

Introduction
Hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis (POIKTMP; Online Mendelian Inheritance in Man number, 615704) is a recently described autosomal dominant condition due to FAM111B mutation. Hallmark cutaneous findings begin in childhood and include poikiloderma, photosensitivity, and bullae on the extremities. Thin hair, sparse eyebrows, and eczematous skin changes are reported. Dermatologists play a crucial role in diagnosing this genetic disorder, which presents with recognizable skin findings in childhood and has known complications of muscular weakness with fatty myopathy, tendon contractures, pancreatic insufficiency, and life-threatening pulmonary fibrosis. We present the case of a girl with a novel FAM111B mutation who, in addition to the previously reported phenotype, suffered from lymphocytic hepatic ductulitis. Transaminitis and hepatomegaly have been described in POIKTMP; we add a histopathologic description of the liver disease observed in this child with POIKTMP.
Case description
A 6-year-old girl of Mexican heritage presented with rare hemorrhagic bullae on the legs and neck and photodistributed dyschromia (Figs 1 and 2). She had sun intolerance from infancy, with facial redness and skin pain within minutes of sun exposure. An examination showed the presence of photodistributed poikiloderma. She exhibited failure to thrive (1.7% weight, 1.4% height), sparse eyebrows, and thin hair. Her mother denied any family history of a similar condition.
Fig 1.
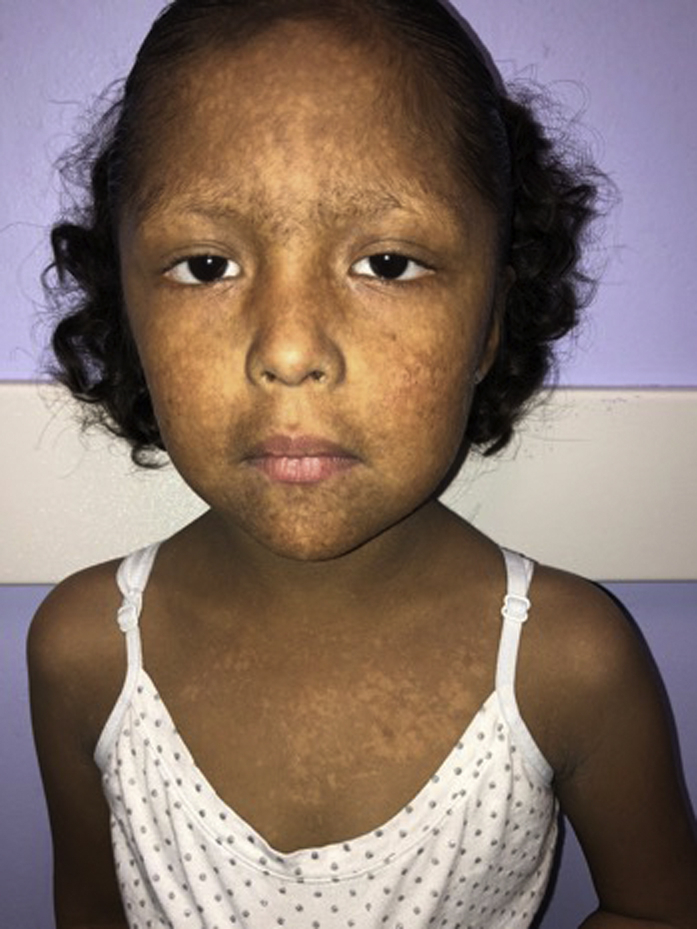
Mottled dyschromia of the face, neck, and sun-exposed chest. Photodistributed poikiloderma, thin hair, and sparse eyebrows.
Fig 2.
