

Abstract
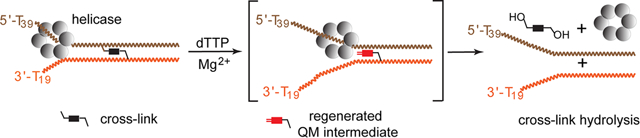
The reversible generation and capture of certain electrophilic quinone methide intermediates support dynamic reactions with DNA that allow for migration and transfer of alkylation and cross-linking. This reversibility also expands the possible consequences that can be envisioned when confronted by DNA repair processes and biological machines. To begin testing the response to such an encounter, quinone methide-based modification of DNA has now been challenged with a helicase (T7 bacteriophage gene protein four, T7gp4) that promotes 5′ to 3′ translocation and unwinding. This model protein was selected based on its widespread application, well characterized mechanism and detailed structural information. Little over one-half of the cross-linking generated by a bisfunctional quinone methide remained stable to T7gp4 and did not suppress its activity. The helicase likely avoids the topological block generated by this fraction of cross-linking by its ability to shift from single- to double-stranded translocation. The remaining fraction of cross-linking was destroyed during T7gp4 catalysis. Thus, this helicase is chemically competent to promote release of the quinone methide from DNA. The ability of T7gp4 to act as a Brownian ratchet for unwinding DNA may block recapture of the QM intermediate by DNA during its transient release from a donor strand. Most surprisingly, T7gp4 releases the quinone methide from both the translocating strand that passes through its central channel and the excluded strand that was typically unaffected by other lesions. The ability of T7gp4 to reverse the cross-link formed by the quinone methide does not extend to that formed irreversibly by the nitrogen mustard mechlorethamine.
Graphical Abstract
INTRODUCTION
DNA alkylation is a common event in biology and derives from a wide range of processes that span from intrinsic regulation to extrinsic chemotherapy. The cellular consequences of such alkylation depend in part on its persistence as a function of repair and innate stability. Many protective mechanisms exist to minimize undesirable modification of DNA, but a significant fraction of electrophiles are still able to form adducts with the nucleophilic centers of DNA. Often, alkylation is the consequence of metabolic activation of xenobiotics acquired from the environment, food, and medicine. Most of the resulting electrophiles react irreversibly and hence generate adducts that are static until repaired. Much of our knowledge on this topic was gained from classic investigations based on benzo[a]pyrene and aflatoxin exposure.1–4
A subset of electrophiles react reversibly on the biological time scale and may spontaneously migrate between nucleophiles to establish a dynamic array of adducts. This unusual property has the potential of supporting two diverging consequences. Reversibility may lead to spontaneous release of an adduct without intervention by the endogenous repair machinery. Alternatively, reversibility may establish an evolving series of interchangeable products that create a complex obstacle for detoxification. The array of adducts generated in this manner may further vary with cellular conditions as well as DNA sequence and conformation.5,6 For example, malondialdehyde, acrolein, and similar electrophiles have the potential to equilibrate between numerous products with DNA that is controlled in part by the variables above.7–9 Similarly, diffusible nucleobase adducts can act as carriers and reservoirs of electrophiles to promote adduct formation in DNA.10 The lifetime of reversible adducts may even persist after their excision from DNA by subsequently regenerating the original electrophile for recapture of DNA.
The time-dependent evolution of DNA adducts formed by a natural product Et-743 illustrates another consequence of reversible reaction.11 In this example, the reversibility of competing adducts dictates the product distribution in a simple model system. This effect is accentuated by the helicase activity of the SV40 large tumor antigen that promotes release of only the most labile adduct. In contrast, a stable adduct effectively blocks the helicase from unwinding duplex DNA. The power of helicases and other biological machines to redistribute reversible adducts has not been addressed beyond this first intriguing study. During replication, helicases are the first to encounter lesions and thus may have a profound influence on subsequent mutation and repair.12–15 Furthermore, helicases are essential for all organisms.16,17 Although their unwinding function has been extensively characterized, research has only begun to appreciate their role in resolving protein–DNA interactions and responding to various lesions.15 This article now describes the ability of a helicase to release interstrand cross-linking formed by the reversible reaction of a bisfunctional quinone methide and equivalent alkylation by a monofunctional quinone methide.
Quinone methides (QM) and related species are biologically relevant electrophiles that alkylate a variety of cellular nucleophiles including the nitrogens of DNA.18–21 These reactive intermediates are typically formed transiently in vivo by oxidative or reductive metabolism of xenobiotics and drugs as illustrated most famously by the food preservative BHT22 and the anticancer drugs mitomycin23 and tamoxifen.24 Synthetic QM precursors (QMP) have been developed for similar activation by oxidation25,26 and reduction,27,28 as well as by light,19,29–31 hydrolysis,32–34 desilylation,35,36 and other mechanisms (Scheme 1). The highly reversible nature of certain QM-DNA conjugates also allows for generation of self-adducts that have been used to cross-link DNA at selected sequences without the need of an external signal.37–39 Similarly, a bisfunctional QMP conjugated to acridine (bisQMP, Scheme 1) forms dynamic intrastrand cross-links that easily transform to interstrand cross-links (Scheme 2).40 Although interstrand cross-links produced by this bisQMP did not appear to migrate between sites within a duplex, a subsequent conjugate that facilitated QM formation and extended its short lifetime was capable of migration within individual duplexes.20,41 This feat was likely achieved by a random walk sustained by the continual regeneration of QMs and their diffusion to neighboring nucleophiles. Within a cell, this stochastic process has the potential to be guided by the vectorial nature of biological machines acting on DNA. The current study tested the ability of a helicase to perturb the dynamic partitioning of the original bisQMP.
Scheme 1.
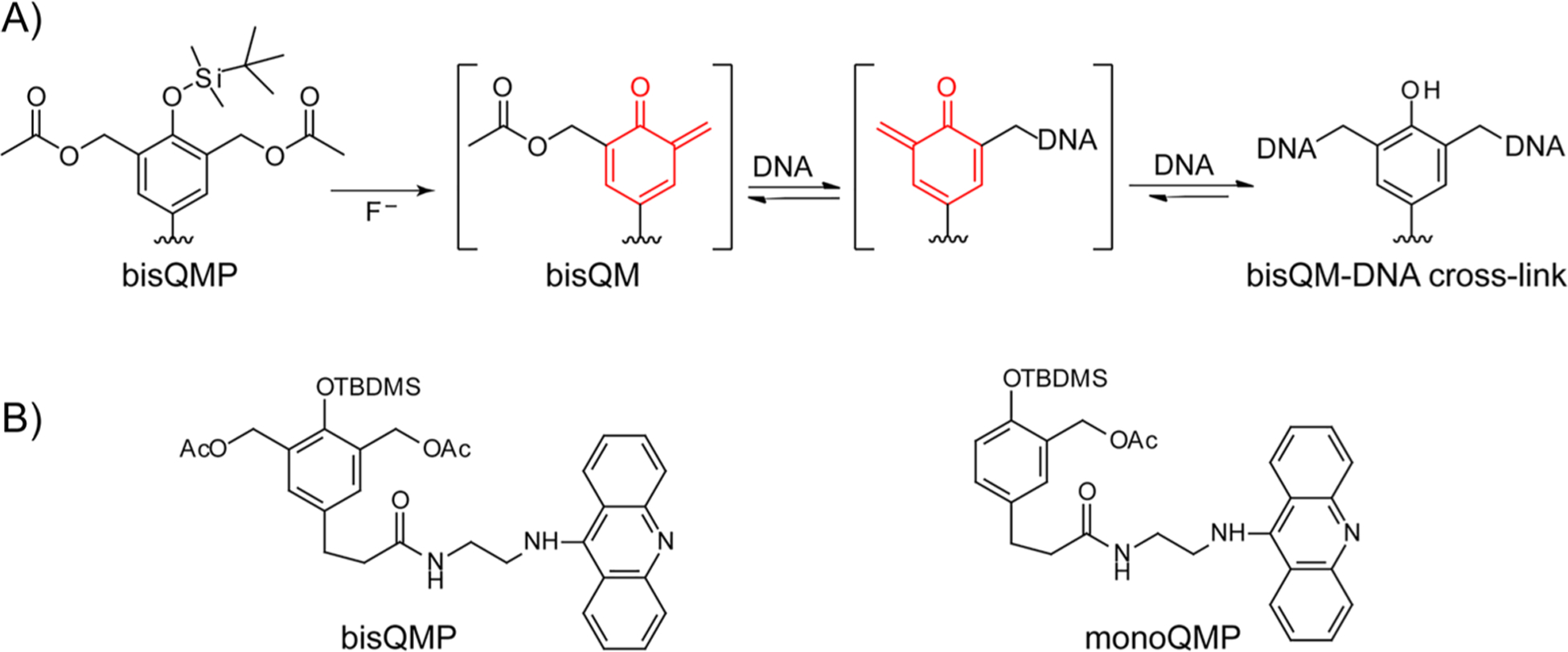
(A) Quinone Methide Deprotection, Generation, and Reversible Reaction with DNA and (B) Bifunctional and Monofunctional Quinone Methide Precursors
Scheme 2.
