

Abstract
Cardiac hypertrophy is a common pathological change in patients with progressive cardiac function failure, which can be caused by hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM) or arterial hypertension. Despite years of study, there is still limited knowledge about the underlying molecular mechanisms for cardiac hypertrophy. NDUFA7, a subunit of NADH:ubiquinone oxidoreductase (complex I), has been reported to be a novel HCM associated gene. However, the biological role of NDUFA7 in heart remains unknown. In this study, we found that NDUFA7 exhibited high expression in the heart, and its level was significantly decreased in mice model of cardiac hypertrophy. Moreover, we demonstrated that ndufa7 knockdown in developing zebrafish embryos resulted in cardiac development and functional defects, associated with increased expression of pathological hypertrophy biomarkers nppa (ANP) and nppb (BNP). Mechanistic study demonstrated that ndufa7 depletion promoted ROS production and calcineurin signalling activation. Moreover, NDUFA7 depletion contributed to cardiac cell hypertrophy. Together, these results report for the first time that ndufa7 is implicated in pathological cardiac hypertrophy.
Keywords: cardiac hypertrophy, ndufa7, nppa, nppb, zebrafish
1. INTRODUCTION
Cardiac hypertrophy is an adaptive response of the heart to haemodynamic and neurohormonal stress to preserve cardiac function. 1 Prolonged cardiac hypertrophy can contribute to functional decompensation, cardiac fibrosis, even progress to heart failure or sudden death, leading to a high mortality worldwide. 2 Cardiac hypertrophy is accompanied by reactivation of a set of cardiac foetal genes, including atrial natriuretic peptide (ANP, nppa), brain natriuretic peptide (BNP, nppb) and β‐myosin heavy chain (β‐MHC), as well as an increase in cell size and protein synthesis, suggesting that molecular events controlling heart development are redeployed to regulate hypertrophic growth. 3 Among many intracellular signalling pathways involved in hypertrophic development, calcineurin/ nuclear factor of activated T cell (NFAT) pathway plays a key role in pathological cardiac hypertrophy. 4 NFAT, resides in the cytoplasm in a hyper‐phosphorylated state, is dephosphorylated by the calcium/calmodulin‐activated phosphatase calcineurin and then translocate to the nucleus where it regulates transcription of hypertrophic genes. 5
The heart is the greatest oxygen‐consuming organ in the body, which requires ATP production from mitochondrial respiration to maintain normal cardiac mechanical function. 6 Therefore, mitochondrial dysfunction plays a key role in the pathogenesis and development of cardiac hypertrophy. 7 Reactive oxygen species (ROS) are a natural by‐product of mitochondrial energy production. 8 ROS are generated during excessive oxidative stress, which has been implicated in the pathogenesis of cardiac hypertrophy and heart failure. 9 Interestingly, it has been reported that calcineurin regulates the pathogenesis of cardiac hypertrophy with accompanying by the intracellular ROS production. 10 It has been demonstrated that ROS stimulates the signal transduction in cardiomyocytes during pathological conditions by activating NFAT. 11
NDUFA7, also known as B14.5, encodes a subunit of NADH:ubiquinone oxidoreductase (complex I) in the mitochondrial respiratory chain. It has been reported that an association is observed between NDUFA7 and rheumatoid arthritis (RA) with severe erosive arthritis. 12 Our previous work of whole exome sequencing analysis on hypertrophic cardiomyopathy patients found that NDUFA7 might be a novel candidate gene associated with cardiomyopathy. 13 However, the biological role of NDUFA7 in cardiac diseases has not been explored. Here we present the first evidence to our knowledge that ndufa7 depletion contributes to pathological cardiac hypertrophy.
2. MATERIALS AND METHODS
2.1. Tissue expression profiles and animal model data
The Genotype‐Tissue Expression database (GTEx, http://www.gtexportal.org/) was used to investigate the expression of candidate genes in multiple human tissues. The zebrafish information network (ZFIN, http://zfin.org/) and the mouse genome database (MGD, http://www.informatics.jax.org/) databases, as well as PubMed, were used to investigate the phenotype of candidate genes in zebrafish and mice. Clustal Omega software (https://www.ebi.ac.uk/Tools/msa/clustalo/) was used for multiple sequence alignment.
2.2. Zebrafish care and breeding
All procedures were performed in accordance with the guidelines of the Canadian Council on Animal Care and approved by the Institutional Animal Care Committee. Wild‐type zebrafish embryos were used for recording heart rates. A transgenic cmlc2::GFP zebrafish line that marks cardiomyocytes with Green Fluorescence Protein (GFP) was used for heart morphology imaging studies or extraction for quantitative RT‐PCR. A transgenic nppb::F‐luc zebrafish line that expresses firefly luciferase under the control of the nppb promoter was employed as in vivo model labelling BNP pathway, a biomarker of heart failure. Wild‐type zebrafish was used for all other experiments. The zebrafish lines were maintained at 28℃ in 14:10 h light/dark conditions. Protocols for experimental procedures were approved by the Research Ethics Board of St. Michael's Hospital (Toronto) (protocol ACC660). Zebrafish were killed by immersion in an ice‐water (4°C or less) bath followed by overdose of clove oil (70%‐95% eugenol) or tricaine methanesulphonate (MS‐222, 200‐300 mg/L). All animal experiments were performed in accordance with the NIH guidelines on the protection of animals used for scientific purposes.
2.3. Morpholinos and expression constructs
Morpholino (MO)‐modified antisense oligonucleotides (Gene Tools) were designed against the splice donor site of zebrafish ndufa7 exon 2 (ndufa7 MO: 5’‐TCCGTTTCTTAACAGCAAGATCTCC‐3’). The morpholinos were injected into zebrafish embryos at the 1‐cell stage. As a negative control, a standard control oligonucleotide (Control MO: 5’‐CCTCTTACCTCAGTTACAATTTATA‐3’) was injected at the same dose.
2.4. RNA isolation, reverse transcription (RT‐PCR) and quantitative real‐time PCR (qPCR)
Total RNA was isolated from whole embryos, larval hearts, cells or tissues with Trizol reagent (Invitrogen) and reverse‐transcribed into cDNA with an Oligo dT primer and SuperScript II reverse transcriptase (Invitrogen). The efficiency of the ndufa7 splice MO was tested by carrying out RT‐PCR from RNA extracted from zebrafish whole embryos at 2dpf with ndufa7 primers. For the analysis of specific gene expression in zebrafish larval hearts or cells, real‐time PCR was performed on all samples in triplicate with a Power SYBR Green PCR Master Mix (Applied Biosystems) as described previously. 14 , 15 The 2-△△Ct method was used to normalize the gene of interest to the endogenous housekeeping gene rpl13a (ribosomal protein L13a) or GAPDH and determine the fold change relative to control. Primers used for experiments were listed in Table 1.
