

Abstract
Acute kidney injury (AKI) and chronic kidney disease (CKD) represent an important challenge for healthcare providers. The identification of new biomarkers/pharmacological targets for kidney disease is required for the development of more effective therapies. Several studies have shown the importance of the endoplasmic reticulum (ER) stress in the pathophysiology of AKI and CKD. ER is a cellular organelle devolved to protein biosynthesis and maturation, and cellular detoxification processes which are activated in response to an insult. This review aimed to dissect the cellular response to ER stress which manifests with activation of the unfolded protein response (UPR) with its major branches, namely PERK, IRE1α, ATF6 and the interplay between ER and mitochondria in the pathophysiology of kidney disease. Further, we will discuss the relationship between mediators of renal injury (with specific focus on vascular growth factors) and ER stress and UPR in the pathophysiology of both AKI and CKD with the aim to propose potential new targets for treatment for kidney disease.
Keywords: acute kidney injury, chronic kidney disease, Endoplasmic reticulum stress, unfolded protein response, vascular growth factors
1. INTRODUCTION
Acute kidney injury (AKI) is characterized by the rapid loss of renal function which, at times, results in partial or full recovery. AKI is an independent risk factor for chronic kidney disease (CKD) as well as for end‐stage renal disease (ESRD). 1 , 2 CKD is characterized by a slow and progressive decline in renal function often secondary to hypertension and/or diabetic mellitus. Both AKI and CKD cases are associated with elevated cardiovascular morbidity and mortality. 3 , 4 , 5 , 6 , 7
AKI prevalence in United States ranges between 1% and 66%, a variation that can be explained by lack of standardized AKI classification and differences between populations. 8 CKD, conversely, is a growing pathology, with a prevalence around 15% (https://www.usrds.org/2019/view/Default.aspx), that is reaching epidemic proportions 9 and appears to be mainly driven by an increase in the diabetic population. 10
2. ENDOPLASMIC RETICULUM, GOLGI APPARATUS AND MITOCHONDRIAL NETWORKS
The endoplasmic reticulum (ER) is a continuum of membranes with tubular and vesicular shape within the cytoplasm of eukaryotic cells. The ER is constituted by the rough ER, which contains ribosomes, and smooth ER, characterized by lacks of ribosomes. 11
In eukaryotic cells, the rough ER has a pivotal function in protein biosynthesis which serves as a checkpoint for the secretory pathway and for protein synthesis/maturation (folding) within the cell. 12 The smooth ER is involved in carbohydrate metabolism, drug detoxification and calcium storage. 13 , 14
The ER could be considered as a signalling platform that responds to stimuli from in and outside the cells, with the aim of maintaining cellular functions and cell survival. The ER is the primary site for synthesis and folding of secreted and membrane‐bound proteins. Proteins are translocated into ER lumen in an unfolded state and require protein chaperones and catalysts of protein folding to assist in proper folding. Properly folded proteins traffic from the ER to the Golgi apparatus; misfolded proteins are targeted to degradation.
A proper protein folding is required to create functional proteins able to execute their function. The ER process that rectifies abnormally folded proteins which need to be physiologically replaced/restored or have been damaged by external insults is defined as 'unfolded protein response' (UPR) characterized by three major pathways: PERK, XBP‐1 and ATF6. 15 UPR provides an adaptive mechanism by which cells can augment protein folding and processing capacities of the ER. If protein misfolding is not resolved, the UPR triggers an apoptotic cascade. 16
The Golgi apparatus is a dynamic organelle constituted by several membranes closely linked with the ER. The Golgi has a crucial function in regulating the trafficking of proteins within the cell, and processing newly synthesized polypeptides, secretory proteins and lipids. 17 Proteins and lipids are delivered from ER to the Golgi apparatus and to the plasma membrane with the auxilium of molecular tags that direct them to their cellular destinations. 18 Conversely, misfolded proteins that escape from the ER are processed by the Golgi or delivered to the lysosome/vacuole for degradation. 19
The mitochondria are double‐membrane‐bound organelles with a highly specialized structure involved in cellular respiration and energy homoeostasis. 20
ER and mitochondria communicate through contact points known as mitochondria‐associated membranes (MAMs). 21 MAMs should not be seen as a static link between ER and mitochondria. On the contrary, the MAMs is represented by a variable collection of proteins that regulate signals between the ER and mitochondria according to the cells’ needs. 22
MAMs are implicated in the calcium transfer from the ER to mitochondria and maintain adequate mitochondria bioenergetics and lipid synthesis and mitochondrial shape and motility. MAMs are essential for the transfer of stress signals from the ER to mitochondria via the activation of UPR. 23 Several MAM connectors can modulate the UPR; of these, mitofusin‐2 protein (Mfn2) not only supports mitochondria‐ER physical interaction, 24 but also participates in the cellular response to ER stress by suppressing PERK activation through direct interaction. In condition of ER stress, loss of Mfn2‐PERK interaction in Mfn2‐deficient cells results in the activation of all three UPR branches (PERK, XBP‐1 and ATF6) resulting in reduced cell apoptosis. 25
Similarly, chaperones, proteins that assist the conformational folding/unfolding and the assembly/disassembly of macromolecular structures in the ER, are important components of MAMs and favour the ER‐mitochondria interaction, and contribute to the regulation of different cellular functions including calcium signalling, energy metabolism and cell survival. 26
The interconnectivity between the ER, the Golgi apparatus and mitochondria has the role to sustain a pro‐survival cellular response to external perturbations 26 such as excess of substrates (eg glucose, lipids), oxidative stress, iron imbalance, viral infections and hypoxia (known as important causes of ER stress). 27
When a pathological insult is sustained, and cell survival is compromised, the ER‐UPR participates in the initiation of apoptosis by at least two main mechanisms. Firstly, apoptosis could be triggered by a progressive accumulation of misfolded protein in the ER when protein ubiquitination and degradation mechanism are overcome. Accumulation of misfolded proteins would trigger a sustained UPR activation and activation of cellular apoptosis mainly via C/EBP homologous protein (CHOP) and IRE1 pathway activation. 26 , 28 Secondly, apoptosis can be induced through the calcium signalling. ER stress‐induced apoptosis is characterized by calcium release from the ER with concomitant increase in calcium within the mitochondria. In mitochondria, calcium leads to inner mitochondrial membrane depolarization and activation of pro‐apoptotic caspase‐mediated pathways. 26 , 29
3. ER‐MITOCHONDRIA INTERPLAY IN RENAL DISEASE
The kidney mitochondrial content is second only to the heart and is crucial for the kidney physiology and pathophysiology of diseases. Mitochondrial dysfunction is universally recognized as a mechanism involved in both AKI and CKD. 30
As discussed above, the communication between ER and the mitochondria has pivotal roles in cellular function such as gene transcription, calcium homoeostasis and cellular redox states. 26
As a response to acute ER stress, UPR (via PERK) drives the dynamic regulation of the mitochondria morphology promoting stress‐induced mitochondrial hyperfusion (SIMH). 31 SIMH is a known pro‐survival mechanism that reduces mitochondrial fragmentation and promotes mitochondrial functions, such as ATP production, to protect cells in response to acute insults. 31
Thus, the PERK arm of the UPR regulates mitochondrial morphology during acute ER stress, 31 confirming that UPR, during an acute perturbation as seen in AKI, has a key protective role.
