

Abstract
Recent advances in high-throughput technologies have enabled the profiling of multiple layers of a biological system, including DNA sequence data (genomics), RNA expression levels (transcriptomics), and metabolite levels (metabolomics). This has led to the generation of vast amounts of biological data that can be integrated in so-called multi-omics studies to examine the complex molecular underpinnings of health and disease. Integrative analysis of such datasets is not straightforward and is particularly complicated by the high dimensionality and heterogeneity of the data and by the lack of universal analysis protocols. Previous reviews have discussed various strategies to address the challenges of data integration, elaborating on specific aspects, such as network inference or feature selection techniques. Thereby, the main focus has been on the integration of two omics layers in their relation to a phenotype of interest. In this review we provide an overview over a typical multi-omics workflow, focusing on integration methods that have the potential to combine metabolomics data with two or more omics. We discuss multiple integration concepts including data-driven, knowledge-based, simultaneous and step-wise approaches. We highlight the application of these methods in recent multi-omics studies, including large-scale integration efforts aiming at a global depiction of the complex relationships within and between different biological layers without focusing on a particular phenotype.
Keywords: multi-omics, data integration, systems biology, metabolomics, lipidomics
1. Introduction
Advances in high-throughput technologies have enabled the generation of vast amounts of data on multiple layers of a biological system, including DNA sequence data (genomics), RNA expression levels (transcriptomics), epigenetic alterations (epigenomics), protein abundances (proteomics), metabolite levels (metabolomics) and more. Considering each of these biological layers separately, numerous omics studies identified genes, proteins, and metabolites that associate with specific diseases or phenotypes of interest. For example, high levels of branched-chain amino acids and their degradation products have been found as hallmarks of type 2 diabetes [1]; in contrast, Alzheimer’s disease associates with low levels of these metabolites [2]. While the identified entities can serve as valuable biomarkers and provide insights into pathways involved in pathomechanisms, single omics studies do not take into account the complex interplay of various biological layers. However, disturbances of cross-omics interactions might play important roles in the development and clinical presentation of a disease [3,4]. Therefore, combining omics data from multiple biological domains (e.g. levels of transcripts, proteins, or metabolites) in multi-omics studies is a promising approach towards a more detailed molecular understanding of health and disease, as well as the chain of cause and effect, which is an essential requirement for guiding novel therapies [5]. For example, results from an integrated analysis of large genetic and metabolomic datasets by Lotta et al. [1] using a Mendelian Randomization approach, were consistent with a causal role of BCAA metabolism in type 2 diabetes and suggested the PPM1K gene (genetic variants therein being specifically associated with levels of BCAA in blood) as a potential drug target. PPM1K encodes the mitochondrial phosphatase that activates the branched-chain alpha-ketoacid dehydrogenase (BCKD) complex, the rate-limiting enzyme in BCAA catabolism, and was only up-regulated in muscles of healthy subjects but not in patients with type 2 diabetes in a validation experiment. Although the availability of multi-omics data does not always allow for direct conclusions on causality, the combination of multiple layers of evidence in a multi-omics study has been demonstrated to provide more reliable results and mitigate the risk of false positive findings [6,7]. Beyond the value of multi-omics approaches for the investigation of particular diseases, large-scale multi-omics studies enable the systematic investigation of inter- (e.g. enzymatic conversion of metabolites) and intra-omics (e.g. protein-protein interactions) relationships independent of a specific phenotype.
In multi-omics studies, metabolomics and its sub-discipline lipidomics occupy a unique position and have received increasing attention in integrative analysis [8]. Metabolites are the downstream output of biological processes, carrying imprints of genomic, epigenomic, and environmental effects. They are often referred to as “the link between genotype and phenotype” [9] and have been implicated in numerous diseases, such as Alzheimer’s Disease [10], type 2 diabetes [11], and various types of cancer [12]. Furthermore, they carry integrated biological and medical signals in easily accessible biofluids (e.g., blood, urine), making them attractive biomarker candidates [13]. Large-scale epidemiological studies have demonstrated the value of integrating metabolomics with other omics layers, such as genomics [14–17], transcriptomics [18] and epigenetics [19], providing insight into metabolic individuality and links to disease mechanisms [20,21]. For example, up to 62 percent of variation in metabolite concentration levels in two population-based cohorts could be explained by common genetic variants [16]. Furthermore, it has been shown that DNA methylation affects metabolism [22]. This effect is partly driven by genetic variation, but further depends on environmental and lifestyle factors, enabling an adaptive response to regular (e.g., food intake) [23,24] and specific (e.g., disease) [25] challenges. Changes in the metabolome can, in turn, modulate the activity of genes and proteins, creating complex feedback mechanisms and interrelationships between omics layers [26]. Therefore, the integration of metabolomics with other omics layers provides exciting opportunities for the study of disease mechanisms and identification of novel therapeutic targets.
To enable the analysis of heterogenous datasets in multi-omics studies, a plethora of data reduction, manipulation, and integration techniques have been developed. Previous review articles have provided comprehensive method summaries for specific integration strategies such as network inference and analysis [27,28] or machine learning techniques [29–32], and have discussed important aspects of metabolite-centered studies [33–35]. However, most reviews concentrate on the integration of two different data types with respect to a specific phenotype of interest. In this review, we will provide an overview over a typical multi-omics workflow, focusing on integration methods that have the potential to combine metabolomics data with more than two omics and highlighting their application in recent multi-omics studies. We will distinguish between integration efforts that build prediction models [36–39] or identify diagnostic and prognostic biomarker candidates [39,40] for a specific disease phenotype or trait of interest, and global integration efforts that are initially not focused on a specific outcome. The latter approaches aim at the systematic integration of multiple omics datasets to provide a basis for generating testable hypotheses and gaining mechanistic insights into the pathophysiology of multiple complex diseases in post-integration analyses [41–43].
The choice of an appropriate integration strategy is not straightforward and heavily depends on the available data and study objective. Data dimensionality, heterogeneity, and lack of universal protocols additionally complicate this task. Generally, two major integration paradigms (Figure 1) have been described in the literature [27,35,44–46] and will be referenced throughout this review; (1) simultaneous and (2) step-wise integration. Simultaneous integration strategies use all available omics data at the same time and perform analysis in a single modeling step. Thereby, complementary information encoded in each omics layer, as well as correlations between the layers, are taken into account. Methods of this category require that the data was derived from the same biological samples or individuals, which poses still a major limitation regarding availability of such data due to funding or technical restrictions. Step-wise integration strategies, on the other hand, analyze omics datasets in isolation or in specific combinations and integrate the results in a subsequent step. This facilitates the integration of data and statistical results from different sources (e.g., different studies or knowledge bases), allowing the large-scale analysis of heterogeneous data in the absence of omics measurements for the same samples.
Figure 1. Multi-omics workflow.
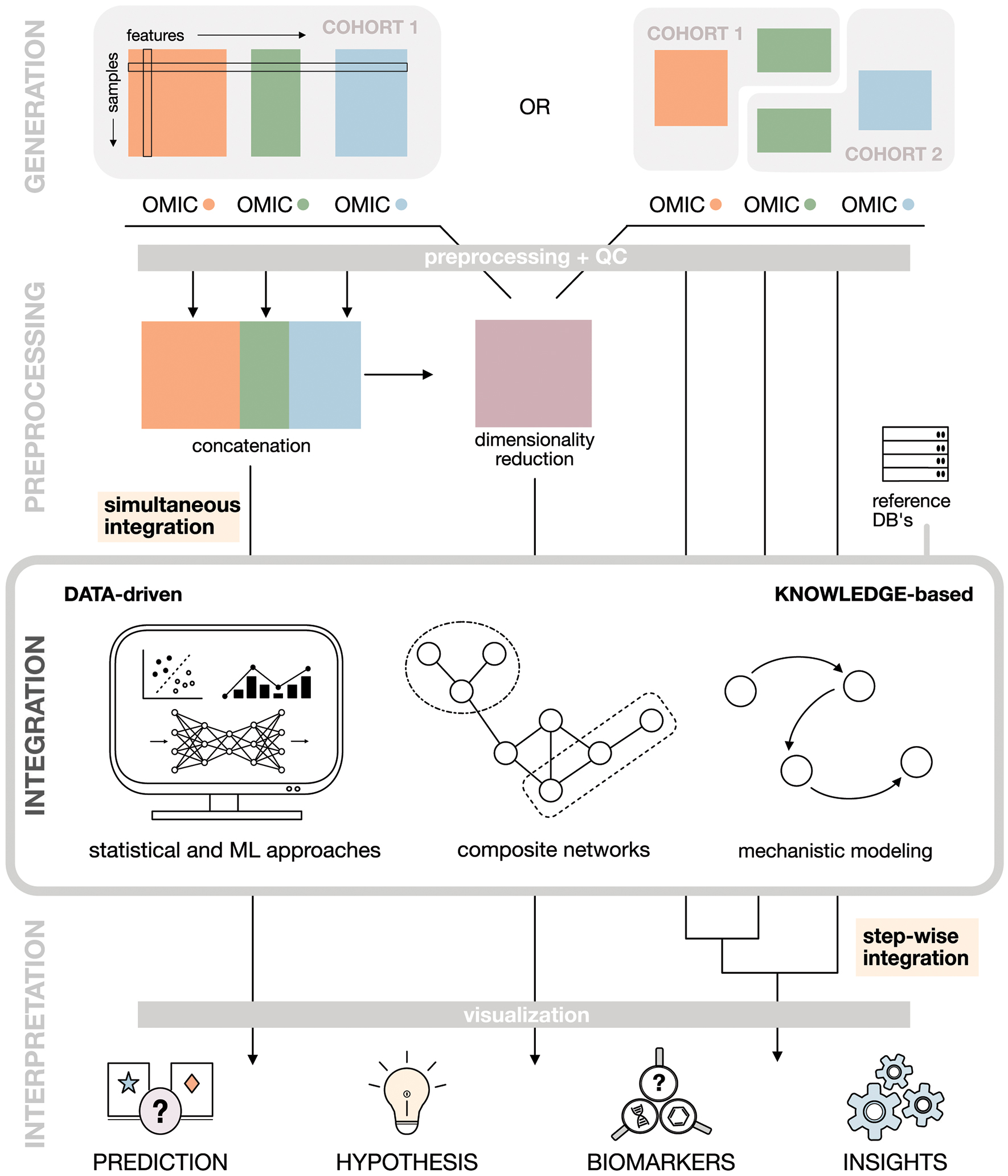
A typical multi-omics analysis can generally be broken down into 4 steps. (i) Data generation. Study design, sample preparation and subsequent data acquisition through high-throughput analytical platforms lead to different data scenarios. (ii) Data preprocessing and dimensionality reduction. Raw data collected on different omics layers is preprocessed appropriately and dimensionality reduction can be applied to reduce the number of variables (measured biological entities). (iii) Data integration. Data from different omics layers are analyzed and integrated using data-driven, knowledge-based or hybrid integration approaches. The choice of method depends on the input data and research question of interest. (iv) Data interpretation. Post-integration visualization and analysis of the integration results (e.g., statistical model or network) can identify novel biomarker candidates, generate testable hypothesis or reveal meaningful biological relationships.
This review will discuss central aspects of a typical multi-omics data integration workflow (Figure 1) and is structured as follows: (i) Data scenarios. Study design, sample preparation and subsequent data acquisition through high-throughput analytical platforms can lead to different data scenarios. (ii) Dimensionality reduction. After appropriate preprocessing of raw data collected on different omics layers, dimensionality reduction is often applied to reduce the number of variables (measured biological entities). (iii) Data integration. Data from different omics layers are analyzed and integrated using a method that is appropriate for the input data and research question of interest. (iv) Data interpretation. Post-integration inspection and further analysis of the integration results (e.g., statistical model or network) enable meaningful biological insights. We conclude with a short outlook on future directions for multi-omics research.
2. Data scenarios
Integrative multi-omics analyses combine several omics measurements, optionally along with additional phenotypes of interest, that are represented by either continuous (e.g., protein levels or metabolite concentrations) or categorical variables (e.g., gender or disease status). Naturally, each dataset comes in a separate data matrix where rows represent individual samples, and columns hold measurements of demographic, clinical, or biological entities (Figure 1). However, depending on the study objective and access to relevant data, there are three different data scenarios: (1) the different datasets are available for the same samples/individuals; (2) the datasets are available for an only partially overlapping set of samples/individuals; (3) omics data is distributed across mostly disjoint sets of samples.
In the first scenario, samples from a study are simultaneously subjected to the same multi-omics screening processes or additional omics technologies are applied to initially collected samples in retrospect. Data from such studies will result in data matrices where the rows in every data matrix correspond to the same samples/individuals and columns hold measurements for each respective omics technology (e.g., metabolomics, transcriptomics, proteomics). This is the optimal scenario, as it allows application of any integration strategy, including simultaneous data integration that requires data matrices with matched samples [47].
However, complete multi-omics profiles are often not available or feasible to get for all samples/study participants. The reasons for this are manifold and include funding limitations, incompatibility of collected samples for certain omics analyses, or depletion of samples preventing application of novel technologies [35,47]. For example, although urine samples have proven very informative in metabolomics studies, they contain limited amounts of proteins and RNA, limiting their use in large-scale proteomics or transcriptomics studies [35]. Furthermore, in long-term studies or studies with rollover participants, both omics and phenotypic screenings applied at baseline may be adapted due to technological advances, falling costs for sample analysis, or evolving study objectives. For instance, the Alzheimer’s Disease Neuroimaging Initiative (ADNI) is a longitudinal, multicenter study launched in 2004 to study biomarkers for early detection of Alzheimer’s Disease (AD) [48]. While large-scale metabolomics and lipidomics profiling is available for the study phases ADNI-1 and -GO/2, up to now (biosamples are still available) proteomics profiling was only applied to a subset of ADNI-1 participants and gene expression profiling is only available for ADNI-GO/2. This leads to differing availabilities of omics profiles for participants across study phases.
Data resulting from such a study will only have partially overlapping samples for multi-omics integration [47]. If the overlap of samples between data types is large enough for a sufficiently powered study, the removal of samples without full omics profiles can still enable simultaneous integration. However, application of such a list-wise deletion of individuals is prone to substantial loss of information [30][47]. In the worst case, this can introduce estimation bias by resulting in a sample set that is unrepresentative of the initial study population [49]. Nevertheless, simultaneous data integration strategies are emerging that can handle a moderate amount of samples with missing omics profiles (see Section 4.2.2).
Due to the restrictions mentioned above, many multi-omics analyses use datasets that have not been collected from the same samples and originate from different sources. A special case of this scenario occurs if the sample sets for each data type were acquired in the same study but have minimal overlap. By integrating such omics measurements, data matrices consequently have mostly unmatched samples and variables as a starting point. For this data scenario, several step-wise integration strategies (discussed in Sections 4.1 and 4.2.1) have been developed that enable both multi-omics analyses in disjoint sample sets and inclusion of preexisting biological data. However, it is important to keep in mind that these types of analyses add another layer of data heterogeneity due to differing sample sizes, study protocols, and study demographics (e.g., age, sex, or ethnicity).
In summary, multi-omics datasets available for the same samples/individuals introduce less unwanted data heterogeneity and enable the application of any integration method. For datasets with only partially overlapping or completely disjoint sets of samples/individuals, the number of applicable integration methods is a bit more limited, but those that are available allow for almost infinite inclusion of data, enabling studies to yield maximal power.
3. Dimensionality reduction
Appropriate preprocessing of the raw data is a key prerequisite for any type of analysis, as technical artifacts and skewed data distributions can distort biological signals [50]. This process typically includes the removal of batch effects, normalization and imputation of missing values for each data type separately before integration [51]. The importance of study design and temporal ordering of sample collection [35,44,51,52], as well as guidelines for appropriate data preprocessing [30,51], have been discussed in previous reviews and are beyond the scope of this review. In the following, we will assume that the data subjected to integrative analyses was appropriately preprocessed and is of high quality.
The curse of dimensionality [53] is a central challenge in single-omics studies and even further aggravated in multi-omics studies, where the number of variables is substantially higher. With increasing dimensions (number of variables), distance measures become meaningless, which is challenging for operations in this high-dimensional space, such as clustering [54,55]. Furthermore, samples are typically significantly outnumbered by measured variables, posing a challenge for most statistical learning methods. This can lead to an underdetermined mathematical system and increases the risk of overfitting classifiers or predictors [27]. Dimensionality reduction (DR) is a way to reduce the complexity of a dataset while increasing prediction stability, boosting statistical power of downstream analyses, and reducing the multiple testing burden. DR is performed by either extracting relevant variables (feature selection) or projecting data onto a lower-dimensional space (feature extraction) [30].
Feature selection often involves prior knowledge or a biological hypothesis that is used to reduce the number of considered variables. Popular approaches are, for example, to limit the analyses to genes, proteins and metabolites involved in certain pathways of interest, or to investigate entities that have been previously associated with a specific trait under study [41]. Such hypothesis-driven DR strategies can significantly boost statistical power but are naturally prone to bias towards biological entities that have been annotated through previous studies. Another knowledge-based approach is to construct new variables that are biologically meaningful, i.e., representative of functional groups such as pathways. For example, metabolites can be analyzed at the pathway-level by aggregating levels of all molecules assigned to a specific pathway (e.g., by using the average z-score of concentrations [56] or first principal component from a PCA [56–58]) to produce new pathway-based variables [59].