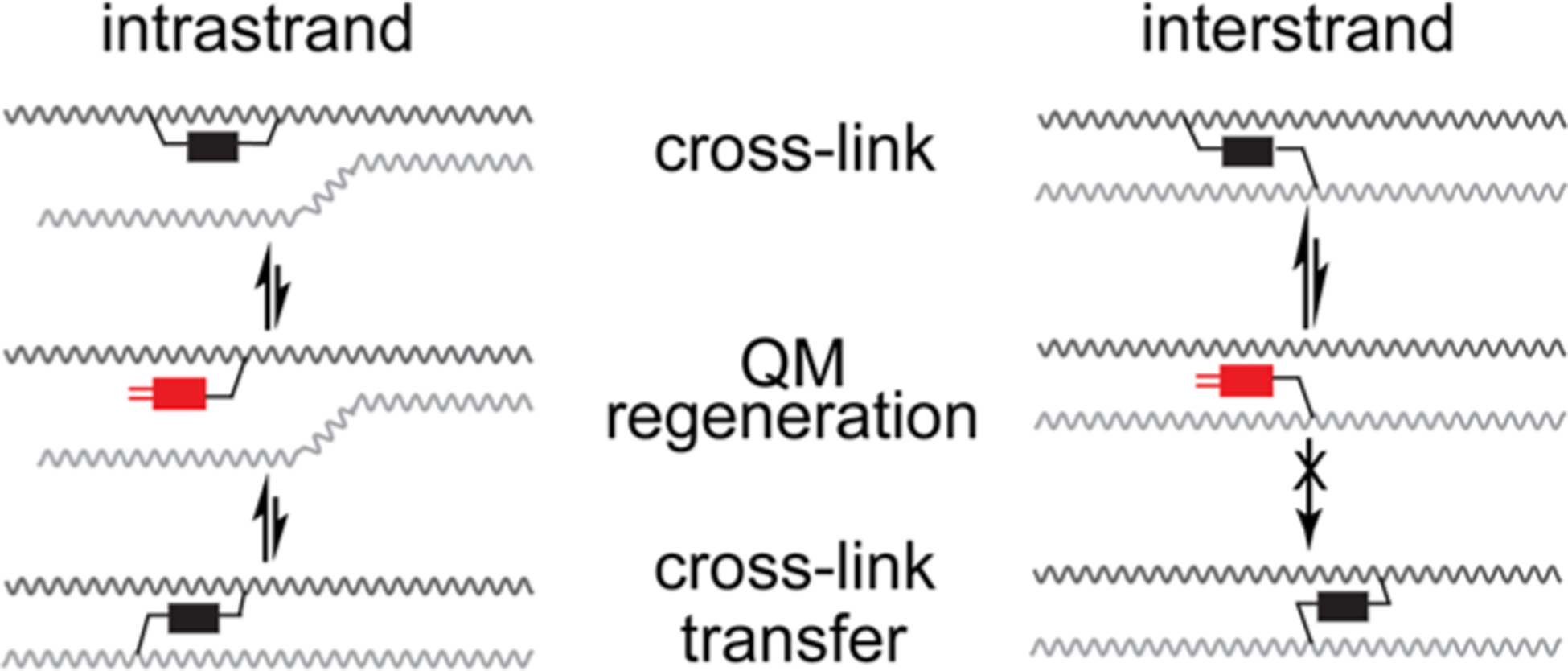
Dynamic Migration of Intra- and Interstrand Cross-Linking by a Bisfunctional and Reversible QM
The potential consequences of challenging a reversible cross-link with a helicase are manyfold. To begin identifying the possibilities, the helicase domain of the bacteriophage T7 gene 4 protein (T7gp4) was selected based on the plethora of catalytic and structural information available.42–46 T7gp4 is a homohexamer that surrounds and travels along one strand of a duplex (the translocation strand) and excludes the complementary strand from its center. DNA is unwound in the 5′ to 3′ direction with concomitant hydrolysis of dTTP in a mechanism that has been described as a Brownian ratchet.47–49 Accordingly, strand dissociation is not driven by destabilization of the duplex but rather by capture of single-stranded residues as they become available through natural breathing of base pairs. Accordingly, unwinding efficiency decreases as duplex stability (G/C content) increases.50,51 The most recent structural analysis indicates that the hexamer forms a spiral lock washer that advances two nucleobases at a time in a “hand-over-hand” mechanism.46 The helicase may also promote transfer of the cross-link by this same process after spontaneous regeneration of the QM intermediate. Subsequent transfer back to DNA could result in vectorial migration of the cross-link. Alternatively, transfer of the QM to protein could generate protein–DNA cross-links and transfer to water, as described below, could release the cross-link from DNA. Such a release was not observed for cross-links formed irreversibly by the nitrogen mustard mechlorethamine.
RESULTS AND DISCUSSION
T7gp4.
Most DNA cross-links investigated to date form irreversibly and create predictable obstacles to biological machines.52 In contrast, obstacles generated by dynamic cross-linking are far from predictable since their product profile may evolve with changing environment and DNA conformation. The effect of a biological machine on such a system has received little attention. For the initial foray into this problem, the hexameric ring helicase T7gp4 was selected due to its possible persistence at cross-links generated by bisQMP. Two previous reports had already suggested that T7gp4 could be trapped at a stable DNA adduct formed by benzo[a]pyrene for sufficient time to experience the reversibility of cross-linking by bisQMP.53,54 Stalling and slow dissociation of T7gp4 was also previously observed with a protein–DNA adduct.55 For simplicity, focus was further narrowed to the helicase function by heterologous expression of T7gp4 lacking its primase domain.56–59 Activity is easily assayed using well-defined oligonucleotide models of a replication fork. For this, the translocation strand contains a (T)n extension on its 5′-terminus (OD1) and the excluded strand contains a (T)m extension on its 3′ terminus (OD2).60 A standard duplex derived from the Richardson laboratory61 provided a positive control for DNA unwinding by T7gp4 in the presence of its preferred triphosphate dTTP using native polyacrylamide gel electrophoresis (PAGE; see the Supporting Information, Figure S1).
Cross-Linking a Model Replication Fork.
Use of poly(T) sequences for mimicking the single-stranded regions of a replication fork is convenient for the studies below since these avoid complications that could have arisen from alkylation or cross-linking of the single-stranded regions. Thymine residues are not sufficiently nucleophilic to react with bisQMP and thus modification is limited to the duplex regions.62 Standard treatment of OD1:OD2 with bisQMP (250 μM) for 2 h and subsequent removal of excess bisQMP and its hydrolysis products yielded the expected cross-link. This is evident by the product’s low mobility relative to single-stranded DNA on denaturing PAGE (Figure 1).63 These conditions represent the minimum necessary to provide the cross-linked duplex without excess modification. As described previously,63 the major site of reaction is guanine N7 as evident from lability to piperidine treatment (10%, 90 °C; Figure S2).41,63 Reaction also likely occurs at adenine N1 and cytosine N3 since these are targets of the simple unconjugated QM. Strands rich in guanine and thymine are still capable of cross-linking with their complementary strands rich in adenine and cytosine.40,62 Thus, the sites of cross-linking may be heterogeneous but all sites maintain the reversibility of QM reaction since all cross-linking can be transferred between strands in an exchange process described previously.40,41 Further characterization of the cross-linked DNA was not attempted since the analytical protocols to identify sites of modification have the potential to perturb the profile of products originally present during incubation with T7gp4.62,64
Figure 1.
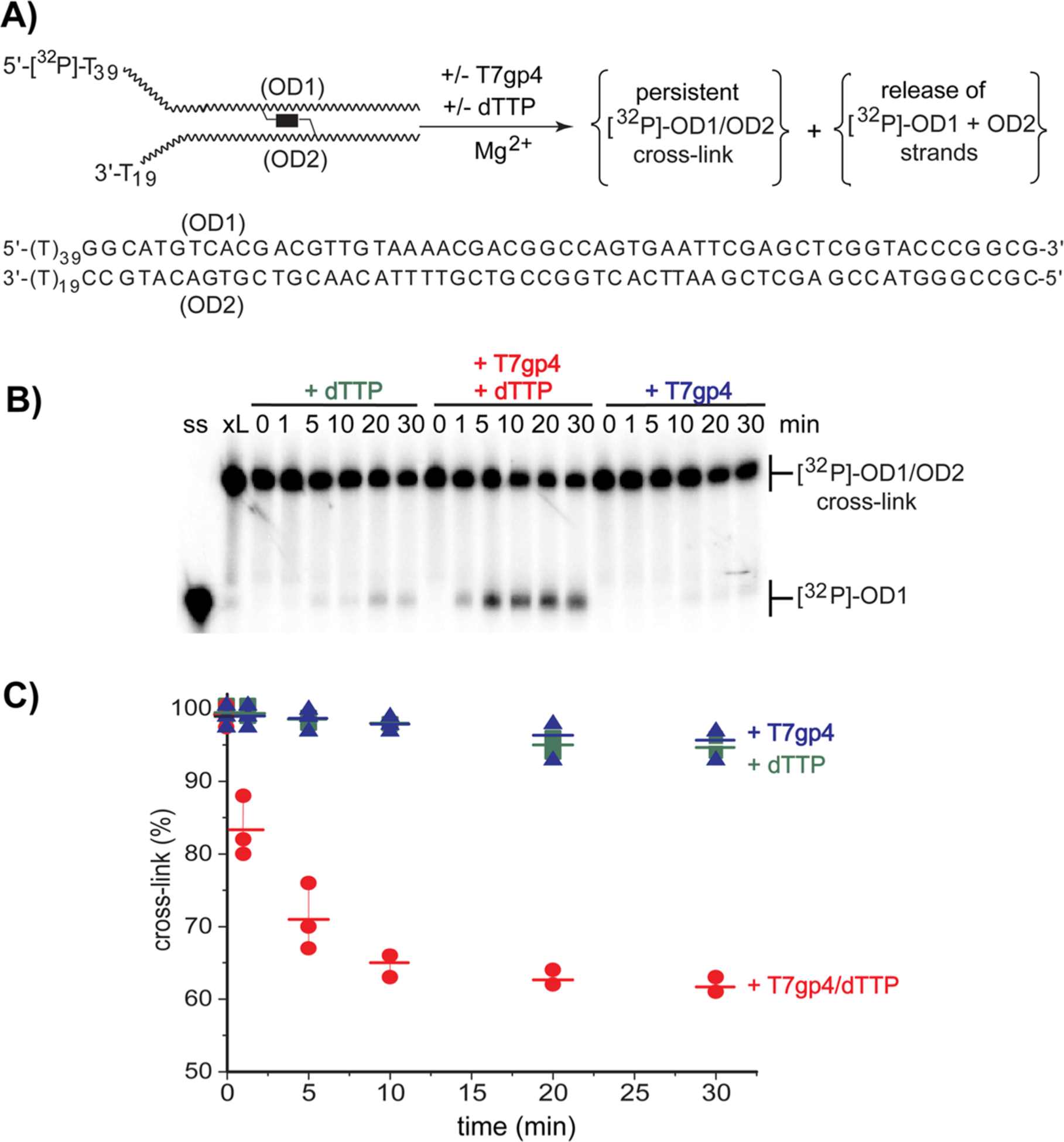
Unwinding DNA and release of a reversible interstrand cross-link. (A) A replication fork model (5′-[32P]-OD1:OD2, 10 nM) containing an interstrand cross-link formed by bisQMP and a radiolabel on the translocated strand was incubated in the alternative and combined presence of T7gp4 (55 nM monomer) and dTTP (1 mM). (B) After incubation at 37 °C for the indicated time, reaction was quenched by addition of EDTA (40 mM) and analyzed by denaturing polyacrylamide gel electrophoresis (PAGE, 10%). As electrophoretic standards, lane ss contains only 5′-[32P]-OD1 and lane xl contains 5′-[32P]-OD1:OD2 cross-linked by bisQMP. (C) The remaining cross-link (% based on total radiolabel) was determined by phosphoimagery in three replicates and their average values are indicated by the cross-bars.