Acute kidney ischaemia is, at times, not followed by organ recovery; this is often paralleled by long‐term morphological and functional damage of renal cells 32 and by a reduction in mitochondrial number. 30 Incomplete kidney recovery after AKI often becomes a predisposing factor towards further renal deterioration and development of CKD. 32
It is well established that mitochondrial homoeostasis can put a brake on the progression of kidney disease and favour a renal recovery from acute insult. 33 On the contrary in CKD, sustained ER stress drives chronic mitochondrial reactive oxygen species generation thus promoting mitochondrial dysfunction. 33 The sustained redox imbalance as seen in CKD contributes to mitochondrial remodelling and cellular damage with no cell recovery, contrarily to what is seen in AKI (transient insult), promoting a progressive chronic renal function decline towards ESRD. 34
It is also worth remembering that mitochondrial biogenesis (process by which cells increase mitochondrial mass) 33 occurs after exposure of cells to external insults and plays a role in UPR activation and positive response to ER stress. 35 Mitochondrial biogenesis requires the synthesis of nuclear and mitochondrial encoded proteins; of note, this augmentation in protein synthesis can, if sustained, disrupt cellular proteostasis by exceeding the protein‐folding capacity of the cell and contributing to ER stress and to a chronic disease process. 36 , 37
4. ER STRESS AND UPR IN KIDNEY DISEASE
ER stress has been implicated in the pathophysiology of both AKI and CKD. 38 Protein misfolding and secondary ER stress have been described in AKI, glomerulonephritis, glomerulopathies associated with genetic mutations, and hypertensive and diabetes‐related CKD. 39
AKI leads to the accumulation of unfolded and misfolded proteins in the ER. The accumulation of non‐functional proteins causes an altered condition of protein homoeostasis that stimulates the UPR to decrease protein translation and increase ER protein‐folding capacity, and activates pathways involved in protein degradation to restore proteostasis. 40
In AKI, renal ischaemic injury such as renal vascular obstruction, cardiac arrest and renal ischaemia‐reperfusion injury in kidney transplantation, is the main driver of ER stress and secondary UPR activation and tissue injury. 39 , 40 Similarly, AKI drug‐induced nephrotoxicity (eg cisplatin induced) is characterized by a significant activation of ER stress, often associated with cell apoptosis. 40 , 41
Inflammation is one of the main mediators of AKI. 42 In AKI different cytokines and chemokines are released by leucocytes and renal tubular cells. The pro‐inflammatory cytokines/chemokines interferon‐γ, interleukin‐2, interleukin‐6 and interleukin‐10, granulocyte‐macrophage colony‐stimulating factor, transforming growth factor‐β 1 (TGF‐β‐1), chemokine (C‐X‐C motif) ligand‐1, macrophage inflammatory protein‐2 and monocyte chemoattractant protein‐1 are increased in the ischaemic kidney. 42 Limited work has described a clear link between ER stress inflammatory cytokines and UPR in the kidney; nevertheless, transcriptional regulators of inflammatory genes such as nuclear factor‐kB (NF‐kB), 43 and cyclic adenosine monophosphate (cAMP)‐responsive element‐binding protein H (CREBH), an ER‐localized transcription factor, 44 have been shown to initiates an acute inflammatory response after activation by ER stress.
It is still unclear why an episode of AKI is at times followed by a partial/full recovery of renal function or, instead, progresses towards ESRD. 7 AKI is a manifestation of different pathological processes which, despite similar ER stress‐mediated UPR activation, can result in different outcomes often difficult to predict. 45 Functionally, under mild‐moderate ER stress, the activation of the UPR to restore proteostasis leads to cell survival. However, in condition of excessive ER stress, the adaptive capacity of UPR is lost resulting in cell apoptosis to eliminate the irreversibly damaged cells and tissue (Figure 1).
FIGURE 1.
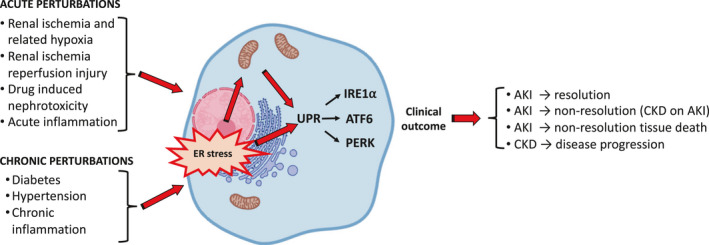
ER stress drives different UPR‐mediated cellular/tissue outcomes in AKI and CKD. Acute and chronic insults to the kidney result in a transient or sustained activation of ER stress/mitochondria and UPR, respectively. ER stress/UPR plays an important role in determining favourable/not favourable clinical outcome of a disease
The different degree of ER stress and UPR stimulation, that either lead to cell survival or death, should not be simply related to a positive or negative outcome. We will need to understand more and better the mechanisms implicated in directing the kidney outcome after AKI. The AKI‐related ER stress likely depends on the magnitude of the insult, the length of the perturbation and the overall condition of the patient; all these variables could contribute to patients’ response to ER stress and prognosis after an episode of AKI. There is certainly an urgent need to investigate ER stress in samples from AKI patients to understand the molecular mechanisms behind a favourable or poor outcome and plan clinical trials to test the effect of drug modulating ER stress and UPR.
