

Abstract
New techniques able to monitor the maturation of tissue engineered constructs over time are needed for a more efficient control of developmental parameters. Here, a label-free fluorescence lifetime imaging (FLIm) approach implemented through a single fiber-optic interface is reported for nondestructive in situ assessment of vascular biomaterials. Recellularization processes of antigen removed bovine pericardium scaffolds with endothelial cells and mesenchymal stem cells were evaluated on the serous and the fibrous sides of the scaffolds, 2 distinct extracellular matrix niches, over the course of a 7 day culture period. Results indicated that fluorescence lifetime successfully report cell presence resolved from extracellular matrix fluorescence. The recellularization process was more rapid on the serous side than on the fibrous side for both cell types, and endothelial cells expanded faster than mesenchymal stem cells on antigen-removed bovine pericardium. Fiber-based FLIm has the potential to become a nondestructive tool for the assessment of tissue maturation by allowing in situ imaging of intraluminal vascular biomaterials.
Keywords: fiber optics imaging, fluorescence lifetime imaging, tissue engineering
Graphical Abstract
1 ∣. INTRODUCTION
Bioengineered tissues have the potential to repair or substitute damaged or diseased human tissues. Developing tissue substitutes is a biologically complex task that requires an orchestrated synchronization of different cell types, nutrient supplies and host scaffolds. The maturation process is thus tightly regulated and benefits from continuous evaluation. Optimized design parameters of tissue recellularization processes lead to successful clinical outcomes on tissue engineering and regenerative medicine applications. For vascular tissue engineering applications, a key step in achieving functional tissue engineered blood vessel analogs or blood vessel patches is the formation of a healthy endothelium [1-3]. A monolayer of endothelial cells (ECs) on the scaffold surface emulates the endothelium that lines the luminal side of the vasculature. These preformed healthy EC layers have the potential to reduce thrombosis formation, inflammatory reactions and intimal hyperplasia [3-5]. Pre-seeding vascular scaffolds with mesenchymal stem cells (MSC) may also improve recruitment of endothelial progenitor cells and circulating ECs for improved endothelialization and may help to reduce the chances of thrombogenicity [6]. Bovine pericardium (BP), mainly composed of collagen (~70% of their dry weight), is a natural biomaterial widely utilized clinically as a vascular scaffold, although current generations of BP require glutaraldehyde fixation to overcome recipient graft-specific immune response. In arterial patches, glutaraldehyde-fixed BP is used as surgical closures, since it supports limited arterial remodeling by endothelialization of the luminal surface [7]. Recent efforts to generate antigen removed BP provide unfixed scaffolds with suppressed antigenicity and maintained structure and function, enhancing its regenerative potential [6]. BP consists of 2 well-differentiated extracellular matrix (ECM) niches that provide distinct environments for cell proliferation and migration [6, 8]. The serous side is lined by a basement membrane, with aligned collagen, fibers that hinder cell penetration while supporting rapid migration of cells along the surface. The fibrous side is composed of loose connective tissue, including thicker collagen bundles and elastic fibers, providing a challenging environment for cells to expand across the surface, but reducing the resistance to penetrate the scaffold (Figure 1A).
FIGURE 1.

Scaffold and experimental timeline. (A) Sketch of the scaffold properties. Bovine pericardium (1-2 mm in thickness) is composed of structural proteins organized in a basement membrane on the serous side, and as loose connective tissue on the fibrous side, providing 2 different extracellular matrix niches for re-seeded cells. (B) The experimental design consists on 3 main steps: (1) antigen removal of the scaffold to generate antigen removed bovine pericardium (AR-BP), (2) seeding the scaffold with either human aortic endothelial cells (hAECs) or human mesenchymal stem cells (hMSCs) and (3) imaging the recellularization process through fiber optic fluorescence lifetime imaging (FLIm) over 7 days after seeding
The current gold standard for tissue recellularization evaluation relies predominantly on histological and immunohistochemical analysis. Although highly informative, these assays require sample destruction preventing continuous monitoring and functional evaluation of the developing tissue construct. Optical imaging techniques have the ability to interrogate tissues nondestructively [9-12]. In particular, autofluorescence imaging stands as a noninvasive and biocompatible source of contrast because of its inherently nondestructive nature. A major challenge in the detection of cellular autofluorescence in biomaterials is posed by the overwhelming fluorescent background provided by the ECM. Like cells, proteins such as collagen and elastin, the main structural proteins of vascular biomaterials, fluoresce upon ultraviolet excitation. Moreover, they have an emission spectrum that strongly overlaps with cellular fluorophores. To overcome these effects, some studies opted to label cells with fluorescent agents such as enhanced green fluorescence protein (eGFP) and use conventional fluorescence imaging to visualize cell growth on synthetic scaffolds with cellular resolution [13, 14]. Although successful at monitoring cellular dynamics in 3D in vitro experiments, the regulatory barriers to the use of eGFP-labeled cells in humans limit the in vivo applications of this approach. Other studies explored how to generate collagenous scaffolds with low autofluorescence levels, by using different crosslinking agents [15], but these alterations may impact cellular activity.
