

Abstract
Novel SARS coronavirus (SARS-CoV-2) has caused a pandemic condition worldwide. It has been declared as a public health emergency of international concern by WHO in a very short span of time. The community transmission of this highly infectious virus has severely affected various parts of China, Italy, Spain, India, and USA, among others. The prophylactic solution against SARS-CoV-2 infection is challenging due to the high mutation rate of its RNA genome. Herein, we exploited a next-generation vaccinology approach to construct a multi-epitope vaccine candidate against SARS-CoV-2 that is predicted to have high antigenicity, safety, and efficacy to combat this deadly infectious agent. The whole proteome was scrutinized for the screening of highly conserved, antigenic, non-allergen, and non-toxic epitopes having high population coverage that can elicit both humoral and cellular mediated immune response against COVID-19 infection. These epitopes along with four different adjuvants, were utilized to construct a multi-epitope-vaccine candidate that can generate strong immunological memory response having high efficacy in humans. Various physiochemical analyses revealed the formation of a stable vaccine product having a high propensity to form a protective solution against the detrimental SARS-CoV-2 strain with high efficacy. The vaccine candidate interacted with immunological receptor TLR3 with a high affinity depicting the generation of innate immunity. Further, the codon optimization and in silico expression show the plausibility of the high expression and easy purification of the vaccine product. Thus, this present study provides an initial platform for the rapid generation of an efficacious protective vaccine for combating COVID-19.
Keywords: Protective vaccine, COVID-19, SARS-CoV-2, Multi-epitope-vaccine, Next generation vaccinology, Antigenic epitopes
Graphical abstract
Highlights
-
•
A multi-epitope vaccine was constructed by scrutinizing whole proteome of SARS-CoV-2 with a propensity to elicit immune response inside the host.
-
•
In-silcio analysis revealed the antigenicity, non-alergencity, and non-toxic nature of the vaccine construct.
-
•
Molecular modeling and simulations showed the formation of stable vaccine construct.
-
•
The vaccine construct forms a energetically favorable Vaccine construct-TLR3 Complex formation.
-
•
The study provides an initial platform for the generation of safe and efficacious vaccine construct against the SARS-CoV-2 virus.
1. Introduction
The most disastrous outbreak of recent days that have put the world into a pandemic threat involves the novel Severe Acute Respiratory Syndrome Coronavirus 2 or the SARS-CoV-2 causing the COVID-19 outbreak. According to the situation report published by the World Health Organization on November 14th, 2020, 53,746,944 confirmed cases had been reported globally, along with 1,309,408 deaths worldwide (https://www.who.int/emergencies/diseases/novel-coronavirus-2019). Due to easy transportation and migration throughout the world, the virus has spread to more than 210 countries or areas from its epicenter, i.e., the city of Wuhan, China (Wells et al., 2020). The United States of America, India, and Brazil are the worst affected countries by the virus, and in the absence of effective drugs or vaccines, the present situation is getting out of control. For this recently emerged highly infectious Coronavirus, preventive measures are the only ways to restrict COVID-19 outcome (Yang et al., 2020).
The surfacing of SARS-CoV-2 in China was reported recently in December 2019, and therefore, very limited and naïve information is available regarding its genomic constituency and pathogenesis. Phylogenetic analyses have shown that bat-CoV-RaTG13 displays 96.3% sequence similarity while bat-SL-CoVZC45 and bat-SL-CoVZXC21 exhibit about 88% identity with the SARS-CoV-2 virus, but the other two pathogenic coronaviruses, SARS-CoV and MERS-CoV have less sequence identity being merely 79% and 50% respectively (Lu et al., 2020). The single-stranded positive-sense RNA genome of SARS-CoV-2 encodes 9860 amino acids translating into several non-structural proteins like replicase, nsp2, nsp3 and accessory proteins (orf3a, orf7a/b) along with structural proteins including Spike (S), Envelope (E), Membrane (M), and Nucleocapsid (N) (Han et al., 2020).
The lessons learned from the SARS-CoV and MERS-CoV outbreak have helped in better understanding of the situation of the new coronavirus epidemic and pushed researchers to develop a vaccine or therapeutic solution to succumb the current situation. An effective vaccine should robustly activate both the humoral and cell-mediated immunity to establish strong protection against the pathogen (Du et al., 2016). The antibody generation as well as acute viral clearance along with virus-specific memory generation by CD8+ T-cells are equally important to develop immunity against coronaviruses (Enjuanes et al., 2016). Vaccines against previously reported SARS-CoV and MERS-CoV majorly focuses on the Spike protein of virus bearing the receptor-binding domain (RBD) of the virus and cell fusion machinery. Mutations in spike protein have been reported to be responsible for the change in host cell tropism (Lu et al., 2015) and result in antibody-dependent enhancement (ADE) (Malone DRaRW, 2020). Other viral proteins that were explored for vaccine development include N, E, and NSP16 (Regla-Nava et al., 2015; Das et al., 2010; Menachery et al., 2017). Almost all vaccine development platforms for SARS-CoV and MERS-CoV have been investigated, including the live-attenuated, recombinant viruses, sub-unit protein vaccines, DNA vaccines, viral vector-based vaccines, nanoparticle-based vaccines, etc. which may form the base for vaccine designing against the newly emerged SARS-CoV-2 (Du et al., 2016; Yong et al., 2019). But these conventional vaccines may take as long as 15 years to develop and be available, often with various undesirable side-effects. Multi-epitope-vaccines offer many advantages over the conventional counterparts as they consist of only antigenic part of the pathogens that can elicit a protective immune response with minimal side effects and have no risk of emergence of virulent pathogens inside the host. Reverse-vaccinology is a novel approach that has shown tremendous growth in vaccine development (Moxon et al., 2019). Reverse-vaccinology-based multi-epitope-vaccine for Neisseria meningitides was approved in 2013 and is used worldwide to immunize against the pathogen (Masignani et al., 2019). After that, various research in recent times have shown the development of various multi-epitope-vaccine constructs in different infectious viruses, including HIV, H1NI, Nipah (Majee et al., 2020), and others (Oli et al., 2020). Reverse-vaccinology has been explored in pathogenic bacteria and protozoans too for vaccine development (Oli et al., 2020). Herein, we applied a reverse-vaccinology approach to design a multi-epitope-based vaccine for SARS-CoV-2, where the entire proteome of the SARS-CoV-2 was maneuvered, and a potential vaccine candidate was conceived. A similar strategy was employed previously for SARS-CoV, and MERS-CoV (Ibrahim and Kafi, 2020; Tahir et al., 2019), as well as certain findings, are reported for the newly emerged SARS-CoV-2 (Baruah and Bose, 2020; Enayatkhani et al., 2020). While immunoinformatics techniques were utilized by groups to predict the B-cell and cytotoxic T-cell epitopes in the SARS-CoV-2 surface glycoprotein and N protein, others have utilized information to design epitope-based vaccine based on spike glycoprotein (Bhattacharya et al., 2020). A recent analysis used Nucleocapsid, ORF3a, and Membrane protein for screening HTL and CTL cell epitopes and constructed a multi-epitope-vaccine and depicted the interaction analysis with the TLR4 (Enayatkhani et al., 2020). Along with structural proteins, utilizing non-structural and accessory proteins for vaccine development can aid in better development of an efficacious vaccine for long term by neutralizing the mutation rate of this RNA virus. In this study, we explored the whole proteome of SARS-CoV-2 to scrutinize the highly conserved antigenic epitopes for construction of a multi-epitope-vaccine candidate that can effectively elicit both humoral and cellular mediated immune response against COVID-19. The constructed vaccine was further analyzed for its binding affinity with the TLR3 that plays a critical role in COVID-19 infection.
