

Abstract
The presence of liver metastases drastically worsens the prognosis of cancer patients. The liver is the second most prevalent metastatic site in cancer patients, but systemic therapeutic opportunities that target liver metastases are still limited. To aid the discovery of novel treatment options for metastatic liver disease, we provide insight into the cellular and molecular steps required for liver colonization. For successful colonization in the liver, adaptation of tumor cells and surrounding stroma is essential. This includes the formation of a pre‐metastatic niche, the creation of a fibrotic and immune suppressive environment, angiogenesis, and adaptation of tumor cells. We illustrate that transforming growth factor β (TGF‐β) is a central cytokine in all these processes. At last, we devise that future research should focus on TGF‐β inhibitory strategies, especially in combination with immunotherapy. This promising systemic treatment strategy has potential to eliminate distant metastases as the efficacy of immunotherapy will be enhanced.
Keywords: cancer, immunotherapy, liver metastasis, targeted therapy, transforming growth factor beta, tumor microenvironment
The liver is a permissive organ for metastasis, but adaptation of both liver environment and tumor cells is necessary for metastatic outgrowth. During liver metastasis formation, the cytokine TGF‐β promotes hepatic colonization by affecting the tumor cells, as well as tumor stroma, vasculature, and immune cells. Targeting TGF‐β, in combination with immunotherapy, has the potential to eliminate liver metastasis.
Abbreviations
- HSC
hepatic stellate cell
- aHSC
activated hepatic stellate cell
- BMDC
bone marrow derived cell
- CAF
cancer associated fibroblasts
- CCL
C‐C motif chemokine ligand
- CCR1
C‐C chemokine receptor type 1
- CRC
colorectal cancer
- CTGF
connective tissue growth factor
- CXCR4
C‐X‐C chemokine receptor type 4
- ECM
extracellular matrix
- EMT
epithelial to mesenchymal transition
- ERK
extracellular signal‐regulated kinases
- FGF
fibroblast growth factor
- FOXp3
Forkhead box p3
- FSP1
fibroblast‐specific protein
- HCC
hepatocellular carcinoma
- HGF
hepatocyte growth factor
- HIF‐1α
hypoxia inducible factor ‐1alpha
- ICAM‐1
intracellular adhesion molecule
- IFN‐γ
interferon gamma
- IL‐6
interleukin 6
- IQGAP1
IQ motif containing GTPase activating protein 1
- JNK
c‐Jun N‐terminal kinases
- LAP
latency associated peptide
- LCC
Lewis lung carcinoma
- LOX
lysyl oxidase
- LSEC
liver sinusoid endothelial cell
- MAPK
mitogen‐activated protein kinase
- MDSC
myeloid derived suppressor cell
- MIF
macrophage migration inhibitory factor
- MMP
matrix metalloprotease
- MSC
mesenchymal stem cell
- NK‐cell
natural killer cell
- PD‐(L)1
programmed cell death (ligand) 1
- PDAC
pancreatic ductal adenocarcinoma
- PDGF
platelet derived growth factor
- PKA
protein kinase A
- Rock
rho‐associated coiled‐coil kinase
- SDF
stromal derived factor
- SMAD
mothers against decapentaplegic homolog
- Smurf
smad ubiquitination regulatory factor
- Spr1
sphingosine‐1‐phosphate receptor 1
- STAT3
signal transducer and activator of transcription
- TAM
tumor associated macrophage
- TGF‐β
transforming growth factor beta
- TRAF
TNF receptor associated factor
- TβRI/II
TGF‐β type I/II receptor
- VEGF‐A
vascular endothelial growth factor‐A
- YAP
yes‐associated protein
- α‐SMA
alpha‐smooth muscle actin
1. INTRODUCTION
Cancer is one of the leading causes of death worldwide. 1 The development of cancer metastasis drastically worsens the prognosis of cancer patients and contributes to the majority of cancer related deaths. 2 Despite many developments in anticancer therapy, metastases remain hard to treat.
The metastatic cascade starts at the primary tumor site, where tumor cells can gain invasive characteristics, allowing penetration of tumor basement membrane and intravasation into the vasculature. Traveling through the vasculature, tumor cells can reach secondary sites, extravasate, and colonize the secondary organ, depicted in Figure 1A. 3 Previous research has shown that many tumor cells disseminate to distant organs, however, very few survive in distant niches. 4 As such, colonization can be seen as the bottleneck for metastasis. Adaptation of the tumor cells as well as the microenvironment is essential to overcome this bottleneck.
FIGURE 1.
