

Abstract
TG‐Interacting Factor 1 (Tgif1) affects proliferation and differentiation of myeloid cells and regulates self‐renewal of haematopoietic stem cells (HSCs). To determine its impact on leukaemic haematopoiesis, we induced acute or chronic myeloid leukaemias (AML or CML) in mice by enforced expression of MLL‐AF9 or BCR‐ABL, respectively, in Tgif1+/+ or Tgif1−/− haematopoietic stem and progenitor cells (HSPCs) and transplanted them into syngeneic recipients. We find that loss of Tgif1 accelerates leukaemic progression and shortens survival in mice with either AML or CML. Leukaemia‐initiating cells (LICs) occur with higher frequency in AML among mice transplanted with MLL‐AF9‐transduced Tgif1−/− HSPCs than with Tgif1+/+ BMCs. Moreover, AML in mice generated with Tgif1−/− HSPCs are chemotherapy resistant and relapse more rapidly than those whose AML arose in Tgif1+/+ HSPCs. Whole transcriptome analysis shows significant alterations in gene expression profiles associated with transforming growth factor‐beta (TGF‐beta) and retinoic acid (RA) signalling pathways because of Tgif1 loss. These findings indicate that Tgif1 has a protective role in myeloid leukaemia initiation and progression, and its anti‐leukaemic contributions are connected to TGF‐beta‐ and RA‐driven functions.
Keywords: acute myeloid leukaemia, bone marrow cells, chronic myeloid leukaemia, leukaemia‐initiating cells, TG‐Interacting Factor 1
1. INTRODUCTION
TG‐Interacting Factor1 (TGIF1) belongs to the three‐amino acid loop extension (TALE) family of homeodomain proteins 1 , 2 , 3 , 4 and functions as a corepressor of retinoic acid (RA) and transforming growth factor‐β (TGF‐β)‐stimulated transcription. It does so by interfering with retinoid X receptor (RXR) binding to DNA and by recruiting general corepressors, 5 histone deacetylases (HDACs) to the TGF‐β signalling intermediate Smad2, 2 , 3 , 4 , 6 respectively. TGIF1 can also inhibit transcription directly through binding to a TG‐rich sequence element via its homeobox domain. Inactivating mutations in TGIF1 cause autosomal dominant holoprosencephaly, 7 , 8 , 9 , 10 the most common inherited defect in forebrain development in humans.
In addition to its role in forebrain development, TGIF1 affects proliferation and differentiation of myeloid lineage cells by regulating cell cycle progression. 11 Also, we have shown that Tgif1 regulates self‐renewal of haematopoietic stem cells (HSCs). Loss of Tgif1 in mice increases quiescence in bone marrow HSCs and enhances long‐term repopulating activity without an effect on steady‐state haematopoiesis. 12 In MLL‐rearranged AML, which is typically aggressive and portends a poor prognosis, TGIF1 expression is decreased compared to AML cases characterized by other molecular lesions. 13 Moreover, enforced expression of TGIF1 in MLL‐AF9‐transduced leukaemia cells influences transcriptional networks regulated by MEIS1, another TALE family member, while the TGIF1:MEIS1 ratio predicts AML survival. These data suggest that altered TGIF1 function could alter outcomes for AML patients.
Here, we present data showing that in addition to its role in normal HSCs, Tgif1 also affects leukaemia‐initiating cell (LIC) function in AML. Loss of Tgif1 increases LIC frequency in both acute and chronic myeloid leukaemia mouse models, resulting in earlier disease relapse, reduced survival and treatment resistance.
2. MATERIALS AND METHODS
2.1. Mice and plasmids
Mice with a Tgif1 null mutation have been previously described. 12 C57BL/6J mice were purchased from the Jackson Laboratories (Bar Harbor, ME). All animal experiments were approved by the Animal Care and Use Committees of Vanderbilt University and University of Virginia. MSCV‐MLL‐AF9‐IRES‐GFP vector was provided by Dr Scott Armstrong (Boston Children's Hospital, Boston, MA).
2.2. Retrovirus production
Retroviral vectors were transfected into Phoenix ecotropic retroviral packaging cells using Lipofectamine 2000 according to the manufacturer's instructions. Forty‐eight hours after transfection, viral supernatants were collected, filtered and stored at −80°C.
2.3. Induction of myeloid leukaemia in mice
The MSCV‐MLL‐AF9‐IRES‐GFP retrovirus was used to induce AML in mice. Bone marrow cells were flushed from the tibias and femurs of Tgif1+/+ or Tgif1−/− mice and lineage marker‐negative (Lin‐) cells were enriched using the Mouse Lineage Cell Depletion kit (Miltenyi Biotec). Lin‐ cells purified from Tgif1+/+ or Tgif1−/− mice were then transduced using low‐speed centrifugation (spinoculation), with MSCV‐MLL‐AF9‐IRES‐GFP retrovirus in the presence of 8 μg/mL polybrene at 1350g × 45 minutes (32°C). Transduced Tgif1+/+ or Tgif1−/− Lin‐ cells were resuspended in phosphate‐buffered saline (PBS), and 1 × 105 cells were injected intravenously into sub‐lethally irradiated (4.5 Gy) Tgif1+/+ or Tgif1−/− recipient mice, respectively. Reconstitution of transduced cells in recipient mice was evaluated by monitoring GFP expression by flow cytometry analysis at 1‐2 weeks intervals following transplantation. Recipient mice were killed for analysis when they developed palpable splenomegaly or appeared ill. Bone marrow and spleen cells from leukaemic mice were isolated and stored frozen for later use.