2. Methodology
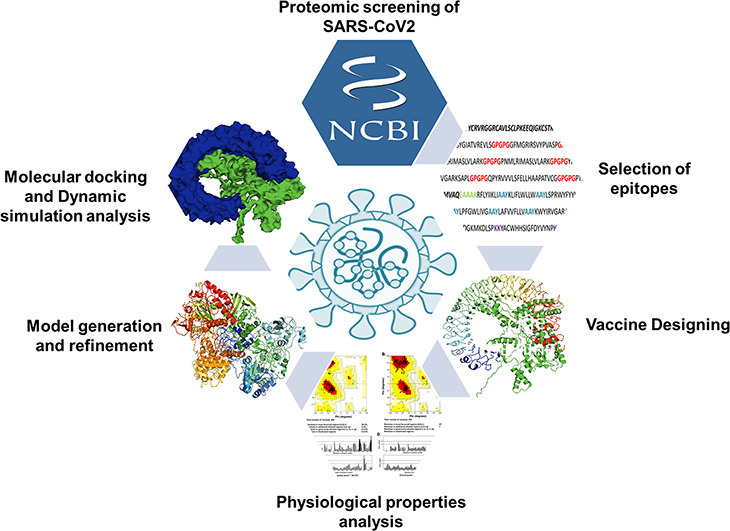
The schematic representation of the methodology applied for constructing a multi-epitope-vaccine construct is depicted in Fig. 1 . For a detailed explanation, please check Supplementary Methods.
Fig. 1.

Schematic representation of next-generation vaccinology approach used for the prediction of multi-epitope-vaccine construct for SARS-CoV-2.
3. Results
3.1. Retrieval of structural and non-structural proteins of SARS-CoV-2 and antigenicity analysis
The complete proteome of the Wuhan seafood market pneumonia virus isolates Wuhan-Hu-1 (accession number - NC_045512.2) was retrieved from the NCBI database. All the protein sequences were analyzed for their antigenic nature by using the VaxiJen server which is based upon auto-cross-covariance (ACC) transformation. The ACC score threshold for the virus model is kept at 0.4. All the SARS-CoV-2 proteins except NSP16 resulted in the ACC score higher than 0.4 depicting the antigenic nature of the viral proteome (Table 1 ).
Table 1.
List of SARS-CoV proteins used for antigenicity prediction using VaxiJen server. The antigenic proteins with Antigenicity Score ≥ 0.4 were taken for epitope screening.