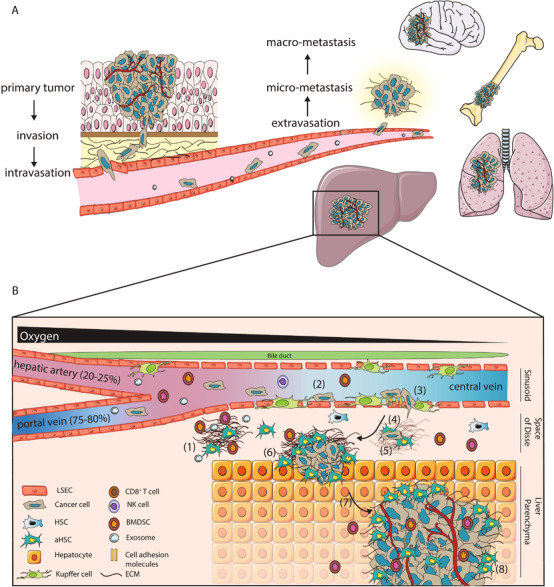
The metastatic cascade and formation of liver metastasis. A, Metastasis is a multistep process starting with invasive cells at the primary tumor site that invade the basement membrane and intravasate into the vasculature. After traveling through the vasculature, tumor cells can extravasate to form micrometastases and eventually macrometastases in secondary tumor sites such as bone, lungs, brain and liver. B, Colonization of the liver can be divided into (at least) eight different steps. Prior to arrival of tumor cells, pre‐metastatic niches can be created to favor tumor cell through secreted factors and extracellular vesicles by primary tumors (1). Tumor cells can arrive in the liver through the portal vein or hepatic artery, where after they arrest in the liver sinusoid (2). Upon extravasation into the Space of Disse (3), niche remodeling (4) is essential for survival and overcoming latency (5). When favorable conditions allow tumor cells to grow out into micrometastasis (6), angiogenesis (7) can allow the formation of macrometastasis (8). Abbreviations: LSEC, liver sinusoid endothelial cell; (a)HSC, (activated) hepatic stellate cell; ECM, extracellular matrix; NK‐cell, natural killer cell; BMDSC, bone marrow derived suppressor cells
Sites of metastasis can be influenced by both anatomical and niche characteristics, as proposed by two theories; Ewing's size constraint theory and Paget's seed and soil theory. 5 , 6 The liver is one of the most prevalent metastatic sites for many cancers, due to its anatomical location and blood supply through the portal vein and the hepatic artery, as well as its permissive microenvironment. 7 , 8 Colonization of the liver can be divided into (at least) eight distinct phases, as shown in Figure 1B, starting with induction of a pre‐metastatic niche prior to tumor cell arrival, to establish liver tropism and promoting later outgrowth of tumor cells (1). 6 , 9 Upon arrival, tumor cells arrest in the liver sinusoids or terminal portal venules (2), followed by extravasation into the Space of Disse (3). When tumor cells resist or evade local anticancer immune responses, a supportive niche needs to be created through remodeling the local microenvironment and recruiting stromal cells (4). Adaptation of the local niche is essential for development into the next phases. After a possible latency period (5), tumor cells have to divide to form micrometastases (6). When angiogenesis is successfully induced (7), full blown macrometastases can be formed (8). 8 , 10
In order to therapeutically target the bottleneck of metastasis, it is essential to understand the underlying signaling processes that allow for niche adaptation and tumor cell adaptation. The regulatory cytokine transforming growth factor beta (TGF‐β) has been shown to be of key importance in metastasis formation, both in tumor cells and different aspects of the tumor niche. 11 , 12 In cancer, TGF‐β can elicit a dual role, suppressing tumor growth in early phases, in contrast to functioning as a tumor promotor in later phases. 12 In addition, TGF‐β is involved in many processes in epithelial, endothelial, and neural tissues, the immune system, and wound repair. 13 All TGF‐β family members have multiple functions and the consequence of their activation is highly dependent on the cellular context. TGF‐β has pivotal roles in maintaining homeostasis in the liver and is expressed in various degrees by cells in the liver. 14 , 15 For example, TGF‐β aids to liver regeneration, however, high levels of TGF‐β signaling can lead to liver fibrosis. 15 Because of this complex multifaceted role of TGF‐β in both liver microenvironmental cells and tumor cells, and because of its expression in the liver by multiple cell types, TGF‐β can have a leading role in liver metastasis formation and can pose as a suitable target for anti‐cancer treatment, tackling multiple facets of metastasis formation.
In this review, we will give an overview of the processes involved in liver metastasis and highlight the current knowledge on the contribution of TGF‐β to liver metastasis formation. Recent progress in TGF‐β targeting, especially in combination with immunomodulatory therapy, will be reflected and put in context of the different layers of complexity of TGF‐β signaling during liver metastasis formation. Taking multiple angles of TGF‐β signaling into account, recommendations will be made for future therapeutic intervention of liver metastasis.
