury, showed a decrease in Bmp2 expression from the mouse SSC to a committed osteoblast30.
As the BMP signalling cascade contains many components, another target of interest to modulate BMP signalling is BMPR type 1A (BMPR1A; also known as ALK3). BMPR1A is present on preosteoblasts and osteoblasts, and constitutively active Bmpr1a has been shown to have a role in cell differentiation135,136. In the context of osteoblasts, Bmpr1a has been shown to have a dual role in restricting preosteoblast proliferation while stimulating osteoblast activity135,137. By deletion of Bmpr1a, an increase in trabecular bone formation was observed owing to an increase in the number of osteoblasts resulting from the hyperproliferation of preosteoblasts136,138. Furthermore, BMPR1A has a key role in determining the mechanical properties of bone. The presence of the receptor is essential for bone quality and mechanical integrity as it leads to increased collagen crosslink maturation in osteoblasts139.
FGF signalling.
Fibroblast growth factors (FGFs) are a family of cell signalling proteins that are crucial for normal development. The growth factors’ activating cell surface receptors are called ‘fibroblast growth factor receptors’ (FGFRs)140. The binding of FGF to the extracellular ligand-binding domain of FGFR results in the phosphorylation of tyrosine residues in the FGFR intracellular domain. Subsequently, this phosphorylation results in the activation of downstream signalling pathways such as the Ras-MAPK, phophoinositide 3-kinase-AKT and protein kinase C pathways141,142. FGF signalling involves crucial elements of normal development, such as mesoderm induction, neural development and limb development. FGF2 is expressed in the developing limb bud and contributes to limb growth and patterning142. Overexpression of human FGF2 in mice results in dwarfism with shortened and flattened long bones143. Deletion of Fgf2 in mice leads to decreased bone mass, bone formation and bone mineralization144 (FIG. 4e).
In addition to FGF2, both FGF8 and FGF10 have a role in limb development. Targeted Fgf8 deletion results in failed limb development in mice, with a substantial reduction in limb bud size and hypoplasia of skeletal elements145,146. A positive-feedback loop exists between FGF8 and FGF10, which is essential for limb development146. Similarly to Fgf8 deletion in mice, Fgf10-knockout mice show a complete lack of forelimb and hindlimb development; limb bud initiation still occurs in these mice but limb bud outgrowth is impaired147–149. FGF8 is also expressed in osteoblasts, specifically in the cortical bone of the calvarium150,151. Addition of FGF8 to bone cell culture medium results in increased differentiation of the cells into osteoblasts and increased in vitro bone formation152. After mandibular distraction, Fgf8 expression increases during the healing process41. Initially, Fgf8 expression in mouse SSCs is upregulated; however, as the cells differentiate into osteoblasts, Fgf8 expression becomes downregulated41. Therefore, FGF8 might regulate the ability of cells to differentiate into osteoblasts and their subsequent osteoblastic activity.
Along with understanding the role of FGF in skeletal development and homeostasis, elucidating the role of FGFRs in these same contexts can give further insight into the role of the signalling pathway. FGFR1 has an important role in limb development. Disruption of FGFR1 in mice at the early stage of development, before limb mesenchyme thickening, results in a severe defect characterized by malformation of the apical ectodermal ridge153. In conditional Fgfr1-knockout mice, in which the disruption of Fgfr1 can be temporally controlled and confined to the late stage of development, the mice show a diminished albeit functional appendicular skeleton and malformed forelimbs and hindlimbs154. FGFR1 also acts as a negative regulator of long bone growth. Conditional deletion of Fgfr1 in osteochondroprogenitor cell lineages leads to an increased height of the hypertrophic zone owing to delayed degradation or maturation of the hypertrophic chondrocytes during endochondral ossification155,156. Furthermore, Fgfr1 deletion studies in mice show an increase in osteoblast proliferation and delay in differentiation and matrix mineralization. Finally, Fgfr1 expression increases during the healing process after mandibular distraction41. Fgfr1 expression is downregulated in mouse SSCs; however, expression levels increase as the cells became more committed to the osteoblast fate41. Therefore, the increase in Fgfr1 expression during osteoblast lineage commitment further indicates the importance of the receptor in differentiation and matrix mineralization to form bone.
FGFR2 is another important receptor in the FGF signalling pathway regulating limb development. FGFR2 is expressed in the condensing mesenchyme of the early limb bud, with expression localized to the perichondrial tissue and periosteal tissue of the long bones and cranial sutures157,158. Selective deletions of the transmembrane domains of FGFR2 result in mouse embryonic lethality. Specifically, deletion of domain III results in the failure of mutant embryos to form limb buds, indicating that Fgfr2 domain III is essential for limb initiation159,160. The role of FGFR2 in cranial suture development has generated several gain-of-function mutant mouse models. One such model is the Fgfr2+/S252W mouse, which develops craniosynostosis, resembling human Apert syndrome161. This model elucidated the role of FGFR2 in the abnormal osteoblast proliferation and differentiation associated with Apert syndrome. Other mutant mouse models, such as the Fgfr2−/− mouse, show delayed differentiation and mineralization of the skull vault and premature coronal suture formation owing to decreased osteoblast differentiation and mineralization159,161. Furthermore, these observations correlate with decreased expression of the osteoblast markers osteopontin and RUNX2 (REF.159).
The role of the niche in bone repair
Osteoblast functionality during bone repair is dependent on the signals of the surrounding niche cells. Niche cells provide a specific microenvironment and integrate signals to mediate the appropriate stem cell response according to the needs of the organism. The niche interacts with osteoprogenitors to maintain their undifferentiated properties during homeostasis. However, after a bone injury, the changing niche environment causes the precursor cells to differentiate into osteoblasts and repair the damaged tissue. Therefore, to understand the molecular regulation of osteoblasts that enables their function in bone repair, the influence of niche cells such as inflammatory, endothelial and Schwann cells must be understood.
Inflammatory cells become activated after acute bone injury, resulting in a cascade initiated by a haematoma (from ruptured blood vessels inside the bone and surrounding soft tissue)162,163. The haematoma, characterized by hypoxia and low pH, acts as a temporary scaffold enabling the active invasion of local tissue macrophages and polymorphonuclear neutrophils. Specifically, in fracture healing, the resident macrophage populations of the endosteal and periosteal surfaces have a pivotal role in intramembranous ossification, while recruited inflammatory macrophages have an important role in endochondral ossification162,164,165. The influx of these inflammatory cells results in the secretion of chemokines such as IL-6 and chemokine ligand 2 (CCL2)166,167. Along with these proinflammatory chemokines, BMP4 and VEGF are released into the microenvironment at the site of the acute bone injury130,165,168. The excreted factors BMP4, VEGF, IL-6 and CCL2 promote osteogenic differentiation by interacting with the nascent osteoprogenitors and commit their fate towards the osteoblast lineage to enable bone repair165–168.