AKI is regarded as a major risk factor for the development of CKD and AKI could influence CKD’s progression. 40 , 46 Important AKI‐related insults could result in a defective repair of the renal tubular compartment with activation of ER stress‐mediated pro‐apoptotic, pro‐inflammatory and pro‐fibrotic pathways to attempt wound repair that results in renal interstitial fibrosis which culminates in progressive loss of renal function. 40 , 47
Our understanding of ER stress and UPR in AKI is quite limited but ER or mitochondrial dysfunction following injury has been implicated in the alteration of mitochondria‐ER crosstalk in the kidney, an event that has been involved in the progression of AKI to CKD. 48
In patient with diabetes mellitus and/or hypertension, chronic perturbations such as hyperglycaemia, dyslipidaemias and haemodynamic insults (elevated systemic‐hypertension and intraglomerular pressure) result in ER stress 49 , 50 and are important factors driving renal decline progression towards CKD and ESRD. 51 , 52
CKD is characterized by a condition of sustained ER stress, mitochondrial dysfunction, sustained UPR‐mediated cell death and autophagy. 39
Importantly, chronic elevated glucose, free fatty acid and advanced glycation end products promote a sustained activation of S‐XBP1 and CHOP, which results in the apoptosis of glomerular endothelial and tubular cells. 49 , 52 Lipid accumulation in renal tubular cells results in mitochondrial dysfunction, increased oxidative stress and inflammation that contributes to extracellular matrix production and tubulointerstitial fibrosis. 53 Importantly, ATF6α, a transcription factor of the UPR, regulates fatty acid metabolism and its overexpression in tubular renal cells results, via peroxisome proliferator‐activated receptor‐a (PPARα) down‐regulation, in cellular lipid accumulation with lipotoxicity‐mediated cellular apoptosis with up‐regulation of pro‐sclerotic cytokines and tubulointerstitial fibrosis. 54
Studies conducted in experimental animal models of diabetes (streptozotocin‐induced diabetes and db/db mice) have shown a sustained activation of UPR, with increased levels of phosphorylated PERK, eIF2α and CHOP, with activation of ATF6 and caspase with secondary glomerular and tubular cells apoptosis. 55 , 56 , 57 Further, mice mutant for glucose‐regulated chaperon protein‐78 (GRP78) show impaired chaperone‐mediated protective effects and develop severe tubulointerstitial lesions with ageing. 58
The sustained UPR activation leads to up‐regulation of inflammatory and fibrotic cytokines that result in progressive tissue injury and fibrosis as a manifestation of wound repair. 59 These processes are even worsened when AKI occurs in patients with CKD; these patients are known to have a severe prognosis and partial or full renal recovery is delayed and often absent. 60
Chronic sustained inflammation as seen in CKD is a promoter for uncontrolled healing and tissue damage. ER stress drives inflammatory signalling that, per se, facilitates fibrotic processes. 59 In this condition, sustained activation of the UPR pathways, such as IRE1α, results in the activation of tumour necrosis factor receptor and the pro‐inflammatory transcription factor AP‐1, which have been shown to promote the activation of pro‐inflammatory pathways, such as NF‐KB, NOD1/2 and RIP‐dependent cascades. 59 Similarly, PERK activation and eIF2α phosphorylation increase NF‐KB stability. ER stress in macrophages supports polarization towards a pro‐inflammatory phenotype driven by NF‐KB. 59 , 61 with increase in pro‐inflammatory cytokines such as interleukin 1‐β and interleukin‐18. Indeed, IRE1α in macrophages has been implicated in promoting macrophages polarization towards a pro‐inflammatory rather than a non‐inflammatory pro‐sclerotic phenotype. 59 , 62 , 63 Further, epithelial cells exposed to ER stress lose their characteristic and undergo an epithelial‐to‐mesenchymal transition that reprogram the cells promoting a fibrotic process that leads to tissue injury and loss of normal renal tissue architecture. 64 , 65
Importantly, TGF‐β‐1 is a pro‐sclerotic cytokine that plays a crucial role in ER stress activation and secondary fibrosis: in cultured renal tubular cells, TGF‐β1 induces the ER stress/UPR pathways which mediate the pro‐fibrotic processes. 59 , 66 , 67
5. VASCULAR GROWTH FACTORS IN THE PATHOPHYSIOLOGY OF AKI AND CKD
Different vascular growth factors have been implicated in the pathophysiology of both AKI and CKD.
5.1. Vascular endothelial growth factor A (VEGFA)
In physiology, VEGFA is constitutively expressed in the glomeruli (podocytes) and its deletion or overexpression result in alteration of the permselective properties of the glomerular filtration barrier. 68
In disease setting, such as AKI, VEGFA supplementation is protective for glomerular and tubulointerstitial injury in different experimental models of ischaemic renal disease 69 and its administration helps recovery. 70
In the initial phase of ischaemia/reperfusion, in a rat experimental model of AKI, VEGFA was initially down‐regulated 71 and recovery was paralleled by a normalization of VEGFA expression. 72 , 73
Rapidly progressive glomerulonephritis characterized by acute loss of renal function is characterized by a protective up‐regulation of VEGFA 74 as blockade of VEGFA results in worsening of the disease. 75
In membranoproliferative glomerulonephritis, studies have demonstrated a protective VEGFA up‐regulation for the endothelium, 76 , 77 suggesting, again, that in acute setting VEGFA therapy could help to increase capillary repair.