2. METASTASIS FORMATION IN THE LIVER
2.1. The liver is a common site of metastasis for different cancer types
Due to the drainage of venous blood from the abdomen to the portal vein, gastrointestinal cancers such as colorectal and pancreatic cancer most frequently metastasize to the liver. 7 , 16 Other cancers regularly presenting liver metastasis are, for example, breast cancer, (uveal) melanomas, pulmonary cancers, and sarcomas. 16 , 17 , 18 , 19 Surgery aiming for curative resection is the main treatment option for resectable liver metastases. 17 , 20 , 21 Additionally, systemic treatment or local treatments such as ablation and liver perfusion with anticancer treatment can be of benefit. 22 , 23 Unfortunately, few patients are eligible for surgical resection, and chances of recurrence can increase after surgery. 24 , 25
2.2. The liver has a complex structure consisting of specialized cell types
The liver has unique anatomical properties consisting of specialized cells to execute the many functions of the liver (Figure 1B). 26 In the liver, oxygen rich blood from the hepatic artery is mixed with venous blood from the portal vein in sinusoids, which is surrounded by the Space of Disse. 26 , 27 Hepatocytes represent the largest cell population in the liver and are responsible for most metabolic and detoxifying functions together with cholangiocytes, the epithelial layer lining bile ducts. Hepatic stellate cells (HSCs) are present in the Space of Disse in a quiescent form and function as storage for retinol esters in lipid droplets. Local damage signals can lead to HSC activation (aHSC), that is, myofibroblasts characterized by alpha smooth muscle actin (α‐SMA) expression, resulting in HSC proliferation and secretion of different cytokines and extracellular matrix (ECM). 28 HSCs are the main source of myofibroblasts, however hepatocytes, portal fibroblasts, bone marrow derived fibroblasts, and cholangiocytes can differentiate into myofibroblasts as well. 29 Specialized liver macrophages, Kupffer cells, represent the majority of the immune cell population in the liver and are located in the sinusoids. Liver sinusoid endothelial cells (LSEC) are specialized to maximize blood flow exchange and contain fenestrae. 26 , 30 , 31 Blood flow is kept low in the sinusoids. The presence of fenestrae and low blood flow, together with the supply of both venous and arterial blood, make the liver anatomically a permissive organ for metastasis. Moreover, the different stromal cell populations in the liver can provide favorable niches by secreting adhesion molecules, cytokines, growth factors, and extracellular matrix (modulating) proteins, promoting tumor cell adhesion, survival, growth, and angiogenesis.
2.3. The different phases of liver colonization
Colonization of the liver can be divided in distinct phases (Figure 1B). 10 , 32 Dynamic interaction between tumor cells and the local microenvironmental during these phases is key for successful colonization. The role of individual cells in the liver microenvironment during colonization is described in detail by 8 , 10 and is summarized below.
2.3.1. Step 1: Pre‐metastatic niche facilitates metastatic outgrowth
Recent research has shown the presence of pre‐metastatic niches that exist at distant sites prior to the formation of metastases. 6 , 9 , 33 Even though the existence of the pre‐metastatic niche is hard to prove in a human setting, it can be recognized as the first step of liver colonization. Primary tumors can secrete cytokines and extracellular vesicles containing, for example, microRNAs, integrins, and cytokines, which can modulate distant niches including the liver, thereby promoting metastatic outgrowth. For example, by promoting fibrosis or recruitment of myeloid or MDSC to the liver. 34 , 35 Integrins in exosomes can influence tropism of metastasis through preferential binding to specific organs. For example, integrin β5‐expressing exosomes were found to specifically adhere to Kupffer cells in fibronectin high liver tissue, where they induce upregulation of pro‐migratory and pro‐inflammatory S100 proteins. 6 Exosome‐derived integrins have also been shown to activate HSCs to induce a pro‐inflammatory environment in the liver, and stimulating stemness and epithelial‐to‐mesenchymal transition (EMT) in later colorectal cancer (CRC) metastases. 36
In conclusion, the liver niche can be adapted to favor tumor cell survival prior to tumor cell arrival through secretion of cytokines and exosomes by the primary tumor.