Revascularization is essential for bone repair, as new vasculature brings nutrients to the site of the injury. These blood vessels are composed of endothelial cells, which interact with osteoprogenitor cells to create a microenvironment supportive of osteoblast lineage fate specification169. Osteogenesis has been linked to type H endothelial cells, a subtype of capillary endothelial cells169–171. The type H cell is found in the metaphysis and endosteum of postnatal long bones171. These cells express PECAM1 and endomucin, which are key endothelial cell surface markers. In addition to promoting angiogenesis, type H cells provide molecular signals that target osteoprogenitor cells through the Notch signalling pathway172. Endothelial Notch signalling regulates osteogenesis, as indicated by the finding that disruption of Notch pathway signalling reduced overall osteogenesis172. On a molecular level, endothelial Notch signalling disruption decreases the expression of Spp1, Runx2 and Osx172. From the findings taken together, Notch signalling mediated by endothelial cells alters the niche at the injury site, enabling osteoprogenitor cells to differentiate into osteoblasts.
Sensory and sympathetic nerves have a crucial role in skeletal homeostasis and bone repair. Nerve growth factor (NGF), a neurotrophin, is involved in the development, maintenance and regeneration of sensory and sympathetic nerves46,173,174. In vivo studies applied distraction osteogenesis in the rabbit mandible; this bone is unique as the inferior alveolar nerve runs through the mandible175. The exogenous application of NGF reduced the consolidation period for bone repair in this model175. Therefore, the presence of NGF, which is natively secreted in the niche by nerves, accelerated osteogenesis. Additionally, the mandibular distraction osteogenesis model found elevated NGF and VEGF expression during the bone repair period175,176. NGF was expressed by cells in the nerve, whereas VEGF was expressed by Schwann cells176. From the findings taken together, the presence of nerve and Schwann cells in the niche supports osteogenic differentiation of osteoprogenitor cells leading to new bone formation. Overall, osteoblast lineage determination depends on the niche to provide the necessary signals to induce vascularization and eventual bone matrix formation to develop new bone.
Dysregulation and clinical relevance
The dysregulation of bone biology in the setting of bone repair occurs on a spectrum from ‘lack of bone’ to ‘excessive bone’, with restoration of ‘normal bone’ as the midpoint (FIG. 5). Thus far, this Review has discussed the development and maintenance of normal bone during homeostasis; however, clinical bone disorders can be found along this continuum. Non-union of a fracture, namely in long bones, occurs when the healing process is interrupted, causing insufficient bone formation within the bone gap177,178. To reduce the occurrence of non-union, clinicians use recombinant human BMP2, resulting in a healing rate of 86.6% for tibial fractures177,179. Safety and efficacy profiling of recombinant human FGF2 has shown a beneficial effect on fracture healing in the tibia. However, clinical studies did not show any significant increases in the healing rate between the therapy group and the control group177,180.
Fig. 5 |. Continuum of bone disorders.
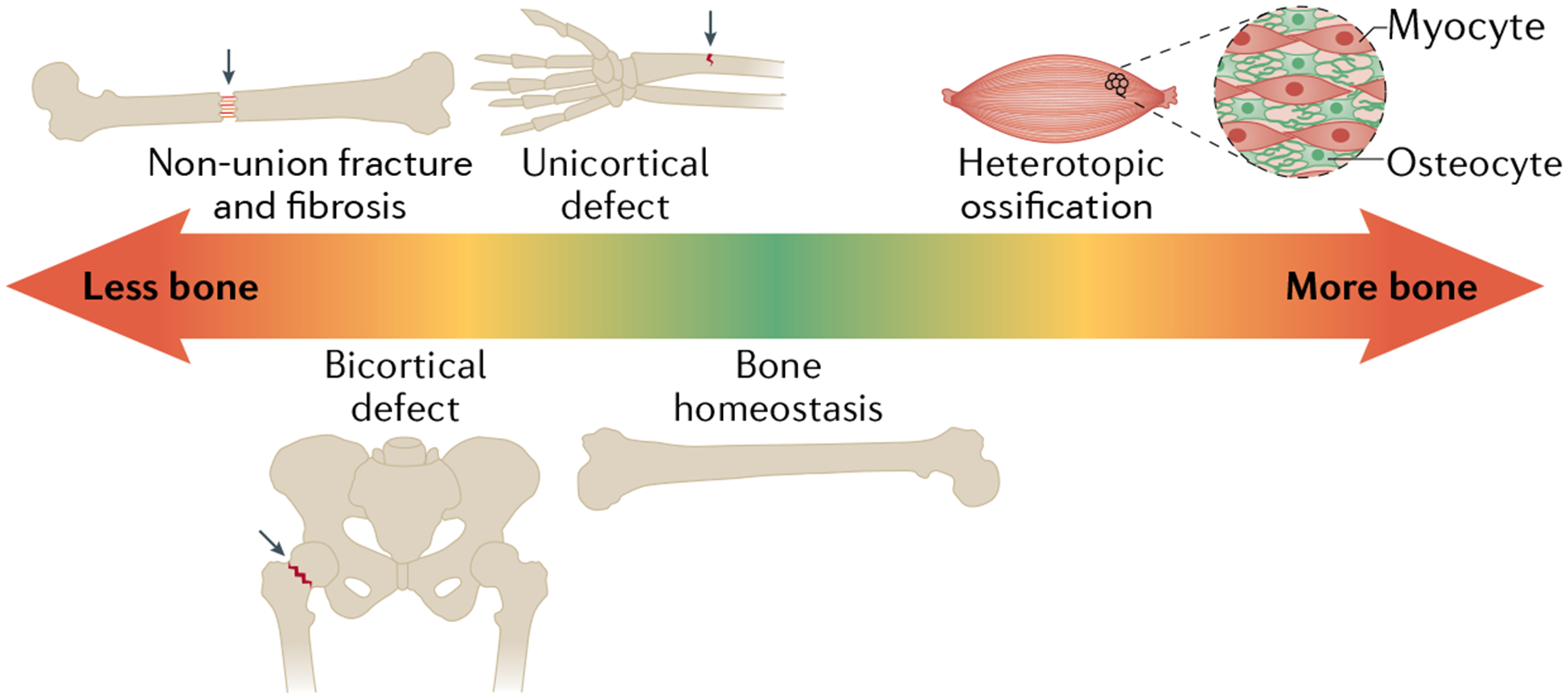
Bone disorders are characterized by a gradient from ‘less bone’ to ‘too much bone’. Beginning on the left side of the spectrum, the non-union fracture with fibrosis within the gap indicates no bone is present. On appropriate healing, the osteotomy site is filled with a new bone regenerate. Next, hip fracture results in a bicortical defect in the femoral head. A greenstick fracture results in a unicortical defect on one side of the bone. In the middle is bone homeostasis, which is represented by a physiologically normal bone. On the right end of the spectrum is too much bone, which is represented by the pathology of heterotopic ossification, wherein extraskeletal bone formation occurs.