Fluorescence lifetime imaging (FLIm) provides a sensitive strategy for nondestructive, label-free monitoring of cells in bioengineered tissues. The metabolic machinery of cells feature nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FAD), 2 cofactors that fluoresce in the visible range upon ultraviolet light excitation (peak at λex ~340 nm and ~385 nm, respectively). In addition to their spectral properties, NADH and FAD have fluorescent decay lifetimes that vary according to their protein-binding state. Consequently, NADH and FAD label-free FLIm microscopy has been extensively utilized for investigations of metabolic signatures of cancerous cells [16, 17], to identify histological features of livers [18], or to probe skin constituents [19]. Conventional approaches to FLIm are based on imaging through microscope objectives, which can achieve high spatial resolution of cells in planar scaffolds or excised tissue, but are not suitable to more challenging geometries such as in situ intravascular imaging. We have previously demonstrated that FLIm through a fiber-optic interface is applicable to clinical settings, where access to the specimen is complex and constrained by the body geometry and the surgical procedure (intravascular, laparoscopy or robot-guided surgery) [12, 20]. Therefore, we anticipate that fiber-based implemented FLIm can be of interest to the tissue engineering community for monitoring recellularization processes of vascular biomaterials.
The principal goal of this study is to demonstrate the use of fiber-based, label-free fluorescence lifetime in resolving the presence of cells or cellular activity on collagenous scaffolds, at tissue level. Particularly, we aim to follow the recellularization process of antigen removed bovine pericardium (AR-BP) with human aortic endothelial cells (hAECs) and human mesenchymal stem cells (hMSCs) over the course of a 7-day culture period, and compare their progression rates on either side of AR-BP scaffolds (Figure 1B).
2 ∣. MATERIALS AND METHODS
2.1 ∣. Bovine pericardium scaffolds
AR-BP scaffolds were generated from adult cattle bovine pericardia (Spear Products, Coopersburg, PA). The native tissue pieces were first stripped of fat and loose connective tissue, then trimmed to 0.2 g pieces, and finally underwent antigen removal through stepwise solubilization consisting of hydrophile solubilization (i.e., using DTT and KCl), lipophile solubilization (ie, using amidosulfobetaine-14, ASB-14; Sigma, St. Louis, MO), nuclear digestions (ie, using DNase/RNase) and washout, as previously described [21]. AR-BP scaffolds were stored in storage solution (Dulbecco's modified eagle medium [DMEM, Sigma] with 15% [vol/vol] dimethyl sulfoxide [Sigma]) at −80°C until experiment was performed. On the day of the seeding, AR-BP pieces were thawed and washed twice in 2 mL culture medium for 30 min on a shaker at 125 rpm.
2.2 ∣. Cell cultures
2.2.1 ∣. Endothelial cells
Human aortic endothelial cells (Lonza, Walkersville, MD, CAT No. CC-2535, LOT No. 0000303583) and eGFP-labeled human aortic endothelial cells (eGFP-hAECs; Angio-Proteomie, Boston, MA, CAT No. cAP0006GFP, LOT No. 201209701SR) were cultured according to the provider's protocol. Briefly, the cells were thawed, plated into T-75 flask precoated with quick coating solution (Angio-Proteomie; cAP-01) at 37°C in EGM-2 media (Lonza) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA) (EGM-2F). After reaching 80% confluency, cells were passaged in 1:4 ratio. All experiments utilized cells between passages 4 and 6. Inspection under bright field and fluorescence microscopy, as well as H&E staining, verified that cells exhibited typical endothelial morphology such as cobblestone-shaped monolayer under confluency, and tubule formation (data not shown).
2.2.2 ∣. Mesenchymal stem cells
Human mesenchymal stem cells and eGFP-hMSCs were obtained as previously described [6]. The cells were cultured in T-75 flask at 37°C in medium consisted of DMEM high glucose, 1% (vol/vol) penicillin-streptomycin (P/S), 1% (vol/vol) L-glutamine 200 mM (Hyclone Laboratories, South Logan, UT) and 20% (vol/vol) FBS (Atlanta Biologicals, Lawrenceville, GA). All experiments utilized cells between passages 4 and 6. Cells exhibited characteristic mesenchymal properties by morphology, phenotype and function, as verified with H&E staining, fluorescence microscopy and previously reported [6].
2.2.3 ∣. FLIm cell characterization
For FLIm characterization of cell cultures, cells were seeded on cell culture treated 8-well chambered coverglass (Thermo Scientific Nunc Lab-Tek II Chambered Coverglass) at 2650 cells/mm2 for hAEC and 1325 cells/mm2 for hMSC, and cultured for 48 h before imaging. Three independent samples were prepared for each case. Cells were rinsed with phosphate-buffered saline (PBS) before the measurements took place.
2.3 ∣. Seeding and imaging protocol
For recellularization experiments, rectangular strips (approximately 8 mm x 20 mm x 700–1200 μm) of AR-BP were cut and placed into a nontissue culture treated 6-well plate (Corning, Corning, NY) containing 2 mL of cell culture medium. Totally, 20 000 hAEC or hMSC were locally seeded into cylinders with inner diameter of 4 mm (Pyrex cloning cylinder, OD x H = 6 mm x 8 mm) onto either the fibrous or serous side of BP scaffolds in a final culture medium volume of 70 μL. Following a 4-h incubation, cylinders and medium were removed and the attached cells were cultured for up to 7 days at 37°C, 5% CO2, with cell medium changed every day. Seeded AR-BP strips were washed in PBS for 5 min prior to imaging. Fluorescence microscopy and FLIm were done every 48 h.