2.3.2. Steps 2 and 3: Tumor cell arrest in liver sinusoids and extravasation
Due to the hostile environment of the blood, including hemodynamic forces and immune cell clearance, most disseminated tumor cells will be eliminated in the circulation. 3 Upon successful arrival of tumor cells in liver sinusoids, cells encounter the first line of defense of the innate immune system of the liver. Kupffer cells can eliminate tumor cells through phagocytosis, which can be promoted by other recruited inflammatory cells, including natural killer (NK) cells. 8 Local NK cells and CD8+ T cells can induce apoptosis of tumor cells through FAS ligand/receptor binding, by forming perforin pores and stimulating granzyme release. 8 , 37 Tumor cells can resist or evade local anticancer immune responses through, for example, downregulation of the major histocompatibility complex class 1 or arriving as tumor cell clumps. 38 Interestingly, when large quantities of tumor cells arrive in the liver, Kupffer cells can switch to a tumor promoting role, secreting different tumor supportive cytokines including hepatocyte growth factor (HGF) and IL‐6. 8 , 39
Next to the local immune system, tumor cells encounter LSECs upon their arrival. LSECs are rich in cell adhesion proteins and surface oligosaccharides, facilitating the adherence of tumor cells, and stimulating the survival and migratory capacity of tumor cells. 40 , 41 Cytokines, secreted by Kupffer cells as well as by arrested tumor cells, can lead to increased adhesion and extravasation of the tumor cells to the sinusoid endothelium, through induced expression of certain adhesion molecules on LSECs, including E‐selectin, vascular cell adhesion molecule‐1, and intracellular adhesion molecule (ICAM)‐1. 39 , 42 , 43 , 44 , 45 , 46 , 47 Thus, conditions upon tumor cell arrival in the liver can be modulated to promote tumor cell survival and extravasation into the organ. 39
2.3.3. Steps 4‐6: Adaptation to local niche leads to micrometastasis formation
Once extravasated, tumor cells arrive in the Space of Disse. Here, a large number of extravasated tumor cells stay in dormancy, although it is not required. 4 Progression to micrometastasis formation is dependent on the evasion of the immune system and the induction of secretion of ECM (remodeling) proteins, growth factors, and cytokines by myofibroblasts. This will promote tumor cell survival, invasion, growth, and create a favorable immune suppressive environment. 28 , 29 , 48 , 49 Through tumor‐stroma crosstalk, tumor cells are able to recruit and activate surrounding liver microenvironmental cells into myofibroblasts. 10 , 28 , 32 , 48 , 50 , 51 TGF‐β signaling is key in this crosstalk. Activation of HSCs into aHSCs leads to secretion of cytokines, including platelet derived growth factor (PDGF), HGF, TGF‐β, stromal derived factor (SDF), ECM proteins fibronectin and collagen, and matrix metalloproteases (MMPs). 28 , 48 , 49 This matrix deposition can induce tumor cell growth and could provide a physical barrier for the immune system, leading to exclusion of CD8+ T cells. 52 , 53 Collagen‐1 deposition has also been shown to be essential to release cells from latency. 54 Secreted cytokines can induce pro‐survival and migration pathways in tumor cells and modulate the immune microenvironment. 49 Thus, activation of stromal cells, mainly HSCs, by tumor cells and tumor‐stroma crosstalk is an important step to adapt the local niche to form micrometastasis.
2.3.4. Steps 7 and 8: Overcoming hypoxia through angiogenesis
To overcome hypoxia induced size restriction of micrometastasis, angiogenesis is required. 55 Interplay between the established niche and tumor cells is important to induce angiogenesis. Hypoxic cells will secrete angiogenic factors including vascular endothelial growth factor (VEGF)A. VEGFA mediates endothelial cell migration, promoting formation of new blood vessels. This could be observed in liver micrometastases containing high numbers of VEGFA‐secreting aHSCs. 56 Indeed, aHSCs were found to mediate LSEC vessel formation in vitro. 57 Recently, a crosstalk between LSECs, HSCs, and tumor cells was elucidated in the context of stroma activation and angiogenesis. 47 ICAM‐1 expressed on LSECs promoted secretion of IL‐6, prostaglandin E2, VEGF, and MMP2 by tumor cells, which in turn induced VEGFA and MMP2 secretion by HSCs. These cytokines stimulated migration and angiogenic potential of LSECs and HSCs. 47 In addition to aHSCs and tumor cells, neutrophils from liver metastases were also shown to express pro‐angiogenic factors including fibroblast growth factor 2 (FGF2). Depletion of FGF2 impaired vascular structure in liver metastases. 58 Thus, a variety of cell types can aid in metastatic colonization by secreting pro‐angiogenic factors. Once new blood vessels are formed, micrometastasis can overcome their growth restraints by hypoxia and develop into macrometastases.