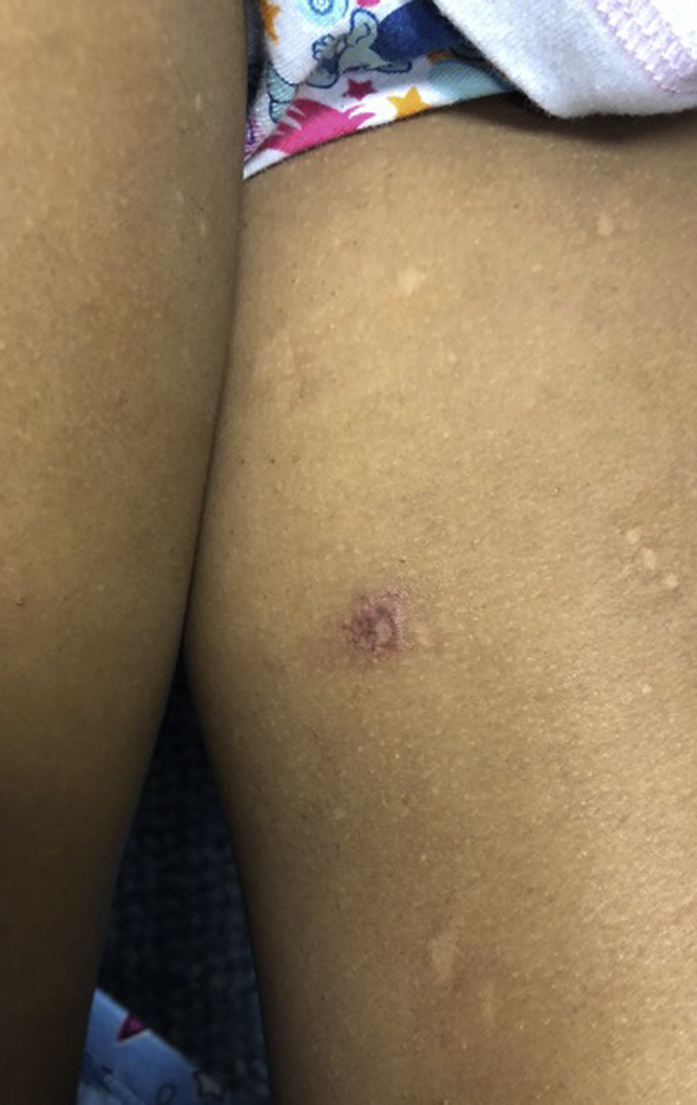
A hemorrhagic bulla on the left leg.
A biopsy of a tense bulla showed a subepidermal vesicle with interface dermatitis, dyskeratotic keratinocytes, pigment incontinence, superficial perivascular lymphocytes, and superficial vascular ectasia (Fig 3). Direct immunofluorescence showed only basement membrane fibrin deposition. Antinuclear antibody and porphyrin blood test results were normal.
Fig 3.
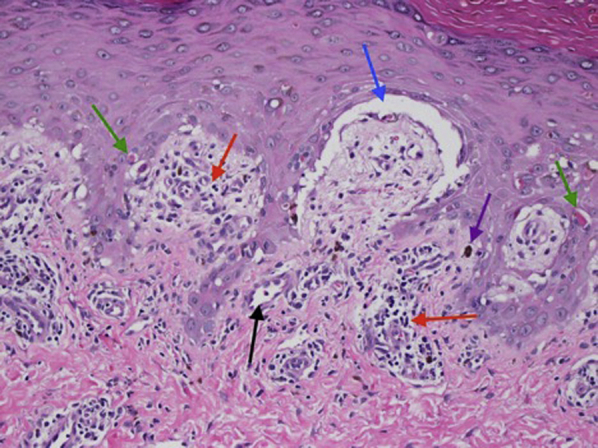
A biopsy of a hemorrhagic blister shows a subepidermal vesicle (blue arrow) on a background of an interface inflammatory reaction (red arrows) with dyskeratotic keratinocytes (green arrows), pigment incontinence (purple arrow), and superficial perivascular lymphocytic inflammation (black arrow). There was no significant thickening or hyalinization of the dermal blood vessel wall. Mucin deposition is not visualized (hematoxylin-eosin stain; ×200).
The patient had transaminitis since she was 10 months old. Around that time, she had scaly eczematous dermatitis on the arms and legs. A skin biopsy revealed interface dermatitis without mucin and was interpreted as neonatal lupus; however, she and her mother tested negative for anti-Ro, anti-La, and anti-RNP antibodies. The dermatitis resolved with corticosteroid treatment and sun protection, but the transaminitis persisted longer than expected for neonatal lupus, calling the diagnosis into question. At the age of 19 months, systemic immunomodulating treatment with azathioprine and budesonide was started for the liver disease.