Fluorescence microscopy images were acquired using a BZ-X710 fluorescence microscope (Keyence, Itasca, IL) with a Plan Fluor 4x/0.13NA objective lens for whole scaffold imaging, and a S Plan Fluor 40x/0.60NA lens for zoomed in images of the cells. eGFP images were acquired with excitation filter at 470/40 nm and emission at 525/50 nm. FLIm was done on cell-seeded scaffolds immersed on PBS, to prevent sample dehydration, with a 10-m long fiber optic installed inside a biosafety cabinet to keep the samples under sterile conditions throughout the entirety of the study.
2.4 ∣. Fluorescence lifetime imaging system
A fiber-based multispectral FLIm system (sketched in Figure S1A in File S1, Supporting Information) was used to map the spatial distribution of vascular tissue autofluorescence intensity and lifetime. The apparatus employs a 355-nm Q-switched microchip-pulsed laser (pulse duration <0.6 ns, pulse energy >2 μJ; TEEM photonics STV-02E, Meylan, France) which is delivered to the sample surface via the 400 μm core of a multimode fiber. The same fiber was used to capture and guide a fraction of the backscattered sample autofluorescence to a custom wavelength selection module (WSM) that is used for multispectral detection. The WSM uses 4 dichroic beamsplitters and bandpass filters to separate tissue autofluorescence into 4 distinct spectral bands (SBs; SB1 = 390/18 nm, SB2 = 435/40 nm, SB3 = 532–553 nm, SB4 = 610/70 nm; SBs were selected to match the fluorescence emission spectra of endogenous fluorophores of interest, see Figure S1B in File S1). Fiber optic delay lines are used to temporally multiplex the SBs on to a single microchannel plate photomultiplier tube (MCP PMT, R3809U-50, Hamamatsu, Japan). A high speed (12.5 GS/s, 3 GHz bandwidth) data acquisition (DAQ, NI PXIe-5185, National Instruments, Austin, TX) board is used to digitize the amplified output of the MCP PMT. Images of fluorescence intensity and lifetime are acquired in a manner analogous to laser scanning microscopy, by raster scanning the location on the sample surface where fluorescence excitation is delivered. In this work, a motorized 3-axis translation stage (MT s50-z8, Thorlabs, Newton, NJ) was used to translate the distal end of the fiber across the sample surface. Fluorescence lifetime images were acquired with a laser repetition rate of 4 kHz, with a rectangular pixel size of 40 μm x 100 μm. Images of 20 mm x 10 mm were acquired in less than 15 min. The instrument response function (IRF) was acquired measuring the decay of 2-DASPI (2-[4-(dimethylamino)styryl]-1-methylpyridinium iodide, Sigma), with an average fluorescence lifetime of 30 ps when dissolved in ethanol [22, 23]. For the present study, SB4 had no significant contribution, and is not considered further.
2.5 ∣. FLIm data processing
Time-correlated single photon counting techniques often require >1 x 105 laser pulses to reconstruct the fluorescence decay for a single pixel. In contrast, our approach referred to as the pulse sampling technique, uses high energy laser pulses (>2 μJ) that generate a sufficiently large number of autofluorescence photons for a complete fluorescence decay to be recorded for each laser pulse. The data acquisition and processing pipeline is graphically presented in Figure S1C in File S1. The emission of a pulse from the laser triggers the high speed DAQ card to begin acquiring data in the form of a voltage vs time waveform. Using a 10-m long fiber, each waveform, which includes the recorded signal of all 4 SBs, consists of 4500 points, which given the 12.5 GS/s sampling rate means that it has a total duration of 360 ns (Figure S1C-i in File S1). To improve signal-to-noise ratio, 16 waveforms are acquired at each location of the sample surface and averaged on the DAQ card prior to being saved. The long fluorescence detection window allows us to temporally resolve background fluorescence excited in the apparatus as the 355 nm laser pulses propagate towards the sample, from tissue autofluorescence. Thus, the background fluorescence generated in all but the last 20 to 30 cm of the multimode fiber interface is well separated from the sample fluorescence. To mitigate the influence of any background fluorescence that is not temporally separated from that of the sample, prior to every image acquisition, a background waveform (Figure S1C-ii in File S1) is recorded by positioning the fiber adjacent to the sample. The first stage of data processing involves the subtraction of this background waveform from all waveforms in the image (Figure S1C-iii in File S1). Next, the different SBs are isolated by segmenting the waveform into 4 sections, each one containing 680 points (total duration of 54.5 ns, Figure S1C-iv in File S1). The IRF of the system (Figure S1C-v in File S1) is then deconvolved from the background subtracted segmented data using a constrained least squares deconvolution with Laguerre expansion [24] (Figure S1C-vi in File S1). Finally, deconvolved decays are parameterized by the average lifetime, which is defined as the expectation value of the probability density function of the decay [25]. The spectral distribution of the sample is reported by the intensity ratio in each SB. The intensity ratio is defined by the autofluorescence intensity in each band divided by the total recorded intensity. These parameters permit us to discriminate sample components according to their fluorescence spectral properties. However, the high degree of spectral overlap between endogenous fluorophores, and fluorescence intensities from scaffolds being stronger than that of cells, complicates the task, stressing the relevance of the additional dimension provided by fluorescence lifetime.