Release of a Reversible Cross-Link.
Many scenarios were possible after confrontation between the bisQM cross-link and the hexameric helicase T7gp4 as summarized in Scheme 3. If QM regeneration was slow relative to T7gp4 turnover, then a stalled complex could form as noted earlier for benzo[a]pyrene adducts.53,54 Dissociation of the helicase from the cross-link was also possible since T7gp4 can shift from single- to double-stranded translocation.50 Alternatively, if QM regeneration is concurrent with T7gp4 translocation three additional results were possible. Trapping of this intermediate with water could release the cross-link. Trapping with DNA could promote cross-link migration and finally trapping with T7gp4 could lead to protein–DNA cross-linking. Each of these possibilities is considered below. To begin, T7gp4 does not appear to facilitate trapping of the regenerated QM by DNA or at least not to force migration away from the replication fork. This would have been evident from a decrease of guanine N7 adducts in the wake of the helicase and a corresponding increase of these adducts preceding DNA unwinding. No such vectorial redistribution of adducts was evident after incubating OD1:[32P]-OD2 in the presence of dTTP and T7gp4 and monitoring the adducts by piperidine treatment (Figure S2). Analysis of either strand was considered sufficient since migration of a cross-link necessarily involves both strands. Moreover, the helicase is shown to process adducts of each helical strand equivalently as described below.
Scheme 3.
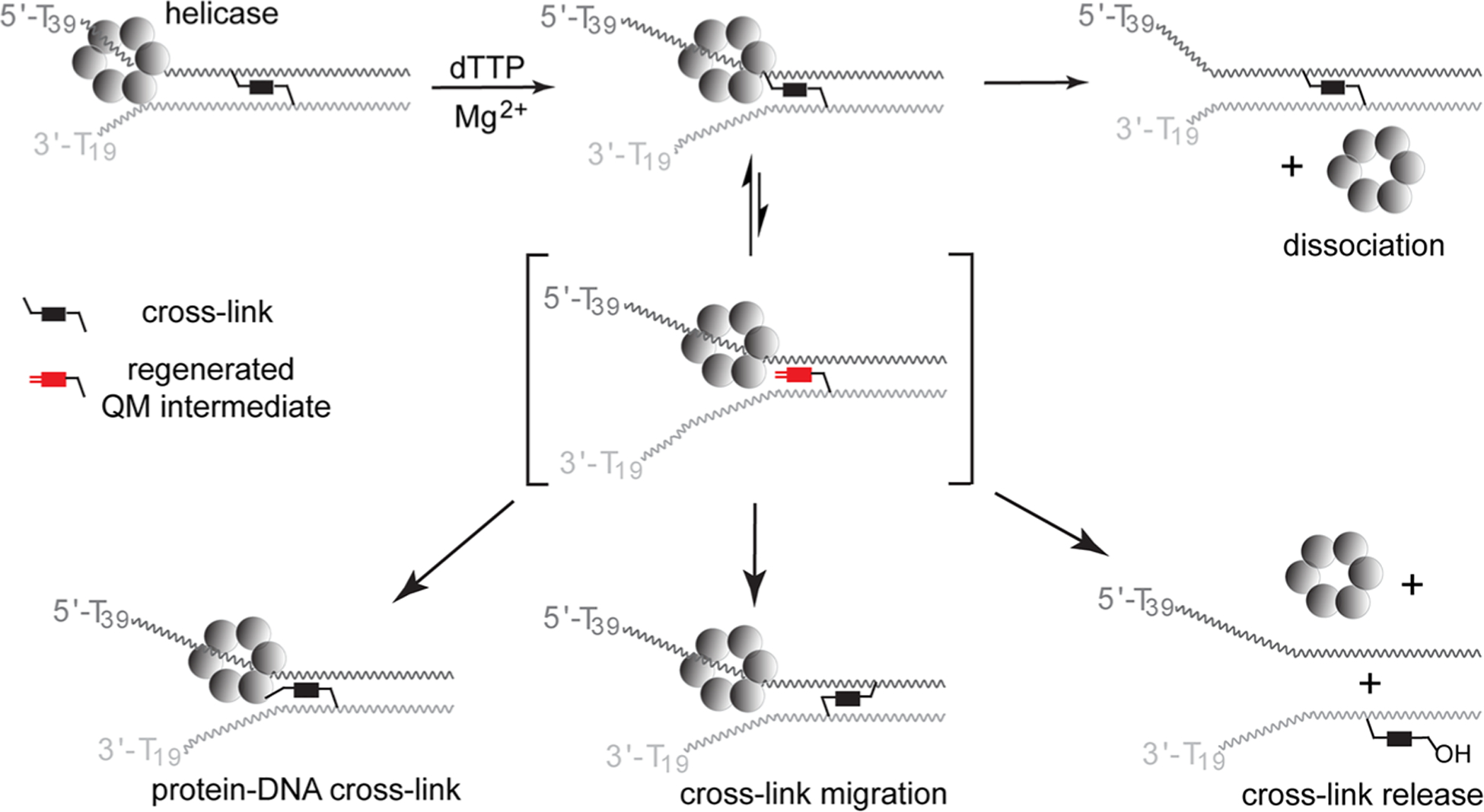
Confrontation between a Helicase and a Reversible Cross-Link May Have Varied Consequences
The helicase most notably promotes release of the QM cross-link as evident by the time-dependent formation of single-stranded [32P]-OD1 in the combined presence of T7gp4 and dTTP (Figure 1). In contrast, little single-stranded DNA was released by hydrolysis in the presence of either dTTP or T7gp4. Thus, this helicase is chemically competent to release the covalent and reversible cross-link although conversion to single-stranded DNA was not complete. Only 35–40% of the cross-links were reversed when added in a slight excess (8%) over the concentration of the active T7gp hexamer (Figure 1C). As expected, equivalent results were observed when the excluded strand (OD2) rather than the translocated strand (OD1) had been labeled (Figure S3). No equivalent release has yet been observed for irreversible modification based on alkylation or photodimerization.13,54,55
A broad range of phenomena were interrogated as possible sources for the heterogeneous response of T7gp4. Partial release of cross-linking was not a function of intercalation by the 9-aminoacridine appendage linked to the bisfunctional QMP. Treatment of OD1/[32P]-OD2 with this acridine moiety rather than its bisQMP conjugate did not inhibit or limit the helicase activity of T7gp4 as evident from a standard uwinding assay monitored by native PAGE (Figure S4). Similarly, partial release of the cross-link was not a function of DNA sequence. Approximately 35–40% of the cross-linking by bisQMP was also released from a second replication fork model (OD3:OD4) that differed in sequence from OD1:OD2. This alternative is based on a guanine-rich duplex that had previously been shown to support spontaneous cross-link migration of a related bisfunctional QMP (Figure S5).41 Again, release of cross-linking required the combined presence of T7gp4 and dTTP and was independent of which strand was monitored by radiolabeling (Figure S6). Common to these sequences is the ability to make a preponderance of guanine N7-adenine N1 and guanine N7-cytosine N3 cross-links. The former is expected to be significantly more labile than the latter by extrapolation from previous studies on alkylation of nucleosides by a related QM.63 Thus, the heterogeneous response of T7gp4 likely reflects a differential lability of cross-link products.
The limited release of cross-linking was not a function of turnover inactivation or substrate depletion. Supplementing the standard assay above with additional T7gp4 and dTTP alone or together after the initial 30 min did not promote further release of cross-linking beyond that caused spontaneous hydrolysis during a second 30 min incubation (Figures 2, S7, and S8). Extended incubation for another 3 also did not affect the fraction of remaining cross-link. Thus, the stable adducts persisted without change during this period and similarly the released strands did not regenerate cross-linking. This latter result would have been possible if helicase had promoted conversion of an inter- to intrastrand cross-link. The intrastrand product was already known preserve the dynamic reactivity of QMs for subsequent regeneration of interstrand cross-linking (Scheme 2).40
Figure 2.