In CKD, when the insult is sustained such as in diabetic nephropathy, VEGFA expression/action is up‐regulated and its inhibition has proven beneficial. 68 , 78 Of importance, VEGFA action should be modulated and not completely suppressed as VEGFA is essential for endothelium survival and renal function. 79 , 80
In immunodeficiency virus (HIV) nephropathy, a progressive condition, up‐regulation of VEGFA appears to be implicated in its pathophysiology. 81 VEGFA up‐regulation has been observed in humans with HIV and in animal experimental models of HIV nephropathy and VEGFA inhibition has been proposed as a potential treatment. 82 , 83
5.2. Angiopoietins (Angpt)
Angpt are vascular growth factors involved in vasculogenesis and vascular repair. Angpt1, by binding to its receptor tyrosine kinase with immunoglobulin and epidermal growth factor homology domain‐2 (Tie2), stabilizes the vessel wall, while Angpt2, by either interacting with integrins or competing with Angpt1/Tie2 receptor binding (inhibiting Angpt1‐mediated Tie2 phosphorylation), promotes vessel wall destabilization and, in the presence of VEGFA, endothelial cell proliferation and new vessel formation. 68
In physiology, Angpt1 is expressed by glomerular cells and pericytes, while Angpt2 expression is absent; increased levels of Angpt2 have been observed in glomerular diseases and appear to be correlated to adverse outcomes. 68
In experimental model of AKI such as ischaemic kidney injury, up‐regulation of Angpt1 attenuates renal damage mainly by promoting endothelium cell survival and vascular repair while down‐regulation is associated with impaired renal recovery. 84
In critically ill patients, Angpt1 circulating levels are associated with a lower risk of AKI; conversely, higher Angpt2 levels were associated with an increased risk for AKI. 85 Of interest, Angpt1 therapy protects against AKI caused by endotoxemia and ischaemia‐reperfusion. 86 , 87
In CKD, an increased ratio of Angpt2/Angpt1 plays a role in the development and progression of glomerular disease in diabetes 88 and correction of this unbalance has been proposed as protective. 89 , 90
As per AKI, a recent study proposed Angpt2 as an independent predictor of adverse renal outcome in chronic kidney disease in both the general and the diabetic population. 91
Angpt2 levels also show a significant independent correlation with proteinuria in another progressive renal disease such as systemic lupus erythematosus. 92
5.3. Fibroblast Growth Factor (FGF)
In AKI, renal ischaemia‐reperfusion injury is associated with a poor renal prognosis. FGFs, important angiogenic inducers, 93 are up‐regulated in AKI. 94 Mainly, FGF2 and FGF10, in averting ischaemia/reperfusion‐induced tubular cell death, have been proposed as potential future targetable pathway and more work is needed to explore their protective role in AKI. 95
In CKD, the role of FGFs is still unclear and more studies are warranted. One of the more studied is FGF23. FGF23 has been proposed to have a protective effect on the vasculature in CKD by reducing vascular calcifications but, despite these findings, high circulating levels of FGF23 have been associated with increased cardiovascular disease independent of CKD. 96
5.4. Epidermal growth factor (EGF)
EGF mediates its effects on cell growth, differentiation and apoptosis by binding to the cell surface tyrosine kinase epidermal growth factor receptor (EGFR). EGFR is expressed in the kidney, both in the glomerular and tubular compartments. EGF effects are either direct or mediated by EGF‐mediated VEGFA up‐regulation. 97
In AKI, EGFR activation enhances renal recovery as suggested by studies in experimental animal model of AKI, where proximal tubule cell deletion of EGFR or treatment with an EGFR inhibitor resulted in impaired recovery from ischaemia‐reperfusion‐induced injury. 98 In rapidly progressive crescentic glomerulonephritis, blockade of EGF is also beneficial. 99
In CKD, as seen in diabetes, EGF is up‐regulated 100 and inhibition of EGF/EGFR system attenuates renal damage in an experimental animal model of diabetes. 101 , 102
Similarly, in experimental model of hypertensive nephropathies, EGF/EGFR system inhibition seems to confer protection to the kidney. 103
6. VASCULAR GROWTH FACTORS AND ENDOPLASMIC RETICULUM STRESS IN KIDNEY DISEASE
Chronic hypoxia plays a crucial role in the development/progression of kidney disease. 104 , 105 In response to hypoxia, the kidney up‐regulates angiogenic factors such as VEGFA to repair/restore the renal capillary network. In CKD, this adaptive repair mechanisms are impaired because of chronic inflammation and tissue scarring that results in progressive capillary rarefaction and kidney tissue damage. 105
Accumulating evidence suggests that ER stress‐dependent or ER stress‐independent activation of UPR modulates vascular growth factors expression/activity and plays an important role in the maintenance and survival of endothelial cells and vasculature. 106 , 107 Other studies have observed that vascular growth factors, per se, can directly signal and modulate the UPR. 108
As discussed, a transient activation of the UPR mostly retains a pro‐survival effect that promotes vascular repair and new vessel formation with tissue recovery; conversely, sustained UPR activation, when tissue damage is prolonged, promotes impaired vascular remodelling cell death and tissue degeneration (Figure 2).
FIGURE 2.
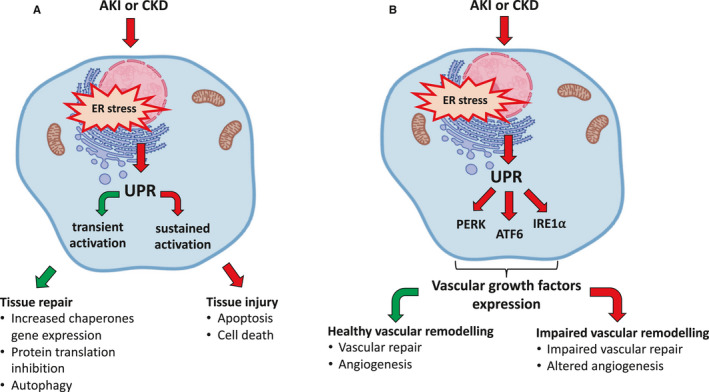
ER stress/UPR transient or sustained activation modulates tissue repair/injury and healthy/impaired vascular remodelling, respectively. A, Acute insults (AKI) stimulate a transient UPR that contributes to tissue repair. Conversely chronic insults, as seen in CKD, stimulate a sustained UPR which promotes tissue injury. B, In condition of ER stress, secondary to AKI or CKD, UPR plays a role in modulating the expression of vascular growth factors. The modulation of vascular growth factor expression drives a healthy or impaired vascular remodelling in condition of transient or sustained UPR activation, respectively
Therefore, modulation of ER stress/UPR may provide attractive novel strategies to ameliorate/correct altered vascular repair and anomalous angiogenesis in diseases without the need of completely blocking vascular growth factors, such as VEGFA or Angpt, known to play many essential roles in kidney tissue homoeostasis. 78 , 109
In a human proximal tubular epithelial cell line (HK2 cells) and in vivo (in a model of renal ischaemia), UPR activation drives the expression of vascular growth factors such as VEGFA and basic fibroblast growth factor via PERK activation. 39 , 110 Indeed, PERK is known to regulate the pro‐angiogenic protein basic FGF 93 expression at both the transcriptional and translational levels. 110 Moreover, transcription factors from all three arms of the UPR (ATF4, spliced XBP‐1 and cleaved ATF6) have consensus sites on the promoter region of VEGFA and have been shown to drive its transcription. 111 , 112
It is also worth remembering that VEGFA signals via ATF6 and PERK. VEGFA‐mediated activation of ATF6 and PERK contributes to the survival effect of VEGFA on endothelial cells by promoting mTORC2‐mediated phosphorylation (and activation) of AKTSer473. 108 UPR pathways constitute important components of VEGFA signalling and endothelial cell survival and angiogenesis, independently of ER stress‐mediated UPR activation 108 (Figure 3 ).
FIGURE 3.