Another example of lack of bone in healing is hip fractures caused by falls in elderly people. Worldwide annually, 1.7 million hip fractures occur, a number that is projected to increase to more than six million by 2050 owing to the ageing population181. Most hip fractures in elderly people occur secondary to underlying osteoporosis.
The final example is a unicortical defect, which occurs frequently in children. Greenstick fractures account for 12% of all paediatric emergency department visits in the USA182. The clinical intervention for this fracture type is to immobilize the bone using a cast, allowing it to naturally heal182. As the fracture still preserves the shape of the bone and the regenerative potential in a child is robust, the fracture can heal quickly over time with minimal strain on the affected bone.
The opposite aspect of the spectrum is ‘excessive bone formation’, exemplified by heterotopic ossification. Heterotopic ossification is defined as ectopic (extraskeletal) bone formation that can occur following soft tissue damage such as a trauma, burn or spinal cord injury. This ectopic bone formation has been shown to require the innate immune response and specifically the response of macrophages, which stimulate prochondrogenic and pro-osteogenic genes and activation of osteoblast progenitor cells in this setting183–185. The inflammatory response activates neutrophils and macrophages, which secrete prochondrogenic factors (such as TGFβ1) and pro-osteogenic factors (such as oncostatin M), driving tissue-resident progenitor cell osteochondral differentiation183,186,187. The inflammatory response induces a cytokine cascade including IL-6 and VEGF, which are known to promote angiogenesis and canonical and non-canonical BMP signalling, as previously discussed74,183,188,189. The elimination of macrophages has been shown to decrease callus formation in fracture healing190 in addition to decreasing traumatic heterotopic ossification187.
Identifying the cell lineage responsible for non-genetic heterotopic ossification has been of significant interest. Lineage tracing analyses have identified tissue-resident cells of the Prx1Cre and ScxCre lineages that mark the presumptive heterotopic ossification progenitor cells191–194. Other Cre alleles that have been shown to mark heterotopic ossification progenitors include PdgfraCre and Gli1Cre195,196. Only a small percentage of heterotopic ossification was shown to express markers consistent with BCSPs197. Similarly to fracture healing, canonical BMP and TGFβ signalling are also central to heterotopic ossification formation and are potential therapeutic targets185,198. Additionally, similarly to fracture repair, mechanotransductive signalling and extracellular matrix properties, such as collagen alignment, have been shown to alter cell fate199. Understanding the cells and pathways responsible for aberrant cell fate in heterotopic ossification might inform additional disease processes such as muscle fibrosis and allow improved targeted therapies for bone repair200. Similarly to bone development and repair, differences exist in heterotopic ossification formation on the basis of age and sex201,202. Overall, clinical bone disorders provide perspective on the regulation of bone biology and the consequences for bone homeostasis when key pathways or molecules become dysregulated.
Conclusions and perspectives
Understanding of the osteoblast lineage has significantly increased through identification of specific cellular and molecular components necessary for bone formation. By application of the physiological implications of the high cellular activity in the growth plate and the use of cell sorting technologies, a stem cell population — SSCs - has been isolated, which is lineage-restricted to bone and has the capability to support haematopoiesis. Through further high-throughput sequencing studies of SSCs, specific genes and chromatin accessibility motifs have been shown to be upregulated, including Runx2, Ihh and Fak (also known as Ptk2). This novel genetic information provides insight into the pathways necessary for the maintenance of SSCs during homeostasis and activation of the cell population during an injury-induced regenerative response. Even with the extensive strides made in the field, more research needs to be conducted to further understand the roles and regulation of the osteoblast lineage in bone development and repair.
First, the relationship, or the lack thereof, between SSCs and MSCs needs to be determined in the context of bone development and disease. Both cell types have been shown to give rise to osteoblasts and chondrocytes. However, further research needs to be conducted to determine if MSCs and SSCs are separate populations or if one cell type is a subset of the other cell type. Understanding this relationship will reconcile the various viewpoints in the field of the origin of the apex osteoblast progenitor cell. Second, determining the role of morphogen gradients in activating the osteoblast differentiation programme during limb bud development is essential. Currently, various developmental signalling pathways are known to be necessary for appropriate limb patterning; however, further research needs to be conducted to determine these signalling interactions and particularly the specific genes committing multipotent cells to an osteoblast lineage. Finally, additional research needs to be performed to determine a possible therapeutic role for guiding stem cells towards osteoblast differentiation. By understanding the nuances of the differentiation pathways and key transcription factors for osteoblast differentiation, therapies can be designed wherein clinicians can harness the endogenous SSCs and/or MSCs within an injured bone to drive osteoblast differentiation, thereby facilitating bone repair. The potential for studying the osteoblast lineage is vast. Driven by the application of new technologies to answer these fundamental questions, an increased understanding of the osteoblast lineage can benefit both science and clinical practice.
Osteon.
A cylindrical structure consisting of a mineralized matrix and osteocytes that transports blood through connected canaliculi.
Long bone growth plate.
An area of differentiating tissue located near the ends of long bones that enables physiological lengthening of the bones.
Axial skeleton.
The portion of the skeleton consisting of the bones of the head and vertebrae.
Appendicular skeleton.
The portion of the skeleton consisting of the bones of the appendages.
Cancellous bone.
Mature adult bone consisting of spongy tissue meshwork typically found in the cores of vertebral bones and the ends of long bones.
Unicortical defect.
A fracture involving only the outer and/or inner cortices on one side of the bone shaft.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Ambrosi TH, Longaker MT & Chan CKF A revised perspective of skeletal stem cell biology. Front. Cell Dev. Biol 7, 189 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy MP et al. The role of skeletal stem cells in the reconstruction of bone defects. J. Craniofac. Surg 28, 1136–1141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long F Building strong bones: molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol 13, 27–38 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Bianco P & Robey PG Skeletal stem cells. Development 142, 1023–1027 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garnero P, Sornay-Rendu E, Chapuy MC & Delmas PD Increased bone turnover in late postmenopausal women is a major determinant of osteoporosis. J. Bone Miner. Res 11, 337–349 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Soltanoff CS, Yang S, Chen W & Li YP Signaling networks that control the lineage commitment and differentiation of bone cells. Crit. Rev. Eukaryot. Gene Expr 19, 1–46 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Compton JT & Lee FY Current concepts review: a review of osteocyte function and the emerging importance of sclerostin. J. Bone Joint Surg. Am 96, 1659–1668 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Bezooijen RL et al. Sclerostin Is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J. Exp. Med 199, 805–814 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robling AG et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J. Biol. Chem 283, 5866–5875 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Tatsumi S et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 5, 464–475 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Jacome-Galarza CE, Lee SK, Lorenzo JA & Aguila HL Identification, characterization, and isolation of a common progenitor for osteoclasts, macrophages, and dendritic cells from murine bone marrow and periphery. J. Bone Miner. Res 28, 1203–1213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kong YY et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397, 315–323 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Dougall WC et al. RANK is essential for osteoclast and lymph node development. Genes. Dev 13, 2412–2424 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu F & Teitelbaum SL Osteoclasts: new insights. Bone Res 1, 11–26 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meyers C et al. Heterotopic ossification: a comprehensive review. JBMR Plus 3, e10172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dallas SL, Xie Y, Shiflett LA & Ueki Y Mouse Cre models for the study of bone diseases. Curr. Osteoporos. Rep 16, 466–477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pittenger MF et al. Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 (1999). [DOI] [PubMed] [Google Scholar]; This work establishes the potential for MSCs to differentiate into bone, cartilage and fat.