MSCV‐BCR/ABL‐IRES‐GFP retrovirus was used to induce CML in mice. Bone marrow cells were flushed from tibias and femurs of Tgif1+/+, Tgif1−/− or Tgif1± mice, and single‐cell suspensions of Lin‐ c‐Kit+ cells were obtained using the Mouse Lineage Cell Depletion kit and CD117 microbeads (Miltenyi Biotec, San Diego, CA). Lin‐ c‐Kit+ cells were spinoculated with MSCV‐BCR/ABL‐IRES‐GFP retrovirus in the presence of 8 μg/mL polybrene for 45 minutes at 1350 g (32°C). Transduced cells were resuspended in PBS, and 8 × 104 cells were injected intravenously into lethally or sub‐lethally irradiated C57BL/6J recipient mice. Reconstitution of transduced cells in recipient mice was evaluated by flow cytometry analysis of GFP expression two weeks after transplantation. Mice were monitored for disease development, at which time they were killed.
2.4. Quantification of leukaemia‐initiating cell frequency
Leukaemia cells from spleens of three Tgif1+/+ or three Tgif1−/− mice with AML induced with MSCV‐MLL‐AF9‐IRES‐GFP retrovirus were pooled, and 1 × 106, 1 × 105, 1 × 104, 1 × 103, 100 and 50 cells were transplanted into sub‐lethally irradiated recipients. Animals were killed when they became visibly ill, and the development of leukaemia was confirmed. LIC frequency was determined by limiting dilution analysis with the ELDA (for Extreme Limiting Dilution Analysis) software package. 14
2.5. Chemotherapy studies in AML mice
For treatment studies, 2 × 103 splenic leukaemia cells from Tgif1+/+ or Tgif1−/− mice were transplanted into sub‐lethally irradiated recipients by tail vein injection. Two weeks after transplant, mice were treated by intraperitoneal injection with 100 mg/kg cytarabine once per day for five days and 3 mg/kg doxorubicin for three days. Mice were monitored closely for disease development, and GFP expression was evaluated weekly by flow cytometry analysis. Animals were killed when they appeared ill.
2.6. RNA sequencing (RNA‐seq) analysis of AML cells
RNA was extracted from sorted populations of GFP‐expressing leukaemia cells isolated from the bone marrow or spleen of Tgif1+/+ or Tgif1−/− mice with AML. RNA‐seq analysis was performed on leukaemia cells from three separate mice for each condition. Sequencing of RNA and subsequent bioinformatic analyses was performed by the Vanderbilt Technologies for Advanced Genomics (or VANTAGE) Shared Resource. Sequencing data were annotated with the University of California, Santa Cruz Genome Browser (https://genome.ucsc.edu/), and the TopHat and Cufflinks software packages were used for transcript alignment and quantification of gene expression, respectively. Gene set enrichment analysis 15 was carried out using the default weighted enrichment statistic to test whether differentially expressed genes (between Tgif1+/+ and Tgif1−/− LSK cells) were randomly distributed or enriched at the top or bottom of the gene list. A false discovery rate of ≤0.25 was considered significant. Gene ontology analysis was performed with Ingenuity Pathway Analysis (http://www.ingenuity.com/) to group functionally related genes and assign them to biological pathways.
3. RESULTS
3.1. Tgif1 gene loss decreased survival in an experimental model of acute myeloid leukaemia
We have previously shown that Tgif1 deletion promotes quiescence and self‐renewal activity in haematopoietic stem cells (HSC) in mice without disturbing steady‐state haematopoiesis. 12 Studies have also shown TGIF1 expression is decreased in MLL‐rearranged AML patients, favouring an anti‐leukaemic role. 13 Here, we sought to determine directly whether Tgif1 expression also affects leukaemic haematopoiesis in mice. We induced AML in mice by introduction of an MLL‐AF9 fusion cDNA in lineage‐negative haematopoietic stem and progenitor cells (Lin‐HSPCs) from Tgif1+/+ and Tgif1−/− mice. Transduced cells were transplanted into sub‐lethally irradiated recipients via tail vein injection. Development and progression of leukaemia in recipient mice was then monitored by flow cytometry analysis for green fluorescent protein (GFP)‐expressing nucleated cells in peripheral blood. Six weeks after transplant, mice that received MLL‐AF9‐transduced Tgif1−/− HSPCs showed higher numbers of GFP+ myeloid (CD11b+ and Gr‐1+) cells (Figure 1A,B) but lower numbers of B cells (Figure 1C) compared to those receiving similarly transduced Tgif1+/+ HSPCs. Necropsy showed that all mice with circulating donor‐derived leukaemia cells had splenomegaly, with massive infiltration of myeloblasts observed in both spleen and bone marrow (data not shown). These data suggest that Tgif1−/− HSPCs are more prone to AML compared to those with Tgif1+/+ HSPCs upon enforced expression of MLL‐AF9 fusion protein.
FIGURE 1.