2.6 ∣. Quantification of cellular area on the scaffolds
For conventional fluorescence microscopy images, the cellular area on the scaffolds was quantified with ImageJ. Images were transformed to 16-bit gray scale, Otsu segmentation was applied to obtain a binary image with black background and white cells. The number of white pixels × the pixel size was used to estimate the area cells occupy on the scaffold at a given time point (see Figure S2A in File S1).
For FLIm images, cellular areas on the scaffolds were estimated by measuring the area of fluorescence lifetime that changes with respect to the acellular scaffold. As indicated by the lifetime measurements of isolated scaffolds (Figure 2B) and cells in a low background environment (Figure 3B), the localized introduction of cells to AR-BP is predicted to decrease the average lifetime of these regions. Thus, the area reporting the effect of cells on the scaffold presents shorter fluorescence lifetime. In addition, the absolute values of fluorescence lifetime change over time in culture, as discussed in Section 3.1. To simplify quantification, fluorescence lifetime maps were normalized to the mean lifetime of the seeded region of each scaffold at each time point (Figure S2A in File S1, white dashed circle), providing relative fluorescence lifetime maps with comparable characteristics across samples and time points. The analysis of the cellular area was done on the relative fluorescence lifetime maps. These show relative lifetimes equal to 1 in the portions of the scaffold with cells, and areas with relative lifetime larger than 1 in those regions of the scaffolds not influenced by cells (Figure S2B in File S1). We estimated the area of the cellular region by contouring an ellipse around the area of relative fluorescence lifetime equal to 1 (see Figure S2B in File S1, black circles).
FIGURE 2.

Antigen removed bovine pericardium (AR-BP) fluorescence lifetime properties. (A) Fluorescence intensity ratio detected at each spectral band (SB) for the fibrous and the serous sides of the scaffolds. (B) Average fluorescence lifetime at each SB for both sides. N =8 scaffolds per side. Paired t test :* P < .05. (C) Representative fluorescence lifetime maps in SB1 of the serous (top) and the fibrous (bottom) side of AR-BP scaffolds in PBS culture for 7 days. Scale bars = 2 mm. (D) Average fluorescence lifetime drift over the 7 days in culture, for both sides, n = 3. Error bars indicate standard deviation
FIGURE 3.

Cellular fluorescence lifetime. (A) Fluorescence intensity ratio and (B) average fluorescence lifetime detected at each spectral band (SB) for human aortic endothelial cells (hAECs) and human mesenchymal stem cells (hMSCs) cultured on glass substrate. N = 3 cultures per cell type. Paired t test: * P < .05. Error bars indicate standard deviation. (C) Representative conventional fluorescence microscopy image of eGFP-hAEC on the serous side of AR-BP after 1 day in culture and (D) corresponding fluorescence lifetime map in SB3. Scale bar = 5 mm. Error bars indicate standard deviation
2.7 ∣. Statistics
Paired t test was used to determine the statistical significance of fluorescence lifetime and intensity ratio measurements on either side of AR-BP (fibrous vs serous), between cell types (hAEC and hMSC), and between eGFP-labeled and -unlabeled cells at each time point and SB. One-way analysis of variance (ANOVA) was used to determine the statistical significance of time on changes in fluorescence lifetime.
3 ∣. RESULTS AND DISCUSSION
3.1 ∣. Characterization of scaffold fluorescence lifetime
We first characterized the autofluorescence of acellular AR-BP scaffolds (Figure 2). Eight individual AR-BP scaffolds were imaged on both sides. Critically, previous studies have shown that the structure, composition and functional properties of both BP ECM niches are maintained following antigen removal [6]. Utilizing a scaffold depleted of antigenicity helps to provide a blank slate for recellularization and becomes crucial to minimize the immune response once the construct is implanted. Figure 2A,B show fluorescence intensity ratios and average fluorescence lifetime acquired from each side of the AR-BP pieces, for each SB. The intensity ratio measurements indicate that the bulk of the fluorescence generated by AR-BP is in SB1 (~70%), for both sides. Indeed, the main contributor to the autofluorescence of AR-BP is collagen, with an emission peak around 390 nm [26-30]. There are, however, significant differences between the intensity ratios on either side of the scaffold for each SB. For SB1, intensity ratio in the serous side (IRS) = 0.757 ± 0.001 a.u., whereas the intensity ratio in the fibrous side (IRF) = 0.675 ± 0.002 a.u. (P < 0.001). Difference are smaller in SB2 (IRS = 0.161 ± 0.001 a.u., IRF = 0.190 ± 0.003 a.u., P < 0.001) and SB3 (IRS = 0.036 ± 0.001 a.u., IRF = 0.059 ± 0.002 a.u., P < 0.001). The average fluorescence lifetime of the collagenous scaffold fluctuates around 6 ns in SB1, but there is a significant difference in lifetime between the 2 sides (6.09 ± 0.05 ns in the serous side, and 5.96 ± 0.04 ns in the fibrous side, P < 0.001). Such difference minimizes in SB2 (5.84 ± 0.05 ns and 5.79 ± 0.05 ns, respectively, P = 0.43), and is significant again in the third SB (3.85 ± 0.05 ns for the serous, 4.29 ± 0.04 ns on the fibrous side, P = .01). Analogous results have been observed for all the scaffold pieces utilized throughout the study. The observed differences in intensity ratio and fluorescence lifetime from either side of the scaffold may be attributed to the different composition and structural properties of the 2 ECM niches of BP [6, 8].