- 18.Chen Q et al. Fate decision of mesenchymal stem cells: adipocytes or osteoblasts? Cell Death Differ. 23, 1128–1139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friedenstein AJ, Chailakhjan RK & Lalykina KS The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Prolif. 3, 393–403 (1970). [DOI] [PubMed] [Google Scholar]
- 20.Friedenstein AJ, Chailakhyan RK & Gerasimov UV Bone marrow osteogenic stem cells: in vitro cultivation and transplantation in diffusion chambers. Cell Prolif. 20, 263–272 (1987). [DOI] [PubMed] [Google Scholar]
- 21.Friedenstein AJ Osteogenic stem cells in the bone marrow. Bone Miner. Res 10.1016/b978-0-444-81371-8.50012-1 (1990). [DOI] [Google Scholar]
- 22.Wei J et al. Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell 161, 1576–1591 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang T, Zhang X & Bikle DD Osteogenic differentiation of periosteal cells during fracture healing. J. Cell Physiol 232, 913–921 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ackema KB & Charité J Mesenchymal stem cells from different organs are characterized by distinct topographic Hox codes. Stem Cell Dev. 17, 979–991 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Rux DR et al. Regionally restricted Hox function in adult bone marrow multipotent mesenchymal stem/stromal cells. Dev. Cell 39, 653–666 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson LT, Rakshit S, Sun H & Wellik DM Generation and expression of a Hoxa11eGFP targeted allele in mice. Dev. Dyn 237, 3410–3416 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swinehart IT, Schlientz AJ, Quintanilla CA, Mortlock DP & Wellik DM Hox11 genes are required for regional patterning and integration of muscle, tendon and bone. Development 140, 4574–4582 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pineault KM, Song JY, Kozloff KM, Lucas D & Wellik DM Hox11 expressing regional skeletal stem cells are progenitors for osteoblasts, chondrocytes and adipocytes throughout life. Nat. Commun 10, 3168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rux DR & Wellik DM Hox genes in the adult skeleton: novel functions beyond embryonic development. Dev. Dyn 246, 310–317 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan CKF et al. Identification and specification of the mouse skeletal stem cell. Cell 160, 285–298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; The work is the first to isolate the SSC in mice, which has the differentiation capacity to be restricted to bone, cartilage,and bone stroma.
- 31.Chan CKF et al. Identification of the human skeletal stem cell. Cell 175, 43–56 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sacchetti B et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 131, 324–336 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Kassem M & Bianco P Skeletal stem cells in space and time. Cell 160, 17–19 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Bianco P Stem cells and bone: a historical perspective. Bone 70, 2–9 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Ueno H & Weissman IL Clonal analysis of mouse development reveals a polyclonal origin for yolk sac blood islands. Dev. Cell 11, 519–533 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Worthley DL et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 160, 269–284 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan CKF et al. Clonal precursor of bone, cartilage, and hematopoietic niche stromal cells. Proc. Natl Acad. Sci. USA 110, 12643–12648 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berendsen AD & Olsen BR Bone development. Bone 80, 14–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marecic O et al. Identification and characterization of an injury-induced skeletal progenitor. Proc. Natl Acad. Sci. USA 112, 9920–9925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tevlin R et al. Pharmacological rescue of diabetic skeletal stem cell niches. Sci. Transl Med 9, eaag2809 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ransom RC et al. Mechanoresponsive stem cells acquire neural crest fate in jaw regeneration. Nature 563, 514–521 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mizuhashi K et al. Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature 563, 254–258 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Debnath S et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 562, 133–139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jia G et al. Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat. Commun 9, 4877 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baker S, Rogerson C, Hayes A, Sharrocks A & Rattray M Classifying cells with Scasat, a single-cell ATAC-seq analysis tool. Nucleic Acids Res. 47, e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Douarin NM & Smith J Development of the peripheral nervous system from the neural crest. Annu. Rev. Cell Biol 4, 375–404 (1988). [DOI] [PubMed] [Google Scholar]
- 47.Long F & Ornitz DM Development of the endochondral skeleton. Cold Spring Harb Perspect. Biol 5, a008334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kronenberg HM Developmental regulation of the growth plate. Nature. 423, 332–336 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Maes C & Kronenberg HM Postnatal bone growth: growth plate biology, bone formation, and remodeling. pediatric. Bone 10.1016/B978-0-12-382040-2.10004-8 (2012). [DOI] [Google Scholar]
- 50.Lefebvr EV & Dvir-Ginzberg M SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect. Tissue Res 58, 2–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lovell-Badge R The early history of the Sox genes. Int. J. Biochem. Cell Biol 42, 378–380 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Bi W, Deng JM, Zhang Z, Behringer RR & De Crombrugghe B Sox9 is required for cartilage formation. Nat. Genet 22, 85–89 (1999). [DOI] [PubMed] [Google Scholar]
- 53.Akiyama H, Chaboissier MC, Martin JF, Schedl A & De Crombrugghe B The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813–2828 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henry SP, Liang S, Akdemir KC & De Crombrugghe B The postnatal role of Sox9 in cartilage. J. Bone Miner. Res 27, 2511–2525 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schafer AJ et al. Campomelic dysplasia with XY sex reversal: diverse phenotypes resulting from mutations in a single gene. Ann. N. Y. Acad. Sci 785, 137–149 (1996). [DOI] [PubMed] [Google Scholar]
- 56.Gentilin B et al. Phenotype of five cases of prenatally diagnosed campomelic dysplasia harboring novel mutations of the SOX9 gene. Ultrasound Obstet. Gynecol 36, 315–323 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Komori T Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. 339, 189–195 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Ducy P, Zhang R, Geoffroy V, Ridall AL & Karsenty G Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89, 747–754 (1997). [DOI] [PubMed] [Google Scholar]
- 59.Harada H et al. Cbfa1 isoforms exert functional differences in osteoblast differentiation. J. Biol. Chem 274, 6972–6978 (1999). [DOI] [PubMed] [Google Scholar]
- 60.Komori T et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89, 755–764 (1997). [DOI] [PubMed] [Google Scholar]; This work establishes RUNX2 as an essential transcription factor for osteoblast differentiation.