| NCBI accession number | SARS-CoV-2 proteins | Antigenicity score | Antigenicity prediction | |
|---|---|---|---|---|
| QHD43415_1 | ORF1ab | Host translation inhibitor (nsp1) | 0.4064 | Probable ANTIGEN |
| QHD43415_2 | Non-structural protein 2 (nsp2) | 0.4034 | Probable ANTIGEN | |
| QHD43415_3 | Papain-like proteinase | 0.5142 | Probable ANTIGEN | |
| QHD43415_4 | Non-structural protein 4 (nsp4) | 0.4575 | Probable ANTIGEN | |
| QHD43415_5 | Proteinase | 0.4159 | Probable ANTIGEN | |
| QHD43415_6 | Non-structural protein 6 (nsp6) | 0.5813 | Probable ANTIGEN | |
| QHD43415_7 | Non-structural protein 7 (nsp7) | 0.4167 | Probable ANTIGEN | |
| QHD43415_8 | Non-structural protein 8 (nsp8) | 0.4008 | Probable ANTIGEN | |
| QHD43415_9 | Non-structural protein 9 (nsp9) | 0.6476 | Probable ANTIGEN | |
| QHD43415_1 | Non-structural protein 10 (nsp10) | 0.4039 | Probable ANTIGEN | |
| QHD43415_1 | RNA-directed RNA polymerase (RdRp) | 0.4064 | Probable ANTIGEN | |
| QHD43415_1 | Helicase (Hel) | 0.448 | Probable ANTIGEN | |
| QHD43415_1 | Guanine-N7 methyltransferase (ExoN) | 0.4138 | Probable ANTIGEN | |
| QHD43415_1 | Uridylate-specific endoribonuclease (NendoU) | 0.5554 | Probable ANTIGEN | |
| QHD43415_1 | 2’-O-methyltransferase (2’-O-MT) | 0.38 | Probable NON-ANTIGEN | |
| QHD43416 | Surface glycoprotein | 0.4646 | Probable ANTIGEN | |
| QHD43417 | ORF3a | 0.4945 | Probable ANTIGEN | |
| QHD43418 | ORF4 (E Protein) | 0.6025 | Probable ANTIGEN | |
| QHD43419 | Membrane Protein | 0.5102 | Probable ANTIGEN | |
| QHD43420 | ORF6 | 0.6131 | Probable ANTIGEN | |
| QHD43421 | ORF7a | 0.6441 | Probable ANTIGEN | |
| QHD43422 | ORF8 | 0.6502 | Probable ANTIGEN | |
| QHD43423 | Nucleocapsid Protein | 0.5059 | Probable ANTIGEN | |
| QHI42199 | ORF10 | 0.7185 | Probable ANTIGEN | |
3.2. Epitope screening revealed the presence of high-affinity HTL, CTL, and B-cell epitopes
T- Lymphocytes play a central role in activating the cell-mediated innate and adaptive immune response against the foreign particles. They are the sole players in generating immunological memory that provides a long-lasting immune response. Thus, the vaccinology approach revolves around the screening of proteome for the high-affinity Helper-T Lymphocytes (HTLs/Th) and Cytotoxic-T Lymphocytes (CTLs/Tc) epitopes that can activate TH and TC cells. The epitopes for the activation of HTLs and CTLs are processed and presented by various human leukocyte antigens (HLA) class I and class II molecules. The HLA-epitope binding steps are crucial and have high specificity and are thus considered as one of the main focus for vaccine designing. HLA shares large diversity due to the presence of several alleles in the human genome, but their occurrence is not uniform worldwide. Few of the alleles are present throughout the world, while some are restricted to only specific regions. Recent reports and literature survey lead us to identify the massively affected areas by COVID-19, namely China, Italy, Spain, Germany, USA, India, Iran, France, South Korea, Switzerland, UK, Netherlands, Austria, Belgium, Norway, Sweden, Denmark, Canada, Malaysia, Australia, Portugal, and Japan. The presence of various epitopes with different HLA-binding specificities leads to greater population coverage as the frequency of HLA allele's expression varies with varying ethnicities worldwide. Henceforth, fourteen HLA-I and twenty HLA-II alleles were selected on the basis of their occurrence in these countries.
HTLs act as the central mediators of immune response and coordinates with B-lymphocytes, TC cells, and macrophages via signaling through cytokines synthesis. Herein, the NetMHCIIPan server was utilized for predicting 15-mer HTL epitopes having a high binding affinity to the selected twenty MHC II alleles. Only strong binders for the respective HLA alleles were selected with a percentile rank of ≤1. The selected epitopes were further analyzed using another tool, namely MHC-II Binding Predictions, available at IEDB and filtered on the basis of percentile rank. Finally, the best HLA-epitope pairs with the highest antigenicity and being non-allergen, and non-toxic was used for multi-epitope-vaccine construct. We received the two best HLA-epitope pairs each for Helicase and RdRp. Three best HLA-epitope pairs were obtained for NSP2 and S protein and one each for M, ORF3a, and ORF8 proteins, respectively. In summary, non-toxic, non-allergenic epitopes with high binding affinity and antigenicity were obtained for fifteen MHC class II alleles, and their respective epitopes were used for multi-epitope-vaccine construct (Supplementary Table S1).
CTLs eliminate foreign particles, thereby helping in pathogen clearance, and are required for maintaining cellular integrity. MHC-I represents the epitopes to the CTLs, which, after activation, perform cytotoxic activities. Here, we used NetMHCpan_v.4 for the mining of CTL epitopes that have a high binding affinity with fourteen HLA-I molecules. The strong binders obtained from the NetMHCpan server were then analyzed for their immunogenicity by using the class I Immunogenicity tool. Depending upon amino acid composition, properties, and position in the epitope, the class I immunogenicity tool predicts the immunogenicity of an MHC class I–epitope complex. A higher score depicts higher immunogenicity and vice-versa. Non-allergen and non-toxic epitopes with the highest antigenicity were selected for the vaccine construction. Proteome mapping revealed 3 HLA-epitope pairs for ORF3a protein, 2 for M, N, and S, while one each for NSP2, NSP4, E, and ORF8 protein (Supplementary Table S2).
Apart from cellular mediated immunity, the humoral immune response mediates pathogen clearance in an antibody-dependent manner. Hence the proteome of SARS-CoV-2 was further scanned for linear B-cell epitopes by using the ABCPred server. For higher selectivity and sensitivity, the threshold of ABCPred was kept at 0.9. The selected epitopes were further screened for their antigenic, non-allergenic and non-toxic nature. On the basis of the above criteria, we received total four B-cell epitopes, one each for N, ExoN, ORF3a, ORF7a, and S protein (Supplementary Table S3). Overall, the antigenic thirteen HTL and twelve CTL epitopes having the highest affinity for the respective HLA alleles and four B-cell epitopes that are non-allergenic, non-toxic, and can generate a potent immune response were selected for incorporation into the multi-epitope-vaccine construct. The locations of the selected epitopes in the respective protein structures are represented in Fig. 2 .
Fig. 2.

HTL, CTL, and BCL epitopes in SARS-CoV-2 proteome. Location of selected Helper T-Lymphocytes epitopes (Blue), Cytotoxic T-Lymphocytes epitopes (Red), and B Cell Lymphocytes epitopes (Purple) in the three-dimensional structures of the antigenic proteins of SARS-CoV-2.
3.3. The screened epitopes shared high conservancy within the coronavirus family
The presence of conserved epitopes in a vaccine can lead to an effective immunization against all the strains of the pathogen. The selected HTL, CTL, and BCL epitopes were analyzed for their conservancy among the various human infecting strains of Coronavirus. Interestingly, all the selected epitopes were 100% conserved throughout the family that may lead to robust vaccine development.
