

Abstract
Small molecule kinase inhibitors that stabilize distinct ATP binding site conformations can differentially modulate the global conformation of Src-family kinases (SFKs). However, it is unclear which specific ATP binding site contacts are responsible for modulating the global conformation of SFKs and whether these inhibitor-mediated allosteric effects generalize to other tyrosine kinases. Here, we describe the development of chemical probes that allow us to deconvolute which features in the ATP binding site are responsible for the allosteric modulation of the global conformation of Src. We find that the ability of an inhibitor to modulate the global conformation of Src’s regulatory domain-catalytic domain module relies mainly on the influence it has on the conformation of a structural element called helix αC. Furthermore, by developing a set of orthogonal probes that target a drug-sensitized Src variant, we show that stabilizing Src’s helix αC in an active conformation is sufficient to promote a Src-mediated, phosphotransferase-independent alteration in cell morphology. Finally, we report that ATP-competitive, conformation-selective inhibitors can influence the global conformation of tyrosine kinases beyond the SFKs, suggesting that the allosteric networks we observe in Src are conserved in kinases that have a similar regulatory architecture. Our study highlights that an ATP-competitive inhibitor’s interactions with helix αC can have a major influence on the global conformation of some tyrosine kinases.
Graphical Abstract
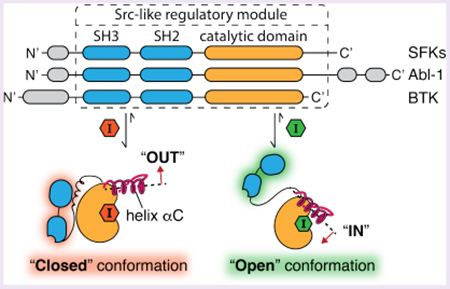
The human genome encodes ~540 eukaryotic protein kinases, an indication of the vast number of kinase-mediated cellular functions.1 All kinases contain at least one highly homologous catalytic domain (CD) that is of similar overall architecture. Almost half of all protein kinases contain at least one auxiliary domain outside of the CD.2 Precise regulation of these multidomain kinases often relies on intramolecular interactions between the CD and auxiliary domains, resulting in the allosteric modulation of kinase phosphotransferase activity.3,4 In many cases, the global conformational changes within these regulatory modules also facilitate important phosphotransferase-independent functions, including regulating protein scaffolding, localization, and gene transcription.5–7
Src-family kinases (SFKs) are one of the best-characterized multidomain kinase families.8–11 SFKs contain a membrane-interacting SH4 domain, a unique domain, and regulatory SH2 and SH3 domains that modulate the phosphotransferase activities of their CDs. Domains outside of the CD also provide additional binding surfaces for facilitating protein–protein interactions (Figure 1A).12,13 In the inactive form, SFKs adopt a closed global conformation, where the phosphotransferase activity of the CD is allosterically suppressed by multiple intramolecular interactions: the SH3 domain’s interaction with the SH2-CD linker, the SH2 domain’s interaction with the phosphorylated C-terminal tail, and the SH4 domain’s interaction with the C-lobe of the CD. Upon activation, these autoinhibitory, intramolecular interactions are released, and SFKs adopt a more open global conformation with enhanced phosphotransferase activity.14,15
Figure 1.
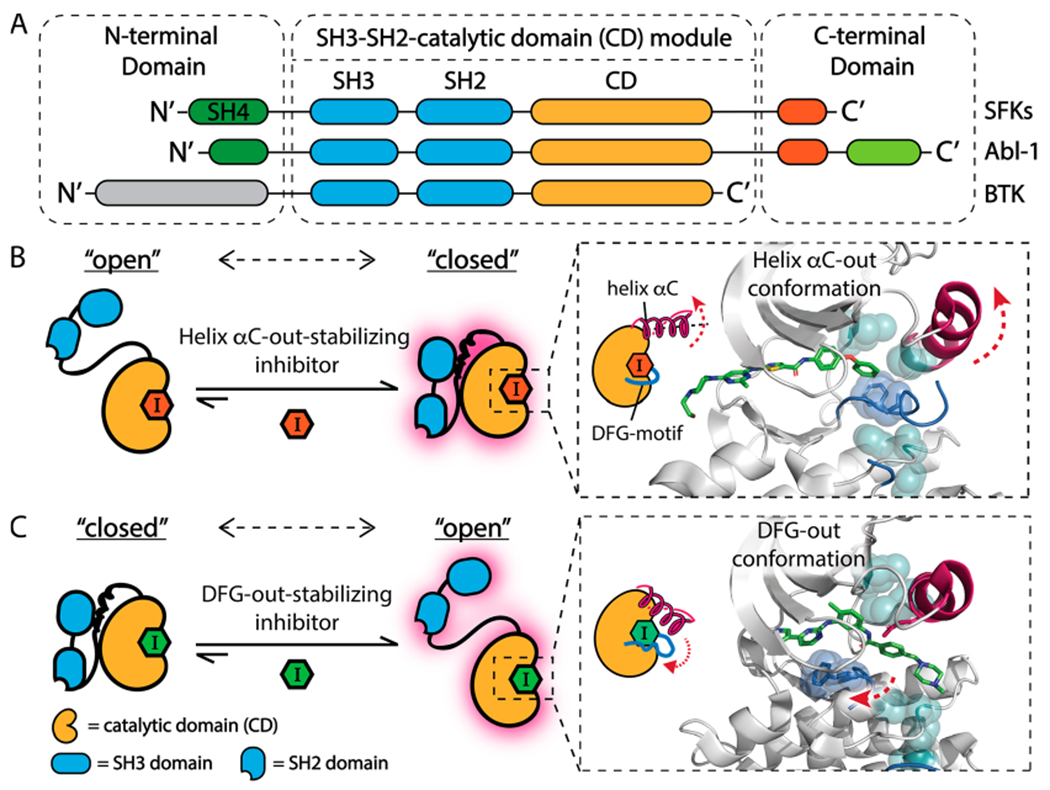
Allosteric modulation of tyrosine kinases that contain an SH3-SH2-CD module. (A) Domain arrangement of SFKs, Abl-1, and BTK. (B, C) ATP-competitive, conformation-selective inhibitors can modulate the global conformation of SFKs. (B) Helix αC-out-stabilizing inhibitors promote a closed global conformation of Src’s SH3-SH2-CD module by stabilizing the ATP binding site in the helix αC-out form (PDB: 4YBK).23 Shown are the catalytic spine (cyan), helix αC (magenta), and the activation loop (blue). (C) DFG-out-stabilizing inhibitors promote an open global conformation of Src’s SH3-SH2-CD module by stabilizing the ATP binding site in the DFG-out conformation (PDB: 2OIQ).24
Interdomain regulatory interactions suppress the phosphotransferase activity of SFKs by allosteric stabilization of an inactive conformation of the ATP binding site, which misaligns catalytic residues required for catalysis.7,16 We and others have shown that ATP-competitive inhibitors that stabilize different ATP binding site conformations can exploit these allosteric networks to modulate the regulatory interactions of the SH3-SH2-CD module of SFKs.14,17–23 Inhibitors that stabilize the helix αC-out inactive conformation of the ATP binding site, which involves outward movement of the catalytically important helix αC (Figure 1B), promote an autoinhibited, closed global conformation of the SH3-SH2-CD module of SFKs. The presence of an extended substituent that occupies the hydrophobic pocket formed by the displacement of helix αC is necessary for inhibitors to stabilize this inactive conformation. In contrast, inhibitors that stabilize the DFG-out inactive ATP binding site conformation, which involves the displacement of the DFG-motif in the activation loop, promote an open, regulatory domain disengaged conformation of the SH3-SH2-CD module (Figure 1C). Despite extensive biochemical and structural characterization, the ATP binding site contacts that are directly responsible for the divergent effects that helix αC-out- and DFG-out-stabilizing inhibitors have on the global conformations of SFKs are not completely known. It is also unclear how general the effects of conformation-selective inhibitors are on kinase conformation beyond the SFKs.
Structural analysis of the Abelson tyrosine kinase 1 (Abl-1), which contains a Src-like SH3-SH2-CD module, has demonstrated that ATP-competitive inhibitors can also modulate the global conformation of Abl-1.25–28 Specifically, the ability of inhibitors to flip the DFG-motif in the activation loop correlates with their abilities to promote an open global conformation of Abl-1’s SH3-SH2-CD module.29 Beyond the SFKs and Abl-1/Arg, there are at least 10 additional tyrosine kinases that contain an SH3-SH2-CD module (Figure S1).30 It is currently unknown whether inhibitors that stabilize the DFG-out and helix αC-out inactive ATP binding site conformations can also allosterically modulate the global conformation of SH3-SH2-CD modules of tyrosine kinases beyond the SFKs.