- 61.Otto F et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89, 765–771 (1997). [DOI] [PubMed] [Google Scholar]
- 62.Inada M et al. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev. Dyn 214, 279–290 (1999). [DOI] [PubMed] [Google Scholar]
- 63.Takarada T et al. An analysis of skeletal development in osteoblast-specific and chondrocyte-specific runt-related transcription factor-2 (Runx2) knockout mice. J. Bone Miner. Res 28, 2064–2069 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Maruyama Z et al. Runx2 determines bone maturity and turnover rate in postnatal bone development and is involved in bone loss in estrogen deficiency. Dev. Dyn 236, 1876–1890 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Sinha KM & Zhou X Genetic and molecular control of osterix in skeletal formation. J. Cell Biochem 114, 975–984 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karsenty G Minireview: tranzscriptional control of osteoblast differentiation. Endocrinology 142, 2731–2733 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Nakashima K & De Crombrugghe B Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet. 19, 458–466 (2003). [DOI] [PubMed] [Google Scholar]
- 68.Nakashima K et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108, 17–29 (2002). [DOI] [PubMed] [Google Scholar]; This work establishes the temporal coordination between OSX and RUNX2 activation for osteoblast differentiation.
- 69.Yang X & Karsenty G Transcription factors in bone: developmental and pathological aspects. Trends Mol. Med 8, 340–345 (2002). [DOI] [PubMed] [Google Scholar]
- 70.Zhou X et al. Multiple functions of osterix are required for bone growth and homeostasis in postnatal mice. Proc. Natl Acad. Sci. USA 107, 12919–12924 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu TM & Lee EH Transcriptional regulatory cascades in Runx2-dependent bone development. Tissue Eng. Part B Rev 19, 254–263 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.St-Arnaud R & Hekmatnejad B Combinatorial control of ATF4-dependent gene transcription in osteoblasts. Ann. N. Y. Acad. Sci 1237, 11–18 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang X et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology: implication for Coffin-Lowry syndrome. Cell 117, 387–398 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Jing D et al. The role of microRNAs in bone remodeling. Int. J. Oral. Sci 7, 131–143 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xiao G et al. Cooperative interactions between activating transcription factor 4 and Runx2/Cbfa1 stimulate osteoblast-specific osteocalcin gene expression. J. Biol. Chem 280, 30689–30696 (2005). [DOI] [PubMed] [Google Scholar]
- 76.Wagner EF Functions of AP1 (Fos/Jun) in bone development. Ann. Rheum. Dis 61, ii40–ii42 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kenner L et al. Mice lacking JunB are osteopenic due to cell-autonomous osteoblast and osteoclast defects. J. Cell Biol 164, 613–623 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zambotti A, Makhluf H, Shen J & Ducy P Characterization of an osteoblast-specific enhancer element in the CBFA1. Gene 277, 41497–41506 (2002). [DOI] [PubMed] [Google Scholar]
- 79.Jochum W et al. Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra-1. Nat. Med 6, 980–984 (2000). [DOI] [PubMed] [Google Scholar]
- 80.Bozec A et al. Fra-2/AP-1 controls bone formation by regulating osteoblast differentiation and collagen production. J. Cell Biol 190, 1093–1106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nüsslein-volhard C & Wieschaus E Mutations affecting segment number and polarity in drosophila. Nature. 287, 795–801 (1980). [DOI] [PubMed] [Google Scholar]
- 82.McMahon AP, Ingham PW & Tabin CJ 1 Developmental roles and clinical significance of Hedgehog signaling. Curr. Top. Dev. Biol 53, 1–114 (2003). [DOI] [PubMed] [Google Scholar]
- 83.Ocbina PJR & Anderson KV Intraflagellar transport, Cilia, and mammalian hedgehog signaling: analysis in mouse embryonic fibroblasts. Dev. Dyn 237, 2030–2038 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Riddle RD, Johnson RL, Laufer E & Tabin C Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 75, 1401–1416 (1993). [DOI] [PubMed] [Google Scholar]
- 85.Rohatgi R, Milenkovic L & Scott MP Patched1 regulates hedgehog signaling at the primary cilium. Science. 317, 372–376 (2007). [DOI] [PubMed] [Google Scholar]
- 86.Corbit KC et al. Vertebrate Smoothened functions at the primary cilium. Nature. 437, 1018–1021 (2005). [DOI] [PubMed] [Google Scholar]
- 87.Towers M, Mahood R, Yin Y & Tickle C Integration of growth and specification in chick wing digit-patterning. Nature 452, 882–886 (2008). [DOI] [PubMed] [Google Scholar]
- 88.Chinnaiya K, Tickle C & Towers M Sonic hedgehog-expressing cells in the developing limb measure time by an intrinsic cell cycle clock. Nat. Commun 5, 4230 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang B, Fallon JF & Beachy PA Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434 (2000). [DOI] [PubMed] [Google Scholar]
- 90.Mo R et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 124, 113–123 (1997). [DOI] [PubMed] [Google Scholar]
- 91.Park H et al. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 127, 1593–1605 (2000). [DOI] [PubMed] [Google Scholar]
- 92.Hojo H et al. Gli1 protein participates in hedgehog-mediated specification of osteoblast lineage during endochondral ossification. J. Biol. Chem 287, 17860–17869 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Amano K, Densmore M, Nishimura R & Lanske B Indian hedgehog signaling regulates transcription and expression of collagen type X via Runx2/Smads interactions. J. Biol. Chem 289, 24898–24910 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jemtland R, Divieti P, Lee K & Segre GV Hedgehog promotes primary osteoblast differentiation and increases PTHrP mRNA expression and iPTHrP secretion. Bone 32, 611–620 (2003). [DOI] [PubMed] [Google Scholar]
- 95.Long F & Linsenmayer TF Regulation of growth region cartilage proliferation and differentiation by perichondrium. Development 125, 1067–1073 (1998). [DOI] [PubMed] [Google Scholar]
- 96.Mak KK, Chen MH, Day TF, Chuang PT & Yang Y Wnt/β-catenin signaling interacts differentially with Ihh signaling in controlling endochondral bone and synovial joint formation. Development 133, 3695–3707 (2006). [DOI] [PubMed] [Google Scholar]
- 97.Day TF & Yang Y Wnt and hedgehog signaling pathways in bone development. J. Bone Joint Surg. Ser. Am 90, 19–24 (2008). [DOI] [PubMed] [Google Scholar]