Fluorescence lifetime of AR-BP scaffolds in SB1 varied over incubation time in PBS. Figure 2C,D show the drift in average fluorescence lifetime in SB1 of a representative AR-BP piece that has been in PBS at 37°C and 5% CO2 for 7 days (only taken out of the incubator during brief imaging periods).
Lifetime shortens by half a nanosecond over this 7-day period for both sides. The same effect was observed when AR-BP scaffolds were incubated in cell culture media, under the same conditions (Figure S3 in File S1). The drift in fluorescence lifetime of AR-BP in SB1 may be a consequence of continued wash out of ECM fragments either not removed during the wash process or newly created due to hydrolysis. Neither PBS nor cell culture media are constituent matched to BP ECM, and therefore additional components may be removed by incubation in either of those media, ultimately affecting the fluorescence lifetime of tissues.
3.2 ∣. Characterization of cellular fluorescence lifetime
Next, we analyzed the fluorescence of the cell lines used in this study, when cultured on glass substrate. Figure 3A shows the fluorescence intensity ratio of hAECs and hMSCs across 3 SBs. The spectral profile of cellular autofluorescence differs from that of the collagenous scaffold. The largest contribution of fluorescence intensity from cells seeded on a glass substrate was detected in SBs 2 and 3, for both cell types. SB2 and SB3 are arranged around the maximum emission of the main cellular fluorophores, NADH and FAD, respectively (Figure S1B in File S1) [1, 5]. Intensity ratios (Figure 3A) display significant differences between hAEC (IRhAEC) and hMSC (IRhMSC) in SB2 (IRhAEC = 0.33 ± 0.04 a.u., IRhMSC = 0.43 ± 0.02 a.u., P = .01) and SB3 (IRhAEC = 0.52 ± 0.05 a.u., IRhMSC = 0.38 ± 0.02 a. u. , P = .01). The measured fluorescence lifetime for both cells types ranges from ~2 to 4 ns in SB2 and SB3 (Figure 3B). On average, hAEC exhibit longer lifetimes than hMSC in all SBs (3.7 ± 0.8 ns and 3.4 ± 0.5 ns, P = .3 for SB1, 3.7 ± 0.4 ns and 2.7 ± 0.2 ns, P < 0.001 for SB2, and 2.8 ± 0.3 ns and 2.4 ± 0.2 ns, P < 0.01 for SB3, respectively). These differences are attributed to differing metabolic states of endothelial and MSCs, which change when cells are resting or proliferating, as well as according to their local environment (eg, oxygen and glucose conditions). ECs are preferentially glycolytic even in resting conditions [31]. MSCs exhibit complex metabolic activity with both glycolysis and oxidative phosphorylation as energy production mechanisms; however, glycolysis is enhanced for proliferating MSCs under normal oxygen conditions [32]. The present study does not address the metabolic state of the cells investigated, but specific excitation of the metabolic cofactors NADH and FAD could be correlated with metabolic activities in further applications.
With scaffolds and cells separately characterized, we proceeded to seedings on AR-BP. Figure 3C shows the conventional fluorescence microscopy image of eGFP-hAEC on the serous side of AR-BP 24 h after seeding. Figure 3D shows the corresponding fluorescence lifetime map in SB3, which exhibits a central area of shorter fluorescence lifetime that matches with the area seeded with cells (dotted circle in Figure 3C). This evidences that fluorescence lifetime reports on tissue changes due to the presence of cells. The change in lifetime from the acellular to the cellular scaffold is ~200 ps. A small, yet significant change allows our fiber-based FLIm system to identify areas of recellularization. The magnitude of the change in lifetime is anticipated based on the nature of our detection scheme. The autofluorescence signal collected by the fiber probe stems from the first ~150 μm of the sample, which is mainly comprised of the collagenous scaffold and a thin layer (~1 μm [33]) of cells, when these are present (see Figure S4B-E in File S1). Therefore, together with the stronger fluorescence yield of collagen fibers, the detected autofluorescence signal is still overwhelmingly dominated by the scaffold contribution. The lifetime component provided by FLIm is key to identify the presence of cells, particularly because the intensity ratio in each SB does not provide conclusive information.
3.3 ∣. Cellular progression on AR-BP
To monitor cellular progression over an extended period of time, we measured the area of the ellipse that contours the region of the scaffold with shorter fluorescence lifetime. Because the scaffold's fluorescence lifetime changes across SBs and across individual samples, and it also drifts over time (Figure 2C,D), we normalize the fluorescence lifetime maps to the mean lifetime of the seeded region in each sample. This provided relative fluorescence lifetime maps with relative lifetime equal to 1 for areas that directly see the effect of cell presence (or cellular areas from now on), and relative lifetime larger than 1 for acellular areas. This process allowed all images to be visualized on the same range of relative lifetime values, and thus a more robust estimation of scaffold recellularization progress. It should be noted here that the normalization step is not strictly necessary to detect the presence of cells, as seen in figure 3D and S2B.i, but rather a convenient step to simplify the comparison of multiple samples at different time points in this particular study. Image analysis tools for direct recognition of recellularized areas based on raw data will need to be developed to apply this approach on more realistic seeding strategies relevant to tissue engineering work-flows.