Feature extraction, on the other hand, is typically achieved by data-driven DR techniques such as Principal Component Analysis (PCA) [30,60]. PCA is classically applied to each omics dataset separately and transforms single-omics variables into a lower-dimensional subspace that maximizes the retained variance within the data by finding orthogonal linear combinations of the original variables. Therefore, PCA enables the use of a reduced set of features with minimal loss of information. Related approaches include clustering techniques (e.g., K-means [61] or hierarchical clustering [62]) followed by replacement of groups of similar variables by a cluster centroid [63]. Here, one popular approach is to cluster correlating biological entities such as metabolites, proteins or transcripts by using weighted gene co-expression network analysis (WGCNA) [64] on each dataset [65,66]. The identified clusters are then summarized by the first principal component from a PCA (“eigengene” or “eigenmetabolite”) on the abundance matrix of each respective cluster that is then used in downstream analyses (e.g. association with a specific phenotype, integration with other omics layers) with a reduced set of features [67]. A limitation of such data-driven approaches is that the interpretation of the derived associations or correlations requires the extracted features to be mapped back onto the original variables.
In summary, DR provides a way to limit the potential for overfitting and significantly reduces the multiple testing burden. Additionally, knowledge-based DR can increase downstream interpretability of analysis outcomes.
4. Data integration
The growing interest in integrative analysis of multi-omics datasets has led to the emergence of various integration frameworks. In the following, we review the major concepts categorized into approaches that take into account external information (knowledge-based approaches) and approaches that primarily rely on intrinsic information (data-driven approaches) to infer dependencies across omics. Finally, we will discuss hybrid approaches (composite networks) that combine knowledge-based and data-driven integration.
4.1. Knowledge-based approaches
Knowledge-based integration strategies use external information from databases or scientific literature to establish relationships between biological entities. Results from previous analyses are either annotated using prior knowledge (e.g., using common functional terms) or mapped onto a reference network that connects different omics layers based on established knowledge. For example, metabolic networks, assembled based on biochemical knowledge, enable the connection of enzymes and metabolites through reactions. By mapping results from single-omics analyses onto such a network, findings can be integrated and interpreted in a multi-omics context, enabling the identification of pathways that are dysregulated at the gene, protein and metabolite level [68]. Furthermore, multi-omics measurements can be integrated into preexisting biological models to make them condition-specific (e.g., deletion of inactive reactions) [69].
Prior knowledge that is used for this type of omics integration includes, but is not limited to, information on functional relationships (e.g., pathways or biological reactions), pharmacogenomic associations, and genome annotations. Depending on the source, this information is either based on experimental data [70], collected from scientific literature (manually or by using automated text-mining techniques) [71], or derived from computational prediction approaches [72]. As knowledge bases typically combine information from multiple sources, they can have varying levels of evidence. For example, STRING [71], a popular protein-protein interaction database, indicates the confidence of functional interactions between proteins by assigning scores that are based on the quality and type of supporting evidence coming from targeted experiments, co-expression analysis, genomic context predictions, or text-mining [73].
While many resources are specific to one omics type, such as STRING or the LIPID MAPS Structure Database (LMSD)[74] for lipid annotations, a number of databases have emerged that cover multiple biological domains (see Table 1). The Kyoto Encyclopedia of Genes and Genomes (KEGG) [75–77] database, for instance, was released in 1995 as one of the first computational resources that linked the genome with higher-order functional information. In KEGG, manually compiled pathway maps enable researchers to view genes and proteins in the context of metabolic networks and pathways, such as sphingolipid metabolism or NF-kappa B signaling. Nearly a decade later, additional curated and pathway-centered resources started emerging, such as Reactome [78,79] and Recon [80–82]. Reactome is a resource that is primarily focused on human biological processes and is built around reactions. Reactions are defined as an event that transforms an input to an output (both being biological entities such as proteins, lipids or nucleotides) and are further grouped into pathways depending on their (temporal) relationships [78]. Taking this concept a step further, Recon3D [80–82] provides a genome-scale metabolic reconstruction that can be used for computational modeling (see Section 4.1.2 on constraint-based metabolic modeling). It also includes three-dimensional (3D) structural data on metabolites and proteins and represents the most comprehensive human metabolic network model to date [82].
Table 1.
A selection of network-based multi-omics knowledge bases, visualization tools and online resources.
| Network visualization | Analysis tools | Project omics data onto network | Biological entities | Implementation | Reference | |
|---|---|---|---|---|---|---|
| BioCyc | x | Enrichment analysis Flux analysis | x | genes proteins metabolites | online | [206] |
| KEGG | x | - | x | genes enzymes metabolites | online KEGGscape+ CytoKegg+ | [76] |
| Reactome | x | Enrichment analysis ID mapping | x | proteins metabolites diseases | online ReactomeFIViz+ | [78] |
| Recon3D | x | x | genes metabolites | online | [82] | |
| PathwayCommons | x | Enrichment analysis | - | proteins metabolites drugs | online R CyPath2+ | [207] |
| WikiPathways | x | - | - | genes proteins metabolites | online WikiPathways+ | [208,209] |
| NDEx | x | Neighborhood search | - | various** | online CyNDEx-2+ | [190,210,211] |
| PaintOmics3 | x | Clustering Correlation analysis Enrichment analysis ID mapping | x | genes proteins metabolites | online | [68] |
| MetaboAnalyst | x | Enrichment analysis ID mapping Shortest path analysis | x | genes metabolites | online R | [84] |
| OmicsNet | x | Clustering Enrichment analysis Shortest path analysis | x | genes proteins TFs miRNAs metabolites | online | [169] |
| MetExplore | x | Enrichment analysis Flux analysis ID mapping Shortest path analysis | x | genes enzymes metabolites | online | [212] |
| ConsensusPathDB | x | Clustering Enrichment analysis Shortest path analysis | x | genes proteins metabolites | online | [167] |
| PathMe Viewer | x | Shortest path analysis | genes proteins metabolites | online | [213] | |
| MetScape | x | Correlation analysis Enrichment analysis | x | genes enzymes metabolites | MetScape+ | [214,215] |
no restrictions
Cytoscape Application [216]
In order to utilize these resources for knowledge-based integration, platform-specific identifiers (IDs) of measured biological entities need to be mapped to the namespace of the respective target database. This task is challenging, as most resources have developed their own internal ID schemes and hierarchies, leading to a plethora of IDs across databases that refer to the same entity. Efforts have been made to enable cross-linking between ID schemes [82] and mapping tools are available online or through R packages, such as biomaRt [83] for genes or MetaboAnalystR [84,85] for metabolites. However, name ambiguities, ID multiplicity and the use of synonyms complicate this task [86] and can lead to significant loss of information if not handled carefully. This is especially challenging for metabolites and lipids due to differences in resolution between platforms and technologies [87]. For example, lipid sidechain composition and configuration are important determinants of the function of phosphatidylcholines (PC). However, many lipidomics techniques cannot distinguish between isobaric species sharing the same nominal mass [88] and annotate PCs at the lipid species level assuming even-numbered fatty acids, as they are more frequent, i.e., PC (731) with m/z 731 will most likely be labeled PC 32:1 and not PC O-33:1, although both are plausible [87].
Knowledge bases are under constant pressure to adapt to technological advances and incorporate novel research findings (e.g., the discovery of various types of regulatory RNA species) to accurately reflect the current state of science, which can lead to further discrepancies. For example, despite the fact that some platforms offer fatty acid side-chain resolving techniques, lipids are often not yet annotated at this level of detail [6] and this information will be lost when matching measured compounds to the namespace of a resource (e.g., PC 16:0_16:1 would simply be mapped to the KEGG identifier C00157 for phosphatidylcholine).
Nevertheless, when correctly employed knowledge bases provide a wealth of valuable information that can be exploited in multi-omics integration.
4.1.1. Set-based enrichment
Set-based enrichment is a commonly used, step-wise results integration strategy. It tests whether certain functional annotations are enriched in a list of interesting (e.g., differentially expressed or abundant) biological entities, which have been identified in preceding omics analysis. Biological entities are assigned to sets (also referred to as annotation terms) using information from knowledge bases to examine whether they are known to participate in the same biological pathways, are significantly changed in a specific disease, or are co-localized (e.g., in the same organelles, tissues or organs) [89]. For example, the annotation term “sphingolipid metabolism” in Reactome [78,79] includes metabolites such as sphingosine 1-phosphate and sphingosine, and genes such as SGPP1 (sphingosine-1-phosphate phosphatase 1) and SPHK1 (Sphingosine Kinase 1). Here, we focus on the most widely used approaches: overrepresentation analysis and functional set enrichment analysis.
Overrepresentation Analysis (ORA) aims at the identification of annotation terms that are overrepresented, i.e. terms that are more frequently assigned to the entities in the input list of interest than expected by chance [89]. This can be statistically tested by using a hypergeometric test such as one-sided Fisher’s exact test with subsequent correction for multiple testing [89]. In order to yield meaningful results, valid definition of the background, i.e., the set of entities that were measured in the analysis and assigned to each annotation term, is a key requirement [52] in order to correct for bias that arises due to unequal annotation coverage of different entities. This is a prominent challenge in metabolomics and lipidomics studies where analytical methods are typically biased towards molecules from certain chemical classes [52,87,88]. For multi-omics integration, ORA is typically performed separately on each omics level. By mapping omics, such as transcriptomics, proteomics or epigenomics, back to the gene-level, multiple omics types can be integrated alongside metabolomics data. The resulting P-values are combined into a joint enrichment P-value for each annotation term using Fisher’s method [90] or Stouffer’s method (unweighted [91] or weighted [92]) as implemented e.g. in the web-resources PaintOmics3 [68], Integrated Molecular Pathway-Level Analysis (IMPaLA) [93], and MetaboAnalyst [84,94]. MetaboAnalyst additionally offers an integrative overrepresentation analysis in which both genes and metabolites are queried together by using annotation terms such as metabolic pathways from KEGG to define sets. A drawback of ORA is that it only considers the subset of measured entities that, for example, showed a significant change in levels between conditions. This makes it sensitive to the chosen significance cutoff, or any other inclusion criterion, that was used to determine the input set of biological entities. At the same time, ORA neglects information on the extent of change (e.g., measured through fold change) between conditions [34].
Functional Set Enrichment Analysis (FSEA) is another set-based enrichment method that addresses these ORA-associated limitations. It was originally developed for the analysis of transcriptomics data in Gene Set Enrichment Analysis (GSEA) [95], but has also been implemented for metabolites (Metabolite Set Enrichment Analysis or MSEA) [89] and lipids (LION/web) [96]. In contrast to ORA, these methods test all measured entities, not just a defined subset, and take into account their quantitative measurements. This enables the identification of annotation terms where only a few entities are significantly changed or where many entities are changed slightly but consistently [89]. Similar to ORA, an integrative analysis of several omics datasets is achieved by calculating a joint P-value from the individual single-omics analyses. This is, for example, implemented in the web-resource IMPaLA which uses Wilcoxon’s signed-rank test to perform FSEA using pathway annotations taken from 11 public databases [93].
The central limitation of both FSEA and ORA is that they are naturally restricted to entities that have been previously annotated. To this end, de novo enrichment methods, such as KeyPathwayMiner [97,98], have been proposed. These methods enable the discovery of uncharacterized pathways by extracting connected subnetworks with a high number of differentially regulated entities from predefined biological networks (e.g., knowledge-based metabolic networks or data-driven correlation networks) [99]. This framework is theoretically applicable to multi-omics data by using pathway annotations or ontologies that include multiple layers of omics. So far, they have been predominantly used in gene-centric studies. For example, Soerensen et al. [100] demonstrated the benefits of using both GSEA and KeyPathwayMiner in an integrative enrichment analysis of genes associated with cognition in both epigenome-wide and transcriptome-wide association analysis. GSEA was able to replicate findings from previous studies by identifying a broad spectrum of enriched biological processes including gene sets involved in neurological functioning and cell cycle control. The use of de novo enrichment identified subnetworks of dysregulated entities that included genes not implicated by GSEA such as Ras And Rab Interactor 3 (RIN3) and Ataxin 2 (ATXN2). Interestingly, this approach also implicated amyloid beta precursor protein (APP) and the nuclear respiratory factor 1 (NRF1), two genes with functions relevant for cognitive health, that were not differentially methylated and expressed in this analysis.
4.1.2. Constraint-based metabolic modeling
Constraint-based metabolic models (CBMMs) enable the in-silico description and prediction of possible metabolic steady states by mathematically representing metabolic reactions in a stoichiometric matrix [101]. The stoichiometric coefficients of these reactions are used to constrain the flow of metabolites through the system, ensuring that, at steady state, the mass of any compound that is being produced must equal the total amount of what was consumed (flux balance) [102]. Genome-wide metabolic models (GEMs), such as Recon3D, are typically constructed in a bottom-up approach [103] using genome annotations to automatically build a draft that contains all enzymatic reactions predicted to be available for an organism considering the proteins encoded in its sequenced genome. This draft is then refined through manual curation and constraint-based modeling (e.g. to identify and fill gaps in the reconstructed metabolic network) [104].
In the context of multi-omics integration, GEMs present comprehensive metabolic networks that can be used to link the results from single-omics analyses to other layers of biological information by projecting high-throughput data (e.g. transcriptomics, proteomics or metabolomics data) onto the network [105], analogously to what we described in Section 4.1.1. For instance, GEMs can be used as the underlying biological network in de novo pathway enrichment analysis to identify subnetworks that are significantly enriched with dysregulated entities [106].
Furthermore, generic GEM drafts can be contextualized to a specific condition, tissue or individual by imposing additional layers of constraints that are inferred from experimental omics data [107,108]. COBRA (Constraint-Based Reconstruction and Analysis) [104,109] is a popular framework that has implemented multiple methods for the integration of omics data, including time-course metabolomics data [110] and transcriptomics and proteomics data [111,112]. Contextualized GEMs provide novel opportunities for metabolic engineering, drug target identification, and personalized therapies [105,107,113]. For example, Agren et al. [114] used proteomics data of hepatocellular carcinoma patients to construct personalized, cell-specific GEMs for the prediction of antimetabolites (drugs that are structural analogs of metabolites) that can prevent tumor growth. The authors identified nearly 150 antimetabolites, one-third of which were specific to individual patients. Despite the small sample size (n=6) and restricting modeling to cellular effects, this study highlights the potential of refining GEMs using experimental omics data for personalized therapies. The recent emergence of whole-body metabolism (WBM) reconstructions [115] that currently model the human metabolism across 20 organs are expected to further advance this important field.
4.2. Data-driven approaches
Data-driven, multi-omics integration approaches use statistical models and machine learning techniques to infer relationships between and within layers of multi-omics data and in some cases a phenotype of interest. Without taking known biological relationships or annotations into account, most approaches rely on the analysis of correlation structures within the data itself. For multi-omics studies focusing on a specific disease or phenotype, common applications of data-driven methods include the training of predictors and classifiers, and identification of multivariate biomarker candidates. Independent of a specific phenotype of interest, the unbiased analysis of relationships between and within omics layers using data-driven approaches enables a global perspective on interactions between biological entities. Using sufficiently large datasets, this approach has the potential to uncover unknown relationships (e.g. not represented in knowledge bases) and to characterize entities with unknown function.
In the following, we review a selection of step-wise and simultaneous integration strategies and highlight their application in metabolomics and lipidomics studies. A list of multi-omics integration methods and frameworks is provided in Table 2.
Table 2.
A selection of multi-omics data integration frameworks and methods.
| Requires matching samples | Integration strategy | Implementation | Reference | Description | |
|---|---|---|---|---|---|
| KNOWLEDGE-BASED | |||||
| IMPaLA | no | enrichment | online | [93] | Integrated Molecular Pathway Level Analysis (IMPaLA) enables joint pathway analysis. |
| COBRA | - | constraint-based modelling | MATLAB Python Julia | [69,104] | The COnstraint-Based Reconstruction and Analysis (COBRA) Toolbox. |
| PathMe | - | composite network | online Python | [213] | Integrates KEGG, Reactome and WikiPathways into a unified abstraction. |
| DATA-DRIVEN | |||||
| KeyPath wayMiner | no | de novo enrichment | online Cytoscape software | [97,98] | Extracts all maximal connected sub-networks which enriched for dysregulated entities. |
| MI-MFA | partially | imputation/ensemble | R code in supplementary | [217] | Uses multiple imputation (MI) to enable the application of multiple factor analysis (MFA) to multi-omics data with partially missing single-omics profiles. |
| MOFA | partially | imputation | R Python | [218,219] | Unsupervised integration framework that infers a low-dimensional data representation and enables the imputation of missing omics profiles. |
| causalMGM | yes | single-block | online | [220] | Learns a causal (i.e., directed) graph using variable selection with subsequent application of a mixed graphical model (MGM) PC-Stable algorithm. |
| omicade4 | yes | single-block | R | [217] | Projection-based method that performs multiple co-inertia analysis. |
| xMWAS | yes | single-block | online R | [221] | Uses (sparse) Partial Least Squares regression to perform pairwise correlation analyses and build a heterogenous network. |
| mixOmics | yes | multi-block | R | [150] | Collection of unsupervised and supervised multivariate methods, including sparse generalized canonical correlation analysis (SGCCA) and Data Integration Analysis for Biomarker discovery using Latent cOmponents (DIABLO). |
| OnPLS | yes | multi-block | Python | [158,159] | Projection-based integration method that decomposes global, local and unique levels of variation. |
4.2.1. Step-wise integration
Step-wise strategies integrate datasets in a sequential manner. Here, individual omics layers are typically analyzed separately or in specific (lower-order) combinations. In subsequent steps, the results from these analyses are integrated into a common framework. The following section will introduce ensemble approaches that are suitable for studying a specific phenotype or outcome of interest, as well as pairwise association-based strategies that enable systematic and large-scale integration without necessarily focusing on a specific disease or phenotype.