- 98.Hojo H et al. Hedgehog-Gli activators direct osteo-chondrogenic function of bone morphogenetic protein toward osteogenesis in the perichondrium. J. Biol. Chem 288, 9924–9932 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schroeter EH, Kisslinger JA & Kopan R Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382–386 (1998). [DOI] [PubMed] [Google Scholar]
- 100.Zanotti S & Canalis E Notch signaling and the skeleton. Endocr. Rev 37, 223–253 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tu X et al. Physiological Notch signaling maintains bone homeostasis via RBPjk and Hey upstream of NFATc1. PLoS Genet. 8, e1002577 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hilton MJ et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat. Med 14, 306–314 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Engin F et al. Dimorphic effects of Notch signaling in bone homeostasis. Nat. Med 14, 299–305 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Canalis E, Parker K, Feng JQ & Zanotti S Osteoblast lineage-specific effects of notch activation in the skeleton. Endocrinology 154, 623–634 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zanotti S & Canalis E Notch1 and Notch2 expression in osteoblast precursors regulates femoral microarchitecture. Bone 62, 22–28 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim JB et al. Bone regeneration is regulated by Wnt signaling. J. Bone Miner. Res 22, 1913–1923 (2007). [DOI] [PubMed] [Google Scholar]
- 107.Huelsken J & Birchmeier W New aspects of Wnt signaling pathways in higher vertebrates. Curr. Opin. Genet. Dev 11, 547–553 (2001). [DOI] [PubMed] [Google Scholar]
- 108.Williams BO & Insogna KL Where Wnts went: the exploding field of Lrp5 and Lrp6 signaling in bone. J. Bone Miner. Res 24, 171–178 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Joiner DM, Ke J, Zhong Z, Xu HE & Williams BO LRP5 and LRP6 in development and disease. Trends Endocrinol. Metab 24, 31–39 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baron R & Kneissel M WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med 19, 179–192 (2013). [DOI] [PubMed] [Google Scholar]
- 111.Boyden LM et al. High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl. J. Med 346, 1513–1521 (2002). [DOI] [PubMed] [Google Scholar]
- 112.Little RD et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am. J. Hum. Genet 70, 11–19 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Houschyar KS et al. Wnt pathway in bone repair and regeneration - what do we know so far. Front. Cell Dev. Biol 6, 170 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Minear S et al. Wnt proteins promote bone regeneration. Sci. Transl Med 2, 29ra30 (2010). [DOI] [PubMed] [Google Scholar]
- 115.Poole KES et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 19, 1842–1844 (2005). [DOI] [PubMed] [Google Scholar]
- 116.Ai M, Holmen SL, Van Hul W, Williams BO & Warman ML Reduced affinity to and inhibition by DKK1 form a common mechanism by which high bone mass-associated missense mutations in LRP5 affect canonical Wnt signaling. Mol. Cell Biol 25, 4946–4955 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brunkow ME et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet 68, 577–589 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Holmen SL et al. Decreased BMD and limb deformities in mice carrying mutations in both Lrp5 and Lrp6. J. Bone Miner. Res 19, 2033–2040 (2004). [DOI] [PubMed] [Google Scholar]
- 119.Kubota T et al. Lrp6 hypomorphic mutation affects bone mass through bone resorption in mice and impairs interaction with Mesd. J. Bone Miner. Res 23, 1661–1671 (2008). [DOI] [PubMed] [Google Scholar]
- 120.Lin GL & Hankenson KD Integration of BMP, Wnt, and notch signaling pathways in osteoblast differentiation. J. Cell Biochem 112, 3491–3501 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wu M, Chen G & Li YP TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 4, 16009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Itasaki N & Hoppler S Crosstalk between Wnt and bone morphogenic protein signaling: a turbulent relationship. Dev. Dyn 239, 16–33 (2010). [DOI] [PubMed] [Google Scholar]
- 123.Luo Q et al. Connective tissue growth factor (CTGF) is regulated by Wnt and bone morphogenetic proteins signaling in osteoblast differentiation of mesenchymal stem cells. J. Biol. Chem 279, 55958–55968 (2004). [DOI] [PubMed] [Google Scholar]
- 124.Si W et al. CCN1/Cyr61 is regulated by the canonical Wnt signal and plays an important role in Wnt3A-induced osteoblast differentiation of mesenchymal stem cells. Mol. Cell Biol 26, 2955–2964 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Boland GM, Perkins G, Hall DJ & Tuan RS Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J. Cell Biochem 93, 1210–1230 (2004). [DOI] [PubMed] [Google Scholar]
- 126.Chen Y et al. β-Catenin signaling pathway is crucial for bone morphogenetic protein 2 to induce new bone formation. J. Biol. Chem 282, 526–533 (2007). [DOI] [PubMed] [Google Scholar]
- 127.Zhang M et al. BMP-2 modulates β-catenin signaling through stimulation of Lrp5 expression and inhibition of β-TrCP expression in osteoblasts. J. Cell Biochem 108, 896–905 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wrana JL et al. TGFβ signals through a heteromeric protein kinase receptor complex. Cell 71, 1003–1014 (1992). [DOI] [PubMed] [Google Scholar]
- 129.Schmierer B & Hill CS TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol 8, 970–982 (2007). [DOI] [PubMed] [Google Scholar]
- 130.Salazar VS, Gamer LW & Rosen V BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol 12, 203–221 (2016). [DOI] [PubMed] [Google Scholar]
- 131.Katagiri T & Watabe T Bone morphogenetic proteins. Cold Spring Harb Perspect. Biol 10.1101/cshperspect.a021899 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Salazar VS et al. Reactivation of a developmental Bmp2 signaling center is required for therapeutic control of the murine periosteal niche. eLife 10.7554/eLife.42386 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bandyopadhyay A et al. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2, 2116–2130 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tsuji K et al. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat. Genet 38, 1424–1429 (2006). [DOI] [PubMed] [Google Scholar]; This work demonstrates the role of reoccurring BMP signalling for limb development and fracture healing of the limb.