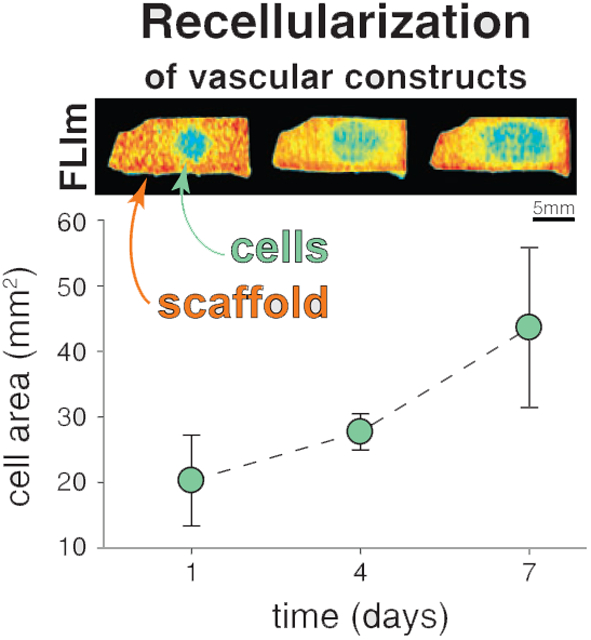
Figure 4 shows a representative AR-BP piece reseeded with eGFP-hAEC in the serous side. Relative fluorescence lifetime maps exhibit a central region with lower relative lifetime in all SBs over the course of 7 days (Figure 4A). The same pieces were imaged with conventional fluorescence microscopy, which show how the cells progress along the surface (Figure 4B). Figure 4C shows zoom-in images into the periphery of the growth area, where we can observe cells alive and active over the 7 days of culture and imaging. Additional immunostaining was performed on day 7 to verify that cells had typical morphology of living cells (Figure S4F in File S1 shows an example of hAEC seeded on the fibrous side of AR-BP). The evolution of the region with relative lifetime equal to 1 in SB3 was found to be highly correlated to the evolution of the cellular area as measured from the fluorescence microscopy images (Figure 4D, E), and less so for SB1 and SB2. SB3 is the least affected by contribution from collagen. Hence, the fluorescence lifetime in SB3 better reports on the presence of cells. Fluorescence lifetime changes observed in SB1 and SB2 carry information on the presence of cells as well as changes to the collagen scaffold as it is remodeled by these cells, which cannot be resolved from one another with our current instrumental setup.
FIGURE 4.

Representative example of eGFP-hAEC recellularization on the serous side of AR-BP. (A) Relative fluorescence lifetime maps for spectral bands (SB) 1, 2 and 3, on days 1, 4 and 7 of culture, scale bar = 5 mm. (B) Conventional fluorescence microscopy images at days 1, 4 and 7 of culture. Dashed white circles indicate seeding area, scale bar = 5 mm and (C) zoom-in areas at the periphery of cellular presence, scale bar =100 μm. (D) Cell area estimated from the fluorescence microscopy images (eGFP) and the relative fluorescence lifetime maps (SB1, SB2 and SB3). (E) Correlation plots and corresponding correlation factors between the cell area measured with FLIm and eGFP images on the serous (top row) and the fibrous (bottom row) side, for hAEC
Utilizing SB3 as proxy, recellularization progress was compared for both cell types (hAEC and hMSC) and for seedings on both sides of AR-BP. Figure 5 shows the temporal evolution of the cellular area for both hAEC and hMSC, in the serous and fibrous sides of AR-BP (n = 4 samples per group). For hAEC, the cellular area significantly increases over time on both sides of the scaffold (on the serous side, P < 0.01; on the fibrous side, P = .01). For hMSC, only growth on the serous side is significant over time (P = .02). Histological evaluation showed that hMSCs preferred to penetrate the scaffold instead of expanding along the surface, especially when seeded on the fibrous side (Figure S4E in File S1), a result previously reported [6].
FIGURE 5.

Representative relative fluorescence lifetime maps of (A) hAEC and (B) hMSC on the serous (top row) and the fibrous (bottom row) side of AR-BP. Images belong to SB3, scale bar = 5 mm. Plots show the average recellularized area in SB3 for (c) hAEC and (d) hMSC, for n = 4 in each group
The serous and the fibrous sides of AR-BP offer 2 ECM niches where cells progress at different rates. As estimated from the FLIm results in SB3, the cellular area on the serous side on day 7 is of the order of 115% larger than on day 1 for hAEC, and of 90% for hMSC. For the fibrous side, the increases from day 1 to day 7 are more moderate, 80% and 8%, respectively. From the fluorescence lifetime maps we can quantify that ECs progress 1.5 times faster on the serous than on the fibrous side. In addition to faster progression rates, as demonstrated with our study and previously reported [6], seeding on the serous as opposed to the fibrous side has other consequences important to the development of vascular constructs. Serous side seeding maintains the cellular ability to remain in a stem state [6], which may ease the assimilation of preseeded AR-BP grafts. Differential modulation of cellular behavior, including cell migration, of hMSC on either side of AR-BP has previously been reported [6]. The generation of an endothelial monolayer (the formation of a monolayer was verified by histological analysis, Figure S4B,C in File S1) on the surface of AR-BP is an important process to monitor as it has the potential to dampen the immune response of the host upon graft reception [3, 4].