3.4. Molecular interaction analysis depicts the strong interaction of T-cell epitopes and HLA molecules
Interaction analysis of the selected CTL and HTL epitopes with their respective HLA alleles revealed favorable interaction of both CTL and HTL epitopes with the HLA allele's epitope binding grooves (Supplementary Fig. S2a & b).
3.5. Population coverage analysis shows the broad range spectrum of the constructed vaccine
Population coverage of vaccines depends upon the interaction of epitopes with the number of HLA alleles. Depending upon the ethnicity, the expression of HLA alleles varies throughout the world. To check the population coverage of the vaccine construct, the presence of selected HLA alleles was analyzed in the 26 severely affected countries by COVID-19 around the world. Individually, MHC class I alleles covered ~91.22% of the world population while MHC class II alleles covered ~99.70% (Supplementary Fig. S3 & S4). The occurrence of MHC-I or MHC-II in combination depicted the coverage of >99% throughout the world (Fig. 3A).
Fig. 3.

Population Coverage and designing of the SARS-CoV-2 multi-epitope vaccine construct. A. Population coverage of the multi-epitope vaccine construct based on the coverages of HLA class I and class II alleles in combination in various countries. B. The sequence of the multi-epitope vaccine construct with HTLs, CTLs, and BCL epitopes along with various adjuvants and linkers. C. Schematic representation of the SARS-CoV-2 multi-epitope vaccine construct.
3.6. Designing of SARS-CoV-2 multi-epitope-vaccine construct
An ideal vaccine should harbor conserved epitopes, have multi-valence, and can elicit both cellular and humoral mediated immune response in the host. A subunit vaccine contains minimal elements that are antigenic and required for the stimulation of prolonged protective immune response. Herein, we constructed a multi-epitope-vaccine candidate by combining thirteen HTL, twelve CTL, and four BCL epitopes that were highly conserved, antigenic, non-toxic, and non-allergens (Fig. 3B & C).
In recent times, various small peptides have been explored that acts as an adjuvant and can potentiate the multi-epitope-vaccine mediated immune response by activating humoral immunity. Taking this into consideration, apart from the conserved epitopes, four adjuvants were also added so as to boost the protective immune response against SARS-CoV-2. β-defensins are antimicrobial peptides that act as a chemoattractant for the immature dendritic cells and T-lymphocytes and induces their maturation (Ferris et al., 2013). A 45 amino acid long β-defensin GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK was added at the vaccine construct. CD4+ T cells are the key regulators of humoral immunity as they assist in B-cells in effective antibody class switching and their affinity maturation. Universal memory T helper cell peptide (TpD), a chimeric peptide derived from tetanus, and diphtheria toxoids have high promiscuity to interact with a wide range of MHC class II alleles. The presence of a TpD in the vaccine construct leads to an effective Th cell activation and enhances the immunogenicity of the vaccine. Similarly, a universal Pan DR epitope (PADRE) has been reported to enhance the potency of long multi-epitope by activating CD4+ T helper cells and helps in generating high titer IgG antibodies (Alexander et al., 2000). Another small peptide CTGKSC, an M cell ligand, enhances the adsorption of oral vaccines from the intestinal membrane barrier (Fievez et al., 2010). The adjuvants and epitopes were arranged in the following order: β-defensins–HTLs–TpD–CTLs–PADRE–BCL–CTGKSC (Fig. 3B & C). To preserve independent immunogenic activities, various linkers were used for combining adjuvants and epitopes. EAAAK was used to join the adjuvants, HTL, CTL, and BCL epitopes with each other as they effectively separate various domains in multi-domain proteins. GPGPG linkers were used to join HTL epitopes, while AAY linkers were used for combining CTLs making it a 602 amino acid long vaccine (Fig. 3B & C). GPGPG and AAY linkers enhances recognition of the vaccine constructs by MHC I and MHC II machineries. Similarly, KK linkers were utilized for combining B-cell epitopes.
3.7. Physiochemical properties, antigenicity, allergenicity, and toxicity analysis revealed the efficacy and safety of the predicted vaccine construct
Analysis of the physicochemical properties of a multi-subunit-vaccine helps in analyzing the optimal immune response of the vaccine and its stability inside the host. The molecular weight of the vaccine construct was observed to be 65.3 kDa, while the pI value was 10.06. Instability index of 31.93 (<40) depicted the stable nature of the vaccine construct while the estimated half-life in mammalian reticulocytes was observed to be >30 h. GRAVY constant of 0.213 depicted the hydrophilicity of the vaccine construct and was predicted to be soluble with a probability of `70% (Table 2 ). Antigenicity is a preliminary requisite of a successful vaccine candidate. A vaccine construct must possess both immunogenicity and antigenicity to elicit the humoral and cell-mediated immune response. Upon analyzing the vaccine construct sequence in VaxiJen server, the construct was found to be antigenic in nature with an overall prediction score of 0.6199. On allergenicity analysis, all the three tools AlgPred, AllergenFP, and AllerTop supported the non-allergenic nature of the vaccine construct while toxicity analysis revealed non-toxic behavior of the construct (Table 2).
Table 2.