Ensemble integration strategies apply multivariate classification or prediction methods, such as k-nearest neighbors [116] or Elastic Net [36] to each dataset individually and then combine the ensemble of results using, e.g., majority voting schemes or stacked generalization to boost performance [117]. Although each dataset is modeled separately, these types of methods require omics data that was collected from the same samples as the predictions are ultimately combined in a global model. For example, Ghaemi et al. [36] built a multivariate model predictive of gestational age on samples from 17 pregnant women at three time points during pregnancy. The datasets included measurements from the immunome, transcriptome, microbiome, proteome and metabolome. Using the Elastic Net algorithm, the authors built multiple predictors (one for each omics dataset) and subsequently used their predictions as input for a final model. This stacked generalization strategy was able to significantly increase performance and ablation analysis [118] gave insights into the respective contribution of each dataset. Furthermore, subsequent analysis of the top predictive features of each individual model, enabled the formulation of multi-omics-informed hypotheses. Among other findings, the authors identified a strong correlation between pregnanolone sulfate and NF-kB signaling in myeloid dendritic cells and regulatory T cells, highlighting a potential regulatory role of this endogenous steroid in the functioning of specific immune cells during pregnancy.
Training the base models in ensemble approaches in an isolated fashion, i.e., on each omics dataset separately, has several consequences. On the one hand, interdependencies between variables of different omics datasets are not fully taken into account such that some cross-omics interactions might be missed. On the other hand, the independence of the base models prevents datasets with a large number of variables from dominating the analysis.
The integration of pairwise association results is another step-wise integration strategy. In contrast to ensemble integration, this approach enables the global analysis of relationships between multiple omics layers by large-scale integration of data from multiple sources. A popular approach, which is centered around the concept of genetic variation as a driver of inter-individual variability, is QTL-based integration [7]. The basis for this integration technique are so-called quantitative trait loci (QTLs) [119]. QTLs are genetic markers (e.g., single nucleotide polymorphisms) that are significantly associated with the variation of quantitative molecular traits (e.g., the transcription level of a particular gene) [120]. They are identified in genome-wide association studies (GWAS) that make use of genome-wide genotypes of a large population of individuals that are tested in univariate analyses for association with molecular traits [120–122]. Besides QTLs of expression levels of genes (eQTLs) [123,124], major examples of investigated traits include abundances of proteins (pQTLs) [125,126] or concentrations of metabolites (mQTLs) [14,127]. For instance, Shin et al. [16] investigated genetic influences on more than 400 human blood metabolites in close to 8,000 individuals from two population-based cohorts. The result is a comprehensive atlas that links genetic variants in 145 loci to biochemical readouts, cataloging mQTLs influencing a wide variety of metabolic pathways.
After association analysis, variant annotation [128] or co-localization analysis [129,130] is used to functionally interlink entities from different omics by identifying overlapping QTLs (Figure 2C). This can be done on a genome-wide scale and with QTLs that have been identified in different studies or cohorts. QTL-based integration has been successfully applied in studies predicting the functional consequences of disease-associated variants, which are often located in non-coding regions of the genome [126,131,132]. For example, Chen et al. [132] systematically overlapped variants associated with autoimmune diseases with eQTLs as well as DNA methylation (meQTL), RNA splicing (sQTL) and histone modification (hQTLs) QTLs to identify cell-specific regulatory effects. Similarly, Suhre et al. [126] demonstrated the power of connecting GWAS-identified risk-variants to disease endpoints via blood proteome-derived pQTLs that overlapped with meQTLs, eQTLs, protein glycosylation QTLs, and mQTLs. Among other findings, this approach revealed a potential link between Alzheimer’s disease (AD) and mRNA splicing through linking protein levels of apolipoprotein E, a gene centrally linked to AD [133], and small ribonucleoprotein F via overlapping QTLs.
Figure 2. Multi-omics integration through composite networks.
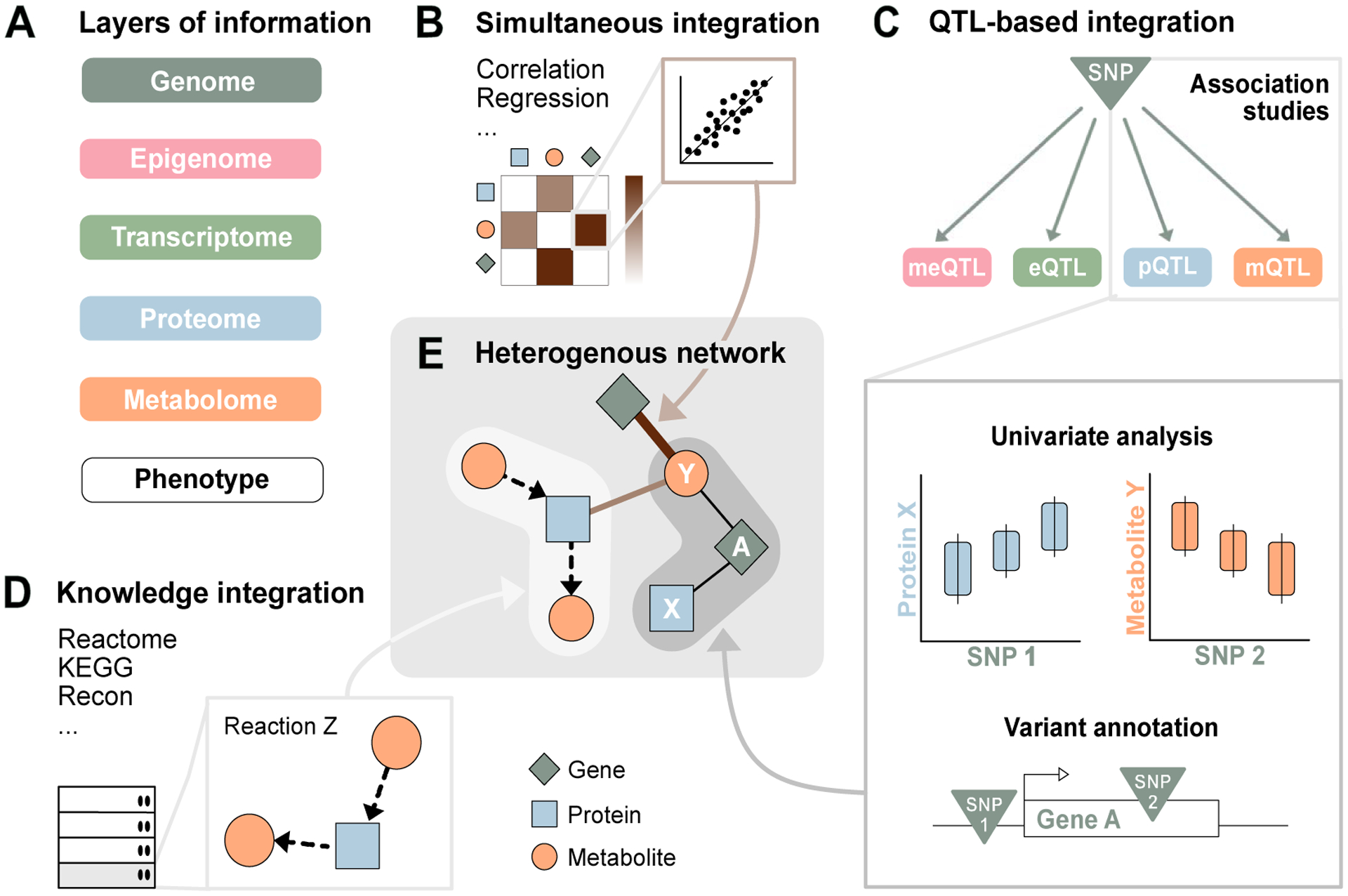
A. Different layers of a biological system that can be profiled using high-throughput technologies and are frequently integrated in multi-omics studies. B. Simultaneous integration. Correlation structures within and across omics datasets are analyzed using statistical methods. C. QTL-based integration. Using the genome as an anchor, quantitative trait loci (QTLs) identified in genome wide association studies (GWASs) are overlaid to establish links between different omics layers. D Knowledge integration. External information from metabolic databases or scientific literature is used to establish relationships between biological entities. E. Composite networks. By merging the networks inferred in (B-D) on common entities, comprehensive multi-omics catalogues can be constructed. These heterogenous networks can be mined in post-integration analysis using established graph algorithms.
Although this integration strategy only takes into account pairwise relationships, it facilitates the large-scale integration of omics datasets from different sources. This is especially valuable in settings where sufficiently large multi-omics studies in the same set of samples are not available. Furthermore, QTL-based integration only requires summary statistics (results of an association study), circumventing data sharing restrictions that may be present on datasets with patient information. Lastly, this approach can integrate results from independent GWASs on the same traits, providing an opportunity to build data confidence by independent replication. Similarly, meta-analysis methods [134] that statistically combine summary statistics from independent association studies on the same traits (e.g., multiple GWASs with metabolic traits) can be used to increase power and reduce false-positive findings. It is important to note that the concept of integrating pairwise-association results is not restricted to using the genome as an anchor but can be centered around any other omics layer, including the metabolome.
4.2.2. Simultaneous integration
Simultaneous integration strategies use all available omics datasets at the same time and integrate the information in a single modeling step. This has the advantage of taking into account correlations between entities within and across omics layers. In the following, we are reviewing approaches by categorizing them into single-block and multi-block strategies. Single-block integration strategies concatenate all available datasets to form one large data matrix (a “single block”) before applying any analysis method without consideration of heterogeneities between omics (e.g. in scale or variance). In contrast, multi-block integration strategies retain and account for the multi-block structure of the data that is defined by the different omics datasets. Both strategies require that full multi-omics profiles are available for the same set of samples/individuals. Some methods enable imputation of missing single-omics profiles for a moderate amount of samples/individuals in a multi-omics context. These include MI-MFA (Multiple Imputation - Multiple Factor Analysis)[47] that uses hot-deck imputation [135] to replace missing omics vectors with observed values from a similar sample, and MOFA (Multi-Omics Factor Analysis) [138,139], a statistical framework that infers a low-dimensional data representation in form of (hidden) factors [136]. However, although imputation can increase power by extending the set of available observations, imputed values can never accurately represent the “true” unobserved measurements and should therefore always be interpreted with caution.
In order to integrate different omics datasets, single-block integration strategies simply concatenate the different data matrices into one large data matrix before applying a statistical analysis method. This enables the direct application of methods that are typically applied to single-omics datasets for tasks such as clustering (e.g., K-means clustering [61]), classification and regression (e.g., Random Forest [137], LASSO regression [138]) or projection (Partial Least Squares Discriminant Analysis (PLSDA) [139,140]). Correlation-based strategies are another popular class of single-block methods, which aim at quantifying the relationships between biological entities by iteratively applying an association measure, such as Pearson’s Correlation Coefficient, to all pairwise combinations of the variables (measured biological entities). However, simple correlation measures cannot distinguish between direct and indirect effects [141]. For example, associations between mRNA levels are quite frequently mediated by transcriptional co-regulation at the gene-level [142]. These confounded associations lead to a drastically inflated number of edges, resulting in dense networks with limited interpretability [41,142,143]. Gaussian Graphical Models (GGMs) [144] circumvent this problem by estimating full-order partial correlation coefficients, i.e., pairwise correlations between variables corrected against all other variables. This measure of conditional independence has been valuable to infer pathway relationships from single omics datasets [57,145,146]. However, GGMs assume multivariate normally distributed data and multi-omics datasets often include variables with different distributions, such as phenotypic data on gender or disease subtype [41,143]. An extension to GGMs that addresses this issue are Mixed Graphical Models (MGMs) [147–149], which can incorporate datasets with mixed distributions (e.g., continuous, discrete, and count variables) [143]. For example, Zierer et al. [41] inferred an MGM from a multi-omics dataset collected from the same individuals, including data on epigenomics, transcriptomics, glycomics, metabolomics, and phenotypic data. The authors used a Graphical Random Forest [149] method for the integration of 144 preselected features and explored the molecular underpinnings of age-related diseases and co-morbidities. They identified seven network modules that reflect distinct aspects of aging, such as lung function, bone density, and renal function. Furthermore, they found that these modules are connected by distinct hubs, highlighting central molecules and potentially linked mechanisms that may drive co-morbidities, such as urate that connects renal disease with body composition and obesity.
Single-block integration ignores heterogeneities between data types which can lead to severe bias and other complications [30,32,150]. For example, metabolomics and transcriptomics data are generated by fundamentally different analytical technologies. This leads to values with different scale and variance as well as different noise distributions [51,151]. When clustering such datasets, the entities within a particular omics type will predominantly cluster together, reflecting intra-, instead of inter-, omics relationships [18,36,41,151]. Similarly, variance maximizing approaches, such as PCA and PLS, will capture these technical differences in their first component [151]. Additionally, the number of variables in each single omics dataset will in most cases be substantially different: a state-of-the-art genomics analysis will provide information on millions of genetic variants, transcriptomics measures tens of thousands of mRNAs, and proteomics and metabolomics technologies usually measure molecules in the range of thousands of molecules [51]. Analyzing such datasets simultaneously without accounting for the diverging numbers of features will introduce bias, as the data type with the most features will drive the results [152].
To circumvent this problem and ensure that every dataset has equal weight, variables can be scaled to unit variance with subsequent block scaling [151] by using, for example, the inverse number of variables in the respective dataset (“block”) to scale each variable. This was implemented in Multiple Factor Analysis [152,153], where data blocks are normalized prior to concatenation by using the inverse of the first squared singular value of a PCA on each data block as weight. However, different methods for variable scaling and block scaling can significantly influence the outcomes [151]. General caution is advised when concatenating datasets from different sources and special care should be taken to identify an integration method that combines and scales data appropriately [7,151].
The need to account for heterogeneities between multi-omics datasets has led to the emergence of multi-block integration strategies that can take the block structure, i.e., groups of omics variables from different sources, into account [154]. Multi-block methods simultaneously model multiple data matrices and provide insights into the relationships between omics (blocks). Many of these approaches are extensions of established multivariate methods, such as Partial Least Squares (PLS). Examples include O2PLS [155,156] for the integration of two omics datasets and Multiple-Block Orthogonal Projections to Latent Structures (OnPLS) [157–159] for the integration of more than two omics datasets. OnPLS decomposes data from multiple omics data matrices into global, local and unique levels of variation [159]. Reinke et al. [160] demonstrated the potential of this approach using a small subset (n=22) of individuals from an asthma cohort. Here, six blocks of data - transcriptomics, metabolomics, three targeted assays (on sphingolipids, oxylipins, and fatty acids), and clinical variables - were integrated using OnPLS. Subsequent variable selection and visualization gave insights into cross-omics interactions, for example, by identifying a potential link between transcript levels of ATP6V1G1, a gene that has been associated with osteoporosis, and multiple metabolites that are dysregulated by inhaled corticoid steroids.
Other popular multi-block integration strategies include unsupervised methods such as regularized generalized canonical correlation analysis (RGCCA) and sparse generalized canonical correlation (SGCCA) [161], as well as the supervised framework Data Integration Analysis for Biomarker discovery using Latent cOmponents (DIABLO). DIABLO [39] is a multivariate classification method that extends SGCCA to a supervised analysis and prediction framework. It can identify key omics variables that drive the discrimination between phenotypic groups of interest and simultaneously builds a predictive model to classify new data [37,40,162–164]. For example, Qui et al. [40] integrated genomic, transcriptomic, epigenomic, and metabolomic datasets from patients with high and low bone mineral density (BMD). Using DIABLO, they identified a multi-omics biomarker panel for osteoporosis that includes 74 differentially expressed genes, 75 differentially methylated CpG sites and 23 differentially abundant metabolites. To gain further mechanistic insights into underlying disease mechanisms, the authors conducted a targeted QTL-based analysis in combination with Mendelian randomization. They were able to identify five biomarkers (ADRA2A, FADS2, FMN1, RABL2A, SPRY1) with a causal effect on levels of BMD. DIABLO and various other projection-based integration methods are implemented in the R package mixOmics [150] which is focused on data exploration, dimensionality reduction and visualization of multi-omics data.
Simultaneous integration strategies have been applied by relatively few studies so far, with mostly small numbers of samples/individuals. This is most likely due to the lack of larger available multi-omics datasets. Nevertheless, simultaneous integration, and especially multi-block methods, are powerful tools that have the potential to fully exploit multi-omics data in integrative analyses.