VEGFA/VEGFR2 system differentially modulates the UPR activation in condition of transient or sustained ER stress. A, In physiology, UPR is activated by the VEGF/VEGFR2 system in the absence of ER stress. The VEGF/VEGFR2 system activates both PERK and ATF6 resulting in cell survival and inhibition of the pro‐apoptotic pathway CHOP favouring a cells’ survival. B, AKI mediates (transient) ER stress and PERK and ATF6 activation, which, in turn, drive an up‐regulation of VEGFA. VEGFA, with an autocrine/paracrine mechanism, activates PERK and ATF6 via VEGFR2 activation. The balance between VEGFA/VEGFR2 system and transient UPR‐mediated PERK and ATF6 activation results in lack of stimulation of CHOP‐mediated cell apoptosis, cell survival and tissue repair. C, CKD after AKI or CKD is characterized by sustained ER stress and UPR stimulation. Sustained ER stress‐mediated UPR activation results in CHOP‐mediated cell apoptosis. The sustained stimulation of PERK up‐regulates the VEGFA/VEGFR2 system resulting in a synergistic (VEGFA/UPR) activation of CHOP and cell apoptosis resulting in tissue injury
VEGFA is crucial in the maintenance of glomerular capillaries homoeostasis 113 and inhibitors of ER stress‐mediated UPR stimulation 114 , 115 suppress VEGFA causing proteinuria. 116 These observations clearly show a close link between UPR activation and VEGFA. It is worth noting that too little VEGFA is not favourable for cell homoeostasis, 68 a concept which seems to apply also to inhibition of adaptive UPR activation.
Other vascular growth factors such as Angpt have been implicated in vascular repair in kidney disease being Angpt1 protective for the vasculature. 68 , 89 , 90 , 117 Angpt1 attenuates ER stress‐induced cellular dysfunction and apoptosis in glomerular endothelial cells. Specifically, Angpt1 is able to prevent/ameliorate the angiotensin‐2–induced expression of UPR proteins and PERK/CHOP activation. 118
FGFs are potent angiogenic inducers 93 and seem to modulate ER stress and UPR. FGF1 treatment suppressed diabetes‐induced ER stress in an experimental model of diabetic nephropathy, 119 and FGF10 significantly attenuates the apoptosis of kidney tissues in AKI caused by renal ischaemia‐reperfusion injury by attenuating UPR. 120
EGF, also involved in angiogenesis, and its receptor EGFR activation have been involved in the development and progression of diabetic nephropathy. Inhibition of EGFR tyrosine kinase activity with erlotinib attenuates renal damage in an experimental animal model of diabetes, partly by inhibition of ER stress. 101 Further studies have suggested a role of EGFR activation and ER stress in angiotensin‐2–mediated vascular remodelling, a phenomena independent from angiotensin‐2–mediated hypertension. 121
7. CONCLUSIONS
The ER stress (external insult) and the UPR are crucial mechanisms that regulate cell survival or death. Vascular growth factors directly regulate UPR and also modulate ER stress‐mediated UPR. Blockade or activation of the UPR in disease setting will not provide a clear‐cut answer to potential treatment for both AKI and CKD. Most importantly, drugs able to modulate the UPR will be extremely important. We will need to learn to consider many variables such as the magnitude and duration of the perturbation and the health‐related conditions of the patient to be able to design the correct modulation of the UPR to result in patient benefit.
More studies are needed, and future work will certainly open new treatment avenues for the treatment of the patient with renal disease.
Funding grant: TF_001_20171120.
8. CONFLICT OF INTERESTS’ STATEMENT
The authors declare no conflict of interest on the topic covered.
AUTHORS’ CONTRIBUTION
Carlo Alberto Ricciardi: Conceptualization (equal); Data curation (equal); Funding acquisition (equal); Investigation (equal); Methodology (equal); Validation (equal); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Luigi Gnudi: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Funding acquisition (equal); Investigation (equal); Methodology (equal); Project administration (equal); Resources (equal); Supervision (equal); Validation (equal); Writing‐review & editing (equal).
Ricciardi CA, Gnudi L. The endoplasmic reticulum stress and the unfolded protein response in kidney disease: Implications for vascular growth factors. J Cell Mol Med. 2020;24:12910–12919. 10.1111/jcmm.15999
REFERENCES
- 1. Fortrie G, de Geus HRH, Betjes MGH. The aftermath of acute kidney injury: A narrative review of long‐term mortality and renal function. Crit Care. 2019;23:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012;380:756–766. [DOI] [PubMed] [Google Scholar]
- 3. Braam B, Joles JA, Danishwar AH, Gaillard CA. Cardiorenal syndrome–current understanding and future perspectives. Nat Rev Nephrol. 2014;10:48–55. [DOI] [PubMed] [Google Scholar]
- 4. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. NEnglJ Med. 2004;351:1296–1305. [DOI] [PubMed] [Google Scholar]
- 5. Doyle JF, Forni LG. Acute kidney injury: Short‐term and long‐term effects. Crit Care. 2016;20:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ali T, Khan I, Simpson W, et al. Incidence and outcomes in acute kidney injury: a comprehensive population‐based study. J Am Soc Nephrol. 2007;18:1292–1298. [DOI] [PubMed] [Google Scholar]
- 7. Murugan R, Kellum JA. Acute kidney injury: What's the prognosis? Nat Rev Nephrol. 2011;7:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hoste EAJ, Kellum JA, Selby NM, et al. Global epidemiology and outcomes of acute kidney injury. Nat Rev Nephrol. 2018;14:607–625. [DOI] [PubMed] [Google Scholar]
- 9. Levin A, Tonelli M, Bonventre J, et al. Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet. 2017;390:1888–1917. [DOI] [PubMed] [Google Scholar]
- 10. Bailey RA, Wang Y, Zhu V, Rupnow MF. Chronic kidney disease in US adults with type 2 diabetes: an updated national estimate of prevalence based on Kidney Disease: Improving Global Outcomes (KDIGO) staging. BMC Res Notes. 2014;7:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci. 2016;73:79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mandon EC, Trueman SF, Gilmore R. Protein translocation across the rough endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2013;5(2):a013342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Missiroli S, Danese A, Iannitti T, et al. Endoplasmic reticulum‐mitochondria Ca(2+) crosstalk in the control of the tumor cell fate. Biochim Biophys Acta Mol Cell Res. 2017;1864:858–864. [DOI] [PubMed] [Google Scholar]