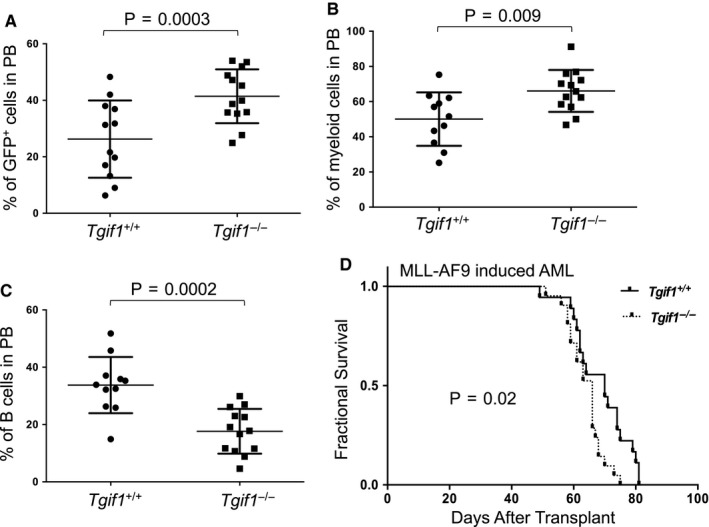
MLL‐AF9‐induced AML in Tgif1−/− mice was more aggressive with a shorter survival time than that in Tgif1+/+ mice. Lin‐ bone marrow cells (Lin‐HSPCs) from Tgif1+/+ or Tgif1−/− mice were transduced with MLL‐AF9‐GFP retrovirus and transduced cells transplanted into sub‐lethally irradiated Tgif1+/+ and Tgif1−/− recipient mice, respectively. Six weeks after transplant, the percentages of A, GFP+ cells; B, myeloid cells (CD11b+ and Gr‐1+) and C, B‐lymphocytes (B220+) in peripheral blood (PB) were analysed by flow cytometry. D, Kaplan‐Meier survival curves for indicated mice in MLL‐AF9‐induced AML
Kaplan‐Meier analysis showed that mice transplanted with MLL‐AF9‐transduced Tgif1−/− HSPCs survived for a shorter time than mice transplanted with transduced Tgif1+/+ HSPCs (Figure 1D). As mice transplanted with either Tgif1+/+ or Tgif1−/− HSPCs succumbed to leukaemia with very short latency, the aggressive nature of this leukaemia almost certainly underestimates the impact of Tgif1 loss on survival. Regardless, these results indicate loss of Tgif1 accelerates progression of MLL‐AF9‐induced AML in mice.
3.1.1. Tgif1 loss increases leukaemia‐initiating cell (LIC) frequency in MLL‐AF9‐induced AML
LICs are crucial for maintenance of AML in vivo. 16 , 17 , 18 Therefore, one possible explanation for shorter survival seen with Tgif1 loss could be increased numbers of LICs. To explore this question, we carried out limiting dilution analysis to quantify LIC functionally by transplanting serial numbers of MLL‐AF9‐transduced Tgif1+/+ or Tgif1−/− spleen cells from primary recipients into non‐conditioned wild‐type C57BL/6J mice. Indeed, our data showed a twofold higher frequency of LICs in Tgif1−/− mice with AML (1 in 125) (95% CI, lower: 1 in 279 and higher: 1 in 56.5) than in Tgif1+/+ mice (1 in 250) (95% CI, lower: 1 in 603 and higher: 1 in 103.6). These data suggest that Tgif1−/− leukaemic populations were enriched in LIC number, function or both. Consistent with this notion, mice transplanted with MLL‐AF9‐transduced Tgif1−/− leukaemia cells over a range of doses had inferior survival compared to those with similarly transplanted Tgif1+/+ cells (Figure 2A‐D).
FIGURE 2.