In this study, we describe the development of chemical probes that allow us to deconvolute which features in the ATP binding site are responsible for the allosteric modulation of the SH3-SH2-CD module of Src. With these probes, we found that the ability of an inhibitor to allosterically modulate the global conformation of Src’s SH3-SH2-CD module depends mainly on its influence on the conformation of helix αC. Thus, the observed ability of DFG-out-stabilizing inhibitors to promote an open global conformation of Src is not due to their influence on the DFG-motif in the activation loop but rather their ability to stabilize an active conformation of helix αC. Using these design principles, we developed new probes that allow us to selectively target a drug-sensitized Src variant and specifically stabilize Src’s helix αC in the active conformation without perturbing its activation loop. Using this chemical genetic approach, we found that stabilizing Src’s helix αC in an active conformation was sufficient to promote a Src-dependent phosphotransferase-independent alteration in cell morphology. Finally, we report that ATP-competitive, conformation-selective inhibitors can divergently modulate the global conformation of the SH3-SH2-CD module of tyrosine kinases beyond the SFKs. Taken together, our biochemical and cellular data strongly suggest that the ability of inhibitors to influence the conformation of helix αC is the main molecular determinant for allosterically modulating the global conformation of tyrosine kinases that contain an SH3-SH2-CD module.
RESULTS AND DISCUSSION
Inhibitors Divergently Modulate the Global Conformation of Tyrosine Kinases That Contain an SH3-SH2-CD Module.
Prior to exploring the molecular details of how inhibitors influence the global conformation of the SH3-SH2-CD module, we first explored whether the divergent allosteric modulation we observed with the SFKs extended beyond this kinase family. To explore the pervasiveness of this effect, we characterized how stabilizing the DFG-out and helix αC-out ATP binding site conformations affect the regulatory domains of Abl-1, the best-characterized member of Abl family kinases, and BTK, a Tec family kinase.
To modulate the ATP binding sites of tyrosine kinases, we generated two inhibitors that stabilize the helix αC-out and DFG-out inactive conformations, respectively. Inhibitor 1 contains an extended 4-phenoxyphenyl substituent at the C-3 position on the pyrazolopyrimidine scaffold that occupies a hydrophobic pocket created by the outward movement of helix αC (Figure 2A, left).14,18,19,31 Inhibitor 2 stabilizes the DFG-out inactive conformation of tyrosine kinases (Figure 2A, right). Inhibitor 2 contains a 3-trifluoromethylbenzamide substituent that occupies the hydrophobic pocket formed by the displacement of the DFG-motif.14,32 The conformationally rigid alkynylphenyl linker of 2 is most likely only compatible with the DFG-out inactive conformation. Because 1 and 2 only differ at their C-3 substituents’ the effects on the regulatory domains of tyrosine kinases should be due to each inhibitor stabilizing the desired inactive ATP binding site conformation.
Figure 2.
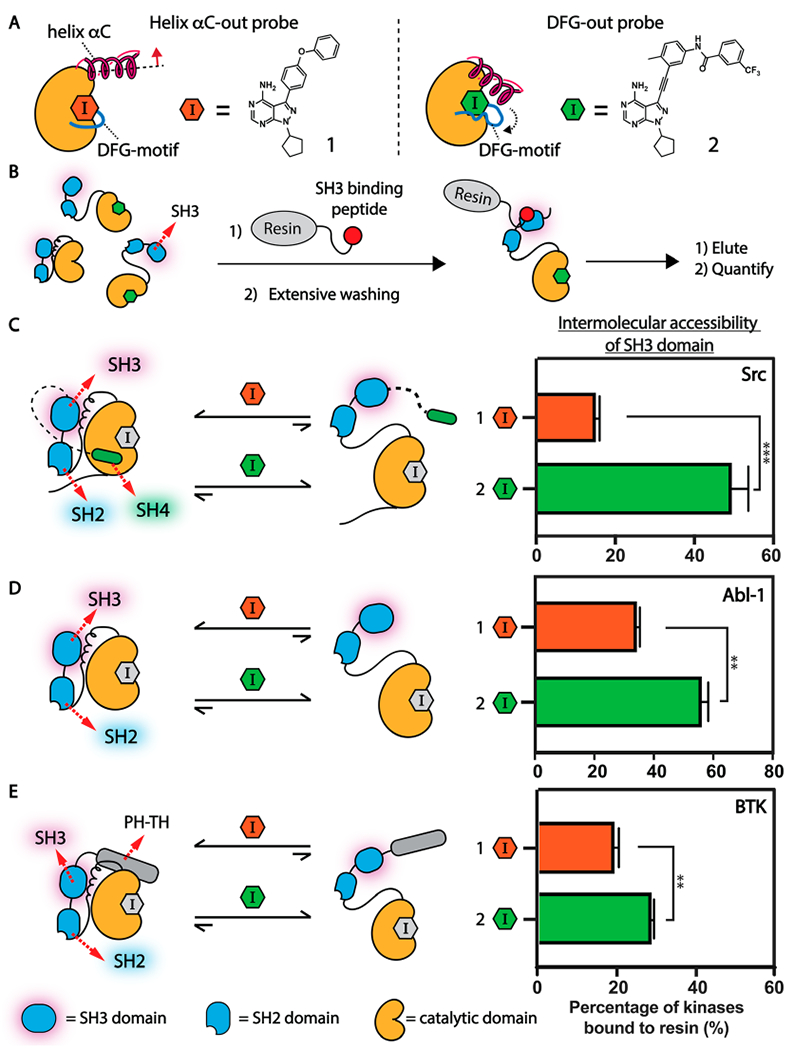
Conformation-selective inhibitors divergently modulate the global conformation of multidomain tyrosine kinases that contain an SH3-SH2-CD module. (A) Helix αC-out-stabilizing inhibitor 1 (left) and DFG-out-stabilizing inhibitor 2 (right). (B) An illustration of the SH3 domain pull-down assay. (C–E) Determination of the global conformation of inhibitor-bound tyrosine kinases that contain the SH3-SH2-CD modules. (C) Allosteric modulation of Src’s SH3-SH2-CD module by conformation-selective inhibitors. Inhibitor 1 promotes a closed SH3-SH2-CD module of Src. Inhibitor 2 promotes an open SH3-SH2-CD module of Src. (D) Allosteric modulation of Abl-1’s SH3-SH2-CD module by conformation-selective inhibitors. Inhibitor 2 promotes a more open conformation of Abl-1’s SH3-SH2-CD module than inhibitor 1. (E) Allosteric modulation of BTK’s SH3-SH2-CD module by conformation-selective inhibitors. Inhibitor 1 promotes a more closed conformation of BTK’s SH3-SH2-CD module than inhibitor 2. All values shown are mean ± SEM (n = 3). p-Values are calculated using a two-tailed t test (* < 0.05, ** < 0.01, *** < 0.001).
We first validated that each inhibitor biochemically modulates Src’s SH3-SH2-CD module as expected by performing SH3 domain pull-down assays with an immobilized SH3 domain ligand and inhibitor-bound Src complexes (Figure 2B). As expected, we found that the SH3 domain of the Src/1 complex was inaccessible to intermolecular engagement with the SH3 domain ligand, consistent with helix αC-out-stabilizing inhibitors promoting a closed, autoinhibited SH3-SH2-CD module (Figure 2C). Conversely, the Src/2 complex was significantly enriched by the immobilized SH3 domain ligand, which is consistent with 2 promoting a regulatory domain disengaged conformation of Src’s SH3-SH2-CD module. Thus, inhibitors 1 and 2 can divergently modulate the Src SH3-SH2-CD module by stabilizing two distinct inactive ATP binding site conformations.