Liver biopsies were performed at 1, 2.5, 4.5, and 5.5 years of age. The biopsy at the age of 1 year showed lymphocytic ductulitis and duct loss (Fig 4). The subsequent biopsies showed mild portal inflammation with variable duct loss. A delicate portal fibrous extension was noted, which correlated with a mildly increased echotexture observed by ultrasound. She had a low body mass index, short stature, low bone mineral density; required vitamin D supplements; and had an elevated international normalized ratio, which normalized with oral vitamin K. A chromosomal microarray and whole-exome sequencing performed at the age of 4 years were reported as negative.
Fig 4.
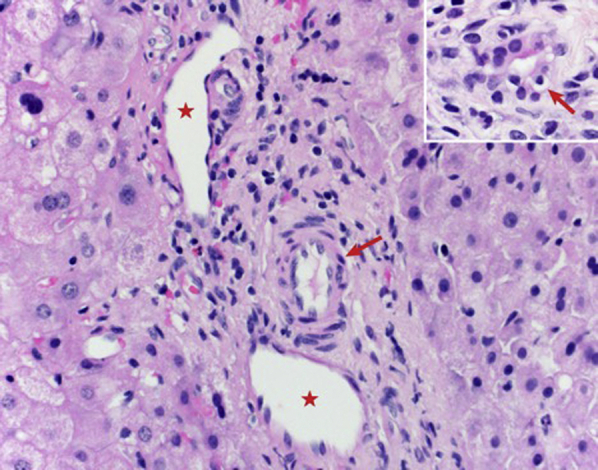
A liver biopsy at the age of 1 year shows a noninflamed portal tract with a portal vein (asterisks) and hepatic artery (red arrows) and the absence of a bile duct (hematoxylin-eosin stain; ×400). The inset shows a reactive bile duct with lymphocytic ductulitis (hematoxylin-eosin stain; ×600).
With the identification of poikiloderma and poor growth in a dermatology clinic at the age of 6 years, Rothmund-Thomson syndrome was considered. However, although the patient had genu valgum and an awkward run, she had no skeletal abnormalities. Kindler syndrome was considered; however, her bullae were sun-induced and rare, and she had no acral hyperkeratosis or acral blistering. Previous whole-exome sequencing data were re-reviewed with poikiloderma as a clinical search term, and a de novo novel mutation in FAM111B (c1881 C>T, p. R672S), a causative of POIKTMP, was detected. A review of a skin biopsy of an eczematous papule during infancy showed dyskeratosis and an interface pattern similar to that of the recent skin biopsy of a bulla, suggesting that the previous dermatitis was related to her genetic condition.
The diagnosis of POIKTMP prompted by her skin findings facilitated referrals for the required subspecialty care. Severe fatty infiltration of the leg musculature was detected on magnetic resonance imaging, and physical therapy was initiated for proximal muscle weakness. Pancreatic insufficiency was diagnosed and treated with pancreatic enzyme therapy. Pulmonary function test results were normal, but continuous monitoring is required as disabling lung fibrosis typically arises during young adulthood in patients with POIKTMP.