Table 1.
Primers for qPCR or ISH analysis
| Use | Gene | Forward primer (5’→3’) | Reverse primer (5’→3’) |
|---|---|---|---|
| qPCR | ndufa7 | ATCCAGAGGCTGAGGAATTATCTGTCAG | TCATGTCTTCACAGAAAGCTCTGACAGC |
| nppa | GATGTACAAGCGCACACGTT | TCTGATGCCTCTTCTGTTGC | |
| nppb | CATGGGTGTTTTAAAGTTTCTCC | CTTCAATATTTGCCGCCTTTAC | |
| rpl13a | TCTGGAGGACTGTAAGAGGTATGC | AGACGCACAATCTTGAGAGCAG | |
| vmhc | TCAGATGGCAGAGTTTGGAG | GCTTCCTTTACAGTTACAGTCTTTC | |
| cmlc2 | GTGATGAAGAGCTGGAGT | GGGTCATTAGCAGCCT | |
| serca | GGATCATCAACATCGGCCAC | CGTTCTTCTTGGCCATACGG | |
| calcineurin | GCCTTTAGGATCTACGACATGG | ATATTCTCCCGTCTCCGTCTTT | |
| NDUFA7 | TACTGTACTCGTGATGGCCG | GACAGCTCCCACCTCTTCAT | |
| ANP | GAGGAGAAGATGCCGGTAG | CAGAGAGGGAGCTAAGTG | |
| BNP | TGATTCTGCTCCTGCTTTTC | GTGGATTGTTCTGGAGACTG | |
| GAPDH | ACAGCAACAGGGTGGTGGAC | TTTGAGGGTGCAGCGAACTT | |
| ISH | ndufa7 | TAATACGACTCACTATAGGGA AAAGAGAAAATCCAGAGGCTGAGGAATTATCTGTCAG | ATTAACCCTCACTAAAGGAAAAGGAGGGCTGTCAGAGCTTTCTGTGAAGACATGA |
| nppa | TAATACGACTCACTATAGGGAAAAGAGAAAGGCAACAGAAGAGGCATCAG | ATTAACCCTCACTAAAGGAAAAGGAGGGGAAGACCCTATGCGATCCA | |
| nppb | TAATACGACTCACTATAGGGA AAAGAGAAAACCGGCGGAAAGAGAAGTAA | ATTAACCCTCACTAAAGGAAAAGGAGGCCCGACTGTGTTACATCCCA |
2.5. Whole‐mount in situ hybridization (ISH)
For all manipulations, embryos at the appropriate developmental stage, 1 cell, 24 hpf (hour post‐fertilization), 48 hpf, 72 hpf and 96 hpf were fixed in 4% paraformaldehyde/PBS. In situ hybridization and immunohistochemistry were performed using standard protocols. Digoxigenin‐labelled riboprobes were prepared from PCR templates with T3 and T7 polymerases as recommended by the supplier (Roche). Staining was visualized using an anti‐DIG‐alkaline phosphatase‐conjugated antibody and NBT/BCIP (Roche). After probe detection, embryos were cleared and photographed in glycerol.
2.6. Cell culture and transfection
H9c2 cells, obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China), were cultured in the Dulbecco's modified Eagle's medium and maintained at 37℃ in a 5% CO2 humidified atmosphere. Rat NDUFA7 siRNA oligonucleotides (siNDUFA7#1: CAACAATTACTACTGTACT, siNDUFA7#2: ATCATCATGTCCTCACAAA) were synthesized (RiboBio) and transfected into cells with Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific) according to manufacturers’ protocols.
2.7. Drug exposures
FK506 (Sigma) were stored in DMSO stocks and diluted into working concentrations (1 μg/ml). The final DMSO concentration was 0.5% for all experiments. Embryos were manually dechorionated at 21 hpf, and FK506 was then applied. 2’,7’‐dichlorofluorescin diacetate (Sigma) was used as an indicator of ROS level. Embryos were stained with 2’,7’‐dichlorofluorescin diacetate at room temperature for an hour at 72 hpf and then imaged under the fluorescence microscope. The intensity of ROS level was examined using ImageJ software (National Institution of Health). For cellular ROS measurement, H9c2 cells were incubated with 2’,7’‐dichlorofluorescin diacetate at 37°C for 30 minutes, washed with PBS for three times and then visualized under the fluorescence microscope.
2.8. Luciferase assay
The nppb::F‐Luc embryos injected with morpholinos or treated with chemicals were placed into a 96‐well microtiter plate with 100 μL buffered embryo water and incubated at 28°C for embryonic development. The plate was removed from the incubator at 72 hpf, and 25 μL of long half‐life firefly luciferase reagent (Perkin Elmer, Steady‐Glo) was added to the well. After incubation for 1 hour in dark, the plate was read in a high‐sensitivity luminescence plate reader (SpetraMax M5e, Molecular Devices).
2.9. Immunoblotting
Protein was extracted in lysis buffer (50 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, 2 mmol/L EDTA, 0.5% Triton X‐100, 50 × Roche protease inhibitor) from mice heart or 3dpf zebrafish embryos. The extracts were then fractionated by SDS‐PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane using a transfer apparatus following the manufacturer's instructions (Bio‐rad). Membranes were blocked in 5% non‐fat milk in TBST (10 mmol/L Tris, pH 8.0, 150 mmol/L NaCl, 0.5% Tween 20) for 2 hours followed by TBST wash for 5 minutes. Then membranes were incubated with primary antibodies against NDUFA7 (Abcam, ab140871), GAPDH (Proteintech, 60004‐1‐Ig), and then horseradish peroxidase‐conjugated secondary antibodies as described previously. 16 All antibodies were diluted in 1:1000 dilution. Membranes were developed with the ECL system (Bio‐rad) following the manufacturer's instructions.
2.10. Microscopy
Live embryos were anesthetized using 0.16 mg/mL tricaine methanesulfonate (Sigma) and embedded in 2.5% methyl cellulose (Sigma). Embryos were imaged in bright field and fluorescent mode using fluorescent microscopy (Leica M205 FA). Images were analysed with LAS FA software (Leica) and ImageJ. For myocardial function analysis, embryos were laterally positioned and taken videos at identical levels of magnification and frame rate. Sequential still frames were used to measure ventricular internal chamber dimensions at end diastole (EDD, end‐diastolic diameter) and end systole (ESD, end‐systolic diameter). Fractional shortening (FS) was calculated using the formula (EDD−ESD)/(EDD).
2.11. Statistical analysis
Data are presented as mean ± SEM using Image GraphPad Prism 5.0 software (GraphPad Software). Analysis of statistical significance was performed by the Student's t test for comparison between two groups. A P value of less than .05 was deemed statistically significant.