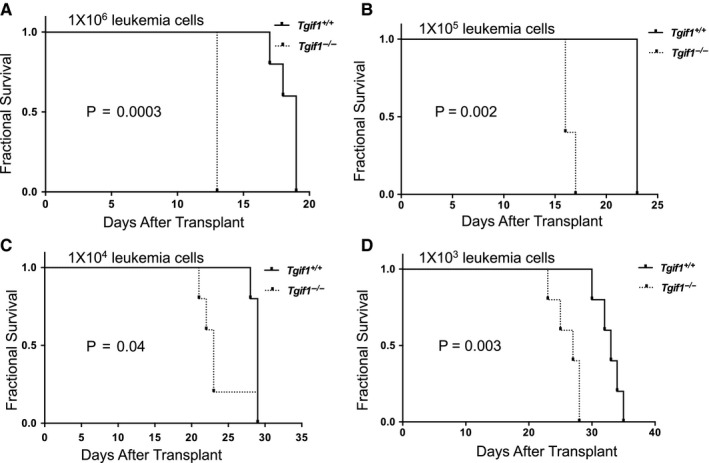
LSC frequency was increased in Tgif1−/− mice with AML compared to Tgif1 +/+ mice. Lin‐ bone marrow cells (Lin‐HSPCs) from Tgif1+/+ and Tgif1−/− mice were transduced with MLL‐AF9‐GFP retrovirus and transduced cells transplanted into sub‐lethally irradiated Tgif1+/+ and Tgif1−/− recipients, respectively. At a predetermined time, serial dilutions of spleen cells from mice with leukaemia then collected and transplanted into secondary recipients. Kaplan‐Meier analysis of survival is plotted as a function of number of transplanted spleen cells. A, 1 × 106; B, 1 × 105; C, 1 × 104; D, 1 × 103
3.1.2. Tgif1 loss results in earlier relapse and more aggressive disease
Next, we sought to investigate whether Tgif1 expression is correlated with time to AML relapse and/or overall survival after treatment with conventional agents used in AML. To simulate the clinical setting, we transplanted Tgif1+/+ and Tgif1−/−, MLL‐AF9‐transformed‐spleen cells from mice with established leukaemia into sub‐lethally irradiated recipients and treated these mice with cytarabine and doxorubicin chemotherapy as described. 19 Our data showed that GFP‐expressing cells appeared earlier and with higher numbers in peripheral blood of mice transplanted with Tgif1−/− leukaemia cells than mice transplanted with Tgif1+/+ leukaemia cells (Figure 3A). While chemotherapy treatment extended survival of both Tgif1+/+ and Tgif1−/− mice with AML (compare Figure 3B to Figure 2D), mice transplanted with Tgif1−/− leukaemia cells still had shorter survival than mice transplanted with Tgif1+/+ leukaemia cells (Figure 3B). Although selection at the level of a stem cell is not proven by these data, the differences in latency of disease and response to chemotherapy, we posit, are because of the higher frequency and/or greater fitness of LICs and more rapid expansion of leukaemia in Tgif1−/− mice.
FIGURE 3.
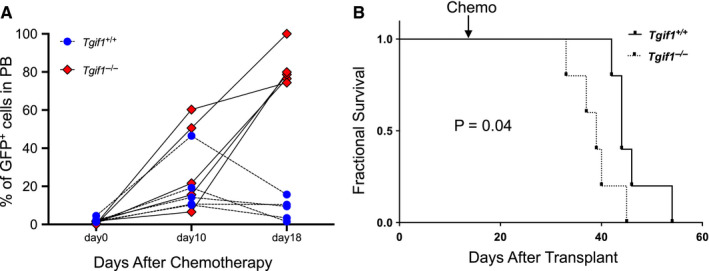
AML in Tgif1−/− mice progressed earlier after chemotherapy than in Tgif1+/+ AML mice. Sub‐lethally irradiated C57BL/6 mice were transplanted with spleen cells from Tgif1+/+ or Tgif1−/− mice with established MLL‐AF9‐induced AML, and two weeks following transplant, recipient mice were treated with doxorubicin for 3 d and cytarabine for 5 d by intraperitoneal injection. A, Percentage of GFP+ cells in peripheral blood were analysed using flow cytometry at day 0, day 10 and day18 after chemotherapy. B, Kaplan‐Meier analysis of survival of Tgif1+/+ and Tgif1−/− mice with AML after chemotherapy
3.1.3. Tgif1 loss decreases survival in a model of chronic myeloid leukaemia
We next investigated whether Tgif1 loss also affects leukaemic progression in a mouse model of CML. CML arguably has the best evidence supporting the existence of a malignant stem cell. To that end, Lin‐ HSPCs from Tgif1+/+ and Tgif1−/− mice were transduced with the BCR‐ABL‐GFP‐expressing retrovirus and transplanted into sub‐lethally or lethally irradiated recipients. After transplant, the kinetics of leukaemic progression in recipient mice was monitored using GFP‐expressing myeloid cells in peripheral blood (PB). Six weeks after transplant, mice receiving BCR‐ABL‐GFP‐expressing Tgif1−/− cells showed higher numbers of GFP+ cells in PB and HSPCs, including the Lin‐Sca1+c‐Kit+ (LSK) population enriched with haematopoietic stem and progenitor cells, compared to Tgif1+/+ mice (Figure 4A‐C, respectively). Kaplan‐Meier analysis revealed that mice transplanted with BCR‐ABL‐GFP‐transduced Tgif1−/− HSPCs showed significantly shorter survival than mice transplanted with BCR‐ABL‐GFP‐transduced Tgif1 +/+ HSPCs (Figure 4D). When recipient mice were conditioned with a lower dose of radiation, leukaemia developed with a longer latency; however, Tgif1 genotype still significantly impacted survival (Figure 4E). Interestingly, mice receiving BCR‐ABL‐transduced heterozygous (Tgif1±) HSPCs also had shorter survival (Figure 4F) paralleling the greater long‐term repopulating ability of Tgif1± HSCs compared to Tgif1+/+ HSCs. 12 Collectively, these data are compatible with mouse AML data and suggest Tgif1 expression impacts survival in CML.
FIGURE 4.