We then investigated how our conformation-selective inhibitors affect Abl-1’s SH3-SH2-CD module. We first compared the SH3 domain accessibility of the Abl-1/1 and Abl-1/2 complexes. Like Src, the Abl-1/2 complex adopts a more open global conformation within the SH3-SH2-CD module, with a more intermolecularly accessible SH3 domain, relative to the Abl-1/1 complex (Figure 2D). To further validate how stabilization of Abl-1 in both the helix αC-out and DFG-out conformations affects the global conformation of its SH3-SH2-CD module, we utilized electrophile-containing analogues of 1 and 2 (1a and 2a, respectively) that potently inhibit a drug-sensitized cysteine mutant of Abl-1 (Abl-1 V256C) (Figure S2A). As expected, we found that the SH3 domain of the Abl-1 V256C/1a complex was largely inaccessible to intermolecular SH3 ligand engagement, which is consistent with helix αC-out-stabilizing 1a promoting a closed conformation of Abl-1 V256C’s SH3-SH2-CD module (Figure S2B and S2C). Conversely, the SH3 domain of Abl-1 V256C/2a complex was efficiently enriched by the immobilized SH3 ligand, consistent with DFG-out-stabilizing inhibitor 2a promoting an open, intramolecularly disengaged Abl-1 SH3-SH2-CD module. Thus, consistent with previous observations,25,29 an inhibitor that stabilizes the DFG-out inactive ATP binding site conformation significantly promotes a regulatory domain-disengaged conformation of Abl-1’s SH3-SH2-CD module. Inhibitors that stabilize the helix αC-out inactive ATP binding site conformation promote a closed Abl-1’s SH3-SH2-CD module, which further highlights the Src-like allosteric communication between Abl-1’s regulatory domains and its ATP binding site.
Next, we explored whether conformation-selective, ATP-competitive inhibitors can also divergently modulate BTK’s SH3-SH2-CD module. We first evaluated how the SH3 domain accessibility of the BTK/1 complex compared to the BTK/2 complex:. Like Src and Abl-1, helix αC-out-stabilizing inhibitor 1 resulted in a closed global conformation of BTICs SH3-SH2-CD module, with a largely intermolecularly inaccessible SH3 domain relative to the BTK/2 complex (Figure 2E). To further validate how stabilization of BTK in both inactive ATP binding site conformations affects the global conformation of its SH3-SH2-CD module, we used a drug-sensitized variant of BTK (BTK V416C) with inhibitors 1a and 2a.13,14 Like wild-type BTK, we found that the SH3-SH2-CD module of the BTK V416C/1a complex adopted a more closed global conformation compared to the BTK/2a complex (Figure S3). Thus, conformation-selective inhibitors are capable of divergently modulating BTK’s SH3-SH2-CD module based on the ATP binding site conformation they stabilize.
Binding Features of Inhibitors That Promote an Open Global Conformation of Src.
While inhibitors that stabilize the DFG-out inactive conformation promote an open global conformation of tyrosine kinases that contain an SH3-SH2-CD architecture,22,23 we have found that other classes of ATP-competitive inhibitors are also capable of promoting allosteric disengagement of regulatory SH2 and SH3 domains.18 To better understand the common features of inhibitors that promote an open global conformation of the SH3-SH2-CD regulatory module, we assembled a panel of 13 inhibitors that contain structurally diversified substituents that make varied ATP binding site contacts and tested them in biochemical assays for the global conformational state of Src (Figure 3A). Inhibitors 1, 3, and 4 contain pharmacophores that should stabilize the helix αC-out inactive conformation of Src,14,17 while 2 and 5 contain substituents that stabilize the DFG-out form.14,32 The other eight inhibitors in our panel (6–13) contain various substituents that project into the ATP-binding pocket but are predicted to not stabilize either inactive conformation of the ATP binding site.
Figure 3.
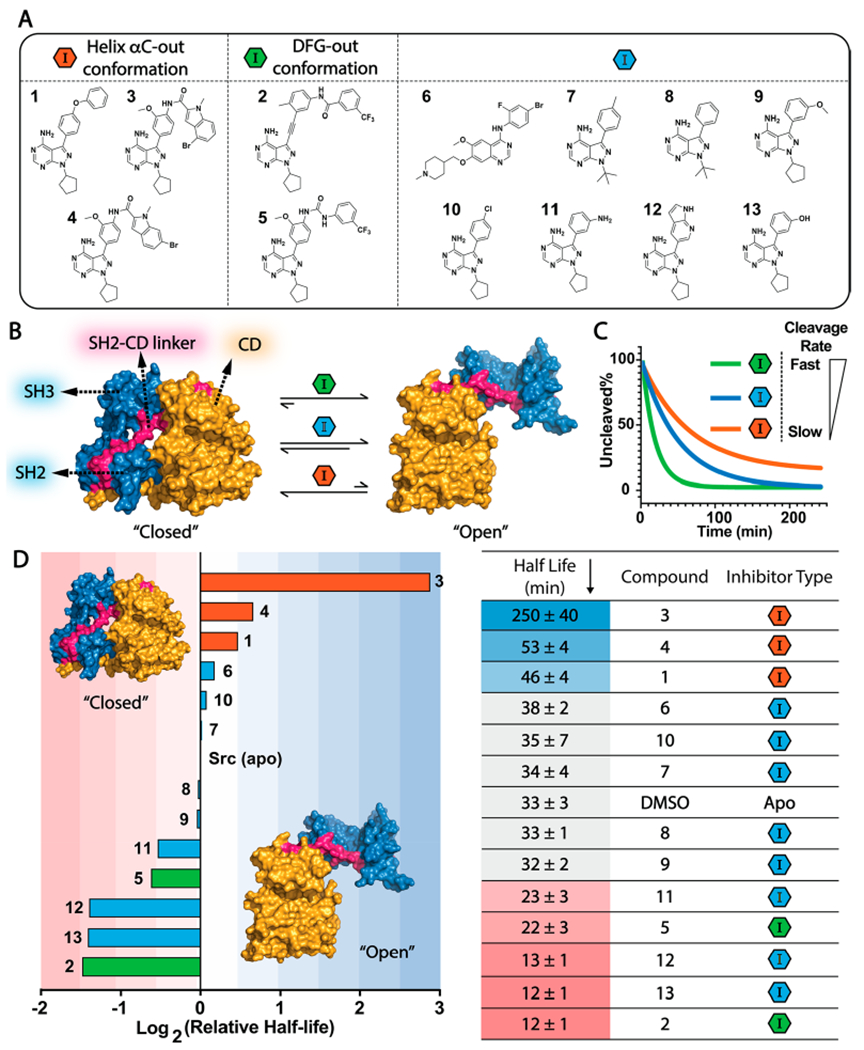
Binding features of inhibitors that promote an open global conformation of Src. (A) A panel of 13 ATP-competitive Src inhibitors. (B) Allosteric modulation of Src’s global conformation with ATP-competitive inhibitors. (C) Thermolysin assay. The rate for proteolytic cleavage of the SH2-CD linker is sensitive to the global conformation of inhibitor-bound Src complexes. Thermolysin more rapidly cleaves the SH2-CD linker (magenta) of inhibitor-bound Src in the open global conformation than the closed global conformation. The catalytic domain (CD) is colored in yellow and regulatory domains in blue. (D) (Left) Ranking of the log2-based relative half-life values of proteolytic cleavage rates of Src’s SH2-CD linker. Relative ratio = log2[half-life(inhibitor-bound Src)/half-life(apo Src)]. The means of the relative ratios are shown. (Right) The mean ± SEM of half-life values are shown (n = 3).
To probe how our panel of inhibitors modulate the global conformation of Src’s SH3-SH2-CD module, we performed limited proteolysis experiments with inhibitor-bound Src complexes (Figure 3B and C).22 It has previously been demonstrated that the metalloprotease thermolysin can selectively cleave the flexible linker that connects Src’s SH2 domain to its CD (SH2-CD linker) and that the cleavage rate is sensitive to Src’s global regulatory conformation (Figure 3B, colored in magenta).22,33 Therefore, we used half-life values for proteolytic cleavage as readouts for the global conformation of Src’s SH3-SH2-CD module. We benchmarked the half-life for cleavage of apo Src as 33 ± 3 min. To assign a global conformation to inhibitor-bound Src complexes, we measured the half-life for their cleavage and used 5σ as the cutoff for significant deviation from apo Src (Figure 3D). Consistent with the ability of DFG-out-stabilizing inhibitors to disengage the regulatory SH2 and SH3 domains from Src’s CD, the SH2-CD linkers of the Src/2 and Src/5 complexes were cleaved more rapidly than apo Src. Conversely, consistent with helix αC-out-stabilizing inhibitors enhancing the regulatory SH2 and SH3 domain engagement with Src’s CD, the SH2-CD linkers of the Src/1, Src/3, and Src/4 complexes were cleaved more slowly than apo Src.