- 135.Lim J et al. Dual function of Bmpr1a signaling in restricting preosteoblast proliferation and stimulating osteoblast activity in mouse. Development 143, 339–347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fujii M et al. Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol. Biol. Cell 10, 3801–3813 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Singhatanadgit W & Olsen I Endogenous BMPR-IB signaling is required for early osteoblast differentiation of human bone cells. Vitr. Cell Dev. Biol. Anim 47, 251–259 (2011). [DOI] [PubMed] [Google Scholar]
- 138.Yoshida Y et al. Negative regulation of BMP/Smad signaling by Tob in osteoblasts. Cell 103, 1085–1097 (2000). [DOI] [PubMed] [Google Scholar]
- 139.Zhang Y et al. Loss of BMP signaling through BMPR1A in osteoblasts leads to greater collagen cross-link maturation and material-level mechanical properties in mouse femoral trabecular compartments. Bone 88, 74–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Johnson DE & Williams LT Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res 60, 1–41 (1992). [DOI] [PubMed] [Google Scholar]
- 141.Ornitz DM et al. Receptor specificity of the fibroblast growth factor family. J. Biol. Chem 271, 15292–15297 (1996). [DOI] [PubMed] [Google Scholar]
- 142.Ornitz DM FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor. Rev 16, 205–213 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Montero A et al. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J. Clin. Invest 105, 1085–1093 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhou M et al. Fibroblast growth factor 2 control of vascular tone. Nat. Med 4, 201–207 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Crossley PH, Minowada G, MacArthur CA & Martin GR Roles for FGF8 in the induction, initiation, and maintenance of chick limb development. Cell 84, 127–136 (1996). [DOI] [PubMed] [Google Scholar]
- 146.Lewandoski M, Sun X & Martin GR Fgf8 signalling from the AER is essential for normal limb development. Nat. Genet 26, 460–463 (2000). [DOI] [PubMed] [Google Scholar]
- 147.Martin GR The roles of FGFs in the early development of vertebrate limbs. Genes Dev. 12, 1571–1586 (1998). [DOI] [PubMed] [Google Scholar]
- 148.Min H et al. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev. 12, 3156–3161 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ohuchi H et al. The mesenchymal factor, FGF10, initiates and maintains the outgrowth of the chick limb bud through interaction with FGF8, an apical ectodermal factor. Development 124, 2235–2244 (1997). [DOI] [PubMed] [Google Scholar]
- 150.Mahmood R et al. A role for FGF-8 in the initiation and maintenance of vertebrate limb bud outgrowth. Curr. Biol 5, 797–806 (1995). [DOI] [PubMed] [Google Scholar]
- 151.Heikinheimo M, Lawshé A, Shackleford GM, Wilson DB & MacArthur CA Fgf-8 expression in the post-gastrulation mouse suggests roles in the development of the face, limbs and central nervous system. Mech. Dev 48, 129–138 (1994). [DOI] [PubMed] [Google Scholar]
- 152.Lin JM et al. Actions of fibroblast growth factor-8 in bone cells in vitro. Am. J. Physiol. Endocrinol. Metab 297, E142–E150 (2009). [DOI] [PubMed] [Google Scholar]
- 153.Yamaguchi TP, Conlon RA & Rossant J Expression of the fibroblast growth factor receptor FGFR-1/flg during gastrulation and segmentation in the mouse embryo. Dev. Biol 152, 75–88 (1992). [DOI] [PubMed] [Google Scholar]
- 154.Deng C et al. Fibroblast growth factor receptor-1 (FGFR-1) is essential for normal neural tube and limb development. Dev. Biol 185, 42–54 (1997). [DOI] [PubMed] [Google Scholar]
- 155.Jacob AL, Smith C, Partanen J & Ornitz DM Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev. Biol 296, 315–328 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Verheyden JM, Lewandoski M, Deng C, Harfe BD & Sun X Conditional inactivation of Fgfr1 in mouse defines its role in limb bud establishment, outgrowth and digit patterning. Development 132, 4235–4245 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Orr-Urtreger A et al. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2). Dev. Biol 158, 475–486 (1993). [DOI] [PubMed] [Google Scholar]
- 158.Li X et al. Fibroblast growth factor signaling and basement membrane assembly are connected during epithelial morphogenesis of the embryoid body. J. Cell Biol 153, 811–822 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Arman E, Haffner-Krausz R, Chen Y, Heath JK & Lonai P Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc. Natl Acad. Sci. USA 95, 5082–5087 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Xu X et al. Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development 125, 753–765 (1998). [DOI] [PubMed] [Google Scholar]
- 161.Wang Y et al. Abnormalities in cartilage and bone development in the Apert syndrome FGFR2(+/S252W) mouse. Development 132, 3537–3548 (2005). [DOI] [PubMed] [Google Scholar]
- 162.Claes L, Recknagel S & Ignatius A Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol 8, 133–143 (2012). [DOI] [PubMed] [Google Scholar]
- 163.Glynne AJ, Andrew SM, Freemont AJ & Marsh DR Inflammatory cells in normal human fracture healing. Acta Orthop. 65, 462–466 (1994). [DOI] [PubMed] [Google Scholar]
- 164.Bolander ME Regulation of fracture repair by growth factors. Exp. Biol. Med 200, 165–170 (1992). [DOI] [PubMed] [Google Scholar]
- 165.Croes M et al. Proinflammatory mediators enhance the osteogenesis of human mesenchymal stem cells after lineage commitment. PLoS ONE 10, e0132781 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lu LY et al. Pro-inflammatory M1 macrophages promote Osteogenesis by mesenchymal stem cells via the COX-2-prostaglandin E2 pathway. J. Orthop. Res 35, 2378–2385 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bernhardsson M & Aspenberg P Osteoblast precursors and inflammatory cells arrive simultaneously to sites of a trabecular-bone injury. Acta Orthop. 89, 457–461 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ono T et al. IL-17-producing γδT cells enhance bone regeneration. Nat. Commun 7, 10928 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows that the presence of the proinflammatory cytokine IL-17, from the niche, aided in bone regrowth after injury.
- 169.Goerke SM, Obermeyer J, Plaha J, Stark GB & Finkenzeller G Endothelial progenitor cells from peripheral blood support bone regeneration by provoking an angiogenic response. Microvasc. Res 98, 40–47 (2015). [DOI] [PubMed] [Google Scholar]
- 170.Langen UH et al. Cell-matrix signals specify bone endothelial cells during developmental osteogenesis. Nat. Cell Biol 19, 189–201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kusumbe AP, Ramasamy SK & Adams RH Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 507, 323–328 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ramasamy SK, Kusumbe AP, Wang L & Adams RH Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature. 507, 376–380 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cao J et al. Sensory nerves affect bone regeneration in rabbit mandibular distraction osteogenesis. Int. J. Med. Sci 16, 831–837 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jones RE et al. Skeletal stem cell-Schwann cell circuitry in Mandibular repair. Cell Rep. 28, 2757–2766.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Park BW, Kim JR, Lee JH & Byun JH Expression of nerve growth factor and vascular endothelial growth factor in the inferior alveolar nerve after distraction osteogenesis. Int. J. Oral Maxillofac. Surg 35, 624–630 (2006). [DOI] [PubMed] [Google Scholar]
- 176.Wang L et al. Locally applied nerve growth factor enhances bone consolidation in a rabbit model of mandibular distraction osteogenesis. J. Orthop. Res 24, 2238–2245 (2006). [DOI] [PubMed] [Google Scholar]
- 177.Emara KM, Diab RA & Emara AK Recent biological trends in management of fracture non-union. World J. Orthop 6, 623–628 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Panteli M, Pountos I, Jones E & Giannoudis PV Biological and molecular profile of fracture non-union tissue: current insights. J. Cell Mol. Med 19, 685–713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jones AL et al. Recombinant human BMP-2 and allograft compared with autogenous bone graft for reconstruction of diaphyseal tibial fractures with cortical defects: a randomized, controlled trial. J. Bone Joint Surg. Ser. Am 88, 1431–1441 (2006). [DOI] [PubMed] [Google Scholar]
- 180.Kawaguchi H et al. Local application of recombinant human fibroblast growth factor-2 on bone repair: a dose-escalation prospective trial on patients with osteotomy. J. Orthop. Res 25, 480–487 (2007). [DOI] [PubMed] [Google Scholar]
- 181.Babcock S & Kellam JF Hip fracture nonunions: diagnosis, treatment, and special considerations in elderly patients. Adv. Orthop 10.1155/2018/1912762 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Atanelov Z & Bentley TP Greenstick fracture. StatPearls (2018). [PubMed] [Google Scholar]
- 183.Kraft CT et al. Trauma-induced heterotopic bone formation and the role of the immune system: a review. J. Trauma. Acute Care Surg 80, 156–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Huang H et al. Relationship between heterotopic ossification and traumatic brain injury: Why severe traumatic brain injury increases the risk of heterotopic ossification. J. Orthop. Transl 12, 16–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sorkin M et al. Regulation of heterotopic ossification by monocytes in a mouse model of aberrant wound healing. Nat Commun. 10.1038/s41467-019-14172-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work determines CD47 activation as a therapeutic approach for heterotopic ossification formation during wound healing.