2.4. TGF‐β can mediate adaptation of tumor and stroma cells to overcome bottleneck of metastasis
The efficiency of liver colonization is dependent on the adaptation of tumor cells and the interaction between tumor cells and liver microenvironmental cells to induce stromal activation, angiogenesis, and immune suppression. 32 While metastases will differ between patients and cancer types, processes leading to overcoming the metastatic bottleneck and establishment of liver metastasis could be shared, and therefore targeted. A key cytokine that is involved in all these processes necessary for efficient hepatic colonization is TGF‐β. 12 , 13 Indeed, liver metastases have been found to be dependent on TGF‐β signaling in liver stroma, while TGF‐β signaling in tumor cells promotes invasion and outgrowth. 59 , 60 , 61 , 62
3. TGF‐β SIGNALING
TGF‐β family members play essential roles in epithelial, endothelial, tissue, and immune cells, where they regulate processes such as cellular proliferation, survival, migration, and differentiation. The TGF‐β family consists of three mammalian TGF‐β isoforms, TGF‐β1, TGF‐β2, and TGF‐β3, and structurally and functionally related proteins. 63 All family members have multiple functions, which are highly dependent on the cellular context. 13 Signaling of TGF‐β cytokines occurs through canonical mothers against decapentaplegic homolog (SMAD) signaling and through crosstalk with a multitude of non‐canonical signaling pathways. Figure 2 gives an overview of the canonical and non‐canonical signaling pathways induced by TGF‐β, as well as its pleiotropic effects in different cell types involved during liver metastasis formation.
FIGURE 2.
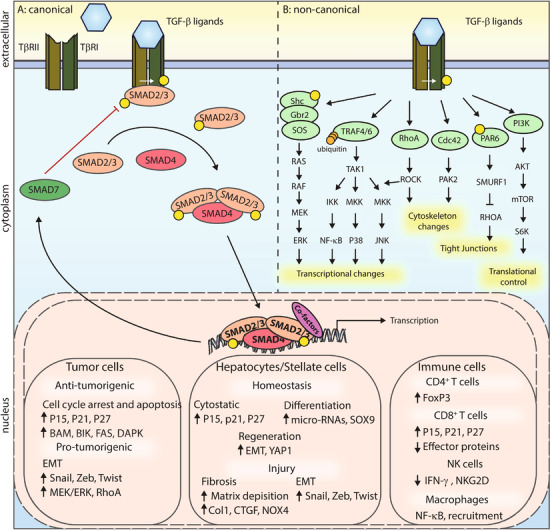
Canonical and non‐canonical TGF‐β signaling. Transforming growth factor (TGF)‐β ligands initially bind to TGF‐β receptor(TβR)II, and thereafter recruit TβRI. A heteromeric TGF‐β receptor complex is formed in which the TβRII kinase transphosphorylates TβRI. A, In canonical TGF‐β signaling, intracellular effectors mothers against decapentaplegic homolog (SMAD)2 and SMAD3 are phosphorylated by TβRI, which thereafter can bind to SMAD4. This complex translocates into the nucleus, and together with other co‐factors, regulates the expression of TGF‐β target genes in a context dependent manner. A negative feedback loop is created through induced expression of inhibitory SMAD7, which by partnering with Smad ubiquitination regulatory factor(SMURF) E3 ubiquitin ligases, targets the TβRI for ubiquitin‐mediated degradation. B, Non‐canonical TGF‐β signaling includes multiple different signaling pathways that are not unique for TGF‐β signaling, an overview is given in this figure. TGF‐β can elicit different responses in different cell types, depending on the cellular context. In tumor cells, TGF‐β/SMAD pathway can induce cytostatic effects through upregulation of cell cycle arrest and apoptosis genes. Pro‐tumorigenic effects of TGF‐β occur through SMAD‐dependent upregulation of EMT transcription factors and also non‐canonical signaling play a role. In hepatic and stellate cells, TGF‐β can induce cytostatic effects, but also differentiation and regeneration. During injury, TGF‐β signaling promotes fibrosis through e.g. matrix deposition and stimulating EMT. In immune cells, TGF‐β dampens immune responses. Cytotoxic effects of CD8+ T cells and natural killer (NK) cells are repressed, while regulatory CD4+ T cells and (immune suppressive) macrophages are promoted
3.1. Canonical and non‐canonical TGF‐β signaling transduction
The canonical TGF‐β signaling pathway is activated upon TGF‐β binding to two serine/threonine kinase receptors TGF‐β type II receptors (TβRII), leading to the recruitment and formation of a heteromeric complex with 2 TGF‐β type I receptors (TβRI). TβRII kinase phosphorylates TβRI, which in turn phosphorylates the SMAD proteins SMAD2 and SMAD3. This induces the assembly into heterodimeric and trimeric complexes with SMAD4, which translocate to the nucleus to regulate expression of TGF‐β target genes. SMAD3 and SMAD4 are able to bind DNA, while SMAD2 cannot bind to DNA. The DNA binding strength of SMADs is relatively weak, making the binding of the complex dependent on additional DNA binding transcription factors that partner with SMADs for specific target gene regulation. 13 SMAD signaling induces a negative feedback loop through SMAD7, which recruits the E3 ubiquitin ligase SMAD ubiquitination regulatory factor (SMURF)2 to the activated TβRI, and thereby mediating its degradation.