3. RESULTS
3.1. The expression level of NDUFA7 is markedly decreased in cardiac hypertrophy
We first checked the expression level of NDUFA7 in tissues using NIH’s Genotype‐Tissue Expression (GTEx) database (https://commonfund.nih.gov/gtex) and found that NDUFA7 displayed relatively high expression in human heart and muscle (Figure 1A). We extracted RNA from different tissues of mice, performed RT‐qPCR analysis and found high expression level of NDUFA7 mRNA in mice heart, which is consistent with GTEx database finding (Figure 1B). We then searched the GEO (gene expression omnibus) database to explore whether NDUFA7 is involved in cardiac diseases. Compared with control group, NDUFA7 expression was significantly decreased in the mice heart with pressure overload left ventricle hypertrophy caused by transverse aortic constriction (TAC; Figure 1C). 17 We further analysed the homology of NDUFA7 by aligning the protein sequences from human, chimpanzee, dog, cattle, mouse, rat, chicken as well as zebrafish and found that NDUFA7 is highly conserved among species (Figure 1D). These data indicate that NDUFA7 displays relatively high expression in heart, and its level decreases in cardiac hypertrophy.
Figure 1.
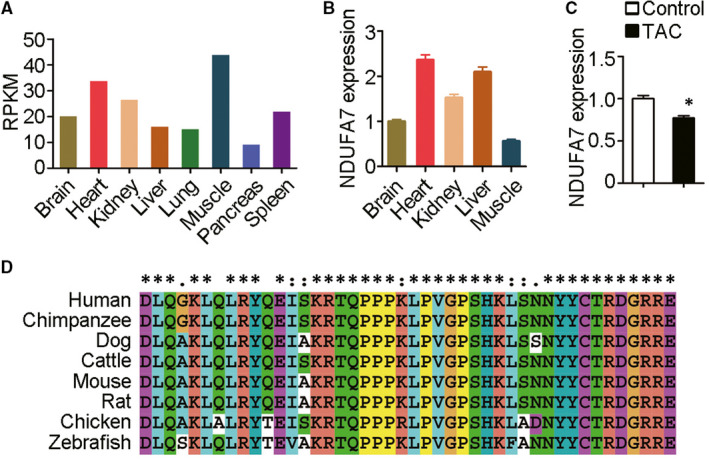
NDUFA7 is involved in cardiac hypertrophy induced by ISO infusion. (A) Tissue expression profiles of NDUFA7 are generated from GTEx database (https://commonfund.nih.gov/gtex). Median RPKM level was shown. (B) RNA was extracted from different tissues of mice, and RT‐qPCR analysis of NDUFA7 and GAPDH expression was then performed. (C) NDUFA7 expression in hearts of mice subjected to cardiac pressure overload by transverse aortic constriction (TAC) or in control group (GSE2459). *P < .05 compared with healthy control. (D) Similarity of Ndufa7 protein sequence from different species including human, chimpanzee, dog, cattle, mouse, rat, chicken and zebrafish
3.2. Knockdown of ndufa7 results in cardiac defect in developing zebrafish embryos
Given the high homology of ndufa7 among species, we studied the function of ndufa7 using zebrafish. To characterize the spatio‐temporal expression pattern of ndufa7, we first performed whole‐mount in situ hybridization with different stages of WT zebrafish embryos. ndufa7 was detected in 1‐cell embryo, suggesting maternal expression of this gene (Figure 2A). We also observed specific somite expression of ndufa7 in 24 hpf embryos, and heart expression in 48 hpf embryos (Figure 2B, C). RT‐PCR analysis of RNA isolated from 2 dpf cmlc2::GFP zebrafish whole embryo, tail and heart further verified the expression of ndufa7 in the somite and heart (Figure 2D).
Figure 2.
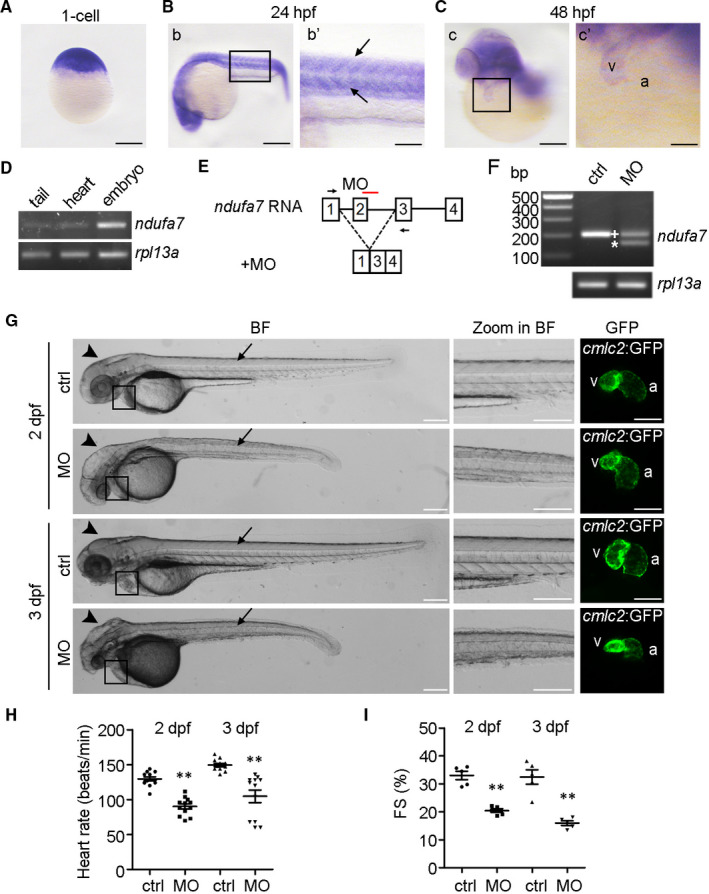
Knockdown of ndufa7 results in cardiac defect in developing zebrafish embryos. (A‐C) Representative images of whole‐mount in situ hybridization using the ndufa7 riboprobe were showed. The experiments were performed in triplicate, with 20 embryos per developmental stage. (A) Zebrafish ndufa7 transcript is broadly expressed at 1‐cell stage. Scale bar, 250 μm. (B) At 24 hpf, ndufa7 is expressed in the somite (box in b, and arrow in b’). Scale bar, 250 μm in b, 100 μm in b’. (C) By 48 hpf, ndufa7 is enriched in the heart (box in c), v represents ventricle, and a represents atrium. Scale bar, 250 μm in c, 100 μm in c’. (D) RT‐PCR of ndufa7 and rpl13a RNA isolated from 2 dpf cmlc2::GFP zebrafish whole embryo, tail and heart. (E) The ndufa7 splice morpholino target site and the aberrant transcript. (F) RT‐PCR amplification of RNA from control MO and ndufa7 MO‐injected zebrafish showing altered splicing of ndufa7 with either the integration of exon 2 (+) or the partial skipping of exon 2 (*). (G) Morphological defects observed in ndufa7 morphants (MO) or controls (ctrl) at 2 dpf and 3 dpf. Arrows and arrowheads showed the defects in somites and head, respectively. The fluorescent image demonstrated heart phenotype from the enlarged box region. Scale bar, 300 μm in bright field (BF) and zoom in BF, 150 μm in GFP field. V represents ventricle, and a represents atrium. The experiments were performed in triplicate, processing 40 embryos per condition. (H) WT embryos were injected with MOs at 1‐cell stage, and heart rate was counted via a recorded video captured with the aid of a microscope. Statistical test: Student's t test. **P < .01 compared with controls. n = 12 measurements per condition. (I) Fractional shortening (FS) of the ventricular chamber in control and ndufa7 morphants was measured at the indicated developmental stages. Statistical test: Student's t test. *P < .05 compared with controls. n = 5 measurements per condition