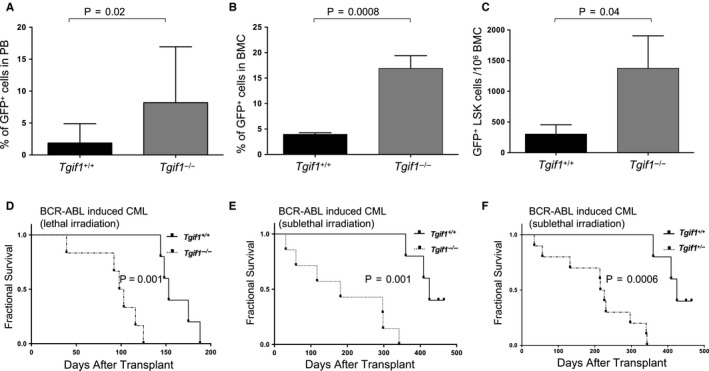
Mice with Tgif1−/− HSPCs developed more aggressive disease and had inferior survival compared to those with Tgif1+/+ HSPCs in BCR‐ABL‐induced CML. Enriched Lin‐ c‐Kit+ cells from Tgif1+/+, Tgif1−/− or Tgif1± bone marrow cells (HSPCs) were transduced with BCR‐ABL‐GFP retrovirus and transduced cells transplanted into lethally or sub‐lethally irradiated C57BL/6J recipients to induce CML. Six weeks after transplant, flow cytometry was used to analyse A, percentage of GFP+ cells in PB; B, percentage of GFP+ cells in bone marrow; C, number of GFP+ LSK cells in bone marrow. Recipient mice were D, lethally irradiated; E and F, sub‐lethally irradiated in fractional survival curves
3.1.4. Tgif1 loss affects multiple transcriptional networks in AML
To gain insight into the biological and molecular pathways affected by Tgif1 gene loss, we compared global gene expression profiles in Tgif1−/− and Tgif1+/+ myeloid leukaemia cells by unbiased mRNA sequencing. We identified at least 45 genes that were differentially expressed in these two populations—33 of these were up‐regulated and 12 were down‐regulated in Tgif1−/− relative to Tgif1+/+ leukaemia cells (Table 1).
TABLE 1.
Differentially expressed genes in Tgif1−/− vs Tgif1+/+ leukaemic cells
| ID | Symbol | Location | Log ratio |
|---|---|---|---|
| Slc15a2 | SLC15A2 | Plasma Membrane | −7.651 |
| Eps8l1 | EPS8L1 | Cytoplasm | −7.587 |
| Dlgap1 | DLGAP1 | Plasma Membrane | −3.719 |
| Gdf3 | GDF3 | Extracellular Space | −3.658 |
| Camk2b | CAMK2B | Cytoplasm | −3.619 |
| Col19a1 | COL19A1 | Extracellular Space | −3.488 |
| Kcnq5 | KCNQ5 | Plasma Membrane | −2.727 |
| Vill | VILL | Cytoplasm | −2.499 |
| Cgnl1 | CGNL1 | Plasma Membrane | −1.938 |
| Cdkn2c | CDKN2C | Nucleus | −1.649 |
| Gm15448 | LILRB3 | Plasma Membrane | −1.362 |
| Selm | SELM | Cytoplasm | −0.761 |
| Ly75 | LY75 | Plasma Membrane | 0.652 |
| Anxa3 | ANXA3 | Cytoplasm | 0.714 |
| Agap1 | AGAP1 | Cytoplasm | 0.742 |
| Ttc21a | TTC21A | Extracellular Space | 0.967 |
| Pip5k1b | PIP5K1B | Cytoplasm | 0.976 |
| B4galt6 | B4GALT6 | Cytoplasm | 1.029 |
| Ptgs1 | PTGS1 | Cytoplasm | 1.045 |
| Olfm4 | OLFM4 | Extracellular Space | 1.123 |
| Dsp | DSP | Plasma Membrane | 1.212 |
| Gfi1 | GFI1 | Nucleus | 1.278 |
| Plekha6 | Plekha6 | Other | 1.291 |
| Sept5 | SEPT5 | Cytoplasm | 1.301 |
| Optn | OPTN | Cytoplasm | 1.325 |
| Kcnh7 | KCNH7 | Plasma Membrane | 1.417 |
| Fcnb | FCN1 | Extracellular Space | 1.427 |
| S100a8 | S100A8 | Cytoplasm | 1.503 |
| Gca | GCA | Cytoplasm | 1.575 |
| Serpine2 | SERPINE2 | Extracellular Space | 1.875 |
| Camp | CAMP | Cytoplasm | 1.940 |
| Serpine1 | SERPINE1 | Extracellular Space | 2.160 |
| Retnlg | Retnlg | Extracellular Space | 2.167 |
| Cd74 | CD74 | Plasma Membrane | 2.609 |
| C1qb | C1QB | Extracellular Space | 3.161 |
| C1qc | C1QC | Extracellular Space | 3.269 |
| Adam12 | ADAM12 | Plasma Membrane | 3.167 |
| C1qa | C1QA | Extracellular Space | 3.503 |
| Mmp14 | MMP14 | Extracellular Space | 4.032 |
| Thbs4 | THBS4 | Extracellular Space | 4.578 |
| Bmp1 | BMP1 | Extracellular Space | 4.577 |
| Npr1 | NPR1 | Plasma Membrane | 5.607 |
| Mtus2 | MTUS2 | Other | 5.713 |
| Syce1 | SYCE1 | Nucleus | 7.905 |
| 4930447C04Rik | C14orf39 | Extracellular Space | 8.430 |
Ingenuity Pathway Analysis (IPA) allowed us to interrogate which upstream regulators contributed to gene expression changes. In this analysis, genes involved in TGF‐β signalling were significantly enriched (Figure 5). Other regulators identified by this analysis included all‐trans retinoic acid (ATRA) (P = 1.02 × 10−8) and serum response factor (SRF) (P = 5.34 × 10−6). Like TGF‐β targets, genes activated by ATRA were up‐regulated by Tgif1 loss, consistent a role for Tgif1 as a corepressor of retinoic acid receptor (RAR)‐dependent transcription.