Besides canonical signaling, TGF‐β can induce various non‐canonical signaling pathways, either indirectly by SMAD‐induced expression of growth factors or directly downstream of the TβRI, recently summarized by Zhang. 64 Depending on cellular context, various non‐canonical signaling pathways can thus be activated, resulting in changes in transcription, the cytoskeleton, tight junctions, and translation. Non‐canonical pathways include, for example, Rho/Rho‐associated coiled‐coil kinase (ROCK) signaling, which promotes motility and cytoskeletal rearrangements, and phosphoinositide 3‐kinase/AKT signaling, which induces translational changes. Multiple mitogen‐activated protein kinases (MAPK) signaling can be activated, including extracellular signal‐regulated kinases (ERK), through Ras activation, and c‐Jun N‐terminal kinases (JNK) and p38, through TNF receptor‐associated factor(TRAF)6 signaling, which promotes proliferation and migration. 65 , 66 , 67 The above‐mentioned pathways are important to overcome TGF‐β induced cytostatic effects and promote growth, migration, and invasion.
3.2. TGF‐β has a dual role in tumor cells
For normal and pre‐malignant cells, TGF‐β is a potent inducer of growth arrest and apoptosis. SMAD signaling results in G1 cell cycle arrest through inducing expression of the cyclin dependent kinases inhibitors p15Ink4, P21CIP1, and P27KIP1 and inhibition of c‐MYC. 11 TGF‐β induced apoptosis can occur through induction of apoptotic regulators. 11 During cancer progression, tumor cells overcome the cytostatic effects of TGF‐β signaling, by acquiring mutations in the canonical signaling components, by adaptation of TGF‐β signaling through non‐canonical signaling and by gain of function of proto‐oncogenes or loss of function of tumor suppressor genes. In particular, mutations in genes encoding TβRII and SMAD4 are frequently found in gastrointestinal cancers. 68 Interestingly, many other cancers have elevated TGF‐β expression and display high activity of the TGF‐β pathway that is associated with worse prognosis. 68 , 69 , 70 , 71 , 72 In later stage tumor cells, the tumor cells can remain responsive to TGF‐β, and collaborate with other pro‐oncogenic pathways promoting cancer progression. A key mechanism in this respect is that TGF‐β can induce EMT, which is a part of the dynamic epithelial‐mesenchymal plasticity of tumor cells. 73 EMT is characterized by the loss of epithelial cell character, cell polarity, and cell‐cell junctions, and the gain of mesenchymal features, resulting in more invasive potential. 11 Moreover, EMT contributes to chemo‐resistance and immune evasion, as summarized by Hao et al. 11 EMT induction by TGF‐β/SMAD signaling is cell and context specific, and can be achieved by inducing the expression of, for example, Snail, Zinc finger E homeobox binding, and Twist transcription factors, together with non‐canonical signaling including ERK and RhoA. 11
3.3. Role of TGF‐β in the immune system
TGF‐β plays an essential role in immune cell homeostasis and suppression. 74 In a metastatic context, TGF‐β contributes to immune evasion through multiple mechanisms, which was recently reviewed by Batlle and Massague. 75 In short, regulatory T cells are stimulated through SMAD‐dependent induction of the forkhead box P3 (FoxP3) transcription factor, resulting in suppressed function of effector T cells. TGF‐β affects T helper 1 cells and CD8+ T cells directly by blocking T helper cell differentiation and blocking T cell receptor‐mediated T cell activation. 75 T cell numbers are reduced through induction of previously mentioned cytostatic and apoptotic pathways. Additionally, SMAD signaling suppresses T cell mediated killing effector genes. 75 In NK cells, TGF‐β can suppress recognition by downregulation of NK‐cell activating receptors, as well as blocking effector cytokines including interferon (IFN)‐γ. 75 Moreover, TGF‐β signaling can promote differentiation towards a pro‐tumorigenic NK cell phenotype. 76 Tumor associated macrophages (TAMs) are important sources of TGF‐β, and result in recruitment and adhesion of additional monocytes to tumor sites. Moreover, TGF‐β could contribute to differentiation of macrophages toward a pro‐tumorigenic M2 phenotype. 75 , 77 Overall, TGF‐β is a strong suppressor of the immune system, acting on both innate and adaptive immune responses.