- 14. Biwer LA, Isakson BE. Endoplasmic reticulum‐mediated signalling in cellular microdomains. Acta Physiol (Oxf). 2017;219:162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69:169–181. [DOI] [PubMed] [Google Scholar]
- 16. Fribley A, Zhang K, Kaufman RJ. Regulation of apoptosis by the unfolded protein response. Methods Mol Biol. 2009;559:191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li J, Ahat E, Wang Y. Golgi structure and function in health, stress, and diseases. Results Probl Cell Differ. 2019;67:441–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kurokawa K, Nakano A. The ER exit sites are specialized ER zones for the transport of cargo proteins from the ER to the Golgi apparatus. J Biochem. 2019;165:109–114. [DOI] [PubMed] [Google Scholar]
- 19. Sun Z, Brodsky JL. Protein quality control in the secretory pathway. J Cell Biol. 2019;218:3171–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fakouri NB, Hansen TL, Desler C, Anugula S, Rasmussen LJ. From powerhouse to perpetrator‐mitochondria in health and disease. Biology (Basel). 2019;8(2):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Vliet AR, Agostinis P. Mitochondria‐associated membranes and ER stress. Curr Top Microbiol Immunol. 2018;414:73–102. [DOI] [PubMed] [Google Scholar]
- 22. Inoue T, Maekawa H, Inagi R. Organelle crosstalk in the kidney. Kidney Int. 2019;95:1318–1325. [DOI] [PubMed] [Google Scholar]
- 23. Saito A, Imaizumi K. Unfolded protein response‐dependent communication and contact among endoplasmic reticulum, mitochondria, and plasma membrane. Int J Mol Sci. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. [DOI] [PubMed] [Google Scholar]
- 25. Munoz JP, Ivanova S, Sanchez‐Wandelmer J, et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013;32:2348–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Malhotra JD, Kaufman RJ. ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harb Perspect Biol. 2011;3:a004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO Rep. 2006;7:880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deniaud A, Sharaf el dein O, Maillier E, et al. Endoplasmic reticulum stress induces calcium‐dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27:285–299. [DOI] [PubMed] [Google Scholar]
- 30. Szeto HH. Pharmacologic approaches to improve mitochondrial function in AKI and CKD. J Am Soc Nephrol. 2017;28:2856–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lebeau J, Saunders JM, Moraes VWR, et al. The PERK arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Rep. 2018;22:2827–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Seigneux S, Martin PY. Preventing the progression of AKI to CKD: The role of mitochondria. J Am Soc Nephrol. 2017;28:1327–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duann P, Lin PH. Mitochondria damage and kidney disease. Adv Exp Med Biol. 2017;982:529–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Galvan DL, Green NH, Danesh FR. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017;92:1051–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40:141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Inagi R, Ishimoto Y, Nangaku M. Proteostasis in endoplasmic reticulum–new mechanisms in kidney disease. Nat Rev Nephrol. 2014;10:369–378. [DOI] [PubMed] [Google Scholar]
- 37. Mesbah Moosavi ZS, Hood DA. The unfolded protein response in relation to mitochondrial biogenesis in skeletal muscle cells. Am J Physiol Cell Physiol. 2017;312:C583–C594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Inagi R. Endoplasmic reticulum stress in the kidney as a novel mediator of kidney injury. Nephron Exp Nephrol. 2009;112:e1–9. [DOI] [PubMed] [Google Scholar]
- 39. Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol. 2017;13:681–696. [DOI] [PubMed] [Google Scholar]
- 40. Yan M, Shu S, Guo C, Tang C, Dong Z. Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury. Ann Med. 2018;50:381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Foufelle F, Fromenty B. Role of endoplasmic reticulum stress in drug‐induced toxicity. Pharmacol Res Perspect. 2016;4:e00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Akcay A, Nguyen Q, Edelstein CL. Mediators of inflammation in acute kidney injury. Mediators Inflamm. 2009;2009:137072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang K, Kaufman RJ. From endoplasmic‐reticulum stress to the inflammatory response. Nature. 2008;454:455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang K, Shen X, Wu J, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. [DOI] [PubMed] [Google Scholar]
- 45. Rosen S, Heyman SN. Difficulties in understanding human "acute tubular necrosis": limited data and flawed animal models. Kidney Int. 2001;60:1220–1224. [DOI] [PubMed] [Google Scholar]
- 46. Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol. 2010;298:F1078–F1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med. 2016;67:293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maekawa H, Inagi R. Pathophysiological role of organelle stress/crosstalk in AKI‐to‐CKD transition. Semin Nephrol. 2019;39:581–588. [DOI] [PubMed] [Google Scholar]
- 49. Cunard R. Endoplasmic reticulum stress in the diabetic kidney, the good, the bad and the ugly. J Clin Med. 2015;4:715–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mohammed‐Ali Z, Lu C, Marway MK, et al. Endoplasmic reticulum stress inhibition attenuates hypertensive chronic kidney disease through reduction in proteinuria. Sci Rep. 2017;7:41572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gnudi L, Gentile G, Ruggenenti P. The patient with diabetes mellitus In: Turner N, Lamiere N, Goldsmith DJ, Wineearls CG, Himmelfarb J, Remuzzi G, eds. Oxford Textbook of Clinical Nephrology. Oxford, UK: Oxford University Press; 2016:1199–1247. [Google Scholar]
- 52. Zhuang A, Forbes JM. Stress in the kidney is the road to pERdition: is endoplasmic reticulum stress a pathogenic mediator of diabetic nephropathy? The Journal of endocrinology. 2014;222:R97–111. [DOI] [PubMed] [Google Scholar]
- 53. Simon N, Hertig A. Alteration of fatty acid oxidation in tubular epithelial cells: From acute kidney injury to renal fibrogenesis. Front Med (Lausanne). 2015;2:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jao TM, Nangaku M, Wu CH, et al. ATF6alpha downregulation of PPARalpha promotes lipotoxicity‐induced tubulointerstitial fibrosis. Kidney Int. 2019;95:577–589. [DOI] [PubMed] [Google Scholar]
- 55. Liu G, Sun Y, Li Z, et al. Apoptosis induced by endoplasmic reticulum stress involved in diabetic kidney disease. Biochem Biophys Res Commun. 2008;370:651–656. [DOI] [PubMed] [Google Scholar]