FIGURE 5.
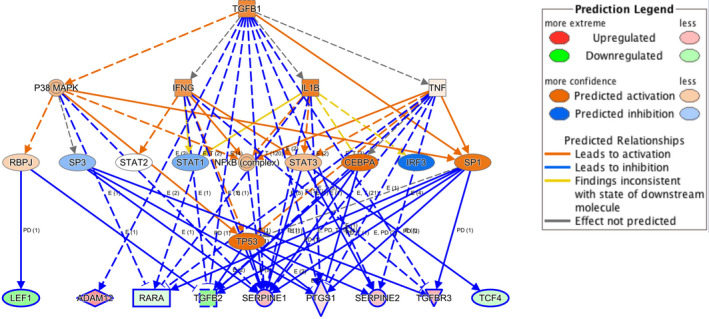
Upstream regulator analysis of differentially expressed genes. This analysis identified TGF‐β as a major regulator of differentially expressed genes (P = 8.52 × 10−10)
Genes important for embryonic stem cell pluripotency were highlighted in IPA canonical pathway analysis (P = 5.88 × 10−4) (Figure 6), while haematological system development and function (P = 1.05 × 10−7) were enriched in IPA physiological functions. Other functions impacted by Tgif1 expression included cellular movement (P = 1.99 × 10−9), leukocyte function (P = 3.87 × 10−8), cell death and survival (P = 5.38 × 10−8), myeloid cell function (P = 3.24 × 10−8) and cancer (P = 2.63 × 10−7). Together, pathway analysis reveals altered regulation of TGF‐β signalling and RA signalling in Tgif1−/− leukaemia cells, with potentially important consequences for LICs function.
FIGURE 6.
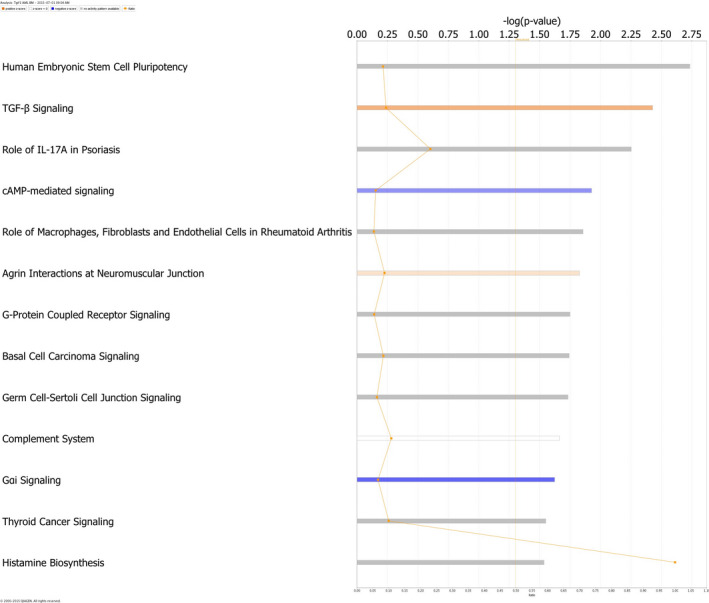
Canonical Pathway Analysis. Depicted are differentially utilized pathways in leukaemia cells from Tgif1−/− mice compared to Tgif1+/+ mice
4. DISCUSSION
We have previously shown that inactivation of the Tgif1 gene in mice, complete or partial, increases quiescence in bone marrow HSCs and enhances long‐term repopulating activity without effecting steady‐state haematopoiesis. 12 We have also shown that TGIF1 affects proliferation and differentiation of myeloid cell lines. 11 Here, we sought to understand the role of Tgif1 in mouse leukaemic haematopoiesis.
We find that loss or haploinsufficiency of Tgif1 accelerates leukaemia development and shortens survival time in mouse models of MLL‐AF9‐induced AML and BCR‐ABL‐induced CML. AML in mice transplanted with oncogene‐transduced Tgif1−/− HSPCs shows an impaired response to conventional chemotherapy agents used to treat myeloid malignancy and correlates with earlier relapse and shorter survival compared to similarly transduced Tgif1+/+ controls. In light of studies demonstrating that LICs frequency at diagnosis in AML correlates with increased minimal residual disease and poor survival, 20 our finding that the Tgif1 knockout enhances LIC functionality by increasing LIC frequency provides an explanation for how Tgif1 expression affects AML recurrence in our model. Our results are well aligned with data showing that enforced expression of TGIF1 decreases human leukaemia cell proliferation, induces terminal differentiation and increases survival in MLL‐AF9 rearranged myeloid leukaemia. 13
In accordance with the transcriptional repression functions of TGIF1, mRNA expression profiling identified a number of genes which were up‐regulated in Tgif1‐null leukaemic cells, with almost half the genes differentially expressed between knockout and wild‐type leukaemia cells involved directly or indirectly in TGF‐β signalling (Figure 5). These results corroborate the enhanced TGF‐β signalling and/or target gene expression noted in epithelial 6 and myeloid leukaemia cells 11 with diminished TGIF1 expression, and align with roles for TGF‐β in HSC self‐renewal, 21 quiescence 22 and enhanced leukaemic stem cell function. 21 , 35
Loss of Tgif1 function also relieves repression of retinoic acid receptor (RAR) target genes, consistent with its ability to act as a corepressor of RAR/RXR‐mediated transcription. Finally, our results do not exclude direct effects of Tgif1 on gene expression or indirect effects through interaction with other TALE homeodomain proteins such as MEIS1 13 As chemical inhibitors are available for TGF‐β and RAR signalling, it should be possible to dissect the unique contributions of these pathways to the altered Tgif1 functions in leukaemia development.