4.3. Composite network approaches
Composite networks aim at capturing relationships between omics layers in heterogeneous networks by merging information from different knowledge-driven and/or data-driven sources. This step-wise integration strategy is gaining increasing popularity due to its scalability and versatile applicability. In order to construct a composite network, the information from each knowledge-based (e.g., STRING, KEGG) or data-driven (e.g., correlation-based) component is stored and interconnected in accessible network structures (graphs) that are merged by overlaying common biological entities (Figure 2B–E). This can be accomplished by simple concatenation of the respective underlying edge lists, provided that there is some degree of overlap between the datasets and/or resources. The resulting network consists of nodes (biological entities such as genes, proteins and metabolites) connected by edges that model pairwise functional, biochemical or physical relationships [165]. Composite networks are per se not bound to a specific phenotype or disease of interest. Once built, they provide a comprehensive catalogue of inter- and intra-omics relationships that can be explored in post-integration analyses to identify and prioritize relevant entities in the neighbourhood of e.g. disease-associated genes within the network or to predict novel associations.
Composite networks can be built in a knowledge-based, data-driven or hybrid fashion. While knowledge-based integration allows the large-scale analysis of vast amounts of published information without requiring additional omics experiments [43], this approach is restricted to entities that have been annotated. Data-driven composite networks merge inferred information from experimental multi-omics data and, in contrast, can naturally only include the biological entities measured by the respective omics technology. By combining these two approaches, for example, by extending data-driven networks (e.g. built through QTL-based integration described in Section 4.2.1) with knowledge-based relationships (e.g., gene-transcript-protein or drug-drug targets relations), it is possible to construct comprehensive multi-layered resources that facilitate the unbiased generation and exploration of multi-omics hypotheses. HENA [166], a heterogeneous network-based dataset for Alzheimer’s disease (AD), is a recent example of this. Sügis et al. integrated data relating to AD, including GWAS results, protein-protein interaction, and gene co-expression networks, from public knowledge databases and experimental datasets. The resulting gene-centric network was subsequently analyzed using graph convolutional networks to identify disease-related genes, highlighting one of the many potential applications of composite networks. Future frameworks that additionally include metabolite data will provide even more comprehensive models for studying molecular mechanisms implicated in AD.
Although conceptually simple, the construction of composite networks is complicated at large due to the discussed challenges of ID mapping and compound identification (see Section 4.1), as well as differing data formats between resources, and considerations regarding statistical cut-offs and weighting of information types. Furthermore, the post-integration analysis of these large and highly complex networks is not straightforward and requires sophisticated algorithms (further discussed in Section 5). Consequently, databases and frameworks that provide access to composite networks are attracting growing interest, such as ConsensusPathDB [167,168] and omicsNet [169,170].
5. Post-integration analysis, visualization and interpretation
Post-integration analysis of inferred networks or multi-omics features through manual inspection or computational algorithms is key to gain biologically relevant insights and fully exploit the potential of multi-omics datasets. So far, a limiting factor has often been the ability to represent, comprehend and reproduce highly complex and multifactorial relationships across multiple biological domains [171].
For studies that are driven by a clear research question, interpretation can be straightforward. For instance, when building a predictor for a specific phenotype of interest, integration methods such as DIABLO (Section 4.2.2) result in a subset of interesting (in a statistical sense, e.g. most predictive, most significant) biological entities. This set of variables can then be subjected to downstream analyses to gain further functional insights or to investigate causality (e.g. via Mendelian randomization). Global integration efforts, on the other hand, enable exploratory analysis by systematically cataloging biological entities and their interactions without focusing on a specific phenotype or disease. Here, post-integration analysis through computational algorithms provides tools to identify patterns in the data and pinpoint interesting entities.
To this end, networks provide a flexible and intuitive mathematical framework to represent, visualize, and analyze these complex relationships [172]. Various techniques have been developed that facilitate the visual representation and exploration of networks in a human-comprehensible form by arranging nodes and edges in specific layouts. For example, by grouping nodes together that are highly connected, modular patterns in the data become more visible [172]. However, with growing complexity and size, networks can quickly become very dense and difficult to comprehend [173]. Alternative representations of large networks, such as structural summary [174] or axis-based node-link representations [175] have been developed to mitigate these challenges and provide scalable layout alternatives [176].
In addition to providing intuitive visualization, networks enable the application of a rich toolbox of established graph algorithms to explore multi-omics networks and extract relevant information in an automated manner [177]. For example, multi-layer networks represent a promising mathematical framework, where layers of nodes (e.g., genes, proteins, metabolites) are connected by different edge types with varying degrees of connectivity (e.g., gene co-expression, trait association and protein co-abundance) [178,179]. Research fields such as graph theory and network science have developed various algorithms that can be applied to such heterogeneous networks, including random walk [43], module identification [180], or meta-path-based techniques [181]. This enables, for example, the prediction of novel edges [181], the identification of key players [182,183], or retrieval of interesting subnetworks (modules) [184–186]. Furthermore, native graph databases, such as Neo4j, represent an attractive framework for post-integration analysis as they enable the efficient storage and analysis of large amounts of semi-structured, diverse and highly connected data [187]. An extensive list of network-based multi-omics visualization tools and online resources is provided in Table 1.
Even after successful identification of interesting entities or modules, the downstream functional interpretation and validation of such complex multi-omics findings is not straightforward. Direct replication as an important tool for identifying false positives [7] is often not an option due to the frequently limited availability of comparable and sufficiently powered omics studies. So far, validation of results has therefore often been performed using prior knowledge [171] to provide functional evidence, for example, through set-based enrichment (Section 4.1.1). However, with growing numbers of large-scale studies and efforts towards standardizing and indexing datasets across sources, such as the Omics Discovery Index (OmicsDI) [188,189], data-driven replication will become increasingly feasible in the future. Beyond that, it is often not possible to describe every finding from a multi-omics study in detail as results can be very complex and numerous. This consequently leads to biased or selective reporting of outcomes that are published [171]. To this end, the sharing of all results in easily accessible data repositories, such as NDEx [190], or dedicated supplemental web-servers [16,20,126], is becoming more popular as it enables the re-use of multi-omics results for further exploration or replication by other researchers.
6. Current trends and future perspectives
As highlighted in this review, various multi-omics integration strategies exist. Developments in research fields such as computer vision and natural language processing offer promising new directions for the unbiased integration of high-dimensional data. Recently, these fields have been transformed by the use of deep learning techniques, such as deep neural networks, which can handle vast amounts of data and are able to discover highly complex and relevant features [191,192]. In deep learning, multiple hidden layers enable the learning of new, highly complex data representations [191]. Furthermore, flexible architectures allow models to be tailored to many different problem domains, providing exciting new possibilities also for multi-omics integration studies [193,194]. For example, variational autoencoders (VAEs) [195] are popular representation learning methods that have been proposed for non-linear dimensionality reduction, unsupervised clustering and denoising of datasets [196,197]. They can be used to encode input data (e.g., different omics datasets) into a low-dimensional embedding, effectively integrating different omics types into a new latent representation [198]. A major limitation of deep learning algorithms, so far, has been their need for vast amounts of high-quality data and the complicated interpretation of model features [192,194,199]. However, the increasing availability of large multi-omics datasets and development of interpretable deep learning methods will enable more and more deep learning applications in the future [191,200].
Besides algorithmic innovations, the ongoing advances of analytical technologies will also provide novel opportunities and challenges for integrative studies. For example, spatial omics profiling has received increasing attention in the past few years due to the advent of high-resolution technologies to generate data in a fine-grained spatial resolution. This is particularly interesting for the cancer field, where there is increasing evidence that the tumor microenvironment, i.e., the collection of all stromal cells surrounding and supporting the tumor cells, plays a major role in prognosis and therapy [201]. For metabolomics, modern “Matrix Assisted Laser Desorption Ionization” (MALDI)-imaging mass spectrometry instruments can acquire metabolite profiles at almost single-cell resolution [202]. This rich new type of data, composed of metabolites, samples, and two or more spatial dimensions, also requires innovative approaches for data processing, integration, and analysis. For example, single-cell metabolic profiles can be assigned and analyzed using the “SpaceM” method, which performs the interpolation of spatial measurement patterns onto microscopy images [203]. Similarly, new technologies and the corresponding computational methods allow for high-resolution protein profiling, e.g., using mass cytometry time of flight (CyTOF) instruments [204], and spatial transcriptomics data can be obtained by a growing number of sequencing and microarray-based techniques [205]. Future applications, where tissue samples or entire organs are analyzed in a sequential fashion with a combination of these techniques to generate spatial multi-omics datasets, promise unprecedented insights into the deep molecular biology of the systems under study.
7. Conclusions
The generation of vast amounts of biological data have generated exciting new opportunities to gain a systems view on molecular wirings across regulatory layers that define health and disease. However, the heterogeneous and high-dimensional nature of multi-omics datasets in combination with differing study objectives and data scenarios make the appropriate data integration strategy a case-by-case choice.
While knowledge-based strategies can guide integrative analysis by harnessing a large body of manually and experimentally validated information from databases and scientific literature, it is restricted to known or previously characterized biological entities and is not applicable for molecules with unknown function or identity. Data-driven methods, on the other hand, use statistical methods such as correlation or association analysis to infer relationships between omics layers. Although this can be prone to identification of spurious associations and success heavily depends on correctly preprocessed, high-quality data, data-driven integration has the potential to discover novel as well as condition-specific interactions. In particular, multi-block integration methods that can simultaneously analyze datasets while taking into account inter-omics heterogeneity show exciting potential to fully exploit multi-omics datasets. To leverage the advantages of both approaches, network-based hybrid integration methods have emerged that enable the combination of knowledge-based and data-driven data integration. This facilitates the generation of highly complex multi-omics interaction catalogues that can be mined in an automated fashion using graph algorithms.
With increasing availability of larger, high-quality datasets paralleled by the development of new omics technologies, the demand for powerful data analysis tools and standardized integration frameworks will continue to grow. The integrative analysis of these multi-omics data, enabled by publishing data in centralized data-repositories adhering to the FAIR Principles (Findable, Accessible, Interoperable and Reusable) [206], will finally allow researchers to promote the usability and reproducibility of their work and has the potential for achieving substantial advances in biomedical research and health care.
Box 1. Glossary of important terms and concepts used throughout this review.
Integration method – A specific method/framework that performs data integration.
Integration strategy – Summary term for multiple data integration methods that follow the same principle.
Knowledge-based integration – Relationships between biological entities across and within omics are established using knowledge bases (extrinsic information).
Data-driven integration – Relationships between biological entities across and within omics are statistically inferred from multi-omics datasets (intrinsic information).
Simultaneous integration – Integration strategies that take into account all available data by merging the data and performing a single method on the concatenated matrix.
Single-block methods – Multivariate methods that perform simultaneous integration and do not take into account heterogeneities between the different omics datasets.
Multi-block methods – Multivariate methods that perform simultaneous integration and can take into account the block structure of multi-omics data by modelling each block separately.
Step-wise integration – Integration strategies that analyze omics datasets separately and integrate the results or models in a subsequent step.
Biological entity – Refers to a measured biological molecule such as protein, metabolite, lipid but also includes single nucleotide polymorphisms (SNP) and epigenetic alterations.
Highlights.
multi-omics studies can unravel the complex molecular underpinnings of diseases
data availability and study aims influence the selection of the integration strategy
knowledge-based integration can enhance the biological interpretability of results
data-driven integration can infer relationships between uncharacterized molecules
network-based, hybrid integration strategies combine the strengths of both
Acknowledgements