Fiber-optic implemented FLIm provides quantitative and nondestructive measures of cellular progression rates on vascular scaffolds. In addition, addressing current limitations of the instrumental design, such as excitation and detection SBs, FLIm has the potential to inform on the metabolic state of the cells undergoing recellularization. Similarly, studies addressing the origin of fluorescence lifetime changes on the scaffold while this is populated by cells, could lead to the identification of ECM remodeling hallmarks useful to monitor the construct development process. Next steps will also benefit from improvements on imaging resolution and from more efficient data processing methods to delineate the effect and presence of the cells on the scaffolds. Therefore, fiber-optic-based FLIm may be a candidate addition to the tool-set for nondestructive quality control of vascular engineered tissues. Other nondestructive approaches to monitor recellularization include endoscopic optical coherent tomography and ultrasound imaging, although neither technique can resolve cell monolayers. OCT provides structural information with depth resolution, useful to assess layer thickness parameters [34, 35], and ultrasound imaging assesses the mechanical properties of the tissue by probing elasticity [36]. FLIm incorporates biochemical information of the surface layers and can ultimately be combined with OCT [37] or ultrasound imaging to add structural information.
3.3.1 ∣. A note on eGFP labeling
Both eGFP-labeled and -unlabeled cells were utilized in this study. We have utilized the eGFP-labeled cells to validate, with conventional fluorescence microscopy, the presence of cells within the scaffold, and to measure their recellularization rate, or progression along the scaffold's surface (Figure 4B-D and Figure 5). We have verified that the use of eGFP-labeled cells does not interfere with the detected autofluorescence signal by using unlabeled counterparts in parallel experiments, and have observed equivalent results in FLIm with both cell types, significance according to ANOVA analysis and relative fluorescence lifetime maps (Figure S5 in File S1) are shown in File S1. In addition, eGFP is excited at 488 nm, far from our 355 nm excitation wavelength.
4 ∣. CONCLUSION
We have successfully identified endothelial and MSCs seeded on collagenous scaffolds using label-free contrast mechanisms with a fiber-optic-based imaging system. The nondestructive nature of this approach allows monitoring of maturation processes of engineered vascular tissues over time. This is the first evidence of cellular identification via autofluorescence on vascular biomaterials. With the current instrument, we are able to show quantifiable and significant differences between cellular progressions on the serous vs the fibrous side of AR-BP, a vascular scaffold frequently used in clinical applications. Further work with cellular cultures on tubular biomaterials that mimic the vascular system will make explicit the benefits of a fiber-based interface, as the lumen surface is not optically accessible via any other approach. Enhanced quantification with FLIm could enable detailed investigations of vascular construct maturation, providing a readout of great interest to the tissue engineering and regenerative medicine community.
Supplementary Material
File S1 Contains supplementary information on the instrument employed for these experiments as well as details on the data processing. It also contains additional data on the fluorescence lifetime drift on AR-BP over time, histology and immunohistochemistry images that demonstrate the presence of cells on the scaffold, and further comparison of the recellularization process with eGFP labeled and unlabeled cells.
ACKNOWLEDGMENTS
The authors would like to acknowledge the funding from the California Institute for Regenerative Medicine (CIRM) (RT3-07879) and the National Institutes of Health (NIH) (5R01HL121068-03). The authors would like to thank Dr. James Mcmasters who assisted with cell culture preparations.