The results of physiochemical properties, antigenicity, allergenicity, and toxicity analysis of the SARS-CoV-2 multi-epitope vaccine construct.
| Antigenicity Prediction using VaxiJen | 0.6199 (Probable ANTIGEN). | ||
|---|---|---|---|
| Allergenicity analysis | Allertop | PROBABLE NON-ALLERGEN The nearest protein is: UniProtKB accession number Q2RB59 defined as non-allergen |
|
| AllergenFP | PROBABLE NON-ALLERGEN The protein with the highest Tanimoto similarity index 0.84 is: UniProtKB accession number P00846 |
||
| AlgPred prediction | Prediction by mapping of IgE epitope | The protein sequence does not contain experimentally proven IgE epitope | |
| MAST RESULT | No Hits found NON ALLERGEN |
||
| Prediction by SVM method based on amino acid composition | Score = −1.0824782 [Threshold = −0.4] NON ALLERGEN |
||
| BLAST Results of ARPS: | No Hits found NON ALLERGEN |
||
| Prediction by Hybrid Approach | NON ALLERGEN | ||
| Toxicity Analysis Using ToxinPred | Non-Toxin | ||
| Physiochemical properties analysis using ProtParam | Number of amino acids | 602 | |
| Molecular weight | 65,323.19 Da | ||
| Theoretical pI | 10.06 | ||
| Estimated half-life | >30 h (mammalian reticulocytes, in vitro). | ||
| >20 h (yeast, in vivo). | |||
| >10 h (Escherichia coli, in vivo). | |||
| Instability index | The instability index (II) is computed to be 31.93. This classifies the protein as stable. | ||
| Grand average of hydropathicity (GRAVY) | 0.213 | ||
| Solubility analysis using SolPro | Predicted Solubility upon Overexpression | SOLUBLE with probability 0.699016 | |
3.8. Structure modeling and validation results in the construction of a feasible 3D structure of the vaccine construct
Secondary and tertiary structures help in the functional annotation of the multi-epitope vaccines. It also helps in analyzing the interaction of vaccine construct with immunological receptors like TLRs. Analysis of secondary structure revealed the presence of ~40% alpha-helix, ~20% β-sheet, ~30% coils, and ~ 6% β turns in the vaccine construct (Fig. 4 and Supplementary Fig. S5). The tertiary structure of the vaccine was predicted by threading based homology modeling. Ramachandran plot analysis of the modeled structure revealed 95.5% residues in the most favored regions and 4.5% in the additionally allowed regions. The quality factor was 90.47 depicting a good modeled structure. To further refine the modeled structure, 3D refine, and GalaxyRefine were utilized that leads to 97.4% residues in the most favored region. The quality factor of the refined structure was increased to 94.29% depicting the improvement of the modeled structure (Fig. 5 ).
Fig. 4.

Secondary structure predictions of SARS-CoV-2 multi-epitope vaccine construct. A. The sequence of the vaccine construct along with the predicted secondary structure. B. The overall percentage of various secondary structures in the vaccine construct as predicted by SOPMA server. C Pictorial representations of various secondary structures in the multi-epitope vaccine construct. D. Propensity of occurrence of various secondary structures according to the residues in the vaccine construct. The secondary structure at a particular residue was predicted by the height of the peak.
Fig. 5.

Tertiary structure and validation of the SARS-CoV-2 multi-epitope vaccine construct. (A) Represents the modeled structure of the vaccine construct where HTL epitopes are depicted by Salmon Pink color, CTL by Orange, BCL epitopes by Cyan, and adjuvants by Blue. (B & C) Ramachandran plot and ERRAT plot generated for the modeled vaccine construct. (D) The modeled (Cyan) and refined structure (Red) of the SARS-CoV-2 multi-epitope vaccine construct depicting the changes in the structure before and after refinement. (E&F) Ramachandran plot and ERRAT plot generated for the refined structure of the vaccine construct.
3.9. Structure dynamics simulation of the vaccine construct depicts the stability of the modeled vaccine construct
The refined structure of the model construct was further checked for its stability by simulating it for 20 ns in a water sphere. 10,000 steps energy minimization was performed to minimize the potential energy of the system. Unnecessary or false geometry of the protein structures are repaired by performing energy minimization resulting in a more stable stoichiometry. Before energy minimization, the potential energy was observed to be −447,451.2005 kcal/mol. After 10,000 steps, the protein was minimized with a potential energy of −676,040.4692 kcal/mol (Fig. 6A). The system was subsequently heated from 0 K to 310 K, and 10 ns molecular dynamic simulation was performed. Upon analyzing the RMSD of the SARS-CoV-2 vaccine construct, it was observed that the system gained equilibrium at ~4 ns and then remained constant till 10 ns depicting the stability of the vaccine construct (Fig. 6C). Furthermore, upon analyzing the change in the kinetic, potential, and total energy of the system, it was observed that after a quick initial change in all the three, they remained constant throughout the simulation, strengthening the stability of the vaccine construct (Fig. 6D–F). Furthermore, the bond energy, VdW energy, dihedral, and improper dihedral energy analysis revealed no change throughout the simulation. Root mean square fluctuation (RMSF) analysis revealed the rigidness of the atoms in vaccine construct with slight mobility observed at ~120 and ~ 501 residues (Supplementary Fig. S6). Thus, simulation analysis revealed the stability of the vaccine construct.
Fig. 6.

Standard molecular Dynamics analysis of SARS-CoV-2 multi-epitope vaccine construct. (A) Representation of the change in potential energy vs time steps (in ns) during energy minimization of the vaccine construct. (B) Temperature Vs Time steps showing the constant temperature throughout the 10 ns simulation study. (C) RMSD Vs TS depicting the root mean square deviation of the atoms of multi-epitope vaccine construct during the 10 ns dynamic simulation. (D – F) Energy plots representing Kinetic, potential, and total energy w.r.t. TS for the Vaccine construct system in a water sphere.
3.10. Interaction of vaccine construct with immunological receptor depicts the strong, feasible binding
Docking of TLR3 and constructed vaccine was carried out using ClusPro server. In total, 30 TLR3-vaccine construct complexes were generated. The fifth model with the lowest energy weighted score of −1427.4 was considered as the best TLR3-vaccine complex (Fig. 7A-B and Supplementary Table S4). The vaccine construct interacted with the ligand-binding groove of TLR-3, generating a strong TLR3-vaccine construct complex. The stability of the complex was further analyzed by performing 10 ns standard molecular dynamics simulation studies using NAMD suite. The energy minimization of the complex leads to the generation of a minimized energy complex with an energy of −756,791.5601 kcal/mol (Fig. 7C). After subsequent heating the system from 0 K to 310 K, the temperature was kept constant throughout (Fig. 7D). Energy plots depicted no major changes (Fig. 7E–G and Supplementary Fig. S7). RMSF analysis of the complex showed a mobility region at ~740th residue and ~ 1100th residue of the TLR3-vaccine complex (Fig. 7H). On trajectory analysis, the constructed RMSD plot revealed the fluctuations during the initial 1.5 ns, thereafter the system remained constant.