Funding: This work was supported by the National Institutes of Health/the National Institute on Aging (NIA) [NIA 1RF1AG057452, NIA 1R01AG059093, NIA 1U01AG061359]. Funding sources had no role in study design, in the collection, analysis and interpretation of data, in the writing of the report, or in the decision to submit this article for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Lotta LA, Scott RA, Sharp SJ, Burgess S, Luan J, Tillin T, Schmidt AF, Imamura F, Stewart ID, Perry JRB, Marney L, Koulman A, Karoly ED, Forouhi NG, Sjögren RJO, Näslund E, Zierath JR, Krook A, Savage DB, Griffin JL, Chaturvedi N, Hingorani AD, Khaw KT, Barroso I, McCarthy MI, O’Rahilly S, Wareham NJ, Langenberg C, Genetic Predisposition to an Impaired Metabolism of the Branched-Chain Amino Acids and Risk of Type 2 Diabetes: A Mendelian Randomisation Analysis, PLoS Med. 13 (2016) 1–22. 10.1371/journal.pmed.1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tynkkynen J, Chouraki V, van der Lee SJ, Hernesniemi J, Yang Q, Li S, Beiser A, Larson MG, Sääksjärvi K, Shipley MJ, Singh-Manoux A, Gerszten RE, Wang TJ, Havulinna AS, Würtz P, Fischer K, Demirkan A, Ikram MA, Amin N, Lehtimäki T, Kähönen M, Perola M, Metspalu A, Kangas AJ, Soininen P, Ala-Korpela M, Vasan RS, Kivimäki M, van Duijn CM, Seshadri S, Salomaa V, Association of branched-chain amino acids and other circulating metabolites with risk of incident dementia and Alzheimer’s disease: A prospective study in eight cohorts, Alzheimer’s Dement. 14 (2018) 723–733. 10.1016/j.jalz.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Civelek M, Lusis AJ, Systems genetics approaches to understand complex traits, Nat. Rev. Genet 15 (2014) 34–48. 10.1038/nrg3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hasin Y, Seldin M, Lusis A, Multi-omics approaches to disease, Genome Biol. 18 (2017) 83 10.1186/s13059-017-1215-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Smith GD, Ebrahim S, “Mendelian randomization”: Can genetic epidemiology contribute to understanding environmental determinants of disease?, Int. J. Epidemiol 32 (2003) 1–22. 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- [6].Kopczynski D, Coman C, Zahedi RP, Lorenz K, Sickmann A, Ahrends R, Multi-OMICS: a critical technical perspective on integrative lipidomics approaches, Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1862 (2017) 808–811. 10.1016/j.bbalip.2017.02.003. [DOI] [PubMed] [Google Scholar]
- [7].Ritchie MD, Holzinger ER, Li R, Pendergrass SA, Kim D, Methods of integrating data to uncover genotype-phenotype interactions, Nat. Rev. Genet 16 (2015) 85–97. 10.1038/nrg3868. [DOI] [PubMed] [Google Scholar]
- [8].Wishart DS, Emerging applications of metabolomics in drug discovery and precision medicine, Nat. Rev. Drug Discov 15 (2016) 473–484. 10.1038/nrd.2016.32. [DOI] [PubMed] [Google Scholar]
- [9].Fiehn O, Metabolomics – the link between genotypes and phenotypes, Plant Mol. Biol 48 (2002) 155–171. [PubMed] [Google Scholar]
- [10].Toledo JB, Arnold M, Kastenmüller G, Chang R, Baillie RA, Han X, Thambisetty M, Tenenbaum JD, Suhre K, Thompson JW, John-Williams LS, MahmoudianDehkordi S, Rotroff DM, Jack JR, Motsinger-Reif A, Risacher SL, Blach C, Lucas JE, Massaro T, Louie G, Zhu H, Dallmann G, Klavins K, Koal T, Kim S, Nho K, Shen L, Casanova R, Varma S, Legido-Quigley C, Moseley MA, Zhu K, Henrion MYR, van der Lee SJ, Harms AC, Demirkan A, Hankemeier T, van Duijn CM, Trojanowski JQ, Shaw LM, Saykin AJ, Weiner MW, Doraiswamy PM, Kaddurah-Daouk R, Metabolic network failures in Alzheimer’s disease: A biochemical road map, Alzheimer’s Dement. 13 (2017) 965–984. 10.1016/j.jalz.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Suhre K, Meisinger C, Döring A, Altmaier E, Belcredi P, Gieger C, Chang D, Milburn MV, Gall WE, Weinberger KM, Mewes HW, Angelis MH, Wichmann HE, Kronenberg F, Adamski J, Illig T, Metabolic footprint of diabetes: A multiplatform metabolomics study in an epidemiological setting, PLoS One. 5 (2010). 10.1371/journal.pone.0013953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yang M, Soga T, Pollard PJ, Yang M, Soga T, Pollard PJ, Oncometabolites : linking altered metabolism with cancer, J. Clin. Invertigation 123 (2013) 3652–3658. 10.1172/JCI67228.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Beger RD, Dunn W, Schmidt MA, Gross SS, Kirwan JA, Cascante M, Brennan L, Wishart DS, Oresic M, Hankemeier T, Broadhurst DI, Lane AN, Suhre K, Kastenmüller G, Sumner SJ, Thiele I, Fiehn O, Kaddurah-Daouk R, for “Precision Medicine, Metabolomics enables precision medicine: “A White Paper, Community Perspective,” Metabolomics. 12 (2016) 149 10.1007/s11306-016-1094-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gieger C, Geistlinger L, Altmaier E, De Angelis MH, Kronenberg F, Meitinger T, Mewes HW, Wichmann HE, Weinberger KM, Adamski J, Illig T, Suhre K, Genetics meets metabolomics: A genome-wide association study of metabolite profiles in human serum, PLoS Genet. 4 (2008). 10.1371/journal.pgen.1000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Draisma HH, Pool R, Kobl M, Jansen R, Petersen AK, Vaarhorst AA, Yet I, Haller T, Demirkan A, Esko T, Zhu G, Böhringer S, Beekman M, Van Klinken JB, Römisch-Margl W, Prehn C, Adamski J, De Craen AJM, Van Leeuwen EM, Amin N, Dharuri H, Westra HJ, Franke L, De Geus EJC, Hottenga JJ, Willemsen G, Henders AK, Montgomery GW, Nyholt DR, Whitfield JB, Penninx BW, Spector TD, Metspalu A, Eline Slagboom P, Van Dijk KW, ‘T Hoen PAC, Strauch K, Martin NG, Van Ommen GJB, Illig T, Bell JT, Mangino M, Suhre K, McCarthy MI, Gieger C, Isaacs A, Van Duijn CM, Boomsma DI, Genome-wide association study identifies novel genetic variants contributing to variation in blood metabolite levels, Nat. Commun 6 (2015) 21 10.1038/ncomms8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shin S-Y, Fauman EB, Petersen A-K, Krumsiek J, Santos R, Huang J, Arnold M, Erte I, Forgetta V, Yang T-P, Walter K, Menni C, Chen L, Vasquez L, Valdes AM, Hyde CL, Wang V, Ziemek D, Roberts P, Xi L, Grundberg E, Waldenberger M, Richards JB, Mohney RP, V Milburn M, John SL, Trimmer J, Theis FJ, Overington JP, Suhre K, Brosnan MJ, Gieger C, Kastenmüller G, Spector TD, Soranzo N, An atlas of genetic influences on human blood metabolites, Nat. Genet 46 (2014) 543–550. 10.1038/ng.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Raffler J, Friedrich N, Arnold M, Kacprowski T, Rueedi R, Altmaier E, Bergmann S, Budde K, Gieger C, Homuth G, Pietzner M, Römisch-Margl W, Strauch K, Völzke H, Waldenberger M, Wallaschofski H, Nauck M, Völker U, Kastenmüller G, Suhre K, Genome-Wide Association Study with Targeted and Non-targeted NMR Metabolomics Identifies 15 Novel Loci of Urinary Human Metabolic Individuality, PLoS Genet 11 (2015). 10.1371/journal.pgen.1005487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bartel J, Krumsiek J, Schramm K, Adamski J, Gieger C, Herder C, Carstensen M, Peters A, Rathmann W, Roden M, Strauch K, Suhre K, Kastenmüller G, Prokisch H, Theis FJ, The Human Blood Metabolome-Transcriptome Interface, PLoS Genet. 11 (2015) 1–32. 10.1371/journal.pgen.1005274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Petersen AK, Zeilinger S, Kastenmüller G, Werner RM, Brugger M, Peters A, Meisinger C, Strauch K, Hengstenberg C, Pagel P, Huber F, Mohney RP, Grallert H, Illig T, Adamski J, Waldenberger M, Gieger C, Suhre K, Epigenetics meets metabolomics: An epigenome-wide association study with blood serum metabolic traits, Hum. Mol. Genet 23 (2014) 534–545. 10.1093/hmg/ddt430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Suhre K, Shin SY, Petersen AK, Mohney RP, Meredith D, Wägele B, Altmaier E, Deloukas P, Erdmann J, Grundberg E, Hammond CJ, De Angelis MH, Kastenmüller G, Köttgen A, Kronenberg F, Mangino M, Meisinger C, Meitinger T, Mewes HW, V Milburn M, Prehn C, Raffler J, Ried JS, Römisch-Margl W, Samani NJ, Small KS, -Erich Wichmann H, Zhai G, Illig T, Spector TD, Adamski J, Soranzo N, Gieger C, Human metabolic individuality in biomedical and pharmaceutical research, Nature. 477 (2011) 54–62. 10.1038/nature10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Suhre K, Gieger C, Genetic variation in metabolic phenotypes: Study designs and applications, Nat. Rev. Genet 13 (2012) 759–769. 10.1038/nrg3314. [DOI] [PubMed] [Google Scholar]
- [22].Petersen AK, Krumsiek J, Wägele B, Theis FJ, Wichmann HE, Gieger C, Suhre K, On the hypothesis-free testing of metabolite ratios in genome-wide and metabolome-wide association studies, BMC Bioinformatics. 13 (2012) 120 10.1186/1471-2105-13-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jaremek M, Yu Z, Mangino M, Mittelstrass K, Prehn C, Singmann P, Xu T, Dahmen N, Weinberger KM, Suhre K, Peters A, Döring A, Hauner H, Adamski J, Illig T, Spector TD, Wang-Sattler R, Alcohol-induced metabolomic differences in humans, Transl. Psychiatry 3 (2013) 1–8. 10.1038/tp.2013.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shaham O, Wei R, Wang TJ, Ricciardi C, Lewis GD, Vasan RS, Carr SA, Thadhani R, Gerszten RE, Mootha VK, Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity, Mol. Syst. Biol 4 (2008) 1–9. 10.1038/msb.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Deberardinis RJ, Thompson CB, Cellular metabolism and disease: What do metabolic outliers teach us?, Cell. 148 (2012) 1132–1144. 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rinschen MM, Ivanisevic J, Giera M, Siuzdak G, Identification of bioactive metabolites using activity metabolomics, Nat. Rev. Mol. Cell Biol 20 (2019) 353–367. 10.1038/s41580-019-0108-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hawe JS, Theis FJ, Heinig M, Inferring interaction networks from multi-omics data, Front. Genet 10 (2019) 1–13. 10.3389/fgene.2019.00535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yan J, Risacher SL, Shen L, Saykin AJ, Network approaches to systems biology analysis of complex disease: Integrative methods for multi-omics data, Brief. Bioinform 19 (2017) 1370–1381. 10.1093/bib/bbx066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Meng C, Zeleznik OA, Thallinger GG, Kuster B, Gholami AM, Culhane AC, Dimension reduction techniques for the integrative analysis of multi-omics data, Brief. Bioinform 17 (2016) 628–641. 10.1093/bib/bbv108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mirza B, Wang W, Wang J, Choi H, Chung NC, Ping P, Machine learning and integrative analysis of biomedical big data, Genes (Basel). 10 (2019). 10.3390/genes10020087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wu C, Zhou F, Ren J, Li X, Jiang Y, Ma S, A Selective Review of Multi-Level Omics Data Integration Using Variable Selection, High-Throughput. 8 (2019) 4 10.3390/ht8010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li Y, Wu FX, Ngom A, A review on machine learning principles for multi-view biological data integration, Brief. Bioinform 19 (2018) 325–340. 10.1093/bib/bbw113. [DOI] [PubMed] [Google Scholar]
- [33].Chu SH, Huang M, Kelly RS, Benedetti E, Siddiqui JK, Zeleznik OA, Pereira A, Herrington D, Wheelock CE, Krumsiek J, Mc Geachie M, Moore SC, Snell RG, Lasky-Su JLS, Integration of Metabolomic and Other Omics Data in Population-Based Study Designs: An Epidemiological Perspective, Metabolites. 9 (2019). 10.3390/metabo9060117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Eicher T, Kinnebrew G, Patt A, Spencer K, Ying K, Ma Q, Machiraju R, and Mathé EA, Metabolomics and Multi-Omics Integration: A Survey of Computational Methods and Resources, Metabolites. 10 (2020) 202 10.3390/metabo10050202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pinu FR, Beale DJ, Paten AM, Kouremenos K, Swarup S, Schirra HJ, Wishart D, Systems biology and multi-omics integration: Viewpoints from the metabolomics research community, Metabolites. 9 (2019) 1–31. 10.3390/metabo9040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ghaemi MS, DiGiulio DB, Contrepois K, Callahan B, Ngo TTM, Lee-Mcmullen B, Lehallier B, Robaczewska A, McIlwain D, Rosenberg-Hasson Y, Wong RJ, Quaintance C, Culos A, Stanley N, Tanada A, Tsai A, Gaudilliere D, Ganio E, Han X, Ando K, McNeil L, Tingle M, Wise P, Maric I, Sirota M, Wyss-Coray T, Winn VD, Druzin ML, Gibbs R, Darmstadt GL, Lewis DB, Partovi Nia V, Agard B, Tibshirani R, Nolan G, Snyder MP, Relman DA, Quake SR, Shaw GM, Stevenson DK, Angst MS, Gaudilliere B, Aghaeepour N, Multiomics modeling of the immunome, transcriptome, microbiome, proteome and metabolome adaptations during human pregnancy, Bioinformatics. 35 (2019) 95–103. 10.1093/bioinformatics/bty537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xicota L, Ichou F, Lejeune FX, Colsch B, Tenenhaus A, Leroy I, Fontaine G, Lhomme M, Bertin H, Habert MO, Epelbaum S, Dubois B, Mochel F, Potier MC, Multi-omics signature of brain amyloid deposition in asymptomatic individuals at-risk for Alzheimer’s disease: The INSIGHT-preAD study, EBioMedicine. 47 (2019) 518–528. 10.1016/j.ebiom.2019.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Borgan E, Sitter B, Lingjærde OC, Johnsen H, Lundgren S, Bathen TF, Sørlie T, Børresen-Dale AL, Gribbestad IS, Merging transcriptomics and metabolomics - advances in breast cancer profiling, BMC Cancer. 10 (2010) 628 10.1186/1471-2407-10-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Singh A, Shannon CP, Gautier B, Rohart F, Vacher M, Tebbutt SJ, Cao KAL, DIABLO: An integrative approach for identifying key molecular drivers from multi-omics assays, Bioinformatics. 35 (2019) 3055–3062. 10.1093/bioinformatics/bty1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Qiu C, Yu F, Su K, Zhao Q, Zhang L, Xu C, Hu W, Wang Z, Zhao L, Tian Q, Wang Y, Deng H, Shen H, Multi-omics Data Integration for Identifying Osteoporosis Biomarkers and Their Biological Interaction and Causal Mechanisms, IScience. 23 (2020) 100847 10.1016/j.isci.2020.100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zierer J, Pallister T, Tsai PC, Krumsiek J, Bell JT, Lauc G, Spector TD, Menni C, Kastenmüller G, Exploring the molecular basis of age-related disease comorbidities using a multi-omics graphical model, Sci. Rep 6 (2016) 1–10. 10.1038/srep37646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Altenbuchinger M, Zacharias HU, Solbrig S, Schäfer A, Büyüközkan M, Schultheiß UT, Kotsis F, Köttgen A, Spang R, Oefner PJ, Krumsiek J, Gronwald W, A multi-source data integration approach reveals novel associations between metabolites and renal outcomes in the German Chronic Kidney Disease study, Sci. Rep 9 (2019) 1–13. 10.1038/s41598-019-50346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yao Q, Xu Y, Yang H, Shang D, Zhang C, Zhang Y, Sun Z, Shi X, Feng L, Han J, Su F, Li C, Li X, Global Prioritization of Disease Candidate Metabolites Based on a Multi-omics Composite Network, Sci. Rep 5 (2015) 1–14. 10.1038/srep17201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chu SH, Huang M, Kelly RS, Benedetti E, Siddiqui JK, Zeleznik OA, Pereira A, Herrington D, Wheelock CE, Krumsiek J, Mc Geachie M, Moore SC, Snell RG, Lasky-Su JLS, Integration of Metabolomic and Other Omics Data in Population-Based Study Designs: An Epidemiological Perspective, Metabolites. 9 (2019). 10.3390/metabo9060117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bersanelli M, Mosca E, Remondini D, Giampieri E, Sala C, Castellani G, Milanesi L, Methods for the integration of multi-omics data: Mathematical aspects, BMC Bioinformatics. 17 (2016) 15 10.1186/s12859-015-0857-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Beale DJ, Kouremenos KA, Palombo EA, Beyond Metabolomics: A Review of Multi-Omics-Based Approaches, Microb. Metabolomics Appl. Clin. Environ. Ind. Microbiol. Chapter 10 (2016) 1–321. 10.1007/978-3-319-46326-1. [DOI] [Google Scholar]
- [47].Voillet V, Besse P, Liaubet L, San Cristobal M, González I, Handling missing rows in multi-omics data integration: Multiple imputation in multiple factor analysis framework, BMC Bioinformatics. 17 (2016) 1–16. 10.1186/s12859-016-1273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Weiner M, ADNI, The ADNI initiative: review of paper published since its inception, Alzheimer Dement: 9 (2013) e111–e194. 10.1016/j.jalz.2013.05.1769.The. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nakagawa S, Freckleton RP, Missing inaction: the dangers of ignoring missing data, Trends Ecol. Evol 23 (2008) 592–596. 10.1016/j.tree.2008.06.014. [DOI] [PubMed] [Google Scholar]