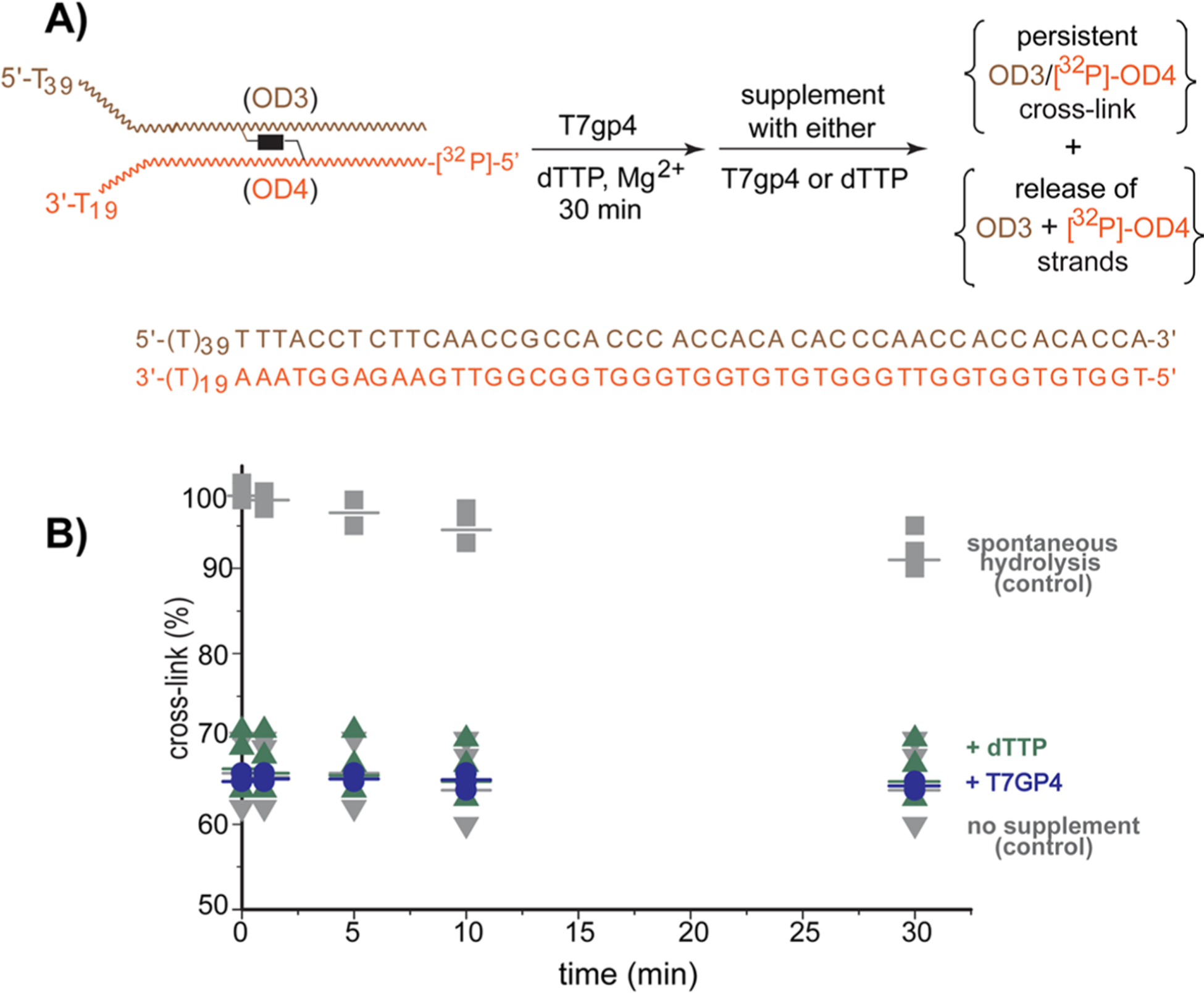
Supplementing conditions after initial unwinding of DNA containing a reversible interstrand cross-link. (A) A second replication fork model (OD3:5′-[32P]-OD4, 10 nM) containing a cross-link formed by bisQMP and a radiolabel on the excluded strand was incubated as described in Figure 1 for 30 min before supplementing with another aliquot of either T7gp4 (55 nM monomer) or dTTP (1 mM). Incubations were continued at 37 °C for the indicated times and then quenched with EDTA (40 mM). (B) Reaction products were detected after separation by denaturing PAGE (10%) and quantified relative to total radiolabel (%). Data represent three replicates and average values are indicated by the cross-bars. The control entitled spontaneous hydrolysis contained no T7gp4 or dTTP throughout the incubations and that entitled no supplement contained the standard concentrations of T7gp4 and dTTP in the initial incubation but no additional T7gp4 or dTTP in the subsequent incubation.
Resistance of an Irreversible Cross-Link.
T7gp4 has been challenged by DNA containing adducts formed by activated metabolites53 and proteins55 as well as by other lesions such as cyclopyrimidine dimers13 and abasic sites,65 there is a paucity of investigations on interstrand cross-links. The lack of attention to this may be blamed on obviousness of the expected response. There is no precedence for T7gp4 to promote cleavage of covalent bonds within DNA or its derivatives and thus irreversible cross-links should remain inert to unwinding and release. However, this prediction now required confirmation since T7gp4 was capable of dissociating a reversible cross-link formed by bisQMP. Accordingly, OD3:OD4 was treated with the nitrogen mustard mechlorethamine to form its well characterized and irreversible cross-link of guanine N7 in duplex DNA.66,67 Note that this common cross-linking reagent is not efficient when compared to that bisQMP. A maximum of ~60% interstrand cross-linking was achieved when the DNA was treated with 5 mM mechlorethamine as is evident by the low mobility species from denaturing PAGE (Figures 3 and S9). In contrast, 100% interstrand cross-linking was achieved with only 250 μM of bisQMP (Figures 1 and S5). Incubation of the mechlorethamine-treated OD3:OD4 with dTTP, T7gp4, or their combination did not change the fraction of cross-linking. Thus, T7gp4 promoted release of the cross-link formed bisQMP is not common to other adducts and is likely dependent on the reversibility of QM formation and subsequent alkylation.40,41,68
Figure 3.
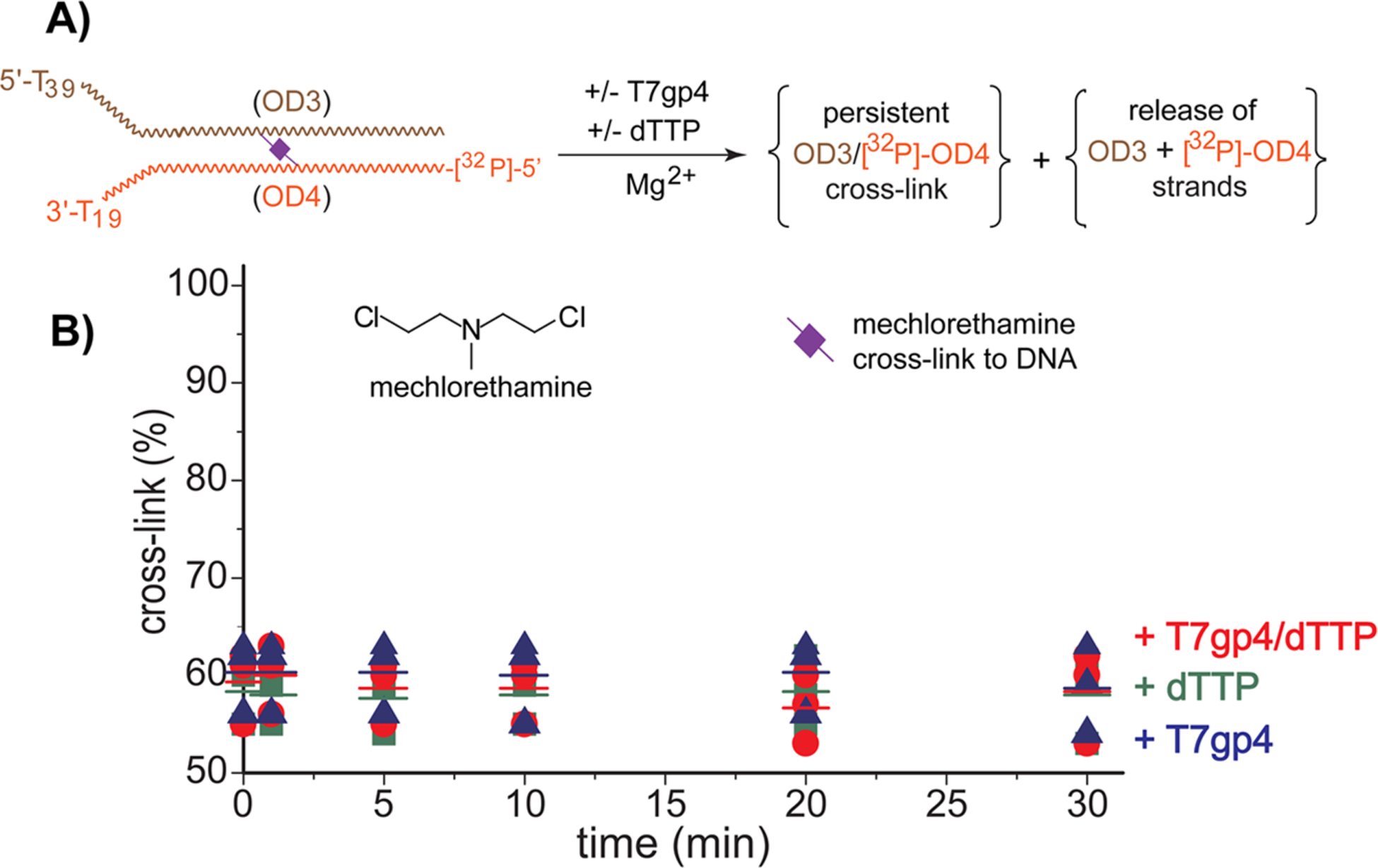
Challenging T7gp4 with an irreversible cross-link. (A) The replication fork model (OD3:5′-[32P]-OD4) containing a cross-link formed by mechlorethamine and a radiolabel on the excluded strand was treated in the alternative and combined presence of T7gp4 and dTTP as described in Figure 1. (B) Reaction products were detected after separation by denaturing PAGE (10%) and quantified relative to total radiolabel (%). Data represent three replicates and average values are indicated by the cross-bars.
Reversible Cross-Links Formed by bisQMP Do Not Trap T7gp4.