3.4. TGF‐β signaling in healthy and diseased liver
In the liver, TGF‐β regulates processes from liver development and regeneration to liver pathologies. In healthy liver, TGF‐β1 is the predominant isoform and is mainly expressed by Kupffer cells and stellate cells, while hepatocytes show absence of TGF‐β expression. In fibrotic conditions, TGF‐β1 levels increase significantly and are found to be expressed by most sinusoid cells. 14 During development, specific spatiotemporal distribution of TGF‐β orchestrates structural organization of the liver and modulates cellular differentiation, through microRNAs and interplay with different signaling pathways. 15 Liver generation is partially controlled by TGF‐β signaling and crosstalk with other pathways in hepatocytes, circumventing the cytostatic effect of TGF‐β. 15 After injury, TGF‐β1 expression is temporarily induced, increasing phosphorylation of SMAD2 and nuclear yes‐associated protein (YAP). 78 These changes promote an EMT‐like response, leading to transdifferentiation of hepatocytes into myofibroblasts. 78 In contrast, in the context of failed regeneration after acute extensive liver injury, it was shown that TGF‐β secreted by local macrophages induced and propagated senescence in hepatocytes. 79 During injury, hepatocytes and Kupffer cells can be important source of TGF‐β. 80 , 81 TGF‐β is a key inducer of fibrosis, activating HSCs into aHSCs and transdifferentiating hepatocytes toward myofibroblasts through an EMT‐like process. 82 TGF‐β increased matrix deposition of HSCs through inducing connective tissue growth factor (CTGF), receptor for activated c kinase 1 and NADPH oxidase 4 signaling, and collagen‐1 secretion. Mild fibrosis could be reversed by among others upregulating quiescent genes and repressing TGF‐β target genes. 82 In conclusion, TGF‐β regulates processes in both healthy and injured liver in a highly spatiotemporal and context dependent manner.
Fibrosis is an essential component of liver metastasis, promoting liver metastasis formation as well as predicting occurrence and relapse of liver metastasis. 83 , 84 , 85 In colorectal cancers, metastases are characterized by high stroma and TGF‐β signaling, resulting in poor prognosis. 86 There is evidence that TGF‐β is not only important in creating the fibrotic niche, but also supports immune evasion and tumor outgrowth in different phases of liver colonization, in both TGF‐β wild‐type and deficient tumors. 11 , 59 , 60 Orchestrating beneficial TGF‐β signaling responses in the different cell types during the different phases of metastasis will contribute to successful outgrowth and thereby pose a potential therapeutic target.
4. TGF‐β IN LIVER METASTASIS
Overcoming the cytostatic effects of TGF‐β is essential in metastasis formation within the liver. Mutations or deletions in genes encoding TGF‐β pathway components can be frequently found, but are not essential for all cancer types. 68 For instance, loss of and mutations in SMAD4 have been associated with poor prognosis and liver metastasis. 87 , 88 In contrast, active TGF‐β signaling and high TGF‐β plasma levels are correlated with aggressive disease, disease relapse to the liver and poor survival. 59 , 70 , 89 This implies that the TGF‐β signaling output is altered (eg, by altered co‐factors or non‐canonical signaling) in aggressive tumor cells or that TGF‐β exerts tumor supporting roles in microenvironmental cells. In the liver, various cell types are a source of TGF‐β, including tumor cells, myofibroblasts, and local immune cells. 14 , 50 , 60 , 90 Below, we will describe the current knowledge of TGF‐β during the different phases of hepatic colonization. A schematic overview of different functions of TGF‐β during liver metastasis is shown in Figure 3.
FIGURE 3.