- 56. Madhusudhan T, Wang H, Dong W, et al. Defective podocyte insulin signalling through p85‐XBP1 promotes ATF6‐dependent maladaptive ER‐stress response in diabetic nephropathy. Nat Commun. 2015;6:6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wu J, Zhang R, Torreggiani M, et al. Induction of diabetes in aged C57B6 mice results in severe nephropathy: an association with oxidative stress, endoplasmic reticulum stress, and inflammation. Am J Pathol. 2010;176:2163–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kimura K, Jin H, Ogawa M, Aoe T. Dysfunction of the ER chaperone BiP accelerates the renal tubular injury. Biochem Biophys Res Commun. 2008;366:1048–1053. [DOI] [PubMed] [Google Scholar]
- 59. Kropski JA, Blackwell TS. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J Clin Invest. 2018;128:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. He L, Wei Q, Liu J, et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017;92:1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Blackwell TS, Christman JW. The role of nuclear factor‐kappa B in cytokine gene regulation. Am J Respir Cell Mol Biol. 1997;17:3–9. [DOI] [PubMed] [Google Scholar]
- 62. Yao Y, Wang Y, Zhang Z, et al. Chop deficiency protects mice against bleomycin‐induced pulmonary fibrosis by attenuating M2 macrophage production. Mol Ther. 2016;24:915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cuevas EP, Eraso P, Mazon MJ, et al. LOXL2 drives epithelial‐mesenchymal transition via activation of IRE1‐XBP1 signalling pathway. Sci Rep. 2017;7:44988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Moon SY, Kim HS, Nho KW, Jang YJ, Lee SK. Endoplasmic reticulum stress induces epithelial‐mesenchymal transition through autophagy via activation of c‐Src kinase. Nephron Exp Nephrol. 2014;126:127–140. [DOI] [PubMed] [Google Scholar]
- 66. Dihazi H, Dihazi GH, Bibi A, et al. Secretion of ERP57 is important for extracellular matrix accumulation and progression of renal fibrosis, and is an early sign of disease onset. J Cell Sci. 2013;126:3649–3663. [DOI] [PubMed] [Google Scholar]
- 67. Liu SH, Yang CC, Chan DC, et al. Chemical chaperon 4‐phenylbutyrate protects against the endoplasmic reticulum stress‐mediated renal fibrosis in vivo and in vitro. Oncotarget. 2016;7:22116–22127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gnudi L, Benedetti S, Woolf AS, Long DA. Vascular growth factors play critical roles in kidney glomeruli. Clin Sci (Lond). 2015;129:1225–1236. [DOI] [PubMed] [Google Scholar]
- 69. Suga S, Kim YG, Joly A, et al. Vascular endothelial growth factor (VEGF121) protects rats from renal infarction in thrombotic microangiopathy. Kidney Int. 2001;60:1297–1308. [DOI] [PubMed] [Google Scholar]
- 70. Kim YG, Suga SI, Kang DH, et al. Vascular endothelial growth factor accelerates renal recovery in experimental thrombotic microangiopathy. Kidney Int. 2000;58:2390–2399. [DOI] [PubMed] [Google Scholar]
- 71. Basile DP, Fredrich K, Chelladurai B, Leonard EC, Parrish AR. Renal ischemia reperfusion inhibits VEGF expression and induces ADAMTS‐1, a novel VEGF inhibitor. Am J Physiol Renal Physiol. 2008;294:F928–F936. [DOI] [PubMed] [Google Scholar]
- 72. Leonard EC, Friedrich JL, Basile DP. VEGF‐121 preserves renal microvessel structure and ameliorates secondary renal disease following acute kidney injury. Am J Physiol Renal Physiol. 2008;295:F1648–F1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chade AR, Kelsen S. Reversal of renal dysfunction by targeted administration of VEGF into the stenotic kidney: a novel potential therapeutic approach. Am J Physiol Renal Physiol. 2012;302:F1342–F1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Nitta K, Uchida K, Kimata N, et al. Increased serum levels of vascular endothelial growth factor in human crescentic glomerulonephritis. Clin Nephrol. 1999;52:76–82. [PubMed] [Google Scholar]
- 75. Hara A, Wada T, Furuichi K, et al. Blockade of VEGF accelerates proteinuria, via decrease in nephrin expression in rat crescentic glomerulonephritis. Kidney Int. 2006;69:1986–1995. [DOI] [PubMed] [Google Scholar]
- 76. Ostendorf T, Kunter U, Eitner F, et al. VEGF(165) mediates glomerular endothelial repair. J Clin Invest. 1999;104:913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Haas CS, Campean V, Kuhlmann A, et al. Analysis of glomerular VEGF mRNA and protein expression in murine mesangioproliferative glomerulonephritis. Virchows Arch. 2007;450:81–92. [DOI] [PubMed] [Google Scholar]
- 78. Dei Cas A, Gnudi L. VEGF and angiopoietins in diabetic glomerulopathy: How far for a new treatment? Metabolism. 2012;61(12):1666–1673. [DOI] [PubMed] [Google Scholar]
- 79. Sivaskandarajah GA, Jeansson M, Maezawa Y, Eremina V, Baelde HJ, Quaggin SE. Vegfa protects the glomerular microvasculature in diabetes. Diabetes. 2012;61:2958–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Eremina V, Baelde HJ, Quaggin SE. Role of the VEGF–a signaling pathway in the glomerulus: Evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol. 2007;106:32–37. [DOI] [PubMed] [Google Scholar]
- 81. Eremina V, Sood M, Haigh J, et al. Glomerular‐specific alterations of VEGF‐A expression lead to distinct congenital and acquired renal diseases. J ClinInvest. 2003;111:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kumar D, Konkimalla S, Yadav A, et al. HIV‐associated nephropathy: role of mammalian target of rapamycin pathway. Am J Pathol. 2010;177:813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Korgaonkar SN, Feng X, Ross MD, et al. HIV‐1 upregulates VEGF in podocytes. J Am Soc Nephrol. 2008;19:877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chiang WC, Huang YC, Fu TI, et al. Angiopoietin 1 influences ischemic reperfusion renal injury via modulating endothelium survival and regeneration. Mol Med. 2019;25:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Robinson‐Cohen C, Katz R, Price BL, et al. Association of markers of endothelial dysregulation Ang1 and Ang2 with acute kidney injury in critically ill patients. Crit Care. 2016;20:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kim DH, Jung YJ, Lee AS, et al. COMP‐angiopoietin‐1 decreases lipopolysaccharide‐induced acute kidney injury. Kidney Int. 2009;76:1180–1191. [DOI] [PubMed] [Google Scholar]
- 87. Jung YJ, Kim DH, Lee AS, et al. Peritubular capillary preservation with COMP‐angiopoietin‐1 decreases ischemia‐reperfusion‐induced acute kidney injury. Am J Physiol Renal Physiol. 2009;297:F952–F960. [DOI] [PubMed] [Google Scholar]
- 88. Gnudi L. Cellular and molecular mechanisms of diabetic glomerulopathy. Nephrol Dial Transplant. 2012;27:2642–2649. [DOI] [PubMed] [Google Scholar]