- [50].van den Berg RA, Hoefsloot HCJ, Westerhuis JA, Smilde AK, van der Werf MJ, Centering, scaling, and transformations: Improving the biological information content of metabolomics data, BMC Genomics. 7 (2006) 1–15. 10.1186/1471-2164-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Misra BB, Langefeld C, Olivier M, Cox LA, Integrated omics: tools, advances and future approaches, J. Mol. Endocrinol (2018) R21–R45. 10.1530/jme-18-0055. [DOI] [PubMed] [Google Scholar]
- [52].Cavill R, Jennen D, Kleinjans J, Briedé JJ, Transcriptomic and metabolomic data integration, Brief. Bioinform 17 (2016) 891–901. 10.1093/bib/bbv090. [DOI] [PubMed] [Google Scholar]
- [53].Bellman RE, Adaptive Control Processes, Princet. Univ. Press; (1961). [Google Scholar]
- [54].Kevin Beyer US, Jonathan Goldstein, Raghu Ramakrishnan, When Is “Nearest Neighbor” Meaningful?, Int. Conf. Database Theory (1999) 217–235. [Google Scholar]
- [55].P. L., H. E., L. H., Subspace Clustering for High Dimensional Data: A Review, SIGKDD Explor. Newsl. ACM Spec. Interes. Gr. Knowl. Discov. Data Min 6 (2004) 90. [Google Scholar]
- [56].Do KT, Rasp DJNP, Kastenmüller G, Suhre K, Krumsiek J, MoDentify: Phenotype-driven module identification in metabolomics networks at different resolutions, Bioinformatics. 35 (2019) 532–534. 10.1093/bioinformatics/bty650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Krumsiek J, Mittelstrass K, Do KT, Stückler F, Ried J, Adamski J, Peters A, Illig T, Kronenberg F, Friedrich N, Nauck M, Pietzner M, Mook-Kanamori DO, Suhre K, Gieger C, Grallert H, Theis FJ, Kastenmüller G, Gender-specific pathway differences in the human serum metabolome, Metabolomics. 11 (2015) 1815–1833. 10.1007/s11306-015-0829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Do KT, Pietzner M, Rasp DJ, Friedrich N, Nauck M, Kocher T, Suhre K, Mook-Kanamori DO, Kastenmüller G, Krumsiek J, Phenotype-driven identification of modules in a hierarchical map of multifluid metabolic correlations, Npj Syst. Biol. Appl 3 (2017). 10.1038/s41540-017-0029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Krumsiek J, Bartel J, Theis FJ, Computational approaches for systems metabolomics, Curr. Opin. Biotechnol 39 (2016) 198–206. 10.1016/j.copbio.2016.04.009. [DOI] [PubMed] [Google Scholar]
- [60].Wold S, Esbensen K, Geladi P, Decret_Du_7_Mai_1993_Fixant_Les_Modalites_D_Application_De_La_Loi_Relative_Aux_Recens ements_Et_Enquetes_Statistiques.Pdf, Chemom. Intell. Lab. Syst 2 (1987) 37–52. 10.1016/0169-7439(87)80084-9. [DOI] [Google Scholar]
- [61].Wong JA, Hartigan MA, A K-Means Clustering Algorithm, J. R. Stat. Soc. Ser. C (Applied Stat. 28 (n.d.) 100–108. 10.9756/bijdm.1106. [DOI] [Google Scholar]
- [62].Johnson SC, Hierarchical clustering schemes, Psychometrika. 32 (1967) 241–254. 10.1007/BF02289588. [DOI] [PubMed] [Google Scholar]
- [63].Guyon I, Elisseeff A, An introduction to variable and feature selection, J. Mach. Learn. Res 3 (2003) 1157–1182. [Google Scholar]
- [64].Langfelder P, Horvath S, WGCNA: An R package for weighted correlation network analysis, BMC Bioinformatics. 9 (2008). 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wahl S, Vogt S, Stückler F, Krumsiek J, Bartel J, Kacprowski T, Schramm K, Carstensen M, Rathmann W, Roden M, Jourdan C, Kangas AJ, Soininen P, Ala-Korpela M, Nöthlings U, Boeing H, Theis FJ, Meisinger C, Waldenberger M, Suhre K, Homuth G, Gieger C, Kastenmüller G, Illig T, Linseisen J, Peters A, Prokisch H, Herder C, Thorand B, Grallert H, Multi-omic signature of body weight change: Results from a population-based cohort study, BMC Med. 13 (2015) 1–17. 10.1186/s12916-015-0282-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Costa RL, Boroni M, Soares MA, Distinct co-expression networks using multi-omic data reveal novel interventional targets in HPV-positive and negative head-and-neck squamous cell cancer, Sci. Rep 8 (2018) 1–13. 10.1038/s41598-018-33498-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pedersen HK, Forslund SK, Gudmundsdottir V, Petersen AØ, Hildebrand F, Hyötyläinen T, Nielsen T, Hansen T, Bork P, Ehrlich SD, Brunak S, Oresic M, Pedersen O, Nielsen HB, A computational framework to integrate high-throughput ‘-omics’ datasets for the identification of potential mechanistic links, Nat. Protoc 13 (2018) 2781–2800. 10.1038/s41596-018-0064-z. [DOI] [PubMed] [Google Scholar]
- [68].Hernández-De-Diego R, Tarazona S, Martínez-Mira C, Balzano-Nogueira L, Furió-Tarí P, Pappas GJ, Conesa A, PaintOmics 3: A web resource for the pathway analysis and visualization of multi-omics data, Nucleic Acids Res. 46 (2018) W503–W509. 10.1093/nar/gky466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Becker SA, Feist AM, Mo ML, Hannum G, Palsson B, Herrgard MJ, Creation and analysis of biochemical constraint-based models: the COBRA Toolbox v3.0, Nat. Protoc 2 (2007) 727–738. 10.1038/nprot.2007.99. [DOI] [PubMed] [Google Scholar]
- [70].Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Sander C, Stuart JM, Chang K, Creighton CJ, Davis C, Donehower L, Drummond J, Wheeler D, Ally A, Balasundaram M, Birol I, Butterfield YSN, Chu A, Chuah E, Chun HJE, Dhalla N, Guin R, Hirst M, Hirst C, Holt RA, Jones SJM, Lee D, Li HI, Marra MA, Mayo M, Moore RA, Mungall AJ, Robertson AG, Schein JE, Sipahimalani P, Tam A, Thiessen N, Varhol RJ, Beroukhim R, Bhatt AS, Brooks AN, Cherniack AD, Freeman SS, Gabriel SB, Helman E, Jung J, Meyerson M, Ojesina AI, Pedamallu CS, Saksena G, Schumacher SE, Tabak B, Zack T, Lander ES, Bristow CA, Hadjipanayis A, Haseley P, Kucherlapati R, Lee S, Lee E, Luquette LJ, Mahadeshwar HS, Pantazi A, Parfenov M, Park PJ, Protopopov A, Ren X, Santoso N, Seidman J, Seth S, Song X, Tang J, Xi R, Xu AW, Yang L, Zeng D, Auman JT, Balu S, Buda E, Fan C, Hoadley KA, Jones CD, Meng S, Mieczkowski PA, Parker JS, Perou CM, Roach J, Shi Y, Silva GO, Tan D, Veluvolu U, Waring S, Wilkerson MD, Wu J, Zhao W, Bodenheimer T, Hayes DN, Hoyle AP, Jeffreys SR, Mose LE, Simons JV, Soloway MG, Baylin SB, Berman BP, Bootwalla MS, Danilova L, Herman JG, Hinoue T, Laird PW, Rhie SK, Shen H, Triche T, Weisenberger DJ, Carter SL, Cibulskis K, Chin L, Zhang J, Sougnez C, Wang M, Getz G, Dinh H, Doddapaneni HV, Gibbs R, Gunaratne P, Han Y, Kalra D, Kovar C, Lewis L, Morgan M, Morton D, Muzny D, Reid J, Xi L, Cho J, Dicara D, Frazer S, Gehlenborg N, Heiman DI, Kim J, Lawrence MS, Lin P, Liu Y, Noble MS, Stojanov P, Voet D, Zhang H, Zou L, Stewart C, Bernard B, Bressler R, Eakin A, Iype L, Knijnenburg T, Kramer R, Kreisberg R, Leinonen K, Lin J, Liu Y, Miller M, Reynolds SM, Rovira H, Shmulevich I, Thorsson V, Yang D, Zhang W, Amin S, Wu CJ, Wu CC, Akbani R, Aldape K, Baggerly KA, Broom B, Casasent TD, Cleland J, Dodda D, Edgerton M, Han L, Herbrich SM, Ju Z, Kim H, Lerner S, Li J, Liang H, Liu W, Lorenzi PL, Lu Y, Melott J, Nguyen L, Su X, Verhaak R, Wang W, Wong A, Yang Y, Yao J, Yao R, Yoshihara K, Yuan Y, Yung AK, Zhang N, Zheng S, Ryan M, Kane DW, Aksoy BA, Ciriello G, Dresdner G, Gao J, Gross B, Jacobsen A, Kahles A, Ladanyi M, Lee W, Van Lehmann K, Miller ML, Ramirez R, Rätsch G, Reva B, Schultz N, Senbabaoglu Y, Shen R, Sinha R, Sumer SO, Sun Y, Taylor BS, Weinhold N, Fei S, Spellman P, Benz C, Carlin D, Cline M, Craft B, Goldman M, Haussler D, Ma S, Ng S, Paull E, Radenbaugh A, Salama S, Sokolov A, Swatloski T, Uzunangelov V, Waltman P, Yau C, Zhu J, Hamilton SR, Abbott S, Abbott R, Dees ND, Delehaunty K, Ding L, Dooling DJ, Eldred JM, Fronick CC, Fulton R, Fulton LL, Kalicki-Veizer J, Kanchi KL, Kandoth C, Koboldt DC, Larson DE, Ley TJ, Lin L, Lu C, Magrini VJ, Mardis ER, McLellan MD, McMichael JF, Miller CA, O’Laughlin M, Pohl C, Schmidt H, Smith SM, Walker J, Wallis JW, Wendl MC, Wilson RK, Wylie T, Zhang Q, Burton R, Jensen MA, Kahn A, Pihl T, Pot D, Wan Y, Levine DA, Black AD, Bowen J, Frick J, Gastier-Foster JM, Harper HA, Helsel C, Leraas KM, Lichtenberg TM, McAllister C, Ramirez NC, Sharpe S, Wise L, Zmuda E, Chanock SJ, Davidsen T, Demchok JA, Eley G, Felau I, Sheth M, Sofia H, Staudt L, Tarnuzzer R, Wang Z, Yang L, Zhang J, Omberg L, Margolin A, Raphael BJ, Vandin F, Wu HT, Leiserson MDM, Benz SC, Vaske CJ, Noushmehr H, Wolf D, Veer LVT, Anastassiou D, Yang THO, Lopez-Bigas N, Gonzalez-Perez A, Tamborero D, Xia Z, Li W, Cho DY, Przytycka T, Hamilton M, McGuire S, Nelander S, Johansson P, Jörnsten R, Kling T, The cancer genome atlas pan-cancer analysis project, Nat. Genet 45 (2013) 1113–1120. 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, Von Mering C, STRING v10: Protein-protein interaction networks, integrated over the tree of life, Nucleic Acids Res. 43 (2015) D447–D452. 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O’Donovan C, Phan I, Pilbout S, Schneider M, The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003, Nucleic Acids Res. 31 (2003) 365–370. 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Von Mering C, STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets, Nucleic Acids Res. 47 (2019) D607–D613. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sud M, Fahy E, Cotter D, Brown A, Dennis EA, Glass CK, Merrill AH, Murphy RC, Raetz CRH, Russell DW, Subramaniam S, LMSD: LIPID MAPS structure database, Nucleic Acids Res. 35 (2007) 527–532. 10.1093/nar/gkl838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kanehisa M, Toward understanding the origin and evolution of cellular organisms, Protein Sci. 28 (2019) 1947–1951. 10.1002/pro.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kanehisa M, Goto S, KEGG: Kyoto Encyclopedia of Genes and Genomes, Nucleic Acids Res. 28 (2000) 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M, New approach for understanding genome variations in KEGG, Nucleic Acids Res. 47 (2019) D590–D595. 10.1093/nar/gky962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Joshi-Tope G, Gillespie M, Vastrik I, D’Eustachio P, Schmidt E, de Bono B, Jassal B, Gopinath GR, Wu GR, Matthews L, Lewis S, Birney E, Stein L, Reactome: A knowledgebase of biological pathways, Nucleic Acids Res. 33 (2005) 428–432. 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, Milacic M, Roca CD, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Viteri G, Weiser J, Wu G, Stein L, Hermjakob H, D’Eustachio P, The Reactome Pathway Knowledgebase, Nucleic Acids Res. 46 (2018) D649–D655. 10.1093/nar/gkx1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Thiele I, Swainston N, Fleming RMT, Hoppe A, Sahoo S, Aurich MK, Haraldsdottir H, Mo ML, Rolfsson O, Stobbe MD, Thorleifsson SG, Agren R, Bölling C, Bordel S, Chavali AK, Dobson P, Dunn WB, Endler L, Hala D, Hucka M, Hull D, Jameson D, Jamshidi N, Jonsson JJ, Juty N, Keating S, Nookaew I, Le Novère N, Malys N, Mazein A, Papin JA, Price ND, Selkov E, Sigurdsson MI, Simeonidis E, Sonnenschein N, Smallbone K, Sorokin A, Van Beek JHGM, Weichart D, Goryanin I, Nielsen J, Westerhoff HV, Kell DB, Mendes P, Palsson BO, A community-driven global reconstruction of human metabolism, Nat. Biotechnol 31 (2013) 419–425. 10.1038/nbt.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R, Palsson B, Global reconstruction of the human metabolic network based on genomic and bibliomic data, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 1777–1782. 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Brunk E, Sahoo S, Zielinski DC, Altunkaya A, Dräger A, Mih N, Gatto F, Nilsson A, Preciat Gonzalez GA, Aurich MK, Prlic A, Sastry A, Danielsdottir AD, Heinken A, Noronha A, Rose PW, Burley SK, Fleming RMT, Nielsen J, Thiele I, Palsson BO, Recon3D enables a three-dimensional view of gene variation in human metabolism, Nat. Biotechnol 36 (2018) 272–281. 10.1038/nbt.4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Durinck S, Spellman PT, Birney E, Huber W, Mapping identifiers for the integration of genomic datasets with the R/ Bioconductor package biomaRt, Nat. Protoc 4 (2009) 1184–1191. 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, Xia J, MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis, Nucleic Acids Res. 46 (2018) W486–W494. 10.1093/nar/gky310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chong J, Xia J, MetaboAnalystR: an R package for flexible and reproducible analysis of metabolomics data, Bioinformatics. 34 (2018) 4313–4314. 10.1093/bioinformatics/bty528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Pham N, van Heck RGA, van Dam JCJ, Schaap PJ, Saccenti E, Suarez-Diez M, Consistency, inconsistency, and ambiguity of metabolite names in biochemical databases used for genome-scale metabolic modelling, Metabolites. 9 (2019). 10.3390/metabo9020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Quell JD, Römisch-Margl W, Haid M, Krumsiek J, Skurk T, Halama A, Stephan N, Adamski J, Hauner H, Mook-Kanamori D, Mohney RP, Daniel H, Suhre K, Kastenmüller G, Characterization of bulk phosphatidylcholine compositions in human Plasma using Side-Chain resolving lipidomics, Metabolites. 9 (2019). 10.3390/metabo9060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Koelmel JP, Ulmer CZ, Jones CM, Yost RA, Bowden JA, Common cases of improper lipid annotation using high-resolution tandem mass spectrometry data and corresponding limitations in biological interpretation, Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1862 (2017) 766–770. 10.1016/j.bbalip.2017.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Xia J, Wishart DS, MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data, Nucleic Acids Res. 38 (2010) 71–77. 10.1093/nar/gkq329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Fisher RA, Statistical methods for research workers., in: Break. Stat., Springer, New York, NY, 1992: pp. 66–70. [Google Scholar]
- [91].Stouffer SA, Suchman EA, DeVinney LC, Star SA, Williams RM Jr, The american soldier: Adjustment during army life.(studies in social psychology in world war ii), vol. 1, (1949). [Google Scholar]
- [92].Lipták T, On the combination of independent tests, Magy. Tud Akad Mat Kut. Int Kozl 3 (1958) 171–197. [Google Scholar]
- [93].Kamburov A, Cavill R, Ebbels TMD, Herwig R, Keun HC, Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA, Bioinformatics. 27 (2011) 2917–2918. 10.1093/bioinformatics/btr499. [DOI] [PubMed] [Google Scholar]
- [94].Xia J, Sinelnikov IV, Han B, Wishart DS, MetaboAnalyst 3.0-making metabolomics more meaningful, Nucleic Acids Res. 43 (2015) W251–W257. 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles, Proc. Natl. Acad. Sci. U. S. A 102 (2005) 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Molenaar MR, Jeucken A, Wassenaar TA, Van De Lest CHA, Brouwers JF, Helms JB, LION/web: A web-based ontology enrichment tool for lipidomic data analysis, Gigascience. 8 (2019) 1–10. 10.1093/gigascience/giz061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].List M, Alcaraz N, Dissing-Hansen M, Ditzel HJ, Mollenhauer J, Baumbach J, KeyPathwayMinerWeb: online multi-omics network enrichment, Nucleic Acids Res. 44 (2016) W98–W104. 10.1093/nar/gkw373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Alcaraz N, Pauling J, Batra R, Barbosa E, Junge A, Christensen AGL, Azevedo V, Ditzel HJ, Baumbach J, KeyPathwayMiner 4.0: Condition-specific pathway analysis by combining multiple omics studies and networks with Cytoscape, BMC Syst. Biol 8 (2014) 4–9. 10.1186/s12918-014-0099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Batra R, Alcaraz N, Gitzhofer K, Pauling J, Ditzel HJ, Hellmuth M, Baumbach J, List M, On the performance of de novo pathway enrichment, Npj Syst. Biol. Appl 3 (2017) 1–7. 10.1038/s41540-017-0007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Soerensen M, Hozakowska-Roszkowska DM, Nygaard M, Larsen MJ, Schwämmle V, Christensen K, Christiansen L, Tan Q, A Genome-Wide Integrative Association Study of DNA Methylation and Gene Expression Data and Later Life Cognitive Functioning in Monozygotic Twins, Front. Neurosci 14 (2020). 10.3389/fnins.2020.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Stalidzans E, Zanin M, Tieri P, Castiglione F, Polster A, Mechanistic Modeling and Multiscale Applications for Precision Medicine : Theory and Practice, 3 (2020) 36–56. 10.1089/nsm.2020.0002. [DOI] [Google Scholar]