A bulky adduct formed by a metabolite of benzo[a]pyrene and located on the translocating strand sequestered the helicase in a stable complex with dTTP.53 Similarly, adducts formed by proteins of greater than 14 kDa also sequestered T7gp4 in a complex when located on the translocating strand.55 In contrast, a small perturbation formed by a cyclopyrimidine dimer in the translocating strand did not stall T7gp413 while an abasic site elicited a heterogeneous response ranging from no interference of unwinding to stalling or dissociating T7gp4 alternatively.65 The inability of a supplementary aliquot of T7gp4 to drive additional release of cross-links formed by bisQMP (Figure 2) suggested that this reaction was not limited by active enzyme. Thus, the cross-link did not likely sequester or inactivate the helicase by stable association through either covalent or noncovalent interactions. Although protein–DNA cross-linking was considered as a possible consequence of QM regeneration (Scheme 3), no evidence supported such transfer to T7gp4. The resulting product of this reaction would have been apparent by its limited electrophoretic mobility similar to that observed previously from treatment of a nucleosome core particle with bisQMP.69 No such species was detected by denaturing PAGE after incubation of T7gp4, dTTP, and DNA containing a bisQMP-generated cross-link (for example, see Figure 1).
Stable complexation of T7gp4 by noncovalent association with a bisQMP cross-link was similarly unlikely. No loss of T7gp4 activity was noticeable after preincubation with dTTP and the cross-linked OD3:OD4 (Figure S10). Under these conditions, the helicase had the opportunity to translocate to the cross-link and form a ternary complex with dTTP similar to that formed by DNA containing the benzo[a]pyrene adduct.53 In the absence of dTTP, T7gp4 would remain independent of the DNA and act as a control for the maximum helicase activity available without sequestration. Cross-link release from OD3:OD4 was experimentally equivalent after preincubation in the presence and absence of dTTP. Thus, T7gp4 dissociation is likely when encountering a DNA cross-link formed by bisQMP (Scheme 3). Escape is possible by slippage of T7gp4 as observed by others during unwinding of DNA under low dTTP concentrations or when dTTP was replaced by ATP.51,70 However, the high concentration of dTTP in the current experiments is expected to minimize this possibility. More likely, T7gp4 switches from tracking on a single strand of DNA to tracking on a double strand of DNA and ultimately dissociating from the duplex terminus.50 Previously, such switching was observed to increase when the duplex was stabilized by high G/C content.50 Cross-linking offers the ultimate stability for duplex DNA and likely induces a similar switch to double-stranded translocation. Another hexameric ring helicase DnaB also demonstrated the ability to translocate past an interstrand DNA cross-link.71 This switch is likely responsible in part for the heterogeneous response of T7gp4 to the cross-links formed by bisQMP (Figure 1). Only the most dynamic cross-links may be processed by T7gp4 prior to its switch to duplex translocation. The remaining fraction of cross-links that regenerate their QM intermediate more slowly may miss the opportunity to be diverted by T7gp4.
Strand Specificity of QM Release by T7gp4.
In principle, a single release of the QM from either the translocated or excluded strand is sufficient to break cross-linking (for example, Figures 1, S3, S5, and S6). However, T7gp4 has previously been shown to respond to only modifications encountered on the translocated strand since this must weave through the central channel created by its homohexamer.13,54,55 In contrast, little perturbation is caused by equivalent modification on the excluded strand. By extrapolation, loss of the QM could be expected from the translocated strand to a much greater extent than the excluded strand. To test this possible dichotomy, OD3:OD4 was alkylated with a monofunctional analogue of bisQMP that contained only one site for generating a QM (monoQMP, Scheme 1 and Figure 4A).72 Alkylation of OD3 after treatment with monoQMP was evident from the change in electrophoretic migration under denaturing conditions (Figure 4B). Incubation of this alkylated duplex with T7gp4 and dTTP under standard conditions released ~50% of the QM to regain a mobility of the original parent OD3 (Figure 4C). This is comparable to or perhaps a little more efficient than the release of the corresponding cross-link (Figure 1). Analogous to the release of cross-links, release of alkylation was not observed in the absence of either T7gp4 or dTTP and was limited to a fraction of the initial population. As described above this heterogeneous response is likely due to competition between T7gp4 translocation and QM regeneration during association with T7gp4 (Scheme 3).
Figure 4.
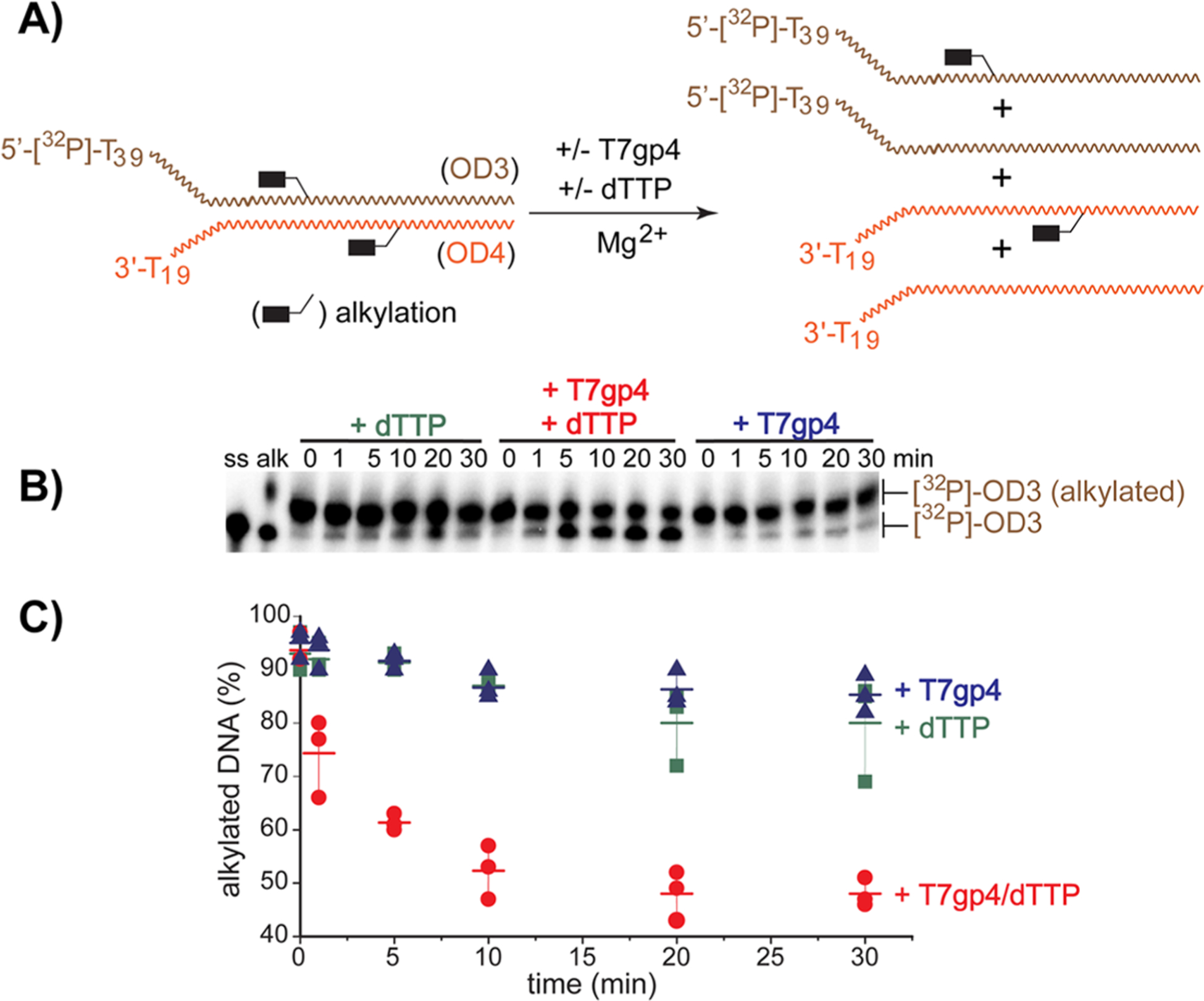
Unwinding DNA and release of a reversible DNA adduct. (A) A model replication fork (5′-[32P]-OD3:OD4, 10 nM) containing reversible adducts formed by monoQMP and a radiolabel on the translocated strand was incubated in the alternative and combined presence of T7gp4 (55 nM monomer) and dTTP (1 mM). (B) After incubation at 37 °C for the indicated time, reaction was quenched by addition of EDTA (40 mM) and analyzed by denaturing PAGE (10%). As electrophoretic standards, lane ss contains only 5′-[32P]-OD3 and lane alk contains OD3 after treatment with monoQMP. (C) The remaining cross-link (% based on total radiolabel) was measured by phosphoimagery in three replicates and their average values are indicated by the cross-bars.
Surprisingly, equivalent release of DNA alkylation was detected from labeled OD4, the excluded strand, after incubating the alkylated OD3:OD4 duplex with T7gp4 and dTTP (Figure S11). Electrophoretic characterization of OD4 was complicated by its high guanine content but time-dependent regeneration of the parent sequence was still confirmed qualitatively. Thus, the helicase was equally competent to promote loss of adducts formed between the QM intermediate and both the translocated and excluded strands of DNA. The conformational constraints of DNA cross-linking were not required to stall T7gp4 for sufficient time to participate in the dynamic partitioning of the QM adducts. Moreover, adducts formed by monoQMP likely represent only a minimal barrier for T7gp4 since they are significantly smaller than many of the protein adducts that were accommodated within the central channel of T7gp4.55 Even less perturbation of T7gp4 can be expected for modifications on the excluded strand.13,54,55 Still, the alkylation products of monoQMP were released from both DNA strands under equivalent conditions used to observe release of cross-links.