- 89. Dessapt‐Baradez C, Woolf AS, White KE, et al. Targeted glomerular angiopoietin‐1 therapy for early diabetic kidney disease. J Am Soc Nephrol. 2014;25(1):33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jeansson M, Gawlik A, Anderson G, et al. Angiopoietin‐1 is essential in mouse vasculature during development and in response to injury. J Clin Invest. 2011;121:2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tsai YC, Chiu YW, Tsai JC, et al. Association of angiopoietin‐2 with renal outcome in chronic kidney disease. PLoS One. 2014;9:e108862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. El‐Banawy HS, Gaber EW, Maharem DA, Matrawy KA. Angiopoietin‐2, endothelial dysfunction and renal involvement in patients with systemic lupus erythematosus. J Nephrol. 2012;25:541–550. [DOI] [PubMed] [Google Scholar]
- 93. Murakami M, Simons M. Fibroblast growth factor regulation of neovascularization. Curr Opin Hematol. 2008;15:215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wai K, Soler‐Garcia AA, Perazzo S, Mattison P, Ray PE. A pilot study of urinary fibroblast growth factor‐2 and epithelial growth factor as potential biomarkers of acute kidney injury in critically ill children. Pediatr Nephrol. 2013;28:2189–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Deng LC, Alinejad T, Bellusci S, Zhang JS. Fibroblast growth factors in the management of acute kidney injury following ischemia‐reperfusion. Front Pharmacol. 2020;11:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yang X, Liaw L, Prudovsky I, et al. Fibroblast growth factor signaling in the vasculature. Curr Atheroscler Rep. 2015;17:509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zepeda‐Orozco D, Wen HM, Hamilton BA, Raikwar NS, Thomas CP. EGF regulation of proximal tubule cell proliferation and VEGF‐A secretion. Physiol Rep. 2017;5(18):e13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chen J, Chen JK, Harris RC. Deletion of the epidermal growth factor receptor in renal proximal tubule epithelial cells delays recovery from acute kidney injury. Kidney Int. 2012;82:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bollee G, Flamant M, Schordan S, et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat Med. 2011;17:1242–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gilbert RE, Cox A, McNally PG, et al. Increased epidermal growth factor in experimental diabetes related kidney growth in rats. Diabetologia. 1997;40:778–785. [DOI] [PubMed] [Google Scholar]
- 101. Zhang MZ, Wang Y, Paueksakon P, Harris RC. Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes. 2014;63:2063–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Advani A, Wiggins KJ, Cox AJ, Zhang Y, Gilbert RE, Kelly DJ. Inhibition of the epidermal growth factor receptor preserves podocytes and attenuates albuminuria in experimental diabetic nephropathy. Nephrology. 2011;16:573–581. [DOI] [PubMed] [Google Scholar]
- 103. Benter IF, Canatan H, Benboubetra M, Yousif MH, Akhtar S. Global upregulation of gene expression associated with renal dysfunction in DOCA‐salt‐induced hypertensive rats occurs via signaling cascades involving epidermal growth factor receptor: a microarray analysis. Vascul Pharmacol. 2009;51:101–109. [DOI] [PubMed] [Google Scholar]
- 104. Fine LG, Norman JT. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: From hypothesis to novel therapeutics. Kidney Int. 2008;74:867–872. [DOI] [PubMed] [Google Scholar]
- 105. Tanaka S, Tanaka T, Nangaku M. Hypoxia and dysregulated angiogenesis in kidney disease. Kidney Dis (Basel). 2015;1:80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Binet F, Sapieha P. ER stress and angiogenesis. Cell Metabolism. 2015;22(4):560–575. [DOI] [PubMed] [Google Scholar]
- 107. Barabutis N. Unfolded protein response supports endothelial barrier function. Biochimie. 2019;165:206–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Karali E, Bellou S, Stellas D, Klinakis A, Murphy C, Fotsis T. VEGF Signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol Cell. 2014;54:559–572. [DOI] [PubMed] [Google Scholar]
- 109. Woolf AS, Gnudi L, Long DA. Roles of angiopoietins in kidney development and disease. J Am SocNephrol. 2009;20:239–244. [DOI] [PubMed] [Google Scholar]
- 110. Bouvier N, Fougeray S, Beaune P, Thervet E, Pallet N. The unfolded protein response regulates an angiogenic response by the kidney epithelium during ischemic stress. J Biol Chem. 2012;287:14557–14568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ghosh R, Lipson KL, Sargent KE, et al. Transcriptional regulation of VEGF‐A by the unfolded protein response pathway. PLoS One. 2010;5:e9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Pereira ER, Liao N, Neale GA, Hendershot LM. Transcriptional and post‐transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS One. 2010;5(9):e12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Park JH, Kim M, Oh JH. Effects of bevacizumab on endoplasmic reticulum stress in hypoxic retinal pigment epithelial cells. PLoS One. 2017;12:e0179048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Li J, Wang JJ, Yu Q, Wang M, Zhang SX. Endoplasmic reticulum stress is implicated in retinal inflammation and diabetic retinopathy. FEBS Lett. 2009;583:1521–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wu S, Kim C, Baer L, Zhu X. Bevacizumab increases risk for severe proteinuria in cancer patients. J Am Soc Nephrol. 2010;21:1381–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Davis B, Dei CA, Long DA, et al. Podocyte‐specific expression of angiopoietin‐2 causes proteinuria and apoptosis of glomerular endothelia. J Am Soc Nephrol. 2007;18:2320–2329. [DOI] [PubMed] [Google Scholar]
- 118. Bi X, Niu J, Ding W, Zhang M, Yang M, Gu Y. Angiopoietin‐1 attenuates angiotensin II‐induced ER stress in glomerular endothelial cells via a Tie2 receptor/ERK1/2‐p38 MAPK‐dependent mechanism. Mol Cell Endocrinol. 2016;428:118–132. [DOI] [PubMed] [Google Scholar]
- 119. Wu Y, Li Y, Jiang T, et al. Reduction of cellular stress is essential for Fibroblast growth factor 1 treatment for diabetic nephropathy. J Cell Mol Med. 2018;22:6294–6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Tan X, Yu L, Yang R, et al. Fibroblast growth factor 10 attenuates renal damage by regulating endoplasmic reticulum stress after ischemia‐reperfusion injury. Front Pharmacol. 2020;11:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Takayanagi T, Kawai T, Forrester SJ, et al. Role of epidermal growth factor receptor and endoplasmic reticulum stress in vascular remodeling induced by angiotensin II. Hypertension. 2015;65:1349–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]