Funding information
California Institute for Regenerative Medicine, Grant/Award Number: RT3-07879
Footnotes
AUTHOR BIOGRAPHIES
Please see Supporting Information online.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- [1].Feugier P, Black RA, Hunt JA, How TV, Biomaterials 2005, 26, 1457. [DOI] [PubMed] [Google Scholar]
- [2].Chlupác J, Filová E, Baáčková L, Physiol. Res 2009, 58, 119. [DOI] [PubMed] [Google Scholar]
- [3].Quint C, Kondo Y, Manson RJ, Lawson JH, Dardik A, Niklason LE, Proc. Natl. Acad. Sci 2011, 108, 9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Baiguera S, Ribatti D, Angiogenesis 2013, 16(1), 1. [DOI] [PubMed] [Google Scholar]
- [5].Davis GE, Senger DR, Circ. Res 2005, 97, 1093. [DOI] [PubMed] [Google Scholar]
- [6].Liu ZZ, Wong ML, Griffiths LG, Sci. Rep 2016, 6, 37089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liu Y, Sakai S, Taya M, Heliyon 2016, 2, e00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Saporito WF, Pires AC, Cardoso SH, Correa JA, de Abreu LC, Valenti VE, Miller LMR, Colombari E, BMC Surg. 2011, 11, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Smith LE, Smallwood ROD, Macneil S, Microsc. Res. Tech 2010, 1133, 1123. [DOI] [PubMed] [Google Scholar]
- [10].O'Sullivan TD, Cerussi AE, Cuccia DJ, Tromberg BJ, J. Biomed. Opt 2012, 17, 071311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Balu M, Kelly KM, Zachary CB, Harris RM, Krasieva TB, Durkin AJ, Tromberg BJ, Cancer Res. 2014, 74, 2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Phipps JE, Gorpas D, Unger J, Darrow M, Bold RJ, Marcu L, Phys. Med. Biol 2017, 63, 015003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hofmann MC, Whited BM, Criswell T, Rylander MN, Rylander CG, Soker S, Wang G, Xu Y, Tissue Eng. C 2012, 18, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hofmann MC, Whited BM, Mitchell J, Vogt WC, Criswell TL, Rylander CG, Rylander MN, Soker S, Wang G, Xu Y, J. Biomed. Opt 2012, 17, 066010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Niu G, Sapoznik E, Lu P, Criswell T, Mohs AM, Wang G, Lee SJ, Xu Y, Soker S, Tissue Eng J. Regen. Med 2016, 10, 955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Skala MC, Riching KM, Gendron-fitzpatrick A, Eickhoff J, Eliceiri KW, White JG, Ramanujam N, Proc. Natl. Acad. Sci 2007, 104, 19494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Berezin MY, Achilefu S, Chem. Rev 2010, 110, 2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang H, Liang X, Mohammed YH, Thomas JA, Bridle KR, Thorling CA, Grice JE, Xu ZP, Liu X, Crawford DHG, Roberts MS, Biomed. Opt. Express 2015, 6, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sanchez WY, Pastore M, Haridass IN, Karsten K, Becker W, Roberts MS, Fluorescence Lifetime Imaging of the Skin. in Advanced Time-Correlated Single Photon Counting Applications Springer Series in Chemical Physics, Vol. 111 (Ed: Becker W), Springer, Cham, Switzerland: 2015. [Google Scholar]
- [20].Bec J, Phipps JE, Gorpas D, Ma D, Fatakdawala H, Margulies KB, Southard JA, Marcu L, Sci. Rep 2017, 7, 8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wong ML, Wong JL, Athanasiou KA, Griffiths LG, Acta Biomater. 2013, 9, 6492. [DOI] [PubMed] [Google Scholar]
- [22].Kim J, Lee M, J. Phys. Chem. A 2000, 104, 1388. [Google Scholar]
- [23].Ma D, Bec J, Yankelevich DR, Gorpas D, Fatakdawala H, Marcu L, J. Biomed. Opt 2014, 19, 066004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu J, Sun Y, Qi J, Marcu L, Phys. Med. Biol 2012, 57, 843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lakowicz JR, Principles of Fluorescence Spectroscopy, 3rd ed. Springer US, Springer, Boston, MA, 2006. [Google Scholar]
- [26].Wagnieres GA, Star WM, Wilson BC, Photochem. Photobiol 1998, 68, 603. [PubMed] [Google Scholar]
- [27].Georgakoudi I, Jacobson BC, Mu MG, Sheets EE, Badizadegan K, Carr-locke DL, Crum CP, Boone CW, Dasari RR, Dam JV, Feld MS, CancerRes. 2002, 62, 682. [PubMed] [Google Scholar]
- [28].Marcu L, French PM, Elson DS, Fluorescence Lifetime Spectroscopy and Imaging: Principles and Applications in Biomedical Diagnostics, CRC Press, Boca Raton, FL, 2014. [Google Scholar]
- [29].Yova T, Dido H, Vladimir T, J. Biomed. Opt 2001, 6, 52. [DOI] [PubMed] [Google Scholar]
- [30].Ashjian P, Elbarbary A, Zuk P, DeUgarte DA, Benhaim P, Marcu L, Hedrick MH, Tissue Eng. 2004, 10, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ghesquière B, Wong BW, Kuchnio A, Carmeliet P, Nature 2014, 511, 167. [DOI] [PubMed] [Google Scholar]
- [32].Pattappa G, Heywood HK, de Bruijn JD, Lee DA, J. Cell. Physiol 2011, 226, 2562. [DOI] [PubMed] [Google Scholar]
- [33].Pries AR, Secomb TW, Gaehtgens P, Eur. J. Physiol 2000, 440, 653. [DOI] [PubMed] [Google Scholar]
- [34].O'Halloran Cardinal K, Bonnema GT, Hofer H, Barton JK, Williams SK, Tissue Eng. 2006, 12, 3431. [DOI] [PubMed] [Google Scholar]
- [35].Bonnema GT, Cardinal KO, Williams SK, Barton JK, J. Biophotonics 2009, 2, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dutta D, Lee KW, Allen RA, Wang Y, Brigham JC, Kim K, Ultra-sound Med. Biol 2013, 39, 2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sherlock BE, Phipps JE, Bec J, Marcu L, Opt. Lett 2017, 42, 3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1 Contains supplementary information on the instrument employed for these experiments as well as details on the data processing. It also contains additional data on the fluorescence lifetime drift on AR-BP over time, histology and immunohistochemistry images that demonstrate the presence of cells on the scaffold, and further comparison of the recellularization process with eGFP labeled and unlabeled cells.