Alkylation of DNA with monoQMP also provided an important electophoretic standard for characterizing the products formed after release of the cross-link by T7gp4. Arguments based on simplicity would contend that only a single attachment of the cross-link would be lost during release of the DNA strands (Scheme 3). However, the ability of T7gp4 to remove adducts of monoQMP from both the translocated and excluded strands suggests an additional possibility of regenerating both original parent strands and full release of the cross-link. By denaturing gel electrophoresis, [32P]-OD4 was clearly regenerated by T7gp4 after release from the cross-linked OD3:[32P]-OD4 and no detectable level of the monoalkylated derivative of [32P]-OD4 was observed (Figure S12). Our focus on OD4, the excluded strand in the model replication fork, was selected since it was least expected to be influenced by the helicase. If release had resulted from a statistical loss of just one attachment from either OD4 or OD3, a mixture of [32P]-OD4 and its alkylated derivative should have been evident. The absence of the alkylated intermediate suggests that release of the cross-link from both strands was promoted by T7gp4 in rapid succession. A subsequent and nonenzymatic release of a monoadduct is too slow to account for regeneration of the parent strand as illustrated by the persistence of the monoadducts in Figure 4.
The QM intermediate regenerated from monoQMP alkylation of duplex DNA was previously shown to diffuse into solution despite the presence of the attached acridine for stabilizing DNA association.72 Once this intermediate escapes the confines of DNA, it can be quenched by water to form its benzyl alcohol derivative that is no longer capable of regenerating a QM under neutral conditions.68 Although regeneration of the monofunctional QM is not likely promoted by T7gp4, subsequent release of this intermediate into solvent is likely facilitated by the unwinding of DNA by T7gp4. Release of the cross-link requires more than dissociation of the transient QM intermediate since it remains covalently anchored to one strand (Scheme 3). The bisQMP is capable of tandem and not concurrent formation of QM intermediates, and thus, the cross-link cannot be expelled from DNA in a concerted manner. Instead, quenching or migration of the QM is a stepwise process.40,41 In the absence of T7gp4, the transient QM formed from a cross-link typically rebounds very efficiently within the DNA to restore cross-linking. Prior attempts to quench this QM intermediate within duplex DNA had limited success even after addition of the highly nucleophilic 2-mercaptoethanol in significant excess (3 mM).37,68 However, the ability of T7gp4 to act as a Brownian ratchet may capture DNA as it is released after QM regeneration. T7gp4 may also subsequently suppress return of the DNA for cross-linking and facilitate water addition to quench the transient QM intermediate. T7gp4 must then ultimately hold the strand containing the remaining QM equivalent in a position that favors quenching of the second QM equivalent when it next forms spontaneously (Scheme 4).
Scheme 4.
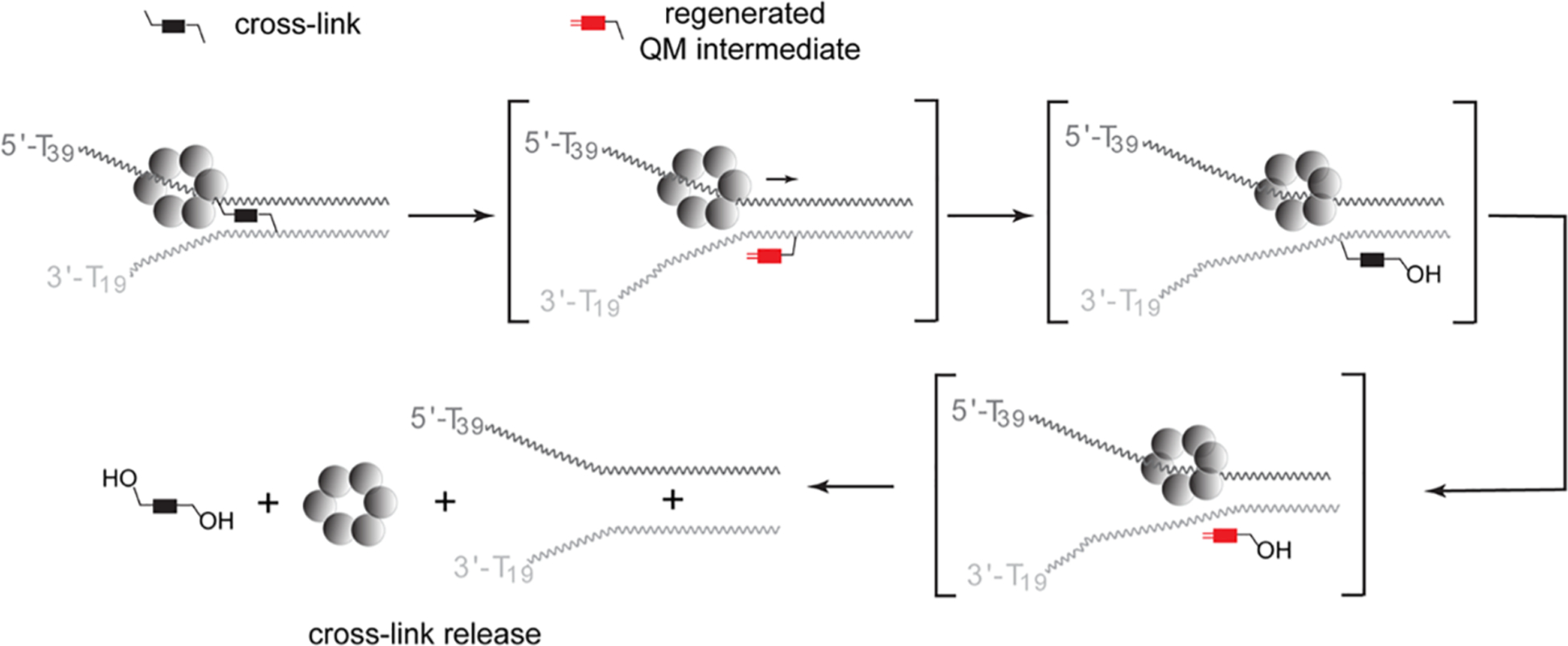
Reversible Alkylation May Persist or Release during DNA Unwinding
Deglycosylation Is Not Responsible for Release of DNA Adducts.
Deglycosylation of the nucleobases modified by bisQMP presents an alternative mechanism for release of cross-linking induced by T7gp4. Although T7gp4 is not known to catalyze deglycosylation, alkylation at such sites as the N7 of guanine greatly facilitates deglycosylation. Such a process is the basis for Maxam–Gilbert sequencing of guanine and is now used frequently for footprinting nucleic acid structures. Guanine N7 is also a predominant target of modification by bisQMP63 and a nucleoside model system suggested an adduct at guanine N7 equally partitions between QM regeneration and deglycosylation.62 DNA alkylation by the monofunctional monoQMP induces deglycosylation as well although this was observed only after incubation times (h) much greater than those reported here (min).72 The potential for deglycosylation to release a QM adduct was examined by the ability of human apurinic/apyrimidinic endonuclease (APE1) to hydrolyze OD3:OD4 after treatment with monoQMP. No fragmentation of the labeled OD4 was detected after the alkylated duplex of OD3:OD4 was incubated at 37 °C for 30 min in the presence or absence of T7gp4 and dTTP prior to treatment with APE1. In contrast, fragmentation of a control sequence containing an abasic site was readily apparent under these conditions (Figure S13). Fragmentation was also observed after the incubation was treated with piperidine to confirm modification at guanine N7. Thus, there is no evidence to suggest that adducts are released by deglycosylation. The dynamic nature of QM alkylation and regeneration remains the most likely source of the T7gp4-dependent loss of cross-linking. Confrontation between this helicase and the dynamic products of the QMs has now demonstrated only two of the many possible responses: (i) release of QM adduct and (ii) dissociation of T7gp4 from the replication forks (Schemes 3 and 4).
CONCLUSION
The ultimate consequences of reversible covalent modification of DNA are likely dictated by the myriad of enzymes that confront such lesions in vivo. Many of these enzymes represent biological machines that translocate along DNA in a single direction while consuming energy. The helicase T7gp4 engages with the reversible reactions of bisQMP to alter cross-linking of DNA. This protein is a member of a large family of biological machines that forms ring structures and hydrolyzes nucleotide triphosphates to drive activities ranging from protein unfolding to nucleic translocation and unwinding.45 Although a small majority of QM-based products remained unaffected by unwinding and translocation, a significant fraction of the reversible cross-linking and alkylation was released by T7gp4 in an energy dependent (dTTP) manner. The ability of this helicase to unwind DNA as a Brownian ratchet and trap adjacent nucleobases after spontaneous breathing of the base pairs may be similarly applicable to strands transiently released after spontaneous QM generation (Scheme 4).47–49 This process can block a rebound reaction with DNA and leave the transient QM exposed for quenching by water. This result is quite distinct from its response to stable DNA adducts that remain unchanged when blocking or passing through the helicase channel.13,53–55,65 Most unusual is the ability of T7gp4 to promote loss of reversible adducts from both the translocated and excluded strand. DNA adducts on the excluded strand, whether large or small, typically do not perturb the unwinding activity of this helicase even though interactions between the excluded strand and T7gp4 are possible in the absence of an associated DNA polymerase.46,61 Incorporating this polymerase and the primase domain of T7gp4 into future studies on cross-link release will provide a useful prelude to further analysis of the more complex hexameric helicases from eukaryotes that act at replication forks. The potential for helicases associated with human disease to release reversible adducts remains to be determined as well. The potential for these helicases and, more generally, other biological machines to act similarly will likely be predicated on the relative kinetics of translocation and lesion reversibility.
EXPERIMENTAL SECTION
General Procedures.