- [102].Orth JD, Thiele I, Palsson BO, What is flux balance analysis?, Nat. Biotechnol 28 (2010) 245–248. 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Thiele I, Palsson B, A protocol for generating a high-quality genome-scale metabolic reconstruction, Nat. Protoc 5 (2010) 93–121. 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Heirendt L, Arreckx S, Pfau T, Mendoza SN, Richelle A, Heinken A, Haraldsdóttir HS, Wachowiak J, Keating SM, Vlasov V, Magnusdóttir S, Ng CY, Preciat G, Žagare A, Chan SHJ, Aurich MK, Clancy CM, Modamio J, Sauls JT, Noronha A, Bordbar A, Cousins B, El Assal DC, Valcarcel LV, Apaolaza I, Ghaderi S, Ahookhosh M, Ben Guebila M, Kostromins A, Sompairac N, Le HM, Ma D, Sun Y, Wang L, Yurkovich JT, Oliveira MAP, Vuong PT, El Assal LP, Kuperstein I, Zinovyev A, Hinton HS, Bryant WA, Aragón Artacho FJ, Planes FJ, Stalidzans E, Maass A, Vempala S, Hucka M, Saunders MA, Maranas CD, Lewis NE, Sauter T, Palsson B, Thiele I, Fleming RMT, Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0, Nat. Protoc 14 (2019) 639–702. 10.1038/s41596-018-0098-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Bordbar A, Monk JM, King ZA, Palsson BO, Constraint-based models predict metabolic and associated cellular functions, Nat. Rev. Genet 15 (2014) 107–120. 10.1038/nrg3643. [DOI] [PubMed] [Google Scholar]
- [106].Patil KR, Nielsen J, Uncovering transcriptional regulation of metabolism by using metabolic network topology, Proc. Natl. Acad. Sci. U. S. A 102 (2005) 2685–2689. 10.1073/pnas.0406811102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Cho JS, Gu C, Han TH, Ryu JY, Lee SY, Reconstruction of context-specific genome-scale metabolic models using multiomics data to study metabolic rewiring, Curr. Opin. Syst. Biol 15 (2019) 1–11. 10.1016/j.coisb.2019.02.009. [DOI] [Google Scholar]
- [108].Reed JL, Shrinking the Metabolic Solution Space Using Experimental Datasets, PLoS Comput. Biol 8 (2012) 1–5. 10.1371/journal.pcbi.1002662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Vlassis N, Pacheco MP, Sauter T, Fast Reconstruction of Compact Context-Specific Metabolic Network Models, PLoS Comput. Biol 10 (2014). 10.1371/journal.pcbi.1003424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Bordbar A, Yurkovich JT, Paglia G, Rolfsson O, Sigurjónsson ÓE, Palsson BO, Elucidating dynamic metabolic physiology through network integration of quantitative time-course metabolomics, Sci. Rep 7 (2017) 1–12. 10.1038/srep46249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Zur H, Ruppin E, Shlomi T, iMAT: An integrative metabolic analysis tool, Bioinformatics. 26 (2010) 3140–3142. 10.1093/bioinformatics/btq602. [DOI] [PubMed] [Google Scholar]
- [112].Becker SA, Palsson BO, Context-specific metabolic networks are consistent with experiments, PLoS Comput. Biol 4 (2008). 10.1371/journal.pcbi.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Saha R, Chowdhury A, Maranas CD, Recent advances in the reconstruction of metabolic models and integration of omics data, Curr. Opin. Biotechnol 29 (2014) 39–45. 10.1016/j.copbio.2014.02.011. [DOI] [PubMed] [Google Scholar]
- [114].Agren R, Mardinoglu A, Asplund A, Kampf C, Uhlen M, Nielsen J, Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling, Mol. Syst. Biol 10 (2014) 1–13. 10.1002/msb.145122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Thiele I, Sahoo S, Heinken A, Heirendt L, Aurich MK, Noronha A, Fleming RMT, When metabolism meets physiology: Harvey and Harvetta, BioRxiv. (2018) 255885 10.1101/255885. [DOI] [Google Scholar]
- [116].Bin Shen H, Chou KC, Ensemble classifier for protein fold pattern recognition, Bioinformatics. 22 (2006) 1717–1722. 10.1093/bioinformatics/btl170. [DOI] [PubMed] [Google Scholar]
- [117].Rokach L, Ensemble-based classifiers, Artif. Intell. Rev 33 (2010) 1–39. 10.1007/s10462-009-9124-7. [DOI] [Google Scholar]
- [118].Fawcett C, Hoos HH, Analysing differences between algorithm configurations through ablation, J. Heuristics 22 (2016) 431–458. 10.1007/s10732-014-9275-9. [DOI] [Google Scholar]
- [119].Miles CM, Wayne M, Quantitative trait locus (QTL) analysis, Nat. Educ 1 208 (2008). [Google Scholar]
- [120].Hirschhorn JN, Daly MJ, Genome-wide association studies for common diseases and complex traits, Nat. Rev. Genet 6 (2005) 95–108. 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- [121].McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JPA, Hirschhorn JN, Genome-wide association studies for complex traits: Consensus, uncertainty and challenges, Nat. Rev. Genet 9 (2008) 356–369. 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- [122].Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D, Benefits and limitations of genome-wide association studies, Nat. Rev. Genet 20 (2019) 467–484. 10.1038/s41576-019-0127-1. [DOI] [PubMed] [Google Scholar]
- [123].Genotype T, Expression T, The GTEx Consortium atlas of genetic regulatory effects across human tissues The Genotype Tissue Expression Consortium, (2019). 10.1101/787903. [DOI] [Google Scholar]
- [124].Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, Gillard B, Ramsey K, Sullivan S, Bridge J, Magazine H, Syron J, Fleming J, Siminoff L, Traino H, Mosavel M, Barker L, Jewell S, Rohrer D, Maxim D, Filkins D, Harbach P, Cortadillo E, Berghuis B, Turner L, Hudson E, Feenstra K, Sobin L, Robb J, Branton P, Korzeniewski G, Shive C, Tabor D, Qi L, Groch K, Nampally S, Buia S, Zimmerman A, Smith A, Burges R, Robinson K, Valentino K, Bradbury D, Cosentino M, Diaz-Mayoral N, Kennedy M, Engel T, Williams P, Erickson K, Ardlie K, Winckler W, Getz G, DeLuca D, Daniel MacArthur M Thomson Kellis, A., Young T, Gelfand E, Donovan M, Meng Y, Grant G, Mash D, Marcus Y, Basile M, Liu J, Zhu J, Tu Z, Cox NJ, Nicolae DL, Gamazon ER, Im HK, Konkashbaev A, Pritchard J, Stevens M, Flutre T, Wen X, Dermitzakis ET, Lappalainen T, Guigo R, Monlong J, Sammeth M, Koller D, Battle A, Mostafavi S, McCarthy M, Rivas M, Maller J, Rusyn I, Nobel A, Wright F, Shabalin A, Feolo M, Sharopova N, Sturcke A, Paschal J, Anderson JM, Wilder EL, Derr LK, Green ED, Struewing JP, Temple G, Volpi S, Boyer JT, Thomson EJ, Guyer MS, Ng C, Abdallah A, Colantuoni D, Insel TR, Koester SE, Roger Little A, Bender PK, Lehner T, Yao Y, Compton CC, Vaught JB, Sawyer S, Lockhart NC, Demchok J, Moore HF, The Genotype-Tissue Expression (GTEx) project, Nat. Genet 45 (2013) 580–585. 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, Burgess S, Jiang T, Paige E, Surendran P, Oliver-Williams C, Kamat MA, Prins BP, Wilcox SK, Zimmerman ES, Chi A, Bansal N, Spain SL, Wood AM, Morrell NW, Bradley JR, Janjic N, Roberts DJ, Ouwehand WH, Todd JA, Soranzo N, Suhre K, Paul DS, Fox CS, Plenge RM, Danesh J, Runz H, Butterworth AS, Genomic atlas of the human plasma proteome, Nature. 558 (2018) 73–79. 10.1038/s41586-018-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Suhre K, Arnold M, Bhagwat AM, Cotton RJ, Engelke R, Raffler J, Sarwath H, Thareja G, Wahl A, Delisle RK, Gold L, Pezer M, Lauc G, Selim MAED, Mook-Kanamori DO, Al-Dous EK, Mohamoud YA, Malek J, Strauch K, Grallert H, Peters A, Kastenmüller G, Gieger C, Graumann J, Connecting genetic risk to disease end points through the human blood plasma proteome, Nat. Commun 8 (2017). 10.1038/ncomms14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Sabatti C, Service SK, Hartikainen AL, Pouta A, Ripatti S, Brodsky J, Jones CG, Zaitlen NA, Varilo T, Kaakinen M, Sovio U, Ruokonen A, Laitinen J, Jakkula E, Coin L, Hoggart C, Collins A, Turunen H, Gabriel S, Elliot P, McCarthy MI, Daly MJ, Järvelin MR, Freimer NB, Peltonen L, Genome-wide association analysis of metabolic traits in a birth cohort from a founder population, Nat. Genet 41 (2009) 35–46. 10.1038/ng.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Arnold M, Raffler J, Pfeufer A, Suhre K, Kastenmü Ller G, SNiPA: an interactive, genetic variant-centered annotation browser, (n.d.) 10.1093/bioinformatics/btu779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Hormozdiari F, van de Bunt M, Segrè AV, Li X, Joo JWJ, Bilow M, Sul JH, Sankararaman S, Pasaniuc B, Eskin E, Colocalization of GWAS and eQTL Signals Detects Target Genes, Am. J. Hum. Genet 99 (2016) 1245–1260. 10.1016/j.ajhg.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V, Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics, PLoS Genet. 10 (2014). 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Bonder MJ, Luijk R, Zhernakova DV, Moed M, Deelen P, Vermaat M, Van Iterson M, Van Dijk F, Van Galen M, Bot J, Slieker RC, Jhamai PM, Verbiest M, Suchiman HED, Verkerk M, Van Der Breggen R, Van Rooij J, Lakenberg N, Arindrarto W, Kielbasa SM, Jonkers I, Van’t Hof P, Nooren I, Beekman M, Deelen J, Van Heemst D, Zhernakova A, Tigchelaar EF, Swertz MA, Hofman A, Uitterlinden AG, Pool R, Van Dongen J, Hottenga JJ, Stehouwer CDA, Van Der Kallen CJH, Schalkwijk CG, Van Den Berg LH, Van Zwet EW, Mei H, Li Y, Lemire M, Hudson TJ, Slagboom PE, Wijmenga C, Veldink JH, Van Greevenbroek MMJ, Van Duijn CM, Boomsma DI, Isaacs A, Jansen R, Van Meurs JBJ, Hoen’t PAC, Franke L, Heijmans BT, Disease variants alter transcription factor levels and methylation of their binding sites, Nat. Genet 49 (2017) 131–138. 10.1038/ng.3721. [DOI] [PubMed] [Google Scholar]
- [132].Chen L, Ge B, Casale FP, Vasquez L, Kwan T, Garrido-Martín D, Watt S, Yan Y, Kundu K, Ecker S, Datta A, Richardson D, Burden F, Mead D, Mann AL, Fernandez JM, Rowlston S, Wilder SP, Farrow S, Shao X, Lambourne JJ, Redensek A, Albers CA, Amstislavskiy V, Ashford S, Berentsen K, Bomba L, Bourque G, Bujold D, Busche S, Caron M, Chen SH, Cheung W, Delaneau O, Dermitzakis ET, Elding H, Colgiu I, Bagger FO, Flicek P, Habibi E, Iotchkova V, Janssen-Megens E, Kim B, Lehrach H, Lowy E, Mandoli A, Matarese F, Maurano MT, Morris JA, Pancaldi V, Pourfarzad F, Rehnstrom K, Rendon A, Risch T, Sharifi N, Simon MM, Sultan M, Valencia A, Walter K, Wang SY, Frontini M, Antonarakis SE, Clarke L, Yaspo ML, Beck S, Guigo R, Rico D, Martens JHA, Ouwehand WH, Kuijpers TW, Paul DS, Stunnenberg HG, Stegle O, Downes K, Pastinen T, Soranzo N, Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells, 2016. 10.1016/j.cell.2016.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL, Alzheimer’s disease, Nat. Rev. Dis. Prim 1 (2015) 1–18. 10.1016/j.med.2019.03.012. [DOI] [PubMed] [Google Scholar]
- [134].Evangelou E, Ioannidis JPA, Meta-analysis methods for genome-wide association studies and beyond, Nat. Rev. Genet 14 (2013) 379–389. 10.1038/nrg3472. [DOI] [PubMed] [Google Scholar]
- [135].Rubin DB, Multiple imputation for nonresponse in surveys, John Wiley & Sons, 2004. [Google Scholar]
- [136].Abdi H, Williams LJ, Valentin D, Bennani-Dosse M, STATIS and DISTATIS: Optimum multitable principal component analysis and three way metric multidimensional scaling, Wiley Interdiscip. Rev. Comput. Stat 4 (2012) 124–167. 10.1002/wics.198. [DOI] [Google Scholar]
- [137].Breiman L, Random forests, Mach. Learn (2001) 5–32. 10.1201/9780367816377-11. [DOI] [Google Scholar]
- [138].Tishbirani R, Regression shrinkage and selection via the Lasso, J. R. Stat. Soc. Ser. B 58 (1996) 267–288. https://statweb.stanford.edu/~tibs/lasso/lasso.pdf. [Google Scholar]
- [139].Nguyen DV, Rocke DM, Tumor classification by partial least squares using microarray gene expression data, Bioinformatics. 18 (2002) 39–50. 10.1093/bioinformatics/18.1.39. [DOI] [PubMed] [Google Scholar]
- [140].Boulesteix A-L, PLS Dimension Reduction for Classification with Microarray Data, Stat. Appl. Genet. Mol. Biol 3 (2005) 1–30. 10.2202/1544-6115.1075. [DOI] [PubMed] [Google Scholar]
- [141].Schäfer J, Strimmer K, An empirical Bayes approach to inferring large-scale gene association networks, Bioinformatics. 21 (2005) 754–764. 10.1093/bioinformatics/bti062. [DOI] [PubMed] [Google Scholar]
- [142].Krumsiek J, Suhre K, Illig T, Adamski J, Theis FJ, Gaussian graphical modeling reconstructs pathway reactions from high-throughput metabolomics data, BMC Syst. Biol 5 (2011) 21 10.1186/1752-0509-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Altenbuchinger M, Weihs A, Quackenbush J, Grabe HJ, Zacharias HU, Gaussian and Mixed Graphical Models as (multi-)omics data analysis tools, Biochim. Biophys. Acta - Gene Regul. Mech 1863 (2020) 194418 10.1016/j.bbagrm.2019.194418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Lauritzen SL, Graphical models, Clarendon Press, 1996. [Google Scholar]
- [145].Mittelstrass K, Ried JS, Yu Z, Krumsiek J, Gieger C, Prehn C, Roemisch-Margl W, Polonikov A, Peters A, Theis FJ, Meitinger T, Kronenberg F, Weidinger S, Wichmann HE, Suhre K, Wang-Sattler R, Adamski J, Illig T, Discovery of sexual dimorphisms in metabolic and genetic biomarkers, PLoS Genet. 7 (2011). 10.1371/journal.pgen.1002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Krumsiek J, Suhre K, Evans AM, Mitchell MW, Mohney RP, V Milburn M, Wägele B, Römisch-Margl W, Illig T, Adamski J, Gieger C, Theis FJ, Kastenmüller G, Mining the Unknown: A Systems Approach to Metabolite Identification Combining Genetic and Metabolic Information, PLoS Genet. 8 (2012). 10.1371/journal.pgen.1003005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Chen S, Witten DM, Shojaie A, Selection and estimation for mixed graphical models, Biometrika. 102 (2015) 47–64. 10.1093/biomet/asu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Lee JD, Hastie TJ, Structure learning of mixed graphical models, J. Mach. Learn. Res 31 (2013) 388–396. [Google Scholar]
- [149].Fellinghauer B, Bühlmann P, Ryffel M, Von Rhein M, Reinhardt JD, Stable graphical model estimation with Random Forests for discrete, continuous, and mixed variables, Comput. Stat. Data Anal 64 (2013) 132–152. 10.1016/j.csda.2013.02.022. [DOI] [Google Scholar]
- [150].Rohart F, Gautier B, Singh A, Lê Cao KA, mixOmics: An R package for ‘omics feature selection and multiple data integration, PLoS Comput. Biol (2017). 10.1371/journal.pcbi.1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Spicker JS, Brunak S, Frederiksen KS, Toft H, Integration of clinical chemistry, expression, and metabolite data leads to better toxicological class separation, Toxicol. Sci 102 (2008) 444–454. 10.1093/toxsci/kfn001. [DOI] [PubMed] [Google Scholar]
- [152].Escofier B, Pagès J, Multiple factor analysis (AFMULT package), Comput. Stat. Data Anal 18 (1994) 121–140. 10.1016/0167-9473(94)90135-X. [DOI] [Google Scholar]
- [153].Abdi H, Williams LJ, Valentin D, Multiple factor analysis: Principal component analysis for multitable and multiblock data sets, Wiley Interdiscip. Rev. Comput. Stat 5 (2013) 149–179. 10.1002/wics.1246. [DOI] [Google Scholar]
- [154].Sun S, A survey of multi-view machine learning, Neural Comput. Appl 23 (2013) 2031–2038. 10.1007/s00521-013-1362-6. [DOI] [Google Scholar]
- [155].Trygg J, O2-PLS for qualitative and quantitative analysis in multivariate calibration, J. Chemom 16 (2002) 283–293. 10.1002/cem.724. [DOI] [Google Scholar]
- [156].Trygg J, Wold S, O2-PLS, a two-block (X-Y) latent variable regression (LVR) method with an integral OSC filter, J. Chemom 17 (2003) 53–64. 10.1002/cem.775. [DOI] [Google Scholar]
- [157].Srivastava V, Obudulu O, Bygdell J, Löfstedt T, Rydén P, Nilsson R, Ahnlund M, Johansson A, Jonsson P, Freyhult E, Qvarnström J, Karlsson J, Melzer M, Moritz T, Trygg J, Hvidsten TR, Wingsle G, OnPLS integration of transcriptomic, proteomic and metabolomic data shows multilevel oxidative stress responses in the cambium of transgenic hipI-superoxide dismutase Populus plants, BMC Genomics. 14 (2013). 10.1186/1471-2164-14-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Löfstedt T, Trygg J, OnPLS-a novel multiblock method for the modelling of predictive and orthogonal variation, J. Chemom 25 (2011) 441–455. 10.1002/cem.1388. [DOI] [Google Scholar]