As expected, we found that the SH2-CD linker of Src bound to inhibitors 6, 7, 8, 9, or 10, which all contain substituents that should make minimal interactions with helix αC or the activation loop, was cleaved at rates similar to Src’s apo form (Figure 3D). However, although 11, 12 (PDB ID: 3EN4),34 and 13 do not contain substituents that stabilize either inactive conformation of Src’s ATP binding site, the SH2-CD linker of Src bound to these inhibitors was cleaved at rates similar to the Src/2 and Src/5 complexes. Thus, 11-13 appear to promote a regulatory domain disengaged conformation of the SH3-SH2-CD module like DFG-out-stabilizing inhibitors. Despite lacking a substituent that promotes a flipped DFG-motif, like DFG-out stabilizing inhibitors 2 and 5, 11–13 contain H-bond donors that we predicted are capable of forming a H-bonding interaction with the side-chain of Glu310 in Src’s helix αC. As most inhibitors that stabilize the DFG-out conformation also contain H-bond donors that form a H-bonding interaction with the side-chain of Glu310 in Src’s helix αC (Figure 4A), we further investigated whether this interaction is responsible for promoting an open global conformation of the SH3-SH2-CD module.
Figure 4.
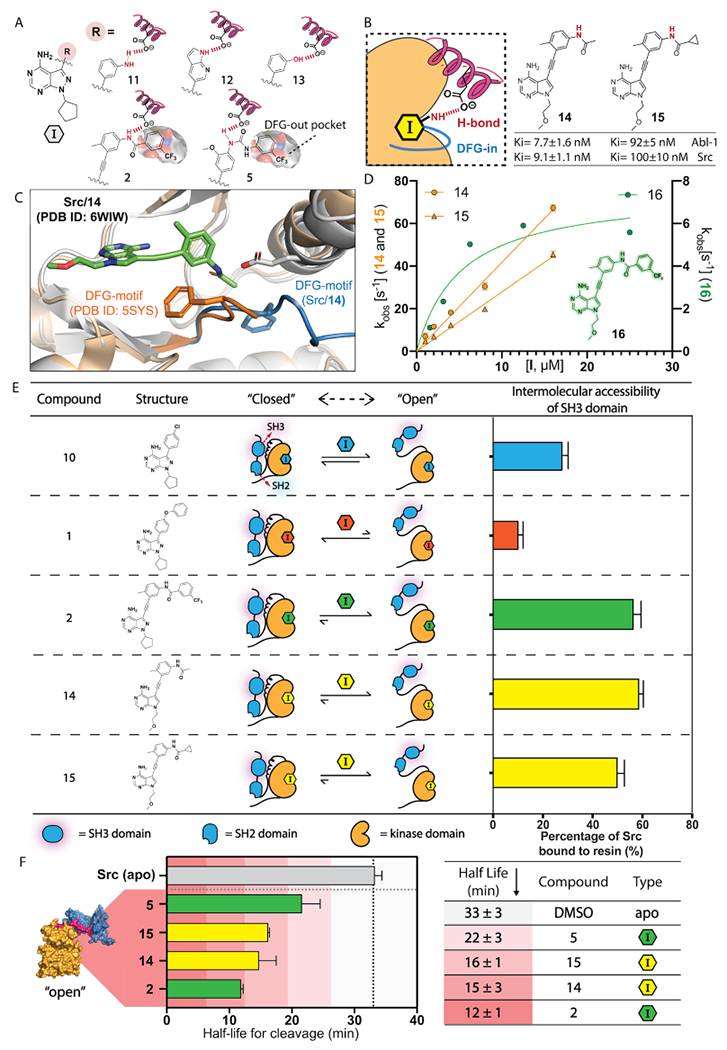
Analysis of how helix αC interactions influence global conformation. (A) ATP binding site contacts of substituents that promote an open global conformation of Src. H-bonding interactions are colored in red. The hydrophobic DFG-out pocket is annotated. (B) Structures and the predicted binding features of inhibitors 14 and 15. Ki values are shown as mean ± SEM (n = 3). (C) An overlay of the X-ray crystal structures of the Src/14 (PDB ID: 6WIW) and Src V284C/2a (PDB ID: 5SYS) complexes. Inhibitor 14 is colored in green. The protein structure of Src/14 is colored in gray, and its activation loop is colored in blue. The protein structure of Src V284C/2a is colored in orange. (D) The observed rate constants for Src binding to 14, 15, or 16 at pH 7.0. Mean ± SEM values are shown (n ≥ 3). (E) SH3 domain intermolecular accessibility of Src bound to inhibitors 10, 1, 2, 14, or 15. Mean ± SEM are shown (n = 3). (F) Thermolysin assays with Src/14 and Src/15 complexes. The half-lives for cleavage of Src’s SH2-CD linker are shown as the mean ± SEM (n = 3).
Analysis of How Helix αC Interactions Influence Global Conformation.
Recent studies have shown that the ability of inhibitors to flip the DFG-motif of Abl-1’s activation loop highly correlates with their promotion of an open global conformation of Abl-1’s SH3-SH2-CD module.29 Our observations in Src made us interested in revisiting the molecular binding features of 14 ligands that have previously been reported to promote an open global conformation of Abl-1, which was determined by characteristic NMR 1H–15N backbone chemical shifts within the SH3 and SH2 domains (Figure S4).29 We found that all inhibitors, including imatinib, nilotinib, ponatinib, rebastinib, and bafetinib, that promote an open global conformation of Abl-1’s SH3-SH2-CD module also form a H-bonding interaction with the side-chain of Abl-1’s Glu286 that stabilizes its helix αC in the active conformation (Figure S5). In contrast, inhibitors that lack the ability to form a H-bonding interaction with Glu286 do not promote an open global conformation of Abl-1 (Figure S4B). Taken together, an inhibitor’s ability to hydrogen bond with the conserved Glu in helix αC highly correlates with the global conformation of Src’s and Abl-1’s SH3-SH2-CD modules. Therefore, we speculate that the influence of an inhibitor on the conformation of helix αC, and not on the DFG-motif, is responsible for its ability to promote an open global conformation of the SH3-SH2-CD module.
To test this notion, we designed inhibitors that possess the same H-bonding pattern as DFG-out-stabilizing inhibitors but lack bulky substituents that flip the DFG motif of the activation loop. We generated two analogues, 14 and 15 (Figure 4B), of DFG-out stabilizing inhibitor 2 that retain the same H-bonding pattern as 2 but contain less bulky alkyl substituents. While, unsurprisingly, 14 and 15 are less potent Src inhibitors than 2 (Ki < 1 nM),14 they both inhibit the phosphotransferase activity of Src and Abl-1 at reasonable concentrations.
To determine whether our new analogues interact with the ATP binding site of Src as designed, we obtained a crystal structure of 14 bound to the CD of Src (Figure 4C and Figure S7). Two molecules of 14-bound Src’s CD were observed per crystallographic asymmetric unit. Inhibitor 14 occupies the ATP binding site of Src and makes the same H-bonding interactions with the hinge region as similar pyrrolopyrimidine-based inhibitors.14,32,34–36 Notably, the Src/14 complex is in the active conformation, which is characterized by an inward position of the helix αC (helix αC-in) in the N-lobe and an unperturbed activation loop. Like a DFG-out stabilizing inhibitor, the amide linker that projects from the alkynylphenyl group of 14 forms a H-bond with Glu310 in the helix αC, presumably stabilizing the active conformation of this structural element (Figure 4C). Although the acetamide group of 14 projects toward the activation loop, Src’s DFG-motif is not flipped. An overlay of the Src/14 complex with a crystal structure of Src in the DFG-out conformation shows that 14’s acetamide group is not large enough to perturb the Phe405 residue of the DFG-motif in the activation loop from an active conformation.
To confirm that inhibitors 14 and 15 have a similar effect on Src’s ATP binding site in solution, we monitored their binding kinetics with Src using a previously reported stopped-flow tryptophan fluorescence assay (Figure 4D).37 For inhibitors that stabilize the DFG-out conformation, the rate-determining event for binding at high inhibitor concentrations is the flipping of the DFG-motif. This leads to an observed rate constant for binding (kobs) that deviates from linearity of otherwise pseudo first-order kinetics.37,38 Consistent with the flipping of the DFG-motif not being required for 14 and 15 to interact with the ATP binding site of Src, we found that the observed rate constants for both inhibitors increased linearly with concentration (Figure 4D, colored in yellow; Figure S8). In contrast, inhibitor 16, which is a direct DFG-out-stabilizing analogue of 14 and 15, demonstrated a nonlinear kobs with increasing inhibitor concentration, consistent with 16 requiring a flipped DFG-motif to be accommodated in Src’s ATP binding site (Figure 4D, colored in green). We also tested 14 and 15 for their binding kinetics with Abl-1 and found that both inhibitors demonstrate a kinetic profile that is similar to their interaction with Src (Figure S9). Taken together, we validated that 14 and 15 retain the same H-bonding pattern as DFG-out-stabilizing inhibitors but do not require flipping of the DFG-motif in the activation loop of tyrosine kinases for binding.