The cross-linking agent bisQMP was prepared as described previously63 and the alkylating reagent monoQMP was a gift from Blessing Deeyaa.72 The plasmid harboring the gene for the 56 kDa fragment of T7gp4 was a gift of Prof. Charles Richardson and this construct was expressed and purified based on procedures published previously.59 Oligonucleotides were synthesized and desalted by Integrated DNA Technologies (IDT). These were further purified by PAGE and radiolabeled as indicated with γ-[32P]-ATP (PerkinElmer) following manufacturer protocols. Complementary oligonucleotides were hybridized by heating at 95 °C for 5 min, followed by slow cooling to room temperature over 2 h. A Typhoon 9410 phosphorimager equipped with ImageQuant TL software was used to detect and quantify radiolabeled materials. Product yields are reported (%) relative to total material labeled in each determination. Oligonucleotide concentration was calculated from absorbance at 260 nm and the extinction coefficients provided by the manufacturer. Protein concentration was determined by a Bradford assay (Bio-Rad).
Cross-Linking and Alkylation of Duplex DNA with the Quinone Methide Reagents.
Equimolar concentrations of oligonucleotides were annealed in MES (10 mM pH 7) and NaF (50 mM). Reaction was initiated by addition of the appropriate bisQMP (250 μM unless stated otherwise) or monoQMP (250 μM unless stated otherwise) in acetonitrile (20% final concentration) under ambient conditions for 2 h. Excess reagent and its low molecular weight products were removed by passing the reaction mixtures through a Bio-Rad P6Micro Bio-Spin column (1000 g, 4 min). The eluant was stored at −20 °C until use.
Cross-Linking Duplex DNA with Mechlorethamine.
OD3 and OD4 (300 nM each) were annealed in potassium phosphate (40 mM, pH 8) and NaCl (10 mM). The reaction was initiated by addition of a fresh solution of mechlorethamine (5 mM final concentration) in potassium phosphate (40 mM, pH 8) and NaCl (10 mM). The reaction was maintained at 37 °C for 3 h, and excess reagent and its low molecular weight products were removed as described above with a Bio-Rad P6Micro Bio-Spin column. The eluant was stored at −20 °C until use.
DNA Unwinding by T7gp4.
61 Cross-linked and alkylated DNA (10 nM) was combined with dTTP (1 mM) in Tris-HCl (40 mM, pH 7.5), MgCl2 (10 mM), and potassium glutamate (50 mM) before reaction was initiated by addition of T7gp4 (55 nM monomer) in potassium phosphate (20 mM pH 7.5) and 50% glycerol. Samples were incubated at 37 °C and quenched at the indicated times with EDTA (40 mM) and formamide loading dye before freezing with dry ice. Samples were then thawed at room temperature for 5 min and analyzed by denaturing PAGE (10%).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Blessing D. Deeyaa for the gift of monoQMP; Charles C. Richardson and Seung-Joo Lee for help, advice, and materials associated with T7gp4. This work was supported in part by the National Science Foundation (CHE-1405123, SER) and National Institute of General Medical Sciences (T32GM080189, SRB).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrestox.0c00413.
General protocols and T7gp4 isolation, additional studies on unwinding of cross-linked DNA, release of alkylation formed by monoQMP, DNA fragmentation after piperidine and APE1 treatment, and Figures S1–S13 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Geacintov NE, Cosman M, Hingerty BE, Amin S, Broyde S, and Patel DJ (1997) NMR solution structures of stereoisomeric covalent polycyclic aromatic carcinogen-DNA adducts: principles, patterns and diversity. Chem. Res. Toxicol 10, 111–146. [DOI] [PubMed] [Google Scholar]
- (2).Guengerich FP (2006) Interactions of carcinogen-bound DNA with individual DNA polymerases. Chem. Rev 106, 420–452. [DOI] [PubMed] [Google Scholar]
- (3).Fedeles BI, and Essigmann JM (2018) Impact of DNA lesion repair, replication and formation on the mutational spectra of environmental carcinogens, aflatoxin B1 as a case study. DNA Repair 71, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).McCullough AK, and Lloyd RS (2019) Mechanisms underlying aflatoxin-associated mutagenesis – implications in carcino-genesis. DNA Repair 77, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Plastaras JP, Dedon PC, and Marnett LJ (2002) Effects of DNA structure on oxopropenylation by the endogenous mutagens malondialdehyde and base propenal. Biochemistry 41, 5033–5042. [DOI] [PubMed] [Google Scholar]
- (6).Riggins JN, Daniels S, Rouzer CA, and Marnett LJ (2004) Kinetic and thermodynamic analysis of the hydrolytic ring-opening of the malondialdehyde-deoxyguanosine adduct, 3-(2′-deoxy-D-erythro-pentofuranosyl)pyrimido[1,2-]Purin-10(3h)-one. J. Am. Chem. Soc 126, 8237–8243. [DOI] [PubMed] [Google Scholar]
- (7).Stone MP, Cho Y-J, Huang H, Kim H-Y, Kozekov ID, Kozekova A, Wang H, Minko IG, Lloyd RS, Harris CM, and Rizzo CJ (2008) Interstrand DNA cross-links induced by α,β-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc. Chem. Res 41, 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Knutson CG, Rubinson EH, Akingbade D, Anderson CS, Stec DF, Petrova KV, Kozekov ID, Guengerich FP, Rizzo CJ, and Marnett LJ (2009) Oxidation and glycolytic cleavage of etheno and propano DNA base adducts. Biochemistry 48, 800–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Codreanu SG, Ullery JC, Zhu J, Tallman KA, Beavers WN, Porter NA, Marnett LJ, Zhang B, and Liebler DC (2014) Alkylation damage by lipid electrophiles targets functional protein systems. Mol. Cell. Proteomics 13, 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dedon PC, Plastaras JP, Rouzer CA, and Marnett LJ (1998) Indirect mutagenesis by oxidative DNA damage: formation of the pyrimidopurinone adduct of deoxyguanosine by base propenal. Proc. Natl. Acad. Sci. U. S. A 95, 11113–11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zewail-Foote M, and Hurley LH (2001) Differential rates of reversibility of ecteinascidin 743-DNA covalent adducts from different sequences lead to migration to favored bonding sites. J. Am. Chem. Soc 123, 6485–6495. [DOI] [PubMed] [Google Scholar]
- (12).Suhasini AN, and Brosh RM (2010) Mechanistic and biological aspects of helicase action on damaged DNA. Cell Cycle 9, 2317–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sun B, Pandey M, Inman JT, Yang Y, Kashlev M, Patel SS, and Wang MD (2015) T7 replisome directly overcomes DNA damage. Nat. Commun 6, 10260 DOI: 10.1038/ncomms10260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Trakselis MA, Seidman MM, and Brosh RM (2017) Mechanistic insights into how CMG helicase facilitates replication past DNA roadblocks. DNA Repair 55, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).O’Donnell ME, and Li H (2018) The ring-shaped hexameric helicases that function at DNA replication forks. Nat. Struct. Mol. Biol 25, 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fairman-Williams ME, Guenther U-P, and Jankowsky E (2010) Sf1 and Sf2 helicases: family matters. Curr. Opin. Struct. Biol 20, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gao Y, and Yang W (2020) Different mechanisms for translocation by monomeric and hexameric helicases. Curr. Opin. Struct. Biol 61, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bolton JL, Turnipseed SB, and Thompson JA (1997) Influence of quinone methide reactivity on the alkylation of thiol and amino groups in proteins: Studies utilizing amino acid and peptide models. Chem.-Biol. Interact. 107, 185–200. [DOI] [PubMed] [Google Scholar]
- (19).Modica E, Zanaletti R, Freccero M, and Mella M (2001) Alkylation of amino acids and glutathione in water by ortho-quinone methides. reactivity and selectivity. J. Org. Chem 66, 41–52. [DOI] [PubMed] [Google Scholar]
- (20).Weinert EE, Dondi R, Colloredo-Melz S, Frankenfield KN, Mitchell CH, Freccero M, and Rokita SE (2006) Substituents on quinone methides strongly modulate formation and stability of their nucleophilic adducts. J. Am. Chem. Soc 128, 11940–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Minard A, Liano D, Wang X, and Di Antonio M (2019) The unexplored potential of quinone methides in chemical biology. Bioorg. Med. Chem 27, 2298–2305. [DOI] [PubMed] [Google Scholar]
- (22).Meier BW, Gomez JD, Zhou A, and Thompson JA (2005) Immunochemical and proteomic analysis of covalent adducts formed by quinone methide tumor promoters in mouse lung epithelial cell lines. Chem. Res. Toxicol 18, 1575–1585. [DOI] [PubMed] [Google Scholar]
- (23).Tomasz M (1995) Mitomycin C: small, fast and deadly (but very selective). Chem. Biol 2, 575–579. [DOI] [PubMed] [Google Scholar]
- (24).Dowers TS, Qin Z-H, Thatcher GRJ, and Bolton JL (2006) Bioactivation of selective estrogen receptor modulators (SERMs). Chem. Res. Toxicol 19, 1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang Y, Fan H, Balakrishnan K, Lin Z, Cao S, Chen W, Fan Y, Guthrie QA, Sun H, Teske KA, Gandhi V, Arnold LA, and Peng X (2017) Hydrogen peroxide activated quinone methide precursors with enhanced DNA cross-linking capability and cytotoxicity towards cancer cells. Eur. Eur. J. Med. Chem 133, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).McLaughlin MF, Massolo E, Cope TA, and Johnson JS (2019) Phenolic oxidation using H2O2 via in situ generated para-quinone methides for the preparation of para-spiroepoxydienones. Org. Lett 21, 6504–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Chatterjee M, and Rokita SE (1991) Sequence specific alkylation of DNA activated by an enzymatic signal. J. Am. Chem. Soc 113, 5116–5117. [Google Scholar]