Fig. 7.

Molecular interaction analysis of SARS-CoV-2 multi-epitope vaccine construct with an immunological receptor Human TLR3. (A & B) Representation of TLR3-vaccine construct in ribbon (A) and surface form (Blue Human TLR3 ad Green- multi-epitope vaccine construct). (C) Representation of the change in potential energy Vs time steps (in ns) of the TLR3-vaccine construct during energy minimization. (D) Temperature Vs Time steps showing the constant temperature throughout 10 ns simulation study. (E – G) Energy plots representing Kinetic, potential, and total energy w.r.t. TS for the Vaccine construct system in a water sphere. (H) RMSF Vs TS for the residues of TLR3-vaccine construct complex (I) RMSD Vs TS depicting the root mean square deviation of the atoms of TLR3-vaccine construct during the 10 ns dynamic simulation.
3.11. cDNA was optimized for the optimal expression of the vaccine product
For the insertion of the vaccine construct into a plasmid vector, the 602 aa construct was reverse translated to cDNA of 1806 nucleotide length. For the optimal expression of the vaccine product in Escherichia coli K12 host, the resultant cDNA was codon-optimized. Also, during optimization, the rho-independent transcription terminator and prokaryotic ribosomal binding sites were avoided in the middle of the cDNA sequence so as to generate an optimal and complete protein expression. The CAI value (codon adaptation index) of the cDNA before adaptation was observed to be 0.5379, with GC content of 59.52%. After adaptation, the CAI score of the improved sequence was increased to 0.946 with 52.54% GC content (Fig. 8A–B and Supplementary data S1). The enhanced CAI score depicts the presence of the most abundantly used codons in Escherichia coli K12.
Fig. 8.

Codon optimization and In silico cloning of the cDNA of SARS-CoV-2 multi-epitope vaccine construct. Representation of the CAI score of the cDNA construct of SARS-CoV-2 multi-epitope vaccine before adaptation (A) and after adaptation (B). (C) Pictorial representation of the SARS-CoV-2-multi-epitope vaccine construct plasmid that can be used for the expression and purification of the vaccine product inside Escherichia coli.
3.12. In silico cloning for expression of multi-epitope-vaccine construct
Escherichia coli K12 strain was selected as a host for cloning purposes because the expression and purification of multi-epitope-vaccines are easier in this bacterium. pET28a(+) expression vector was cleaved using BamHI, and HindIII restriction enzyme, and the cDNA was inserted near the ribosome binding site using SnapGene. 6× histidine tag was added at the 3′ end for the isolation and purification of the vaccine construct (Fig. 8C).
4. Discussion
SARS-CoV-2 that causes COVID-19 has recently emerged as one of the deadliest pathogens severely affecting humans worldwide. The SARS-CoV-2 is a highly contagious virus with a high mortality rate, especially in immunocompromised and elderly persons. Vaccines are the utmost need of the time to succumb the rising infections due to COVID-19 and represent the best way to combat infectious diseases in the community. The conventional vaccine development methods are time-consuming, laborious, and expensive. For the development of a conventional vaccine, the pathogens are needed to be grown in-vitro, purified antigens are required in large quantities, and also, they often manifest several undesired side-effects due to the use of whole pathogens. A successful vaccine must be safe and able to elicit long-lasting protection against the diversified strains of the pathogen. Also, such a vaccine candidate must have a broad-spectrum range worldwide and should be produced easily and cost-effectively. The computationally assisted next-generation vaccinology approach helps in combating some of these necessities and has proved to be a boon (De Groot et al., 2009). It overcomes the risk of using whole pathogens, provides a scope to engineer and modify the epitopes to increase the efficacy and stability of the vaccine candidate, and allows choosing the most immuno-dominant epitopes and adding adjuvants and linkers to elicit better immunogenicity. Moreover, the availability of a large number of efficient tools to predict the immuno-determinants, and a large database of information facilitates and accelerates the process of vaccine designing (Soria-Guerra et al., 2015). Multi-epitope-vaccines are designed by using the antigenic conserved part of the pathogen's proteins that makes them more effective and can help in combating the high mutation rate in RNA viruses. These epitope-based vaccines have added advantages of being safe, stable, highly specific, cost-curtailing, easy to produce in bulk, and provides an option to manipulate the epitopes for designing a better vaccine candidate (Li et al., 2015). As well, one of the major concerns regarding the development of a successful vaccine for Coronavirus is the antibody-dependent enhancement (ADE) effect, which can be minimized using multi-epitope-vaccine. Antibody-dependent enhancement is mainly caused by the generation of non-neutralizing antibodies that binds to the virus and increases the cellular uptake of it. In Coronaviruses, it has been reported to occur by the peptides of spike protein, where a high mutation rate is observed. Herein, for the multi-epitope-vaccine construct designed, only the antigenic epitope regions of spike proteins that were highly conserved in the coronavirus family were taken into consideration. Also, the addition of antigenic epitopes from various other SARS-CoV-2 proteins makes it more efficacious and safer as these proteins cannot generate neutralizing antibodies but can induce the generation of specific antibodies and cellular immune response against the virus.