Although rare loss‐of‐function variants in TGIF1 are associated with holoprosencephaly, 9 , 10 these same TGIF1 variants have not been observed in AML patients. Furthermore, individuals with holoprosencephaly and Tgif1‐null mice are not at increased risk for development of myeloid leukaemia, all of which argues against a function for this protein as a tumour suppressor. Interestingly Means‐Powell et al reported that TGIF1 levels were an independent predictor of survival in AML, with lower levels associated with earlier relapse and poor survival 36 (personal communication). Taken together, these data may suggest that TGIF1 acts as a prototypical stem cell modifier gene, operating not in the initiation of leukaemia but in disease progression and persistence.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTION
Ling Yan: Data curation (lead); Investigation (lead); Methodology (lead); Project administration (lead); Writing‐original draft (lead); Writing‐review & editing (equal). Utpal Dave: Investigation (supporting); Methodology (supporting); Writing‐review & editing (supporting). Michael Engel: Data curation (supporting); Methodology (supporting); Writing‐review & editing (supporting). Stephen Brandt: Data curation (supporting); Project administration (supporting); Writing‐review & editing (supporting). Rizwan Hamid: Data curation (supporting); Funding acquisition (lead); Project administration (supporting); Supervision (lead); Writing‐review & editing (lead).
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health grants K08 HL089903 (to RH) and P30 CA068485 (to Vanderbilt‐Ingram Cancer Center) and American Cancer Society Scholar Award RSG LIB‐118462 (to R.H).
Yan L, Davé UP, Engel M, Brandt SJ, Hamid R. Loss of TG‐Interacting Factor 1 decreases survival in mouse models of myeloid leukaemia. J Cell Mol Med. 2020;24:13472–13480. 10.1111/jcmm.15977
REFERENCES
- 1. Hamid R, Patterson J, Brandt SJ. Genomic structure, alternative splicing and expression of TG‐interacting factor, in human myeloid leukemia blasts and cell lines. Biochim Biophys Acta. 2008;1779(5):347‐355. [DOI] [PubMed] [Google Scholar]
- 2. Wotton D, Knoepfler PS, Laherty CD, Eisenman RN, Massague J. The Smad transcriptional corepressor TGIF recruits mSin3. Cell Growth Differ. 2001;12(9):457‐463. [PubMed] [Google Scholar]
- 3. Wotton D, Lo RS, Lee S, Massague J. A Smad transcriptional corepressor. Cell. 1999;97(1):29‐39. [DOI] [PubMed] [Google Scholar]
- 4. Wotton D, Lo RS, Swaby LA, Massague J. Multiple modes of repression by the Smad transcriptional corepressor TGIF. J Biol Chem. 1999;274(52):37105‐37110. [DOI] [PubMed] [Google Scholar]
- 5. Bertolino E, Reimund B, Wildt‐Perinic D, Clerc RG. A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid‐responsive motif. J Biol Chem. 1995;270(52):31178‐31188. [DOI] [PubMed] [Google Scholar]
- 6. Seo SR, Lallemand F, Ferrand N, et al. The novel E3 ubiquitin ligase Tiul1 associates with TGIF to target Smad2 for degradation. EMBO J. 2004;23(19):3780‐3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ming JE, Kaupas ME, Roessler E, et al. Mutations in PATCHED‐1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet. 2002;110(4):297‐301. [DOI] [PubMed] [Google Scholar]
- 8. Ming JE, Muenke M. Multiple hits during early embryonic development: digenic diseases and holoprosencephaly. Am J Hum Genet. 2002;71(5):1017‐1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wallis D, Muenke M. Mutations in holoprosencephaly. Hum Mutat. 2000;16(2):99‐108. [DOI] [PubMed] [Google Scholar]
- 10. Wallis DE, Muenke M. Molecular mechanisms of holoprosencephaly. Mol Genet Metab. 1999;68(2):126‐138. [DOI] [PubMed] [Google Scholar]
- 11. Hamid R, Brandt SJ. Transforming growth‐interacting factor (TGIF) regulates proliferation and differentiation of human myeloid leukemia cells. Mol Oncol. 2009;3(5‐6):451‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yan L, Womack B, Wotton D, et al. Tgif1 regulates quiescence and self‐renewal of hematopoietic stem cells. Mol Cell Biol. 2013;33(24):4824‐4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Willer A, Jakobsen JS, Ohlsson E, et al. TGIF1 is a negative regulator of MLL‐rearranged acute myeloid leukemia. Leukemia. 2015;29(5):1018‐1031. [DOI] [PubMed] [Google Scholar]
- 14. Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1‐2):70‐78. [DOI] [PubMed] [Google Scholar]