Inhibitors 14 and 15 Promote an Open Global Conformation of Src.
To determine whether our new helix αC-in-stabilizing inhibitors 14 and 15 promote an open global conformation of Src’s SH3-SH2-CD module, we performed SH3 domain pull-down assays and limited proteolysis experiments with inhibitor-bound Src complexes. We found that both the Src/14 and Src/15 complexes were efficiently enriched by an immobilized SH3 ligand, consistent with 14 and 15 promoting an open, regulatory domain-disengaged conformation of Src (Figure 4E). Notably, the SH3 regulatory domains of both the Src/14 and Src/15 complexes had similar intermolecular accessibilities as the SH3 domain of Src bound to DFG-out stabilizing inhibitor 2. We also found that the half-lives for cleavage of the SH2-CD linkers of the Src/14 (t1/2 = 15 ± 3 min) and Src/15 (t1/2 = 16 ± 1 min) complexes by thermolysin was within the range of Src bound to DFG-out-stabilizing inhibitors 2 and 5 (Figure 4F). Thus, the observed capability of DFG-out-stabilizing inhibitors to promote an open global conformation of Src appears to rely on their ability to stabilize an active conformation of helix αC, rather than their ability to perturb the activation loop.
A Chemical Genetic Strategy for Investigating the Effects of Stabilizing the Helix αC-in Conformation in Cells.
We recently demonstrated that modulating Src’s global conformation in cells with conformation-selective inhibitors can influence the phosphotransferase-independent functions of its regulatory domains.13,14 To determine how stabilizing the helix αC-in conformation (characterized by an inward position of the helix αC in the N-lobe and an unperturbed activation loop) of Src affects its phosphotransferase-independent functions in cells compared to alternative ATP binding site conformations, we pursued a chemical genetic strategy for sensitizing kinases to specific inhibitors.35,36,39,40 Our strategy, which we call Cysteine Installation for Modulating Allostery and Targeted Inhibition of Kinases (CystIMATIK) (Figure 5A),14 involves the introduction of a cysteine residue at the β2 strand of the N-terminal lobe of the catalytic domains of kinases that provides sensitivity to pyrrolopyrimidine-based inhibitors that contain a Michael acceptor at the C-6 position (Figure 5B). To implement the CystIMATIK strategy for Src, we generated electrophile-containing analogues of 14 and 15 (14a and 15a, Figure 5C) and tested them for inhibition of drug-sensitized Src (Src V284C). We found that both 14a and 15a provided potent inhibition of Src V284C but not the wild-type kinase (Figure 5C).
Figure 5.
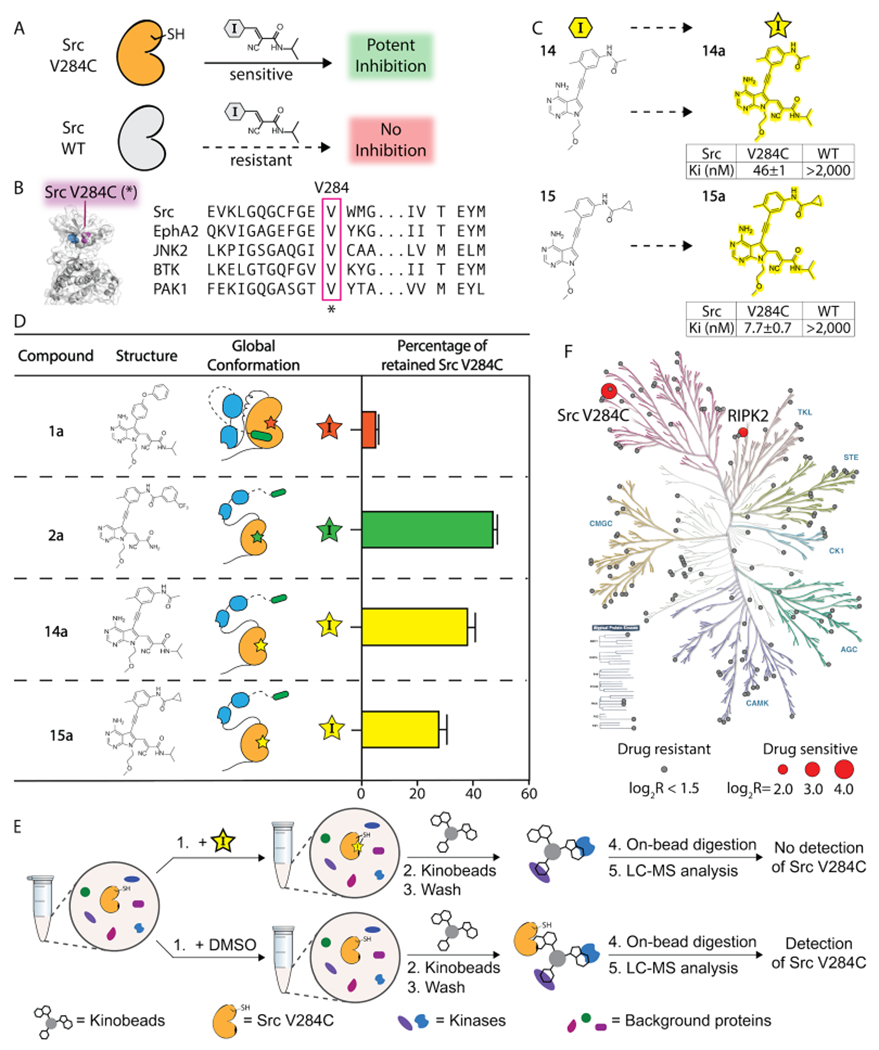
Chemical genetic strategy for investigating the effects of stabilizing the helix αC-in conformation of Src in cells. (A) CystIMATIK probes provide potent inhibition of drug-sensitized kinase variants, while wild-type kinases are largely resistant to CystIMATIK probes. (B) Sensitization of Src to CystIMATIK probes through the introduction of a cysteine at V284. The sequence alignment shows a conserved valine at the position equivalent to V284 of Src. (C) Electrophile-containing analogues of 14 and 15. The Ki values of 14a (top) and 15a (bottom) for wild-type Src and Src V284C are shown as mean ± SEM (n = 3). (D) SH3 domain pull-down assays for characterizing the global conformation of the 14a/Src V284C and 15a/ Src V284C complexes. Values shown are the mean ± SEM (n = 3). (E) Kinobead-based profiling method for determining the kinome-wide selectivity of 14a and 15a. Src V284C HeLa cell lysates were incubated with DMSO, 14a, or 15a and an immobilized matrix of nonselective kinase inhibitors (kinobeads). Captured kinases were subjected to the proteomics workflow described in the Supporting Information. (F) Phylogenetic trees showing the selectivity of 14a. All profiled kinases are represented by circles. Drug-sensitive kinases are shown as red circles with the size corresponding to the level of competition (larger circle, more competed). Drug-resistant kinases are shown as gray circles. Kinases reported as being drug-sensitive (Log2R > 1.5) were also required to show significance (−Log10P value > 1.5) from a two-sample t test with an FDR of 0.05 (n = 3).
To validate that both 14a and 15a modulate the global conformation of Src like their nonelectrophilic counterparts (Figure 5D), we performed SH3 domain pull-down assays with Src V284C bound to 14a, 15a, and electrophile-containing analogues of 1 and 2, 1a and 2a. As expected, we found that the SH3 domain of the Src V284C/1a complex was largely inaccessible to an immobilized SH3 ligand, while the Src V284C/2a complex’s SH3 domain readily participated in intermolecular interactions. Like the Src V284C/2a complex, we observed that both the Src V284C/14a and Src V284C/15a complexes were efficiently enriched by the immobilized SH3 ligand, which is consistent with their ability to promote an open global conformation of Src V284C’s SH3-SH2-CD module.