Herein, we applied a reverse-vaccinology approach for designing a multi-epitope-vaccine that can efficiently elicit a humoral and cellular mediated immune response against SARS-CoV-2 infection. The main focus for the vaccine construction to date for the coronavirus family, including SARS-CoV-2 is based on the exploitation of Spike protein (He et al., 2004a; Chen et al., 2020). But a high mutation rate in the spike protein may act as a drawback for universal vaccine designing. Various other structural and non-structural proteins like N (He et al., 2004b), NSP1 (Züst et al., 2007), NSP16, and M of SARS-CoV and MERS-CoV have shown to elicit a high immune response in the animal models (He et al., 2005). Though these viral proteins cannot elicit neutralizing antibodies, they may induce specific antibody and cellular immune response. Here, antigenic viral proteins were screened for the most immunogenic and antigenic epitopes that could efficiently evoke the immune response. For the generation of an efficacious vaccine, successful recognition of epitopes by various HLA alleles is a preliminary requisite. Therefore, fourteen HLA I and twenty HLA II alleles were selected that cover the maximum population throughout the world. The selected epitopes covered fifteen various antigenic proteins of SARS-CoV-2, including various non-structural structural and accessory proteins. The inclusion of these proteins made it a potent and effective vaccine candidate and was observed to be antigenic, non-allergenic, and non-toxic, making it a potent vaccine candidate against SARS-CoV-2.
TLR3 acts as a nucleic acid sensor and helps in recognizing RNA and DNA viruses inside the human body and establishes a protective role against these infectious agents (Lester and Li, 2014). In SARS-CoV infection, TLR3 signaling contributes to the innate immune response against viral infection and its activation during immunization significantly increases the innate immune response by the activation of type I interferons and NF-kappa β (Totura et al., 2015). Hence, the binding affinity of the constructed multi-epitope vaccine construct was checked with the antigenic recognition receptor of TLR-3, which activates during immunization. Molecular interaction analysis of the vaccine construct with the TLR3 resulted in a stable complex formation. RSMD analysis of the vaccine construct, and the vaccine-TLR3 complex depicted the stable nature of both the systems.
In summary, a rigorous reverse-vaccinology approach has been applied for the construction of a multi-epitope-vaccine construct that can elicit a long-term immune response and have broad-spectrum coverage.
5. Conclusion
SARS-CoV-2 has become one of the most detrimental pathogens worldwide. Herein, we explored the reverse- vaccinology approach for designing a multi-epitope-vaccine construct. The proteins of SARS-CoV-2 were screened for highly conserved, antigenic, non-allergen, and non-toxic HTL, CTL, and BCL epitopes. Molecular interaction analysis revealed the high propensity of the interaction of the predicted epitopes with HLA alleles. Physiochemical analyses revealed the formation of a stable vaccine candidate with population coverage of >99% worldwide. Thus, the present study scrutinizes the antigenic proteins of SARS-CoV-2 for designing a highly efficacious protective vaccine candidate eliciting both humoral and cellular mediated memory response and provides a starting platform for the development of a prophylactic solution against COVID-19.
Author contributions
Data conceptualization and methodology was performed by AK. In-silico prediction were performed by NJ, US, and PM. Analysis was performed by NJ. NJ and PM collectively wrote the manuscript. A.K. did the review and editing.
Credit author statement
Amit Kumar: Data conceptualization and methodology; Neha Jain, Uma Shankar, Prativa Majee: In-silico prediction; Neha Jain, Uma Shankar, Prativa Majee: Analysis and manuscript drafting. Amit Kumar: Data review and manuscript editing. I ensure for the description mentioned and all authors agreed to it.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors acknowledge the Computer server facility provided by Dr. Parimal Kar at IIT, Indore. NJ acknowledges the Council for Scientific and Industrial Research, Govt. of India, New Delhi, while US and PM acknowledge the Ministry of Education, Govt. of India, New Delhi for respective Ph.D. research fellowships.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.meegid.2020.104648.
Appendix A. Supplementary data
Supplementary Method, Figures S1-S6; Table S1-S4
References
- Alexander J., del Guercio M.-F., Maewal A., Qiao L., Fikes J., Chesnut R.W. Linear PADRE T helper epitope and carbohydrate B cell epitope conjugates induce specific high titer IgG antibody responses. J. Immunol. 2000;164:1625–1633. doi: 10.4049/jimmunol.164.3.1625. [DOI] [PubMed] [Google Scholar]
- Baruah V., Bose S. Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. J. Med. Virol. 2020;92:495–500. doi: 10.1002/jmv.25698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya M., Sharma A.R., Patra P., Ghosh P., Sharma G., Patra B.C. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. J. Med. Virol. 2020;92(6):618–631. doi: 10.1002/jmv.25736. n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.-H., Strych U., Hotez P.J., Bottazzi M.E. The SARS-CoV-2 vaccine pipeline: an overview. Curr. Trop. Med. Rep. 2020;7:1–4. doi: 10.1007/s40475-020-00201-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D., Kammila S., Suresh M.R. Development, characterization, and application of monoclonal antibodies against severe acute respiratory syndrome coronavirus nucleocapsid protein. CVI. 2010;17:2033–2036. doi: 10.1128/CVI.00293-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot A.S., Moise L., McMurry J.A., Martin W. Springer; Clinical Applications of Immunomics: 2009. Epitope-Based Immunome-Derived Vaccines: A Strategy for Improved Design and Safety; pp. 39–69. [Google Scholar]