- 186.Agarwal S et al. Disruption of neutrophil extracellular traps (NETs) links mechanical strain to post-traumatic inflammation. Front. Immunol 10, 2148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Torossian F et al. Macrophage-derived oncostatin M contributes to human and mouse neurogenic heterotopic ossifications. JCI Insight 2, e96034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hwang C et al. Mesenchymal VEGFA induces aberrant differentiation in heterotopic ossification. Bone Res. 7, 36 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hsieh HHS et al. Coordinating tissue regeneration through transforming growth factor-β activated kinase 1 inactivation and reactivation. Stem Cells 37, 766–778 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Raggatt LJ et al. Fracture healing via periosteal callus formation requires macrophages for both initiation and progression of early endochondral ossification. Am. J. Pathol 184, 3192–3204 (2014). [DOI] [PubMed] [Google Scholar]
- 191.Agarwal S et al. Inhibition of Hif1α prevents both trauma-induced and genetic heterotopic ossification. Proc. Natl Acad. Sci. USA 113, E338–E347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Agarwal S et al. Scleraxis-lineage cells contribute to ectopic bone formation in muscle and tendon. Stem Cells 35, 705–710 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Loder SJ et al. Characterizing the circulating cell populations in traumatic heterotopic ossification. Am. J. Pathol 188, 2464–2473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Dey D et al. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci. Transl Med 10.1126/scitranslmed.aaf1090 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kan C et al. Gli1-labeled adult mesenchymal stem/progenitor cells and hedgehog signaling contribute to endochondral heterotopic ossification. Bone 109, 71–79 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Eisner C et al. Murine tissue-resident PDGFRα+ fibro-adipogenic progenitors spontaneously acquire osteogenic phenotype in an altered inflammatory environment. J. Bone Miner. Res (2020). [DOI] [PubMed] [Google Scholar]
- 197.Agarwal S et al. Analysis of bone-cartilage-stromal progenitor populations in trauma induced and genetic models of heterotopic ossification. Stem Cells 34, 1692–1701 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Agarwal S et al. Strategic targeting of multiple BMP receptors prevents trauma-induced heterotopic ossification. Mol. Ther 25, 1974–1987 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Huber AK et al. Immobilization after injury alters extracellular matrix and stem cell fate. J. Clin. Invest 10.1172/JCI136142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Stepien DM et al. Tuning macrophage phenotype to mitigate skeletal muscle fibrosis. J. Immunol 204, 2203–2215 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Peterson JR et al. Effects of aging on osteogenic response and heterotopic ossification following burn injury in mice. Stem Cell Dev. 24, 205–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ranganathan K et al. Role of gender in burn-induced heterotopic ossification and mesenchymal cell osteogenic differentiation. Plast. Reconstr. Surg 135, 1631–1641 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Akiyama H et al. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc. Natl Acad. Sci. USA 102, 14665–14670 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Maes C et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell 19, 329–344 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Greenbaum A et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 495, 227–230 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Xiong J et al. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLoS ONE 10.1371/journal.pone.0138189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Pineault KM et al. Hox11 genes regulate postnatal longitudinal bone growth and growth plate proliferation. Biol. Open 4, 1538–1548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yu VWC et al. FIAT represses ATF4-mediated transcription to regulate bone mass in transgenic mice. J. Cell Biol 169, 591–601 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ambrogini E et al. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 11, 136–146 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shimoyama A et al. Ihh/Gli2 signaling promotes osteoblast differentiation by regulating Runx2 expression and function. Mol. Biol. Cell 18, 2411–2418 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Li J et al. Suppressor of fused restraint of hedgehog activity level is critical for osteogenic proliferation and differentiation during calvarial bone development. J. Biol. Chem 292, 15814–15825 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Funato N et al. Hand2 controls osteoblast differentiation in the branchial arch by inhibiting DNA binding of Runx2. Development 136, 615–625 (2009). [DOI] [PubMed] [Google Scholar]
- 213.Kanzler B, Kuschert SJ, Liu YH & Mallo M Hoxa-2 restricts the chondrogenic domain and inhibits bone formation during development of the branchial area. Development 125, 2587–2597 (1998). [DOI] [PubMed] [Google Scholar]
- 214.Komori T Regulation of osteoblast differentiation by runx2. Adv. Exp. Med. Biol 658, 43–49 (2010). [DOI] [PubMed] [Google Scholar]
- 215.Hong JH et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 309, 1074–1078 (2005). [DOI] [PubMed] [Google Scholar]
- 216.Bialek P et al. A twist code determines the onset of osteoblast differentiation. Dev. Cell 6, 423–435 (2004). [DOI] [PubMed] [Google Scholar]
- 217.Cancela L, Hsieh CL & Francke UPP Molecular structure, chromosome assignment, and promoter organization of the human matrix Gla protein gene. J. Biol. Chem 265, 15040–15048 (1990). [PubMed] [Google Scholar]
- 218.Karsenty G & Park RW Regulation of type I collagen genes expression. Int. Rev. Immunol 12, 177–185 (1995). [DOI] [PubMed] [Google Scholar]
- 219.Pinzone JJ et al. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood 113, 517–525 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kim JB et al. Reconciling the roles of FAK in osteoblast differentiation, osteoclast remodeling, and bone regeneration. Bone 41, 39–51 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Li X et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem 280, 19883–19887 (2005). [DOI] [PubMed] [Google Scholar]