To study the role of ndufa7 in cardiac development, we employed morpholino (MO)‐based technology to knockdown the ndufa7 gene in developing zebrafish embryos. Microinjection of splicing MO (e2i2) targeting the splicing donor of exon 2 resulted in aberrant splicing and in turn a frameshift in the resulting ndufa7 protein (Figure 2E). RT‐PCR analysis revealed an extra band resulting from the aberrant splicing (Figure 2F). We then injected control or ndufa7 MO into 1‐cell cmlc2::GFP transgenic zebrafish embryos and found that ndufa7 morphants displayed a slightly curved, short and roughed tail, as well as a small head at both 2 dpf and 3 dpf stages (Figure 2G). Furthermore, a slight deformed ventricle was observed in 2 dpf embryos with an even more severe heart phenotype presented in 3 dpf embryos. To further investigate the role that ndufa7 plays in myocardial function, the heart‐beating videos in live embryos were taken. We found that the heart rate of ndufa7 morphant was markedly reduced compared to that of control group (Figure 2H). Moreover, fractional shortening (FS) measurements showed that ndufa7 depletion leads to a significant reduction in ventricular function, decreasing from around 34% to 20% at 48 hpf and from approximately 33% to 14% at 72 hpf (Figure 2I). These data demonstrate that ndufa7 depletion contributes to myocardial dysfunction in zebrafish embryos.
3.3. ndufa7 inhibition contributes to cardiac structural defects
To investigate whether the ndufa7 MO‐induced cardiac dysfunction is due to the structure defect, we evaluated the cardiac phenotype with heart‐chamber marker vmhc (ventricular myosin heavy chain). Whole‐mount ISH performed on 3 dpf zebrafish embryos showed that vmhc mRNA is specifically expressed in the skeletal muscle of the trunk as well as in cardiac ventricular muscle (Figure 3A). Compared with the control group, ndufa7 morphants exhibited a small head and disorganized somite structure missing the paralleled V shape (Figure 3A‐a,b,a’,b’). The dorsal view of the embryo hearts showed that ndufa7 morphants exhibit an enlarged ventricle and wide outflow track (Figure 3A‐a",b"). The size of the ventricle in ndufa7 morphant group was around 1.9 times that of the control group (Figure 3B). We then injected cmlc2::GFP embryos with ndufa7 MO or control MO and extracted RNA from collected embryo hearts at 3 dpf. The qPCR analysis showed that depletion of ndufa7 significantly increases the expression level of vmhc in the heart (Figure 3C).
Figure 3.
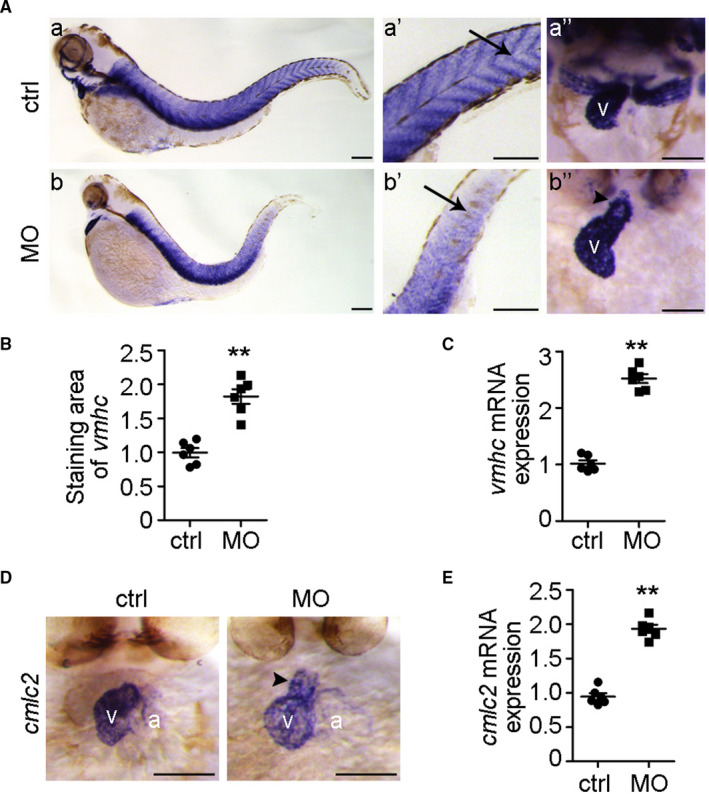
ndufa7 inhibition contributes to cardiac structural defects. (A) Wild‐type zebrafish embryos were injected with ndufa7 MO or control MO at 1‐cell stage and then subjected to whole‐mount ISH with riboprobes against vmhc at 3 dpf. Lateral views of endogenous vmhc mRNA expression pattern were visualized under the microscope (a, b). The expression pattern of vmhc mRNA transcripts was expressed in the trunk muscle (a’, b’), as well as in the ventricle under the dorsal views (a’’, b’’). Arrows and arrowheads indicate the muscle structure and outflow track, respectively, and v represents ventricle. Scale bar, 75 μm. The experiments were performed in triplicate, with 20 individuals per condition. (B) Experiments were performed as in (A), and the staining area of vmhc in the heart was examined using ImageJ software. Statistical test: Student's t test. **P < .01 compared with control MO group, n = 6 measurements per condition. (C) cmlc2::GFP embryos were injected with ndufa7 MO or control MO at 1‐cell stage, and RNA was then extracted from harvested embryo hearts at 3 dpf. The expression level of vmhc in the hearts was examined by RT‐qPCR. Statistical test: Student's t test. **P < .01 compared with control MO group, n = 6 measurements per condition. (D) Wild‐type zebrafish embryos were injected with ndufa7 MO or control MO at 1‐cell stage, and then subjected to whole‐mount ISH with riboprobes against cmlc2 at 3 dpf. Dorsal views of endogenous cmlc2 mRNA expression pattern were observed. Arrowheads indicate the outflow track, v represents ventricle and a represents atrium. Scale bar, 75 μm. The experiments were performed in triplicate, with 20 individuals per condition. (E) cmlc2::GFP embryos were injected with ndufa7 MO or control MO at 1‐cell stage, and RNA was then extracted from embryo hearts at 3 dpf for RT‐qPCR. Statistical test: Student's t test. *P < .05 compared with control MO group, n = 6 measurements per condition
To verify the above result, we further performed whole‐mount ISH with another heart‐chamber marker cmlc2 (cardiac myosin light chain 2). cmlc2 was shown to be predominantly expressed in the ventricle and was slightly expressed in the outcurvature of the atrium (Figure 3D). Similarly, compared with the control group, ndufa7 morphants displayed a swelling ventricle with wide outcurvature, as well as elongated outflow track. Furthermore, the expression level of cmlc2 in the heart was significantly upregulated upon ndufa7 knockdown as measure by qPCR (Figure 3E). These data show that inhibition of ndufa7 leads to altered cardiac structure.