- (28).Zhou F, Fu T, Huang Q, Kuai H, Mo L, Liu H, Wang Q, Peng Y, Han D, Zhao Z, Fang X, and Tan W (2019) Hypoxia-activated pegylated conditional aptamer/antibody for cancer imaging with improved specificity. J. Am. Chem. Soc 141, 18421–18427. [DOI] [PubMed] [Google Scholar]
- (29).Wang P, Liu R, Wu X, Ma H, Cao X, Zhou P, Zhang J, Weng X, Zhang X-L, Qi J, Zhou X, and Weng L (2003) A potent, water-soluble and photoinducible DNA cross-linking agent. J. Am. Chem. Soc 125, 1116–1117. [DOI] [PubMed] [Google Scholar]
- (30).Kralj M, Uzelac L, Wang Y-H, Wan P, Tireli M, Mlinarić-Majerski K, Piantanida I, and Basarić N (2015) Enhancement of antiproliferative activity by phototautomerization of anthrylphenols. Photochem. Photobiol. Sci 14, 1082–1092. [DOI] [PubMed] [Google Scholar]
- (31).Perez-Ruiz R, Molins-Molina O, Lence E, Gonzalez-Bello C, Miranda MA, and Jimenez MC (2018) Photogeneration of quinone methides as latent electrophiles for lysine targeting. J. Org. Chem 83, 13019–13029. [DOI] [PubMed] [Google Scholar]
- (32).Dunlap T, Piyankarage SC, Wijewickrama GT, Abdul-Hay S, Vanni M, Litosh V, Luo J, and Thatcher GRJ (2012) Quinone-induced activation of Keap1/Nrf2 signaling by aspirin prodrugs masquerading as nitric oxide. Chem. Res. Toxicol 25, 2725–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Weinschenk L, Schols D, Balzarini J, and Meier C (2015) Nucleoside diphosphate prodrugs: nonsymmetric dippro-nucleotides. J. Med. Chem 58, 6114–6130. [DOI] [PubMed] [Google Scholar]
- (34).Yoo D, Jung E, Noh J, Hyun H, Seon S, Hong S, Kim D, and Lee D (2019) Glutathione-depleting pro-oxidant as a selective anticancer therapeutic agent. ACS Omega 4, 10070–10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lönnberg T, Hutchinson M, and Rokita SE (2015) Selective alkylation of C-rich bulge motifs in nucleic acids by quinone methide derivatives. Chem. -Eur. J 21, 13127–13186. [DOI] [PubMed] [Google Scholar]
- (36).Hutchinson MA, Deeyaa B, Byrne SR, Williams SJ, and Rokita SE (2020) Directing quinone methide-dependent alkylation and cross-linking of nucleic acids with quaternary amines. Bioconjugate Chem. 31, 1486–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhou Q, and Rokita SE (2003) A general strategy for target-promoted alkylation in biological systems. Proc. Natl. Acad. Sci. U. S. A 100, 15452–15457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Rossiter CS, Modica E, Kumar D, and Rokita SE (2011) Few constraints limit the design of quinone methide-oligonucleotide self-adducts for directing DNA alkylation. Chem. Commun 47, 1476–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Huang C, Liu Y, and Rokita SE (2016) Targeting duplex DNA with the reversible reactivity of quinone methides. Signal Transd, Target Ther. 1, 16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wang H, and Rokita SE (2010) Dynamic cross-linking is retained in duplex DNA after multiple exchange of strands. Angew. Chem., Int. Ed 49, 5957–5960. [DOI] [PubMed] [Google Scholar]
- (41).Fakhari F, and Rokita SE (2014) A walk along DNA using bipedal migration of a dynamic and covalent cross-linker. Nat. Commun 5, 5591. [DOI] [PubMed] [Google Scholar]
- (42).Donmez I, and Patel SS (2006) Mechanisms of a ring shaped helicase. Nucleic Acids Res. 34, 4216–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Patel SS, and Donmez I (2006) Mechanisms of helicases. J. Biol. Chem 281, 18265–18268. [DOI] [PubMed] [Google Scholar]
- (44).Lee S-J, and Richardson CC (2011) Choreography of bacteriophage T7 replication. Curr. Opin. Chem. Biol 15, 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lyubimov AY, Strycharska M, and Berger JM (2011) The nuts and bolts of ring-translocase structure and mechanism. Curr. Opin. Struct. Biol 21, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gao Y, Cui Y, Fox T, Lin S, Wang H, De Val N, Zhou ZH, and Yang W (2019) Structures and operating principles of the replisome. Science 363, eaav7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Jeong Y-J, Levin MK, and Patel SS (2004) The DNA-unwinding mechanism of the ring helicase of bacteriophage T7. Proc. Natl. Acad. Sci. U. S. A 101, 7264–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Rasnik I, Jeong Y-J, McKinney SA, Rajagopal V, Patel SS, and Ha T (2008) Branch migration enzyme as a Brownian ratchet. EMBO J. 27, 1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Lee S-J, and Richardson CS (2010) Molecular basis for recognition of nucleoside triphosphate by gene 4 helicase of bacteriophage T7. J. Biol. Chem 285, 31462–31471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Jeong Y-J, Rajagopal V, and Patel SS (2013) Switching from single-stranded to double-stranded DNA limits the unwinding processivity of ring-shaped T7 DNA helicase. Nucleic Acids Res. 41, 4219–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Syed S, Pandey M, Patel SS, and Ha T (2014) Single-molecule fluorescence reveals the unwinding stepping mechanism of replicative helicase. Cell Rep. 6, 1037–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Rycenga HB, and Long DT (2018) The evolving role of DNA inter-strand crosslinks in chemotherapy. Curr. Opin. Pharmacol 41, 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Brown WC, and Romano LJ (1989) Benzo[a]pyrene-DNA adducts inhibit translocation by the gene 4 protein of bacteriophage T7. J. Biol. Chem 264, 6748–6754. [PubMed] [Google Scholar]
- (54).Yong Y, and Romano LJ (1996) Benzo[a]pyrene-DNA adducts inhibit the DNA helicase activity of the bacteriophage T7 gene 4 protein. Chem. Res. Toxicol 9, 179–187. [DOI] [PubMed] [Google Scholar]
- (55).Nakano T, Miyamoto-Matsubara M, Shoulkamy MI, Salem AMH, Pack SP, Ishimi Y, and Ide H (2013) Translocation and stability of replicative DNA helicases upon encountering DNA-protein cross-links. J. Biol. Chem 288, 4649–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Bernstein JA, and Richardson CS (1988) Purification of a 56-kDa domponent of the bacteriophage T7 primase/helicase and characterization of its nucleoside 5′-triphosphatase activity. J. Biol. Chem 263, 14891–14899. [PubMed] [Google Scholar]
- (57).Bernstein JA, and Richardson CS (1988) A 7-kDa region of the bacteriophage T7 gene 4 protein is required for primase but not helicase activity. Proc. Natl. Acad. Sci. U. S. A 85, 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Guo S, Tabor S, and Richardson CS (1999) The linker region between the helicase and primase domains of the bacteriophage T7 gene 4 protein is critical for hexamer formation. J. Biol. Chem 274, 30303–30309. [DOI] [PubMed] [Google Scholar]
- (59).Zhu B, Lee SJ, and Richardson CS (2009) An in trans interaction at the interface of the helicase and primase domains of the hexameric gene 4 protein of bacteriophage T7 modulates their activities. J. Biol. Chem 284, 23842–23851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Ahnert P, and Patel SS (1997) Asymmetric interactions of hexameric bacteriophage T7 DNA helicase with the 5′- and 3′-tails of the forked DNA substrate. J. Biol. Chem 272, 32267–32273. [DOI] [PubMed] [Google Scholar]
- (61).Satapathy AK, Kulczyk AW, Ghosh S, van Oijen AM, and Richardson CS (2011) Coupling dTTP hydrolysis with DNA unwinding by the DNA helicase of bacteriophage T7. J. Biol. Chem 286, 34468–34478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Weinert EE, Frankenfield KN, and Rokita SE (2005) Time-dependent evolution of adducts formed between deoxynucleo-sides and a model quinone methide. Chem. Res. Toxicol 18, 1364–1370. [DOI] [PubMed] [Google Scholar]
- (63).Veldhuyzen WF, Pande P, and Rokita SE (2003) A transient product of DNA alkylation can be stabilized by binding localization. J. Am. Chem. Soc 125, 14005–14013. [DOI] [PubMed] [Google Scholar]
- (64).Pande P, Shearer J, Yang J, Greenberg WA, and Rokita SE (1999) Alkylation of nucleic acids by a model quinone methide. J. Am. Chem. Soc 121, 6773–6779. [Google Scholar]
- (65).Ma J-B, Chen Z, Xu C-X, Huang X-Y, Jia Q, Zou Z-Y, Mi C-Y, Ma D-F, Lu Y, Zhang H-D, and Li M (2020) Dynamic structural insights into the molecular mechanism of DNA unwinding by the bacteriophage T7 helicase. Nucleic Acids Res. 48, 3156–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Mattes WB, Hartley JA, and Kohn KW (1986) DNA sequence selectivity of guanine N-7 alkylation by nitrogen mustards. Nucleic Acids Res. 14, 2971–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Millard JT, Spencer RJ, and Hopkins PB (1998) Effect of nucleosome structure on DNA interstrand cross-linking reactions. Biochemistry 37, 5211–5219. [DOI] [PubMed] [Google Scholar]
- (68).Wang H, Wahi MS, and Rokita SE (2008) Immortalizing a transient electrophile for DNA cross-linking. Angew. Chem., Int. Ed 47, 1291–1293. [DOI] [PubMed] [Google Scholar]
- (69).Byrne SR, Yang K, and Rokita SE (2019) Effect of nucleosome assembly on alkylation by a dynamic electrophile. Chem. Res. Toxicol 32, 917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Sun B, Johnson DS, Patel G, Smith BY, Pandey M, Patel SS, and Wang MD (2011) ATP-induced helicase slippage reveals highly coordinated subunits. Nature 478, 132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Bastia D, Zzaman S, Krings G, Saxena M, Peng X, and Greenberg MM (2008) Replication termination mechanism as revealed by Tus-mediated polar arrest of a sliding helicase. Proc. Natl. Acad. Sci. U. S. A 105 (35), 12831–12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Deeyaa BD, and Rokita SE (2020) Migratory ability of quinone methide-generating acridine conjugates in DNA. Org. Biomol. Chem 18, 1671–1678. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.