- 15. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kirstetter P, Schuster MB, Bereshchenko O, et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia‐initiating cells. Cancer Cell. 2008;13(4):299‐310. [DOI] [PubMed] [Google Scholar]
- 17. Shlush LI, Zandi S, Mitchell A, et al. Identification of pre‐leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taussig DC, Vargaftig J, Miraki‐Moud F, et al. Leukemia‐initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(‐) fraction. Blood. 2010;115(10):1976‐1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zuber J, Radtke I, Pardee TS, et al. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009;23(7):877‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Rhenen A, Feller N, Kelder A, et al. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res. 2005;11(18):6520‐6527. [DOI] [PubMed] [Google Scholar]
- 21. Pierelli L, Marone M, Bonanno G, et al. Transforming growth factor‐beta1 causes transcriptional activation of CD34 and preserves haematopoietic stem/progenitor cell activity. Br J Haematol. 2002;118(2):627‐637. [DOI] [PubMed] [Google Scholar]
- 22. Yamazaki S, Iwama A, Takayanagi S, Eto K, Ema H, Nakauchi H. TGF‐beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood. 2009;113(6):1250‐1256. [DOI] [PubMed] [Google Scholar]
- 23. Falk LA, De Benedetti F, Lohrey N, et al. Induction of transforming growth factor‐beta 1 (TGF‐beta 1), receptor expression and TGF‐beta 1 protein production in retinoic acid‐treated HL‐60 cells: possible TGF‐beta 1‐mediated autocrine inhibition. Blood. 1991;77(6):1248‐1255. [PubMed] [Google Scholar]
- 24. Imai Y, Kurokawa M, Izutsu K, et al. Mutations of the Smad4 gene in acute myelogeneous leukemia and their functional implications in leukemogenesis. Oncogene. 2001;20(1):88‐96. [DOI] [PubMed] [Google Scholar]
- 25. Kim SJ, Letterio J. Transforming growth factor‐beta signaling in normal and malignant hematopoiesis. Leukemia. 2003;17(9):1731‐1737. [DOI] [PubMed] [Google Scholar]
- 26. Krause DS, Fulzele K, Catic A, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med. 2013;19(11):1513‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kurokawa M, Mitani K, Imai Y, Ogawa S, Yazaki Y, Hirai H. The t(3;21) fusion product, AML1/Evi‐1, interacts with Smad3 and blocks transforming growth factor‐beta‐mediated growth inhibition of myeloid cells. Blood. 1998;92(11):4003‐4012. [PubMed] [Google Scholar]
- 28. Kurokawa M, Mitani K, Irie K, et al. The oncoprotein Evi‐1 represses TGF‐beta signalling by inhibiting Smad3. Nature. 1998;394(6688):92‐96. [DOI] [PubMed] [Google Scholar]
- 29. Marone M, Scambia G, Bonanno G, et al. Transforming growth factor‐beta1 transcriptionally activates CD34 and prevents induced differentiation of TF‐1 cells in the absence of any cell‐cycle effects. Leukemia. 2002;16(1):94‐105. [DOI] [PubMed] [Google Scholar]
- 30. Murohashi I, Endho K, Nishida S, et al. Differential effects of TGF‐beta 1 on normal and leukemic human hematopoietic cell proliferation. Exp Hematol. 1995;23(9):970‐977. [PubMed] [Google Scholar]
- 31. Naka K, Hoshii T, Muraguchi T, et al. TGF‐beta‐FOXO signalling maintains leukaemia‐initiating cells in chronic myeloid leukaemia. Nature. 2010;463(7281):676‐680. [DOI] [PubMed] [Google Scholar]
- 32. Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF‐beta in homeostasis and cancer. Nat Rev Cancer. 2003;3(11):807‐821. [DOI] [PubMed] [Google Scholar]
- 33. Sitnicka E, Ruscetti FW, Priestley GV, Wolf NS, Bartelmez SH. Transforming growth factor beta 1 directly and reversibly inhibits the initial cell divisions of long‐term repopulating hematopoietic stem cells. Blood. 1996;88(1):82‐88. [PubMed] [Google Scholar]
- 34. Soucek K, Pachernik J, Kubala L, Vondracek J, Hofmanova J, Kozubik A. Transforming growth factor‐beta1 inhibits all‐trans retinoic acid‐induced apoptosis. Leuk Res. 2006;30(5):607‐623. [DOI] [PubMed] [Google Scholar]
- 35. Tabe Y, Shi YX, Zeng Z, et al. TGF‐beta‐Neutralizing Antibody 1D11 Enhances Cytarabine‐Induced Apoptosis in AML Cells in the Bone Marrow Microenvironment. PLoS One. 2013;8(6):e62785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Means‐Powell JA, Kravtsov VSY, Levy SE, Greer JP, Koury MJ, Brandt S. Expression of homeobox gene TG‐interaction factor is an independent predictor of survival in acute myelogenous leukemia. 45th Am Soc Hematol Meet. 2003;764:p128a. [Google Scholar]