3.4. The cardiac hypertrophy biomarkers nppb and nppa are upregulated by ndufa7 depletion
To examine whether natriuretic peptide signalling is implicated in ndufa7‐MO‐induced cardiac hypertrophy, we first performed whole‐mount ISH to examine the alteration of nppb upon ndufa7 depletion. As shown in Figure 4A, nppb was mainly expressed in ventricle, with its expression level decreased during the development in the control group. In contrast, the nppb expression level in ndufa7 morphants remained high and elevated during the development. In particular, the ventricle chamber of ndufa7 morphant was enlarged compared to the control group at 3 dpf. Moreover, the qPCR analysis showed that ndufa7 morphants exhibited a significantly higher nppb mRNA expression, with an average 1‐fold increase at 3 dpf and 2.8‐fold increase at 4 dpf (Figure 4B). We then performed a luciferase assay with nppb::F‐Luc transgenic zebrafish line, a genetic model to study HCM signalling. In comparison with the control group, depletion of ndufa7 significantly increased the nppb promoter activity, increasing luciferase activity by around 1.5‐fold (Figure 4C).
Figure 4.
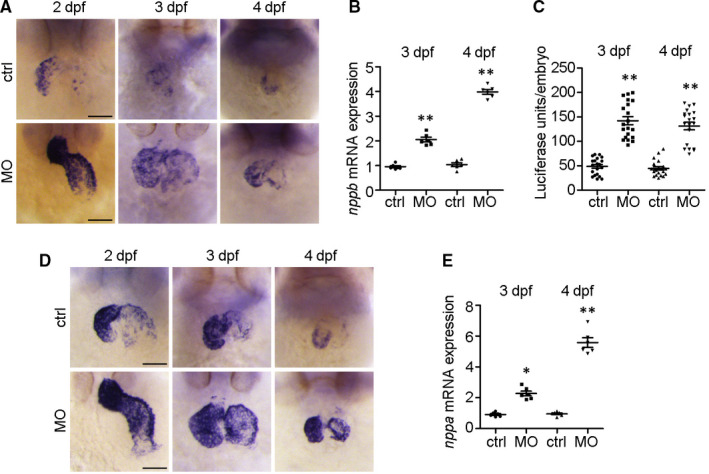
The cardiac hypertrophy biomarkers nppb and nppa are upregulated by ndufa7 depletion. (A) WT zebrafish embryos were injected with ndufa7 MO or control MO and then subjected to whole‐mount in situ hybridization with riboprobes against nppb. Scale bar, 75 μm. The experiments were performed in triplicate, with 20 individuals per condition. (B) cmlc2::GFP embryos were injected with ndufa7 MO or control MO at 1‐cell stage, and RNA was then extracted from harvested embryo hearts at 3 dpf or 4 dpf for RT‐qPCR. The experiments were performed in triplicate, with 60 individuals per condition. Statistical test: Student's t test. **P < .01 compared with control MO group, n = 6 measurements per condition. (C) nppb::F‐luc embryos were injected with ndufa7 splice‐blocking MO or control MO at 1‐cell stage and collected at 3 dpf and 4 dpf for the luciferase assay. The experiments were performed in triplicate, with n = 30 individuals per condition. Statistical test: Student's t test. **P < .01 compared with control MO group, n = 20 measurements per condition. (D) Wild‐type zebrafish embryos were injected with ndufa7 MO or control MO and then subjected to whole‐mount in situ hybridization with riboprobes against nppa probe. Scale bar, 75 μm. The experiments were performed in triplicate, with 20 individuals per condition. (E) cmlc2::GFP embryos were injected with ndufa7 MO or control MO at 1‐cell stage, and RNA was then extracted from harvested embryo hearts at 3 dpf or 4 dpf for RT‐qPCR. The experiments were performed in triplicate, with 60 individuals per condition. Statistical test: Student's t test. **P < .01, *P < .05 compared with control MO group, n = 6 measurements per condition
To further confirm the role of ndufa7 in cardiac hypertrophy, we performed whole‐mount ISH with another hypertrophic marker nppa. It has been shown that nppa is expressed both in ventricle and atrium. Similar to nppb, its expression level decreased during development in the control group and the expression level of nppa in ndufa7 morphants was increased compared with control group (Figure 4D). Interestingly, the staining pattern of nppa at 3 dpf larval zebrafish also indicated that the ventricle chamber of ndufa7 morphant is enlarged as compared with control. We also showed that knockdown of ndufa7 enhances expression level of nppa in the heart by qPCR analysis, with an average 1.4‐fold increase at 3 dpf and 5.6‐fold increase at 4 dpf (Figure 4E). These results show that ndufa7 depletion contributes to increased expression of pathological hypertrophy markers.
3.5. Calcineurin signalling is involved in ndufa7 inhibition induced cardiac hypertrophy
To gain mechanistic insight into the role of ndufa7 in cardiac hypertrophy, we first examined the impact of ndufa7 deficiency on ROS level by staining embryos with 2’,7’‐dichlorofluorescin diacetate. A significant increase in ROS level especially in zebrafish heart was observed in ndufa7 morphant compared with control group (Figure 5A, B). It has been reported that calcineurin signalling regulates cardiac hypertrophy with accompanying by the intracellular ROS production. 18 We further examined several important genes in calcineurin signalling and found that ndufa7 MO significantly reduces serca2 level and increase calcineurin level in embryonic hearts (Figure 5C).
Figure 5.