Prior to studying how 14a and 15a affect Src’s phosphotransferase-independent functions in cells, we determined their selectivity for Src V284C with a lysate profiling method (Figure 5E).41–43 To do this, we measured the ability of 14a and 15a to compete for binding to a mixture of resin-immobilized nonselective ATP-competitive inhibitors (kinobeads) with lysate kinases. The binding of an inhibitor being profiled prevents enrichment of lysate kinases by kinobeads, which allows us to profile inhibitors against a large number of human kinases using quantitative mass spectrometry. Specifically, inhibitor selectivity can be determined by measuring the loss of relative enrichment of kinases in inhibitor-treated lysates versus lysate treated with a vehicle control (DMSO). 14a and 15a were both profiled at a single high concentration (20 μM) in Src V284C-expressing HeLa lysates, and the relative enrichment of ~150 kinases was quantified. We found that Src V284C was the most competed target of 14a, with Receptor Interacting Serine/Threonine Kinase 2 (RIPK2) being the only significant off-target kinase (Figure 5F). 15a demonstrated a similar selectivity profile, except Src V284C and RIPK2 were competed at similar levels (Figure S10). Thus, Src V284C appears to be the primary target for 14a and 15a with RIPK2 being the only off-target for both inhibitors in HeLa lysates.
Stabilization of Src in the Helix αC-in Conformation Is Sufficient to Promote a Phosphotransferase-Independent Alteration in Cell Morphology.
We recently showed that Src can promote nonapoptotic membrane blebs, which are characterized by localized disruption of the actin-myosin cortex, through a phosphotransferase-independent mechanism and that conformation-selective inhibitors can differentially modulate this phenotype (Figure 6A).14,44 We found that the expression of drug-sensitive Src V284C and wild-type Src (WT) in HeLa cells yielded a basal number of cells with membrane blebs (~10%), which was negligibly influenced by treatment with helix αC-out-stabilizing 1a (Figure 6B). In contrast, treatment with DFG-out-stabilizing inhibitor 2a significantly induced membrane blebs in HeLas expressing Src V284C (~55%) but not in Src WT-expressing HeLa cells (Figure 6B).14
Figure 6.
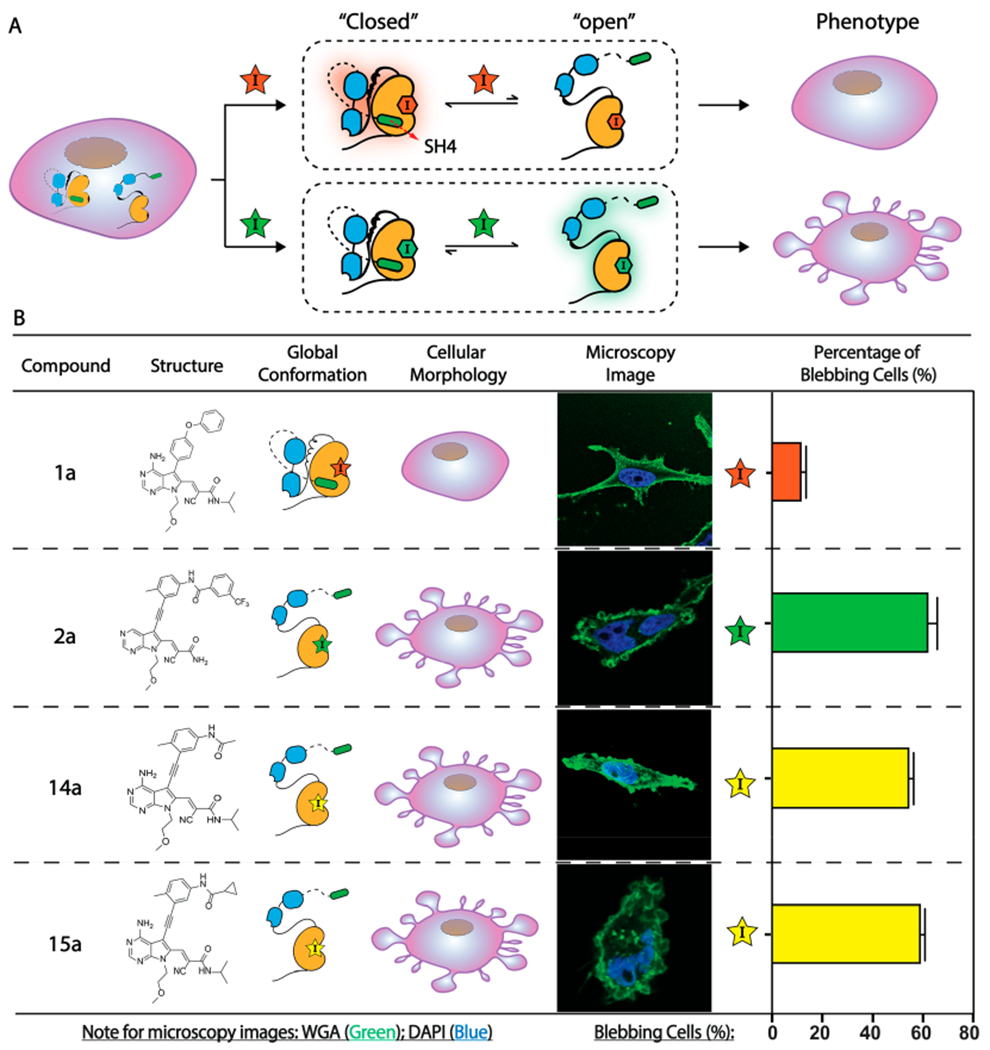
Stabilization of Src’s helix αC in an active conformation (helix αC-in) is sufficient to promote a Src-mediated, phosphotransferase-independent alteration in cell morphology. (A) CystIMATIK probes that enforce an open global conformation of Src V284C promote nonapoptotic membrane blebs in HeLas expressing drug-sensitized Src V284C. (B) Levels of membrane blebs in Src V284C-expressing HeLas that are treated with CystIMATIK probes. Percentages of blebbing cells are shown as mean ± SEM (n = 3). Nuclei were stained with DAPI (blue) and membranes were stained with Wheat Germ Agglutinin (WGA)-Alexa488 (green). For each replicate, 17 to 32 images were obtained and the percentage of blebbing cells from all imaged cells was quantified. Each image contained 1–3 stained cells. The number of blebbing cells was scored based on the criteria described in the Supporting Information.
Our observations with 14 and 15 in vitro made us interested in investigating whether stabilizing Src in the helix αC-in conformation in cells is sufficient to mimic the phenotype promoted by DFG-out-stabilizing inhibitor 2a. Therefore, we tested the influence of 14a and 15a on the cell morphology of Src V284C-expressing HeLas. We found that treatment of Src V284C-expressing HeLas with 14a and 15a yielded a significant increase in the percentage of cells with membrane blebs (~60%) (Figure 6B), while Src WT-expressing HeLas remained at the basal level when treated with these inhibitors (Figure S11). Notably, Src V284C-expressing HeLas that are treated with 14a and 15a showed levels of blebbing comparable to cells treated with 2a. Thus, stabilization of Src in the helix αC-in conformation is sufficient to induce membrane blebs. Therefore, the observed ability of a DFG-out-stabilizing inhibitor to promote membrane blebs is not due to its ability to perturb Src’s activation loop but rather its ability to stabilize Src’s helix αC in an active conformation.
CONCLUSIONS
Interdomain regulation is a defining characteristic of many multidomain kinases. Almost half of the human kinases contain at least one auxiliary domain which, in many cases, are implicated in phosphotransferase-independent functions within cells.5 Although a growing body of evidence strongly suggests that conformation-selective, ATP-competitive inhibitors can modulate regions distal to a kinase’s CD, the overall generality and molecular determinants of this phenomenon are not completely known. In this study, we have demonstrated that inhibitors which stabilize two different inactive ATP binding site conformations can divergently modulate the global conformation of Src, Abl-1, and the Tec family kinase BTK. Despite structural differences in the N- and C-termini of SFKs, Abl-1, and BTK, the allosteric communication between their ATP binding sites and regulatory SH3 and SH2 domains remain conserved. This observation suggests that the effects of conformation-selective inhibitors on global conformation are likely conserved in other tyrosine kinases that contain a homologous SH3-SH2-CD module.
By systematically analyzing the common features of inhibitors that promote an open global conformation of Src, we were able to develop chemical probes that allowed us to isolate the effects of specific ATP binding site interactions. With these probes, we found that an inhibitor’s ability to promote an open global conformation of Src’s SH3-SH2-CD module mainly relies on its ability to stabilize an active conformation of the helix αC. Thus, the noted ability of DFG-out-stabilizing inhibitors to promote an open global conformation in Src and Abl-1 is most likely due to their interactions with the helix αC, rather than perturbation of the DFG-motif in the activation loop (Figure 7). Furthermore, we have determined that stabilization of Src’s helix αC in the active conformation (helix αC-in) is sufficient to promote a Src-mediated, phosphotransferase-independent alteration in cell morphology, a phenotypic effect consistent with releasing the otherwise intramolecularly sequestered N-terminal SH4 domain of Src. Like Src’s SH4 domain, Abl-1’s N-terminal myristate and BTK’s N-terminal PH-TH domain not only suppress phosphotransferase activity by interacting with their CDs26,27,45,46 but also interact with the plasma membrane and additional organelles when intermolecularly accessible.27,45,47–49 Based on our observation with Src, inhibitors that stabilize or perturb the helix αCs of Abl-1 and BTK likely influence the localization and other phosphotransferase-independent functions of these kinases by allosterically modulating global conformation.50,51
Figure 7.