Discussion
POIKTMP is a recently described genetic condition with early-onset photosensitivity and poikiloderma due to mutations in FAM111B, which encodes a trypsin-like cysteine/serine peptidase. Photo-exacerbated erythema of the face frequently develops before the age of 6 months.1,2 Additional cutaneous findings include nonscarring alopecia, sparse eyebrows, hypopigmentation, hypohidrosis, bullae, and eczematous lesions.1,2 The skin biopsy results vary, with eczematous lesions indicating interface dermatitis and scleroderma-like lesions indicating dermal sclerosis with or without elastic fiber abnormalities.3
Patients with POIKTMP develop muscle contractures, fatty myopathy, pancreatic insufficiency, and life-threatening pulmonary fibrosis.3,4 The fatty infiltration of the leg muscles can be seen using magnetic resonance imaging, and a muscle biopsy reveals the heterogeneous degeneration of fragmented muscle fascicles.5 The muscle and tendon contractures result in gait abnormalities, and many patients require surgical Achilles tendon lengthening.4,6 Pancreatic exocrine insufficiency is common in these patients and may manifest with diarrhea, fatty stools, vitamin K deficiency, and an increased international normalized ratio.6,7 Pancreatic cancer has been reported, suggesting that mutations in FAM111B may predispose the individuals to cancerous pancreatic lesions.2 Lung involvement typically manifests during the second decade of life, with dyspnea and pulmonary fibrosis, progressing to a restrictive lung impairment and death.4,5
Hepatomegaly and transaminitis have been reported in patients with POIKTMP.2,8 Our patient with persistent transaminitis underwent liver biopsies and received systemic immunosuppressants for the lymphocytic ductulitis, a reminiscent of that seen in immune-mediated bile duct injury processes, such as graft-versus-host disease or acute cellular rejection.9 The duct loss was not associated with lobular cholestasis, jaundice, or elevated conjugated bilirubin. Our patient showed a mild portal fibrous extension and hepatic vein thickening since she was 2.5 years old. Similar portal and hepatic vein abnormalities have been described in connective tissue disorders, including scleroderma (another fibrotic disease).9 Currently, at the age of 8 years, our patient has no portal hypertension and normal liver stiffness, yet her liver disease continues to be monitored.
Our patient has a novel pathogenic mutation in the FAM111B gene, Arg627Ser. Some variability in the phenotypic manifestations of POIKTMP exists in the literature, but for this rare condition, it is not clear if specific mutations are predictive of the clinical outcomes.
Dermatologists should consider POIKTMP as a cause of childhood photosensitivity and poikiloderma as its recognition may facilitate early diagnosis and facilitate subspecialty patient care and genetic counseling for parents. This case provides a histopathologic insight into liver disease in our patient with POIKTMP.
Footnotes
Funding sources: None.
Conflicts of interest: None disclosed.
References
- 1.Chasseuil E., Mcgrath J.A., Seo A. Dermatological manifestations of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis (POIKTMP): a case series of 28 patients. Br J Dermatol. 2019;181(4):862–864. doi: 10.1111/bjd.17996. [DOI] [PubMed] [Google Scholar]
- 2.Goussot R., Prasad M., Stoetzel C., Lenormand C., Dollfus H., Lipsker D. Expanding phenotype of hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis caused by FAM111B mutations: report of an additional family raising the question of cancer predisposition and a short review of early-onset poikiloderma. JAAD Case Rep. 2017;3(2):143–150. doi: 10.1016/j.jdcr.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kazlouskaya V., Feldman E., Jakus J., Heilman E., Glick S. A case of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis (POIKTMP) with the emphasis on cutaneous histopathological findings. J Eur Acad Dermatol Venereol. 2018;32(12):e443–e445. doi: 10.1111/jdv.14968. [DOI] [PubMed] [Google Scholar]
- 4.Khumalo N.P., Pillay K., Beighton P. Poikiloderma, tendon contracture and pulmonary fibrosis: a new autosomal dominant syndrome? Br J Dermatol. 2006;155(5):1057–1061. doi: 10.1111/j.1365-2133.2006.07473.x. [DOI] [PubMed] [Google Scholar]
- 5.Mercier S., Küry S., Shaboodien G. Mutations in FAM111B cause hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Am J Hum Genet. 2013;93(6):1100–1107. doi: 10.1016/j.ajhg.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mercier S., Küry S., Salort-Campana E. Expanding the clinical spectrum of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis due to FAM111B mutations. Orphanet J Rare Dis. 2015;10(1):135. doi: 10.1186/s13023-015-0352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seo A., Walsh T., Lee M.K. FAM111B mutation is associated with inherited exocrine pancreatic dysfunction. Pancreas. 2016;45(6):858–862. doi: 10.1097/MPA.0000000000000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeichi T., Nanda A., Yang H.-S. Syndromic inherited poikiloderma due to a de novo mutation in FAM111B. Br J Dermatol. 2016;176(2):534–536. doi: 10.1111/bjd.14845. [DOI] [PubMed] [Google Scholar]
- 9.Youssef W.I., Tavill A.S. Connective tissue diseases and the liver. J Clin Gastroenterol. 2002;35(4):345–349. doi: 10.1097/00004836-200210000-00012. [DOI] [PubMed] [Google Scholar]