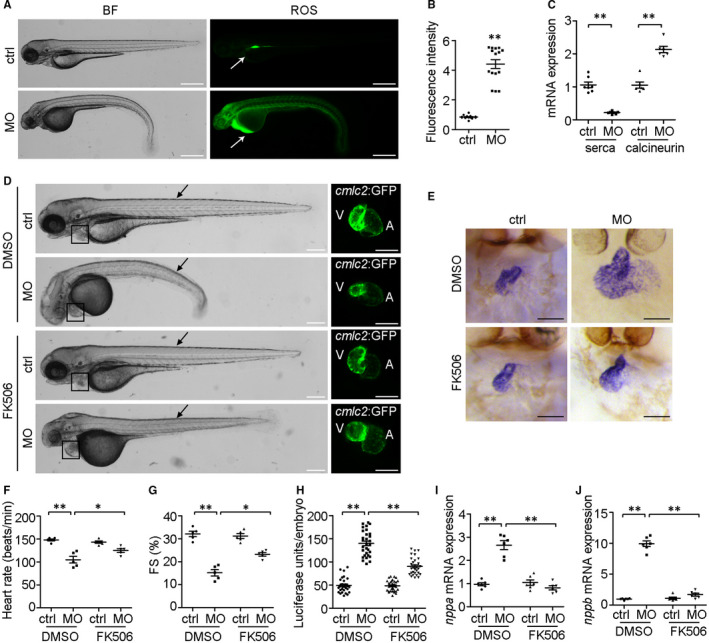
Calcium signalling is involved in ndufa7‐depletion induced cardiac hypertrophy. (A) WT zebrafish embryos were injected with MO at 1‐cell stage and stained for ROS using 2’,7’‐dichlorofluorescin diacetate. Arrows indicate the ROS staining in the heart. Scale bar, 500 μm. (B) Experiments were performed as in (A), and the intensity of ROS level was analysed. **P < .01 compared with control MO group. (C) cmlc2::GFP embryos were injected with ndufa7 MO or control MO at 1‐cell stage, and RNA was then extracted from harvested embryo hearts at 3 dpf for RT‐qPCR. The expression level of serca and calcineurin were examined. The experiments were performed in triplicate, with 60 individuals per condition. Statistical test: Student's t test. **P < .01 compared with control MO group. (D) MO microinjections were performed at 1‐cell stage, 0.5% DMSO (vehicle control) or FK506 (in 0.5% DMSO) were added to embryo water at 21 hpf and cardiac fluorescent imaging was performed on cmlc2::GFP embryos at 3 dpf. Black arrow indicates somite defects, and black box shows cardiac defects. Scale bar, 300 μm in bright field, 150 μm in GFP field. The experiments were performed in triplicate, processing 30 embryos per condition. (E) Wild‐type zebrafish embryos were injected with MO at 1‐cell stage, treated with DMSO or FK506 at 21 hpf, and then subjected to whole‐mount ISH with riboprobes against cmlc2 at 3 dpf. Dorsal views of endogenous cmlc2 mRNA expression pattern were observed. Scale bar, 75 μm. (F‐G) Experiments were performed as in (D), heart rate and fractional shortening (FS) of the ventricular chamber were measured. **P < .01, *P < .05 compared with controls. (H) nppb::F‐luc embryos were injected with ndufa7 splice‐blocking MO or control MO at 1‐cell stage, treated with FK506 or 0.5% DMSO at 21 hpf, and collected at 3 dpf for the luciferase assay. The experiments were performed in triplicate, with 50 individuals per condition. Statistical test: Student's t test. **P < .01 compared with control MO group, n = 30 measurements per condition. (I‐J) cmlc2::GFP embryos were injected with ndufa7 MO or control MO at 1‐cell stage, and treated with 0.5% DMSO or FK506. RNA was extracted from harvested embryo hearts at 3 dpf to examine the mRNA expression level of nppa and nppb. The experiments were performed in triplicate, with 60 individuals per condition. Statistical test: Student's t test. **P < .01 compared with control MO group or DMSO vehicle group, n = 6 measurements per condition
To further verify our hypothesis, we treated ndufa7‐MO‐injected embryos with the calcineurin inhibitor, FK506. In the vehicle (DMSO) treated group, ndufa7 morphant presented a curved and roughed tail and evidently defected ventricle (Figure 5D). However, ndufa7 morphant treated with FK506 exhibited a straight tail and less defected ventricle. Besides, whole‐mount ISH analysis with cmlc2 showed that FK506 treatment restored ndufa7 depletion induced swelling ventricle and wide outcurvature to an extent (Figure 5E). Moreover, FK506 treatment partially rescued defects in heart rate and ventricle function in ndufa7 morphant (Figure 5F, G). To further investigate whether calcineurin signalling is involved in ndufa7 inhibition induced cardiac hypertrophy, we performed a luciferase assay with nppb:Luc zebrafish embryos treated with FK506. As shown in Figure 5H, FK506 treatment restored the ndufa7 MO‐induced upregulation of nppb. Moreover, we found that inhibition of calcineurin restored the expression level of both nppa and nppb in embryo hearts by qPCR analysis (Figure 5I, J). These results show that calcineurin signalling is involved in ndufa7 induced cardiac hypertrophy.
3.6. Depletion of NDUFA7 leads to cardiac cell hypertrophy
To verify the above findings in zebrafish, we further examined the role of NDUFA7 in cardiac cells. H9c2 cells, derived from embryonic rat heart tissue, have been used as a cardiac cell model in many cardiac hypertrophy studies. 19 , 20 Immunoblotting revealed that NDUFA7 siRNAs decreased the protein level of NDUFA7 effectively (Figure 6A). Moreover, NDUFA7 depletion promotes ROS generation, which is consistent with the phenotype in zebrafish heart (Figure 6B, C). We further examined expression of cardiac hypertrophy markers by qPCR analysis and found that NDUFA7 inhibition markedly increase the mRNA level of ANP and BNP, indicating that depletion of NDUFA7 leads to cardiac hypertrophy (Figure 6D). We proposed that depletion of ndufa7 promoted ROS production and calcineurin signalling activation, leading to the expression of cardiac hypertrophy genes, thus contributing to cardiac hypertrophy (Figure 6E).
Figure 6.