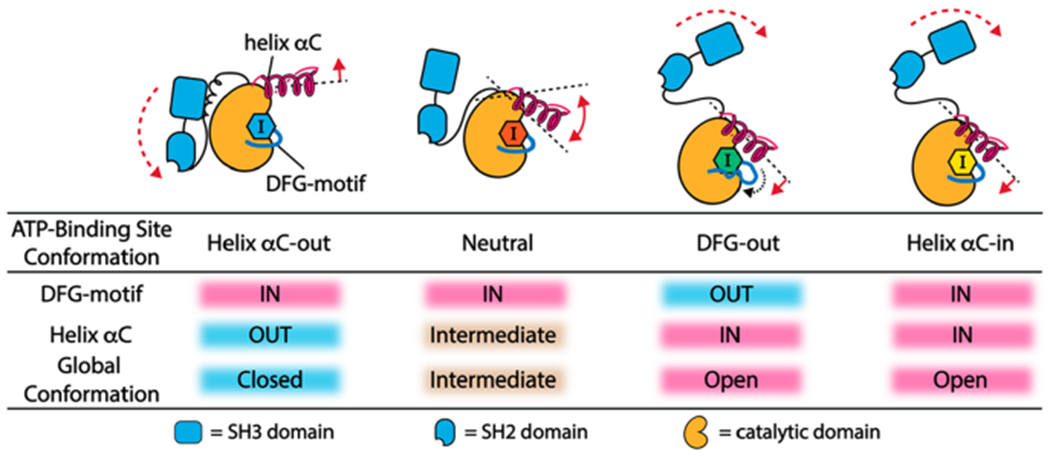
ATP binding site contacts of inhibitors that allosterically modulate the SH3-SH2-CD module. An inhibitor’s capability of stabilizing helix αC in an active conformation (helix αC-in) is the main determinant of its ability to promote an open global conformation of the SH3-SH2-CD module.
Would we expect a DFG-out stabilizing inhibitor and an inhibitor that stabilizes the helix αC-in conformation without flipping the DFG-motif to differentially modulate the cellular function of kinases that contain a Src-like regulatory module? At this point in time, we do not know the answer to this question. In addition to their ability to completely suppress the phosphotransferase activity of Src, we have observed that both classes of inhibitors have similar effects on Src’s regulatory domain apparatus. While both classes of inhibitors similarly influence one Src-mediated phenotypic effect, we cannot exclude the possibility that there are specific cellular interactions that are influenced by the conformation of the DFG-motif. Differential modulation of these cellular interactions could result in divergences in Src’s phosphotransferase-independent functions. Comparative analyses of how 14a and 15a differ from DFG-out-stabilizing inhibitor 2a in their ability to modulate the interactome and cellular functions of drug-sensitized Src (or other drug-sensitized tyrosine kinases) will provide insight into this question.
Taken together, our results have direct implications for targeting the helix αC in the ATP binding site of multidomain tyrosine kinases with inhibitors to modulate kinase functional surfaces distal to their CDs. Although our results highlight the effects of conformation-selective inhibitors on the subset of tyrosine kinases that contain an SH3-SH2-CD module, it is likely that multidomain kinases with alternative regulatory architectures can also be allosterically modulated using similar principles. The conceptual framework and probes that we describe in this report may help facilitate these studies.
Supplementary Material
ACKNOWLEDGMENTS
We thank N. Peters at the W.M. Keck Center for Advanced Studies in Neural Signaling for microscopy assistance. We thank P. von Haller at the University of Washington Proteomics Resource (UWPR) and M. Sadilek for mass spectrometry assistance. We thank S. Taylor for providing the Flp-In T-Rex HeLa cell line. We thank the NSLS2 facility of U.S. Department of Energy (DOE) Office operated by the Brookhaven National Laboratory under Contract No. DESC0012704. This work was supported by NIH R01 GM086858 (DM.), NIH R35 GM119437 (M.S.), and R25 GM103962 (J.V.-B.).
ABBREVIATIONS
- SFKs
Src-family kinases
- CD
catalytic domain
- 3D
three domain kinase containing CD, SH2, and SH3 domains
- CystIMATIK
Cysteine Installation for Modulating Allostery and Targeted Inhibition of Kinases
- DMSO
dimethyl sulfoxide
- NMR
nuclear magnetic resonance
- MS
mass spectrometry
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.0c00429
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00429.
Supplementary figures, experimental details, and synthesis of all compounds used in the study (PDF)
Supplementary Excel file containing the MaxQuant output data for inhibitor profiling of 14a and 15a (XLSX)
The authors declare no competing financial interest.
Contributor Information
Linglan Fang, Departments of Chemistry, University of Washington, Seattle, Washington 98195, United States.
Jessica Vilas-Boas, Department of Pharmacological Sciences, Stony Brook University, Stony Brook, New York 11794-8651, United States.
Sujata Chakraborty, Departments of Chemistry, University of Washington, Seattle, Washington 98195, United States.
Zachary E. Potter, Departments of Chemistry, University of Washington, Seattle, Washington 98195, United States
Ames C. Register, Departments of Chemistry, University of Washington, Seattle, Washington 98195, United States
Markus A. Seeliger, Department of Pharmacological Sciences, Stony Brook University, Stony Brook, New York 11794-8651, United States.
Dustin J. Maly, Departments of Chemistry and Biochemistry, University of Washington, Seattle, Washington 98195, United States.
REFERENCES
- (1).Taylor SS, Keshwani MM, Steichen JM, and Kornev AP (2012) Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos. Trans. R. Sac., B 367, 2517–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Manning G, Whyte DB, Martinez R, Hunter T, and Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- (3).Kuriyan J, and Eisenberg D (2007) The origin of protein interactions and allostery in colocalization. Nature 450, 983–990. [DOI] [PubMed] [Google Scholar]
- (4).Pellicena P, and Kuriyan J (2006) Protein-protein interactions in the allosteric regulation of protein kinases. Curr. Opin. Struct. Biol 16, 702–709. [DOI] [PubMed] [Google Scholar]
- (5).Kung JE, and Jura N (2016) Structural Basis for the Non-catalytic Functions of Protein Kinases. Structure 24, 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Huse M, and Kuriyan J (2002) The conformational plasticity of protein kinases. Cell 109, 275–282. [DOI] [PubMed] [Google Scholar]
- (7).Taylor SS, and Kornev AP (2011) Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci 36, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Engen JR, Wales TE, Hochrein JM, Meyn MA 3rd, Banu Ozkan S, Bahar I, and Smithgall TE (2008) Structure and dynamic regulation of Src-family kinases. Cell. Mol. Life Sci 65, 3058–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sicheri F, and Kuriyan J (1997) Structures of Src-family tyrosine kinases. Curr. Opin. Struct. Biol 7, 777–785. [DOI] [PubMed] [Google Scholar]
- (10).Kim LC, Song L, and Haura EB (2009) Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol 6, 587–595. [DOI] [PubMed] [Google Scholar]
- (11).Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, and Kuriyan J (2011) Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 42, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Boggon TJ, and Eck MJ (2004) Structure and regulation of Src family kinases. Oncogene 23, 7918–7927. [DOI] [PubMed] [Google Scholar]
- (13).Fang L, Chakraborty S, Dieter EM, Potter ZE, Lombard CK, and Maly DJ (2019) Chemoproteomic Method for Profiling Inhibitor-Bound Kinase Complexes. J. Am. Chem. Soc 141, 11912–11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ahler E, Register AC, Chakraborty S, Fang L, Dieter EM, Sitko KA, Vidadala RSR, Trevillian BM, Golkowski M, Gelman H, Stephany JJ, Rubin AF, Merritt EA, Fowler DM, and Maly DJ (2019) A Combined Approach Reveals a Regulatory Mechanism Coupling Src’s Kinase Activity, Localization, and Phosphotransferase-Independent Functions. Mol. Cell 74, 393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Young MA, Gonfloni S, Superti-Furga G, Roux B, and Kuriyan J (2001) Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell 105, 115–126. [DOI] [PubMed] [Google Scholar]
- (16).Xu W, Doshi A, Lei M, Eck MJ, and Harrison SC (1999) Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell 3, 629–638. [DOI] [PubMed] [Google Scholar]