- Du L., Tai W., Zhou Y., Jiang S. Vaccines for the prevention against the threat of MERS-CoV. Expert Rev. Vacc. 2016;15:1123–1134. doi: 10.1586/14760584.2016.1167603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enayatkhani M., Hasaniazad M., Faezi S., Gouklani H., Davoodian P., Ahmadi N. Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: an in silico study. J. Biomol. Struct. Dyn. 2020:1–16. doi: 10.1080/07391102.2020.1756411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enjuanes L., Zuñiga S., Castaño-Rodriguez C., Gutierrez-Alvarez J., Canton J. Sola I. Chapter eight - molecular basis of coronavirus virulence and vaccine development. In: Ziebuhr J., editor. Advances in Virus Research. Academic Press; 2016. pp. 245–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris L.K., Mburu Y.K., Mathers A.R., Fluharty E.R., Larregina A.T., Ferris R.L. Human beta-defensin 3 induces maturation of human Langerhans cell–like dendritic cells: an antimicrobial peptide that functions as an endogenous adjuvant. J. Investig. Dermatol. 2013;133:460–468. doi: 10.1038/jid.2012.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fievez V., Plapied L., Plaideau C., Legendre D., Des Rieux A., Pourcelle V. In vitro identification of targeting ligands of human M cells by phage display. Int. J. Pharm. 2010;394:35–42. doi: 10.1016/j.ijpharm.2010.04.023. [DOI] [PubMed] [Google Scholar]
- Han Q., Lin Q., Jin S., You L. Coronavirus 2019-nCoV: a brief perspective from the front line. J. Inf. Secur. 2020;80(4):373–377. doi: 10.1016/j.jinf.2020.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y., Zhou Y., Siddiqui P., Jiang S. Inactivated SARS-CoV vaccine elicits high titers of spike protein-specific antibodies that block receptor binding and virus entry. Biochem. Biophys. Res. Commun. 2004;325:445–452. doi: 10.1016/j.bbrc.2004.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y., Zhou Y., Wu H., Kou Z., Liu S., Jiang S. Mapping of antigenic sites on the nucleocapsid protein of the severe acute respiratory syndrome coronavirus. J. Clin. Microbiol. 2004;42:5309–5314. doi: 10.1128/JCM.42.11.5309-5314.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y., Zhou Y., Siddiqui P., Niu J., Jiang S. Identification of immunodominant epitopes on the membrane protein of the severe acute respiratory syndrome-associated coronavirus. J. Clin. Microbiol. 2005;43:3718–3726. doi: 10.1128/JCM.43.8.3718-3726.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim H.S., Kafi S.K. A computational vaccine designing approach for MERS-CoV infections. Methods Mol. Biol. (Clifton, NJ) 2020;2131:39–145. doi: 10.1007/978-1-0716-0389-5_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester S.N., Li K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014;426:1246–1264. doi: 10.1016/j.jmb.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Guo L., Kong M., Su X., Yang D., Zou M. Design and evaluation of a multi-epitope peptide of human metapneumovirus. 2015;58:403–412. doi: 10.1159/000445059. [DOI] [PubMed] [Google Scholar]
- Lu G., Wang Q., Gao G.F. Bat-to-human: spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015;23:468–478. doi: 10.1016/j.tim.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majee P., Jain N., Kumar A. Designing of a multi-epitope vaccine candidate against Nipah virus by in silico approach: a putative prophylactic solution for the deadly virus. J. Biomol. Struct. Dyn. 2020:1–20. doi: 10.1080/07391102.2020.1734088. [DOI] [PubMed] [Google Scholar]
- Malone DRaRW Medical countermeasures analysis of 2019-nCoV and vaccine risks for antibody-dependent enhancement (ADE) Preprints Lancet. 2020 [Google Scholar]
- Masignani V., Pizza M., Moxon E.R. The development of a vaccine against meningococcus B using reverse vaccinology. Front. Immunol. 2019;10 doi: 10.3389/fimmu.2019.00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V.D., Gralinski L.E., Mitchell H.D., Dinnon K.H., 3rd, Leist S.R., Yount B.L., Jr. Middle East respiratory syndrome coronavirus nonstructural protein 16 is necessary for interferon resistance and viral pathogenesis. mSphere. 2017;2 doi: 10.1128/mSphere.00346-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moxon R., Reche P.A., Rappuoli R. Editorial: reverse vaccinology. Front. Immunol. 2019;10 doi: 10.3389/fimmu.2019.02776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oli A.N., Obialor W.O., Ifeanyichukwu M.O., Odimegwu D.C., Okoyeh J.N., Emechebe G.O. Immunoinformatics and vaccine development: an overview. Immunotargets Ther. 2020;9:13–30. doi: 10.2147/ITT.S241064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regla-Nava J.A., Nieto-Torres J.L., Jimenez-Guardeno J.M., Fernandez-Delgado R., Fett C., Castano-Rodriguez C. Severe acute respiratory syndrome coronaviruses with mutations in the E protein are attenuated and promising vaccine candidates. J. Virol. 2015;89:3870–3887. doi: 10.1128/JVI.03566-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria-Guerra R.E., Nieto-Gomez R., Govea-Alonso D.O., Rosales-Mendoza S. An overview of bioinformatics tools for epitope prediction: implications on vaccine development. J. Biomed. Inform. 2015;53:405–414. doi: 10.1016/j.jbi.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Tahir U.L., Qamar M., Saleem S., Ashfaq U.A., Bari A., Anwar F., Alqahtani S. Epitope-based peptide vaccine design and target site depiction against Middle East Respiratory Syndrome Coronavirus: an immune-informatics study. J. Transl. Med. 2019;17:362. doi: 10.1186/s12967-019-2116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totura A.L., Whitmore A., Agnihothram S., Schäfer A., Katze M.G., Heise M.T. Toll-Like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio. 2015;6 doi: 10.1128/mBio.00638-15. e00638-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells C.R., Sah P., Moghadas S.M., Pandey A., Shoukat A., Wang Y. Impact of international travel and border control measures on the global spread of the novel 2019 coronavirus outbreak. Proc. Natl. Acad. Sci. U. S. A. 2020;117(13):7504–7509. doi: 10.1073/pnas.2002616117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Peng F., Wang R., Guan K., Jiang T., Xu G. The deadly coronaviruses: the 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J. Autoimmun. 2020:102434. doi: 10.1016/j.jaut.2020.102434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong C.Y., Ong H.K., Yeap S.K., Ho K.L., Tan W.S. Recent advances in the vaccine development against Middle East respiratory syndrome-coronavirus. Front. Microbiol. 2019;10:1781. doi: 10.3389/fmicb.2019.01781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züst R., Cervantes-Barragán L., Kuri T., Blakqori G., Weber F., Ludewig B. Coronavirus non-structural protein 1 is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog. 2007;3 doi: 10.1371/journal.ppat.0030109. e109-e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Method, Figures S1-S6; Table S1-S4