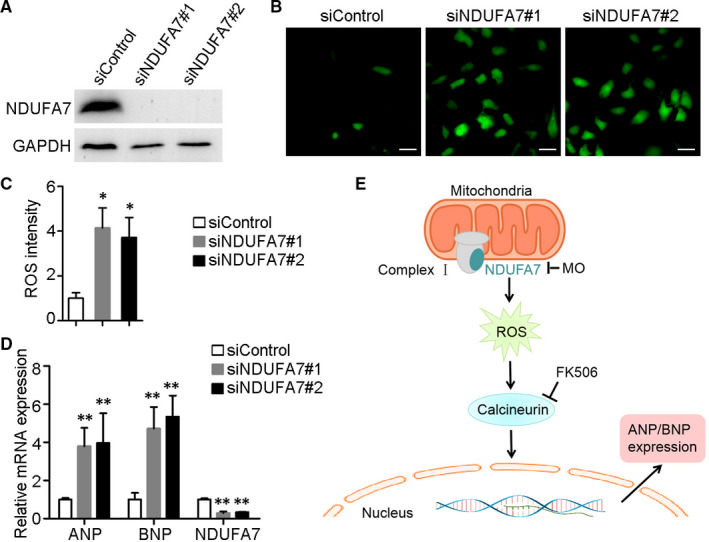
Depletion of NDUFA7 leads to cardiac hypertrophy in H9c2 cells. (A) H9c2 cells were transfected with control or NDUFA7 siRNAs for 72 hours, and expression level of NDUFA7 and GAPDH were examined. (B) H9c2 cells transfected with control or NDUFA7 siRNAs were stained for ROS using 2’,7’‐dichlorofluorescin diacetate, and then visualized under the fluorescence microscope. Scale bar, 50 µm. (C) Experiments were performed as in (B), and the ROS intensity in each group were examined by ImageJ. *P < .05 compared with control group. (D) H9c2 cells transfected with control or NDUFA7 siRNAs for 72 h, RNA was then extracted for RT‐qPCR analysis. **P < .01 compared with control group. (E) Schematic representation of the proposed mechanism. Depletion of ndufa7 triggered ROS production and subsequent calcineurin signalling activation, which further led to the expression of cardiac hypertrophy genes
4. DISCUSSION
Due to the embryo transparency and the ease of direct embryonic manipulation, the zebrafish has emerged as a promising research tool to model cardiovascular diseases including cardiac hypertrophy, congenital heart defects, arrhythmia, as well as cardiomyopathy. For example, a zebrafish model of human cardiac troponin T (TNNT2) mutation known to cause HCM has been generated. 21 The morphant zebrafish embryos with tnnt2 knockdown displayed sarcomere disarray and a robust induction of myocardial hypertrophic pathways, which is similar to humans with HCM. 22 Moreover, zebrafish with pickwick m171 mutation demonstrated reduced cardiac contractile function and pericardial oedema. 23 The mutation was attributed to the gene ttn, encoding the protein Titin, which is an important cause of human idiopathic dilated cardiomyopathy. 24 Besides, the establishment of zebrafish models to dissect ANP/BNP signalling pathway further prompts the functional studies of cardiac hypertrophy pathogenesis, disease mechanisms and potentially drug screens. 25 , 26 , 27
NDUFA7 has been reported to be associated with rheumatoid arthritis (RA) with severe erosive arthritis. 12 Till now, the biological functions of NDUFA7 remain unknown. In this study, we employed zebrafish models extensively to explore ndufa7 function in pathological cardiac hypertrophy. Consistent with tissue expression database, zebrafish ndufa7 is expressed in the heart and muscle during embryonic development. We further provide several lines of evidence showing the important role of ndufa7 in cardiac hypertrophy: Knockdown of ndufa7 leads to cardiac defect in developing zebrafish embryos; ndufa7 depletion contributes to the elevated expression of hypertrophic biomarkers nppb and nppa; calcineurin signalling is involved in ndufa7 inhibition induced cardiac hypertrophy. These findings were further verified in the model of cardiac cells. It is possible that ndufa7 depletion activates calcineurin signalling, which allows the dephosphorylated NFAT to be imported into the nucleus, thus leading to the expression of cardiac hypertrophy genes. It will be interesting to explore in further studies, which will deepen our understanding of the biological function of ndufa7. Given the high density of mitochondria in cardiomyocytes, it is not surprising that ndufa7 plays an important role in cardiac function. 28
Mitochondrial function is essential for proper heart function by producing a constant supply of energy to accomplish complex cellular processes including continuous repetitive contraction and maintenance of Ca2+ homeostasis. 29 Oxidative stress, characterized by the accumulation of ROS, is known to exert detrimental influence on the myocardium such as the induction of apoptotic cell death, hypertrophy and dysfunction. Increasing evidence suggests that cardiac hypertrophy induced by mechanical left ventricular wall stress is partially triggered by ROS generation. 30 , 31 It has been reported that calcineurin regulates the pathogenesis of cardiac hypertrophy with accompanying by the intracellular ROS production. 10 Mitochondrial respiratory complex I has been considered to be the major ROS generation site because large changes in the potential energy of the electrons occur in the sites. 32 In a recent study, it is indicated that ROS might involve in the NDUFA7 induced rheumatoid arthritis. 12 In this study, we demonstrate that depletion of ndufa7 promotes ROS production and calcineurin signalling activation, leading to cardiac hypertrophy. Furthermore, alterations in mitochondrial metabolism have been reported in pathological cardiac hypertrophy, including dysfunction of the electron transport chain and reduced capacity of ATP synthesis. 33 It is possible that ndufa7 might be associated with cardiac hypertrophy by regulating the activity of mitochondria complex I, as well as the ATP generation of mitochondrial oxidative phosphorylation. Elucidation of this will help to fully understand the role of ndufa7 in the pathogenesis of cardiac hypertrophy.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTION
Xingjuan Shi: Conceptualization (lead); Data curation (lead); Formal analysis (lead); Funding acquisition (lead); Investigation (lead); Methodology (lead); Supervision (lead); Validation (lead); Visualization (lead); Writing‐original draft (lead); Writing‐review & editing (lead). Yu Zhang: Data curation (supporting); Investigation (supporting). Ru Chen: Data curation (supporting); Investigation (supporting); Methodology (supporting). Yijie Gong: Data curation (supporting); Investigation (supporting); Methodology (supporting). Mingming Zhang: Methodology (supporting). Rui Guan: Validation (supporting); Visualization (supporting). Ori D. Rotstein: Resources (supporting). Xiangdong Liu: Funding acquisition (supporting); Resources (supporting); Supervision (supporting). Xiao‐Yan Wen: Conceptualization (supporting); Funding acquisition (supporting); Resources (lead).
ACKNOWLEDGEMENTS
We would like to thank Koroboshka Brand‐Arzamendi (St. Michael's Hospital), Dr Kim A Connelly (St. Michael's Hospital) for technical help and Dr Peter P. Liu (University of Ottawa Heart Institute) for critical comments on the manuscript.
Shi X, Zhang Y, Chen R, et al. ndufa7 plays a critical role in cardiac hypertrophy. J. Cell. Mol. Med.. 2020;24:13151–13162. 10.1111/jcmm.15921
Funding information
This work was supported by grants from the National Natural Science Foundation of China grants 81770381 (XS) and 81270295 (XL), Zhishan Youth Scholar Program of SEU (XS), the Fundamental Research Funds for the Central Universities (XS), Natural Sciences and Engineering Research Council of Canada (NSERC, #RGPIN 05389‐14 to X‐YW), Brain Canada Foundation/Health Canada (#PSG14‐3505 to X‐YW) and Canada Foundation for Innovation (CFI, #26233 to X‐YW).