- (17).Chakraborty S, Inukai T, Fang L, Golkowski M, and Maly DJ (2019) Targeting Dynamic ATP-Binding Site Features Allows Discrimination between Highly Homologous Protein Kinases. ACS Chem. Biol 14, 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Leonard SE, Register AC, Krishnamurty R, Brighty GJ, and Maly DJ (2014) Divergent modulation of Src-family kinase regulatory interactions with ATP-competitive inhibitors. ACS Chem. Biol 9, 1894–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Krishnamurty R, Brigham JL, Leonard SE, Ranjitkar P, Larson ET, Dale EJ, Merritt EA, and Maly DJ (2013) Active site profiling reveals coupling between domains in SRC-family kinases. Nat. Chem. Biol 9, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Register AC, Leonard SE, and Maly DJ (2014) SH2-catalytic domain linker heterogeneity influences allosteric coupling across the SFK family. Biochemistry 53, 6910–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tong M, Pelton JG, Gill ML, Zhang W, Picart F, and Seeliger MA (2017) Survey of solution dynamics in Src kinase reveals allosteric cross talk between the ligand binding and regulatory sites. Nat. Commun 8, 2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Agius MP, Ko KS, Johnson TK, Kwarcinski FE, Phadke S, Lachacz EJ, and Soellner MB (2019) Selective Proteolysis to Study the Global Conformation and Regulatory Mechanisms of c-Src Kinase. ACS Chem. Biol 14, 1556–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kwarcinski FE, Brandvold KR, Phadke S, Beleh OM, Johnson TK, Meagher JL, Seeliger MA, Stuckey JA, and Soellner MB (2016) Conformation-Selective Analogues of Dasatinib Reveal Insight into Kinase Inhibitor Binding and Selectivity. ACS Chem. Biol 11, 1296–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Seeliger MA, Nagar B, Frank F, Cao X, Henderson MN, and Kuriyan J (2007) c-Src binds to the cancer drug imatinib with an inactive Abl/c-Kit conformation and a distributed thermodynamic penalty. Structure 15, 299–311. [DOI] [PubMed] [Google Scholar]
- (25).Skora L, Mestan J, Fabbro D, Jahnke W, and Grzesiek S (2013) NMR reveals the allosteric opening and closing of Abelson tyrosine kinase by ATP-site and myristoyl pocket inhibitors. Proc. Natl. Acad. Sci. U. S. A 110, E4437–4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hantschel O, Nagar B, Guettler S, Kretzschmar J, Dorey K, Kuriyan J, and Superti-Furga G (2003) A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 112, 845–857. [DOI] [PubMed] [Google Scholar]
- (27).Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, Clarkson B, Superti-Furga G, and Kuriyan J (2003) Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 112, 859–871. [DOI] [PubMed] [Google Scholar]
- (28).Nagar B, Hantschel O, Seeliger M, Davies JM, Weis WI, Superti-Furga G, and Kuriyan J (2006) Organization of the SH3-SH2 unit in active and inactive forms of the c-Abl tyrosine kinase. Mol. Cell 21, 787–798. [DOI] [PubMed] [Google Scholar]
- (29).Sonti R, Hertel-Hering I, Lamontanara AJ, Hantschel O, and Grzesiek S (2018) ATP Site Ligands Determine the Assembly State of the Abelson Kinase Regulatory Core via the Activation Loop Conformation. J. Am. Chem. Soc 140, 1863–1869. [DOI] [PubMed] [Google Scholar]
- (30).Amatya N, Lin DY, and Andreotti AH (2019) Dynamic regulatory features of the protein tyrosine kinases. Biochem. Soc. Trans 47, 1101–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tong M, and Seeliger MA (2015) Targeting conformational plasticity of protein kinases. ACS Chem. Biol 10, 190–200. [DOI] [PubMed] [Google Scholar]
- (32).Dar AC, Lopez MS, and Shokat KM (2008) Small molecule recognition of c-Src via the Imatinib-binding conformation. Chem. Biol 15, 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).MacAuley A, and Cooper JA (1989) Structural differences between repressed and derepressed forms of p60c-src. Mol. Cell. Biol 9, 2648–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, and Knight ZA (2008) Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol 4, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Garske AL, Peters U, Cortesi AT, Perez JL, and Shokat KM (2011) Chemical genetic strategy for targeting protein kinases based on covalent complementarity. Proc. Natl. Acad. Sci. U. S. A 108, 15046–15052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Zhang C, Lopez MS, Dar AC, Ladow E, Finkbeiner S, Yun CH, Eck MJ, and Shokat KM (2013) Structure-guided inhibitor design expands the scope of analog-sensitive kinase technology. ACS Chem. Biol 8, 1931–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Shan Y, Seeliger MA, Eastwood MP, Frank F, Xu H, Jensen MO, Dror RO, Kuriyan J, and Shaw DE (2009) A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. U. S. A 106, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Seeliger MA, Ranjitkar P, Kasap C, Shan Y, Shaw DE, Shah NP, Kuriyan J, and Maly DJ (2009) Equally potent inhibition of c-Src and Abl by compounds that recognize inactive kinase conformations. Cancer Res. 69, 2384–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kung A, Schimpl M, Ekanayake A, Chen YC, Overman R, and Zhang C (2017) A Chemical-Genetic Approach to Generate Selective Covalent Inhibitors of Protein Kinases. ACS Chem. Biol 12, 1499–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhang C, Kenski DM, Paulson JL, Bonshtien A, Sessa G, Cross JV, Templeton DJ, and Shokat KM (2005) A second-site suppressor strategy for chemical genetic analysis of diverse protein kinases. Nat. Methods 2, 435–441. [DOI] [PubMed] [Google Scholar]
- (41).Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, and Drewes G (2007) Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol 25, 1035–1044. [DOI] [PubMed] [Google Scholar]
- (42).Golkowski M, Vidadala RS, Lombard CK, Suh HW, Maly DJ, and Ong SE (2017) Kinobead and Single-Shot LC-MS Profiling Identifies Selective PKD Inhibitors. J. Proteome Res 16 (3), 1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Golkowski M, Perera GK, Vidadala VN, Ojo KK, Van Voorhis WC, Maly DJ, and Ong SE (2018) Kinome chemoproteomics characterization of pyrrolo[3,4-c]pyrazoles as potent and selective inhibitors of glycogen synthase kinase 3. Mol. Omics 14, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Charras GT (2008) A short history of blebbing. J. Microsc 231, 466–478. [DOI] [PubMed] [Google Scholar]
- (45).Wang Q, Pechersky Y, Sagawa S, Pan AC, and Shaw DE (2019) Structural mechanism for Bruton’s tyrosine kinase activation at the cell membrane. Proc. Natl. Acad. Sci. U. S. A 116, 9390–9399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wang Q, Vogan EM, Nocka LM, Rosen CE, Zorn JA, Harrison SC, and Kuriyan J (2015) Autoinhibition of Bruton’s tyrosine kinase (Btk) and activation by soluble inositol hexaki-sphosphate. eLife 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Choi Y, Seeliger MA, Panjarian SB, Kim H, Deng X, Sim T, Couch B, Koleske AJ, Smithgall TE, and Gray NS (2009) N-myristoylated c-Abl tyrosine kinase localizes to the endoplasmic reticulum upon binding to an allosteric inhibitor. J. Biol. Chem 284, 29005–29014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Hantschel O (2012) Structure, regulation, signaling, and targeting of abl kinases in cancer. Genes Cancer 3, 436–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Varnai P, Rother KI, and Balla T (1999) Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton’s tyrosine kinase pleckstrin homology domain visualized in single living cells. J. Biol. Chem 274, 10983–10989. [DOI] [PubMed] [Google Scholar]
- (50).Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, Mehdizadeh M, Ghosh R, Wang L, Colon-Negron K, Meza-Acevedo R, Backes BJ, Maly DJ, Bluestone JA, and Papa FR (2017) Targeting ABL-IRE1alpha Signaling Spares ER-Stressed Pancreatic beta Cells to Reverse Autoimmune Diabetes. Cell Metab. 25, 883–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Saito K, Tolias KF, Saci A, Koon HB, Humphries LA, Scharenberg A, Rawlings DJ, Kinet JP, and Carpenter CL (2003) BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity 19, 669–678. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.