- [159].Löfstedt T, Hoffman D, Trygg J, Global, local and unique decompositions in OnPLS for multiblock data analysis, Anal. Chim. Acta 791 (2013) 13–24. 10.1016/j.aca.2013.06.026. [DOI] [PubMed] [Google Scholar]
- [160].Reinke SN, Galindo-Prieto B, Skotare T, Broadhurst DI, Singhania A, Horowitz D, Djukanović R, Hinks TSC, Geladi P, Trygg J, Wheelock CE, OnPLS-Based Multi-Block Data Integration: A Multivariate Approach to Interrogating Biological Interactions in Asthma, Anal. Chem 90 (2018) 13400–13408. 10.1021/acs.analchem.8b03205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Tenenhaus A, Tenenhaus M, Regularized Generalized Canonical Correlation Analysis, Psychometrika. 76 (2011) 257–284. 10.1007/s11336-011-9206-8. [DOI] [PubMed] [Google Scholar]
- [162].Langenberg MCC, Hoogerwerf MA, Koopman JPR, Janse JJ, Kos-van Oosterhoud J, Feijt C, Jochems SP, de Dood CJ, van Schuijlenburg R, Ozir-Fazalalikhan A, Manurung MD, Sartono E, van der Beek MT, Winkel BMF, Verbeek-Menken PH, Stam KA, van Leeuwen FWB, Meij P, van Diepen A, van Lieshout L, van Dam GJ, Corstjens PLAM, Hokke CH, Yazdanbakhsh M, Visser LG, Roestenberg M, A controlled human Schistosoma mansoni infection model to advance novel drugs, vaccines and diagnostics, Nat. Med 26 (2020) 326–332. 10.1038/s41591-020-0759-x. [DOI] [PubMed] [Google Scholar]
- [163].Cano-Sancho G, Alexandre-Gouabau MC, Moyon T, Royer AL, Guitton Y, Billard H, Darmaun D, Rozé JC, Boquien CY, Le Bizec B, Antignac JP, Simultaneous exploration of nutrients and pollutants in human milk and their impact on preterm infant growth: An integrative cross-platform approach, Environ. Res 182 (2020) 109018 10.1016/j.envres.2019.109018. [DOI] [PubMed] [Google Scholar]
- [164].Pekmez CT, Larsson MW, Lind MV, Vazquez Manjarrez N, Yonemitsu C, Larnkjær A, Bode L, Mølgaard C, Michaelsen KF, Dragsted LO, Breastmilk Lipids and Oligosaccharides Influence Branched Short-Chain Fatty Acid Concentrations in Infants with Excessive Weight Gain, Mol. Nutr. Food Res 64 (2020) 1–10. 10.1002/mnfr.201900977. [DOI] [PubMed] [Google Scholar]
- [165].Vidal M, Cusick ME, Barabási AL, Interactome networks and human disease, Cell. 144 (2011) 986–998. 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Sügis E, Dauvillier J, Leontjeva A, Adler P, Hindie V, Moncion T, Collura V, Daudin R, Loe-Mie Y, Herault Y, Lambert J-C, Hermjakob H, Pupko T, Rain J-C, Xenarios I, Vilo J, Simonneau M, Peterson H, HENA, heterogeneous network-based data set for Alzheimer’s disease, Sci. Data 6 (2019) 151 10.1038/s41597-019-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Kamburov A, Wierling C, Lehrach H, Herwig R, ConsensusPathDB - A database for integrating human functional interaction networks, Nucleic Acids Res. 37 (2009) 623–628. 10.1093/nar/gkn698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Herwig R, Hardt C, Lienhard M, Kamburov A, Analyzing and interpreting genome data at the network level with ConsensusPathDB, Nat. Protoc 11 (2016) 1889–1907. 10.1038/nprot.2016.117. [DOI] [PubMed] [Google Scholar]
- [169].Zhou G, Xia J, Using OmicsNet for Network Integration and 3D Visualization, Curr. Protoc. Bioinforma 65 (2019) 1–26. 10.1002/cpbi.69. [DOI] [PubMed] [Google Scholar]
- [170].Zhou G, Xia J, OmicsNet: A web-based tool for creation and visual analysis of biological networks in 3D space, Nucleic Acids Res. 46 (2018) W514–W522. 10.1093/nar/gky510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Haas R, Zelezniak A, Iacovacci J, Kamrad S, Townsend SJ, Ralser M, Designing and interpreting “multi-omic” experiments that may change our understanding of biology, Curr. Opin. Syst. Biol 6 (2017) 37–45. 10.1016/j.coisb.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Merico D, Gfeller D, Bader GD, How to visually interpret biological data using networks, Nat. Biotechnol 27 (2009) 921–924. 10.1038/nbt.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Yoghourdjian V, Archambault D, Diehl S, Dwyer T, Klein K, Purchase HC, Wu HY, Exploring the limits of complexity: A survey of empirical studies on graph visualisation, Vis. Informatics 2 (2018) 264–282. 10.1016/j.visinf.2018.12.006. [DOI] [Google Scholar]
- [174].Yoghourdjian V, Dwyer T, Klein K, Marriott K, Wybrow M, Graph Thumbnails: Identifying and Comparing Multiple Graphs at a Glance, IEEE Trans. Vis. Comput. Graph 24 (2018) 3081–3095. 10.1109/TVCG.2018.2790961. [DOI] [PubMed] [Google Scholar]
- [175].Krzywinski M, Birol I, Jones SJ, Marra MA, Hive plots-rational approach to visualizing networks, Brief. Bioinform 13 (2012) 627–644. 10.1093/bib/bbr069. [DOI] [PubMed] [Google Scholar]
- [176].McGee F, Ghoniem M, Melançon G, Otjacques B, Pinaud B, The State of the Art in Multilayer Network Visualization, Comput. Graph. Forum 38 (2019) 125–149. 10.1111/cgf.13610. [DOI] [Google Scholar]
- [177].Barabási AL, Oltvai ZN, Network biology: Understanding the cell’s functional organization, Nat. Rev. Genet 5 (2004) 101–113. 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- [178].Boccaletti S, Bianconi G, Criado R, del Genio CI, Gómez-Gardeñes J, Romance M, Sendiña-Nadal I, Wang Z, Zanin M, The structure and dynamics of multilayer networks, Phys. Rep 544 (2014) 1–122. 10.1016/j.physrep.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Kivelä M, Arenas A, Barthelemy M, Gleeson JP, Moreno Y, Porter MA, Multilayer networks, J. Complex Networks 2 (2014) 203–271. 10.1093/comnet/cnu016. [DOI] [Google Scholar]
- [180].Zachariou M, Minadakis G, Oulas A, Afxenti S, Spyrou GM, Integrating multi-source information on a single network to detect disease-related clusters of molecular mechanisms, J. Proteomics 188 (2018) 15–29. 10.1016/j.jprot.2018.03.009. [DOI] [PubMed] [Google Scholar]
- [181].Himmelstein DS, Baranzini SE, Heterogeneous Network Edge Prediction: A Data Integration Approach to Prioritize Disease-Associated Genes, PLoS Comput. Biol 11 (2015) 1–27. 10.1371/journal.pcbi.1004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].De Domenico M, Solé-Ribalta A, Omodei E, Gómez S, Arenas A, Ranking in interconnected multilayer networks reveals versatile nodes, Nat. Commun 6 (2015) 1–6. 10.1038/ncomms7868. [DOI] [PubMed] [Google Scholar]
- [183].Halu A, Mondragón RJ, Panzarasa P, Bianconi G, Multiplex PageRank, PLoS One. 8 (2013) 1–10. 10.1371/journal.pone.0078293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Edler D, Bohlin L, Rosvall M, Mapping higher-order network flows in memory and multilayer networks with infomap, Algorithms. 10 (2017) 1–23. 10.3390/a10040112. [DOI] [Google Scholar]
- [185].De Domenico M, Lancichinetti A, Arenas A, Rosvall M, Identifying modular flows on multilayer networks reveals highly overlapping organization in interconnected systems, Phys. Rev. X 5 (2015) 1–14. 10.1103/PhysRevX.5.011027. [DOI] [Google Scholar]
- [186].Teran Hidalgo SJ, Ma S, Clustering multilayer omics data using MuNCut, BMC Genomics. 19 (2018) 1–13. 10.1186/s12864-018-4580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Lysenko A, RoznovǍţ IA, Saqi M, Mazein A, Rawlings CJ, Auffray C, Representing and querying disease networks using graph databases, BioData Min. 9 (2016) 23 10.1186/s13040-016-0102-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Perez-Riverol Y, Bai M, Da Veiga Leprevost F, Squizzato S, Park YM, Haug K, Carroll AJ, Spalding D, Paschall J, Wang M, Del-Toro N, Ternent T, Zhang P, Buso N, Bandeira N, Deutsch EW, Campbell DS, Beavis RC, Salek RM, Sarkans U, Petryszak R, Keays M, Fahy E, Sud M, Subramaniam S, Barbera A, Jiménez RC, Nesvizhskii AI, Sansone SA, Steinbeck C, Lopez R, Vizcaíno JA, Ping P, Hermjakob H, Discovering and linking public omics data sets using the Omics Discovery Index, Nat. Biotechnol 35 (2017) 406–409. 10.1038/nbt.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Perez-Riverol Y, Zorin A, Dass G, Vu MT, Xu P, Glont M, Vizcaíno JA, Jarnuczak AF, Petryszak R, Ping P, Hermjakob H, Quantifying the impact of public omics data, Nat. Commun 10 (2019). 10.1038/s41467-019-11461-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Pratt D, Chen J, Welker D, Rivas R, Pillich R, Rynkov V, Ono K, Miello C, Hicks L, Szalma S, Stojmirovic A, Dobrin R, Braxenthaler M, Kuentzer J, Demchak B, Ideker T, NDEx, the Network Data Exchange, Cell Syst. 1 (2015) 302–305. 10.1016/j.cels.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Eraslan G, Avsec Ž, Gagneur J, Theis FJ, Deep learning: new computational modelling techniques for genomics, Nat. Rev. Genet (2019). 10.1038/s41576-019-0122-6. [DOI] [PubMed] [Google Scholar]
- [192].Ching T, Himmelstein DS, Beaulieu-Jones BK, Kalinin AA, Do BT, Way GP, Ferrero E, Agapow PM, Zietz M, Hoffman MM, Xie W, Rosen GL, Lengerich BJ, Israeli J, Lanchantin J, Woloszynek S, Carpenter AE, Shrikumar A, Xu J, Cofer EM, Lavender CA, Turaga SC, Alexandari AM, Lu Z, Harris DJ, Decaprio D, Qi Y, Kundaje A, Peng Y, Wiley LK, Segler MHS, Boca SM, Swamidass SJ, Huang A, Gitter A, Greene CS, Opportunities and obstacles for deep learning in biology and medicine, 2018. 10.1098/rsif.2017.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Zhang Z, Zhao Y, Liao X, Shi W, Li K, Zou Q, Peng S, Deep learning in omics: a survey and guideline, Brief. Funct. Genomics 18 (2018) 41–57. 10.1093/bfgp/ely030. [DOI] [PubMed] [Google Scholar]
- [194].Camacho DM, Collins KM, Powers RK, Costello JC, Collins JJ, Next-Generation Machine Learning for Biological Networks, Cell. 173 (2018) 1581–1592. 10.1016/j.cell.2018.05.015. [DOI] [PubMed] [Google Scholar]
- [195].Hinton GE, Salakhutdinov RR, Reducing the dimensionality of data with neural networks, Science (80-.) 313 (2006) 504–507. 10.1126/science.1127647. [DOI] [PubMed] [Google Scholar]
- [196].Gomes T, Teichmann SA, Talavera-López C, Immunology Driven by Large-Scale Single-Cell Sequencing, Trends Immunol. 40 (2019) 1011–1021. 10.1016/j.it.2019.09.004. [DOI] [PubMed] [Google Scholar]
- [197].Chung NC, Mirza B, Choi H, Wang J, Wang D, Ping P, Wang W, Unsupervised classification of multi-omics data during cardiac remodeling using deep learning, Methods. 166 (2019) 66–73. 10.1016/j.ymeth.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Zhang X, Zhang J, Sun K, Yang X, Dai C, Guo Y, Integrated Multi-omics Analysis Using Variational Autoencoders: Application to Pan-cancer Classification, Proc. - 2019 IEEE Int. Conf. Bioinforma. Biomed. BIBM 2019 (2019) 765–769. 10.1109/BIBM47256.2019.8983228. [DOI] [Google Scholar]
- [199].Webb S, Deep learning for biology, Nat. 2018 5547693. 554 (2018) 555–557. 10.1038/d41586-018-02174-z. [DOI] [PubMed] [Google Scholar]
- [200].Webb S, Deep learning for biology, Nature. 554 (2018) 555–557. 10.1038/d41586-018-02174-z. [DOI] [PubMed] [Google Scholar]
- [201].Xiao Z, Locasale JW, Dai Z, Metabolism in the tumor microenvironment: insights from single-cell analysis, Oncoimmunology. 9 (2020). 10.1080/2162402X.2020.1726556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Alexandrov T, Spatial Metabolomics and Imaging Mass Spectrometry in the Age of Artificial Intelligence, Annu. Rev. Biomed. Data Sci (2020). 10.1146/annurev-biodatasci-011420-031537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Rappez L, Stadler M, Triana S, Phapale P, Heikenwalder M, Alexandrov T, Spatial single-cell profiling of intracellular metabolomes in situ, BioRxiv. (2019) 510222 10.1101/510222. [DOI] [Google Scholar]
- [204].Palii CG, Cheng Q, Gillespie MA, Shannon P, Mazurczyk M, Napolitani G, Price ND, Ranish JA, Morrissey E, Higgs DR, Brand M, Single-Cell Proteomics Reveal that Quantitative Changes in Co-expressed Lineage-Specific Transcription Factors Determine Cell Fate, Cell Stem Cell. 24 (2019) 812–820.e5. 10.1016/j.stem.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Burgess DJ, Spatial transcriptomics coming of age, Nat. Rev. Genet 20 (2019) 317 10.1038/s41576-019-0129-z. [DOI] [PubMed] [Google Scholar]
- [206].Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Subhraveti P, Weaver DS, Karp PD, The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases, Nucleic Acids Res. 44 (2016) D471–D480. 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Rodchenkov I, Babur O, Luna A, Aksoy BA, Wong JV, Fong D, Franz M, Siper MC, Cheung M, Wrana M, Mistry H, Mosier L, Dlin J, Wen Q, O’Callaghan C, Li W, Elder G, Smith PT, Dallago C, Cerami E, Gross B, Dogrusoz U, Demir E, Bader GD, Sander C, Pathway Commons 2019 Update: Integration, analysis and exploration of pathway data, Nucleic Acids Res. 48 (2020) D489–D497. 10.1093/nar/gkz946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Slenter DN, Kutmon M, Hanspers K, Riutta A, Windsor J, Nunes N, Mélius J, Cirillo E, Coort SL, DIgles D, Ehrhart F, Giesbertz P, Kalafati M, Martens M, Miller R, Nishida K, Rieswijk L, Waagmeester A, Eijssen LMT, Evelo CT, Pico AR, Willighagen EL, WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research, Nucleic Acids Res. 46 (2018) D661–D667. 10.1093/nar/gkx1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Kelder T, Pico AR, Hanspers K, Van Iersel MP, Evelo C, Conklin BR, Mining biological pathways using WikiPathways web services, PLoS One. 4 (2009) 2–5. 10.1371/journal.pone.0006447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Pillich RT, Chen J, Rynkov V, Welker D, Pratt D, NDEx: a community resource for sharing and publishing of biological networks, in: Protein Bioinforma, Springer, 2017: pp. 271–301. [DOI] [PubMed] [Google Scholar]
- [211].Pratt D, Chen J, Pillich R, Rynkov V, Gary A, Demchak B, Ideker T, NDEx 2.0: A clearinghouse for research on cancer pathways, Cancer Res. 77 (2017) e58–e61. 10.1158/0008-5472.CAN-17-0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Cottret L, Frainay C, Chazalviel M, Cabanettes F, Gloaguen Y, Camenen E, Merlet B, Heux S, Portais JC, Poupin N, Vinson F, Jourdan F, MetExplore: Collaborative edition and exploration of metabolic networks, Nucleic Acids Res. 46 (2018) W495–W502. 10.1093/nar/gky301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Domingo-Fernández D, Mubeen S, Marín-Llaó J, Hoyt CT, Hofmann-Apitius M, PathMe: Merging and exploring mechanistic pathway knowledge, BMC Bioinformatics. 20 (2019) 1–12. 10.1186/s12859-019-2863-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Karnovsky A, Weymouth T, Hull T, Glenn Tarcea V, Scardoni G, Laudanna C, Sartor MA, Stringer KA, Jagadish HV, Burant C, Athey B, Omenn GS, Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data, Bioinformatics. 28 (2012) 373–380. 10.1093/bioinformatics/btr661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Basu S, Duren W, Evans CR, Burant CF, Michailidis G, Karnovsky A, Sparse network modeling and metscape-based visualization methods for the analysis of large-scale metabolomics data, Bioinformatics. 33 (2017) 1545–1553. 10.1093/bioinformatics/btx012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T, Cytoscape: A software Environment for integrated models of biomolecular interaction networks, Genome Res. (2003). 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Meng C, Kuster B, Culhane AC, Gholami AM, A multivariate approach to the integration of multi-omics datasets, BMC Bioinformatics. 15 (2014) 1–13. 10.1186/1471-2105-15-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Argelaguet R, Velten B, Arnol D, Dietrich S, Zenz T, Marioni JC, Buettner F, Huber W, Stegle O, Multi-Omics Factor Analysis—a framework for unsupervised integration of multi-omics data sets, Mol. Syst. Biol 14 (2018) 1–13. 10.15252/msb.20178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Argelaguet R, Arnol D, Bredikhin D, Deloro Y, Velten B, Marioni JC, Stegle O, MOFA+: a probabilistic framework for comprehensive integration of structured single-cell data, BioRxiv. (2019) 837104 10.1101/837104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Ge X, Raghu VK, Chrysanthis PK, Benos PV, CausalMGM: an interactive web-based causal discovery tool, Nucleic Acids Res. 48 (2020) W597–W602. 10.1093/nar/gkaa350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Uppal K, Ma C, Go YM, Jones DP, XMWAS: A data-driven integration and differential network analysis tool, Bioinformatics. 34 (2018) 701–702. 10.1093/bioinformatics/btx656. [DOI] [PMC free article] [PubMed] [Google Scholar]