Contributor Information
Xingjuan Shi, Email: xingjuanshi@seu.edu.cn.
Xiangdong Liu, Email: xiangdongliu@seu.edu.cn.
Xiao‐Yan Wen, Email: x.wen@utoronto.ca.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245‐262. [DOI] [PubMed] [Google Scholar]
- 2. Bisping E, Wakula P, Poteser M, Heinzel FR. Targeting cardiac hypertrophy: toward a causal heart failure therapy. J Cardiovasc Pharmacol. 2014;64:293‐305. [DOI] [PubMed] [Google Scholar]
- 3. Huang ZP, Chen J, Seok HY, et al. MicroRNA‐22 regulates cardiac hypertrophy and remodeling in response to stress. Circ Res. 2013;112:1234‐1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sarikhani M, Maity S, Mishra S, et al. SIRT2 deacetylase represses NFAT transcription factor to maintain cardiac homeostasis. The Journal of biological chemistry. 2018;293:5281‐5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hullmann JE, Grisanti LA, Makarewich CA, et al. GRK5‐mediated exacerbation of pathological cardiac hypertrophy involves facilitation of nuclear NFAT activity. Circ Res. 2014;115:976‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ventura‐Clapier R, Garnier A, Veksler V. Energy metabolism in heart failure. J Physiol. 2004;555:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rosca MG, Tandler B, Hoppel CL. Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell Cardiol. 2013;55:31‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191‐227. [DOI] [PubMed] [Google Scholar]
- 9. McMurray J, Chopra M, Abdullah I, Smith WE, Dargie HJ. Evidence of oxidative stress in chronic heart failure in humans. Eur Heart J. 1993;14:1493‐1498. [DOI] [PubMed] [Google Scholar]
- 10. Gooch JL, Gorin Y, Zhang BX, Abboud HE. Involvement of calcineurin in transforming growth factor‐beta‐mediated regulation of extracellular matrix accumulation. J Biol Chem. 2004;279:15561‐15570. [DOI] [PubMed] [Google Scholar]
- 11. Kalivendi SV, Konorev EA, Cunningham S, et al. Doxorubicin activates nuclear factor of activated T‐lymphocytes and Fas ligand transcription: role of mitochondrial reactive oxygen species and calcium. Biochem J. 2005;389:527‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mitsunaga S, Hosomichi K, Okudaira Y, et al. Aggregation of rare/low‐frequency variants of the mitochondria respiratory chain‐related proteins in rheumatoid arthritis patients. J Hum Genet. 2015;60:449‐454. [DOI] [PubMed] [Google Scholar]
- 13. Xu J, Li Z, Ren X, et al. Investigation of Pathogenic Genes in Chinese sporadic Hypertrophic Cardiomyopathy Patients by Whole Exome Sequencing. Sci Rep. 2015;5:16609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shi X, Li D, Wang Y, et al. Discovery of centrosomal protein 70 as an important player in the development and progression of breast cancer. Am J Pathol. 2017;187:679‐688. [DOI] [PubMed] [Google Scholar]
- 15. Shi X, Wang Y, Sun X, et al. Centrosomal protein 70 Is a mediator of paclitaxel sensitivity. Int J Mol Sci. 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shi X, Yao Y, Wang Y, et al. Cep70 regulates microtubule stability by interacting with HDAC6. FEBS Lett. 2015;589:1771‐1777. [DOI] [PubMed] [Google Scholar]
- 17. Mirotsou M, Dzau VJ, Pratt RE, Weinberg EO. Physiological genomics of cardiac disease: quantitative relationships between gene expression and left ventricular hypertrophy. Physiol Genomics. 2006;27:86‐94. [DOI] [PubMed] [Google Scholar]
- 18. Hasan P, Saotome M, Ikoma T, et al. Mitochondrial fission protein, dynamin‐related protein 1, contributes to the promotion of hypertensive cardiac hypertrophy and fibrosis in Dahl‐salt sensitive rats. J Mol Cell Cardiol. 2018;121:103‐106. [DOI] [PubMed] [Google Scholar]
- 19. Su H, Pistolozzi M, Shi X, Sun X, Tan W. Alterations in NO/ROS ratio and expression of Trx1 and Prdx2 in isoproterenol‐induced cardiac hypertrophy. Acta Biochim Biophys Sin. 2017;49:1022‐1028. [DOI] [PubMed] [Google Scholar]
- 20. Guan XH, Hong X, Zhao N, et al. CD38 promotes angiotensin II‐induced cardiac hypertrophy. J Cell Mol Med. 2017;21:1492‐1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stainier DY, Fouquet B, Chen JN, et al. Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development. 1996;123:285‐292. [DOI] [PubMed] [Google Scholar]
- 22. Becker JR, Deo RC, Werdich AA, Panakova D, Coy S, MacRae CA. Human cardiomyopathy mutations induce myocyte hyperplasia and activate hypertrophic pathways during cardiogenesis in zebrafish. Dis Model Mech. 2011;4:400‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Driever W, Solnica‐Krezel L, Schier AF, et al. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996;123:37‐46. [DOI] [PubMed] [Google Scholar]
- 24. Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619‐628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Becker JR, Robinson TY, Sachidanandan C, et al. In vivo natriuretic peptide reporter assay identifies chemical modifiers of hypertrophic cardiomyopathy signalling. Cardiovasc Res. 2012;93:463‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shi X, Verma S, Yun J, et al. Effect of empagliflozin on cardiac biomarkers in a zebrafish model of heart failure: clues to the EMPA‐REG OUTCOME trial? Mol Cell Biochem. 2017;433:97‐102. [DOI] [PubMed] [Google Scholar]
- 27. Shi X, Chen R, Zhang Y, et al. Zebrafish heart failure models: opportunities and challenges. Amino Acids. 2018;50:787‐798. [DOI] [PubMed] [Google Scholar]
- 28. Vasquez‐Trincado C, Garcia‐Carvajal I, Pennanen C, et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. The Journal of physiology. 2016;594:509‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nan J, Zhu W, Rahman MS, et al. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim Biophys Acta Mol Cell Res. 2017;1864:1260‐1273. [DOI] [PubMed] [Google Scholar]
- 30. Nagarajan N, Oka S, Sadoshima J. Modulation of signaling mechanisms in the heart by thioredoxin 1. Free Radic Biol Med. 2017;109:125‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med. 2019;51:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483‐495. [DOI] [PubMed] [Google Scholar]
- 33. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. 2007;356:1140‐1151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.