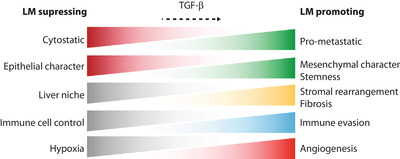
TGF‐β promotes liver metastasis at multiple angles. Transforming growth factor (TGF)‐β can influence multiple facets of liver metastases formation, promoting metastatic outgrowth. During liver metastasis, cytostatic TGF‐β signaling is suppressed in tumor cells, while pro‐metastatic signaling is promoted. Moreover, TGF‐β can promote the loss of the epithelial character and the increase of mesenchymal and stemness character of tumor cells. Through signaling in microenvironmental cells, TGF‐β can induce alterations in the liver niche to promote tumor outgrowth, through, for example, stromal rearrangement and induction of fibrosis. In immune cells, TGF‐β aids in evading immune responses. Angiogenic process promoted by TGF‐β signaling in cells promotes the influx of oxygen and nutrients in the growing liver metastasis. Abbreviation: LM, liver metastasis
4.1. TGF‐β signaling in liver stroma induced by the primary tumor facilitates metastatic outgrowth
TGF‐β signaling has been linked to the creation of a permissive niche prior to arrival of tumor cells because of its key role in HSCs activation, matrix remodeling, and creation of an immune suppressive environment (Figure 4). TGF‐β signaling in liver stroma can be triggered after uptake of different cytokines or extracellular vesicles secreted by the primary tumor. For example, Costa‐Silva et al. demonstrated an essential role for PDAC‐derived exosomes in TGF‐β‐mediated pre‐metastatic niche formation. 9 Cancer exosomes from primary PDAC cells were found to be taken up by Kupffer cells in the liver. Macrophage migration inhibitory factor (MIF) present in these exosomes stimulated TGF‐β secretion by Kupffer cells, which in turn activated HSCs leading to fibronectin and collagen‐1 deposition. 9 This fibrotic environment increased recruitment of bone marrow‐derived macrophages and granulocytes. Upon treatment with these exosomes prior to liver metastasis induction, metastatic load was increased. Thus, MIF present in exosomes triggered crosstalk between multiple stromal cell types in the liver resulting in pre‐metastatic niche formation and increased metastatic outgrowth. 9 Besides tumor cells, LSECs were also found to secrete MIF. 41 In vitro, LSEC‐derived MIF served as a chemoattractant for CRC cells and increased primary tumor growth and metastasis formation in vivo. 41 In CRC patients, primary tumor MIF levels were not correlated with distant metastasis, while higher liver MIF levels did correlate with larger liver metastasis. 41 Overall, this supports MIF as an important pre‐metastatic niche factor that might mediate (part of) its effect through TGF‐β.
FIGURE 4.
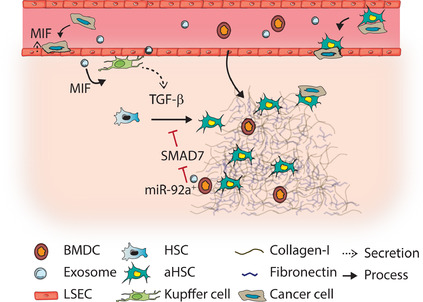
TGF‐β signaling in the pre‐metastatic niche. Primary tumors secrete cytokines and exosomes that home to the liver and trigger transforming growth factor (TGF)‐β signaling in liver cells. Exosomes containing migration inhibitory factor (MIF) triggered TGF‐β release by kupffer cells, resulting hepatic stellate cell (HSC) activation. MIF can also be secreted by liver sinusoid endothelial cells (LSECs), which increases tumor cell adhesion to LSECs. miR‐92a, present in exosomes secreted by bone marrow derived cells, represses mothers against decapentaplegic homolog(SMAD)7 in HSCs, sensitizing HSCs toward TGF‐β mediated activation. The result of TGF‐β induced HSC activation is a fibrotic niche of collagen‐1 and fibronectin, which promotes recruitment of suppressive bone marrow derived cells (BMDCs), and metastatic outgrowth. Migration of tumor cells toward the liver can be increased by TGF‐β induced interaction between tumor cells and fibroblasts
In pre‐metastatic livers of mice bearing Lewis lung carcinoma (LCC), HSCs were found to be more sensitive to TGF‐β activation through the uptake of miR‐92a in exosomes derived from bone‐marrow derived cells (BMDCs). 91 miR‐92a targets the 3′UTR of SMAD7, and as a consequence inhibits SMAD7 expression, removing the negative feedback on the TGF‐β pathway. This resulted in increased aHSC and collagen‐1 in mouse livers, promoting LCC adhesion and transendothelial migration of immune suppressive granulocytic myeloid derived cells. In lung cancer patients, increased miR‐92a levels were identified in circulating extracellular vesicles, which in vitro indeed activated HSCs. 91 This suggests that miR‐92 containing exosomes might sensitize HSCs to TGF‐β, which results in HSC activation and pre‐metastatic niche formation.
In conclusion, different mechanisms resulting in enhanced TGF‐β signaling in the liver have been identified for tumor cells to promote pre‐metastatic niche formation. These mechanisms are induced by exosomes secreted by tumor cells or bone marrow derived cells. The resulting fibrotic niche promotes recruitment of immune suppressing cells, attachment of disseminated tumor cells, and metastasis formation.
4.2. TGF‐β induced migration of tumor cells toward the liver
TGF‐β can mediate adhesion between CRC cells and isolated cancer associated fibroblasts (CAF) or endothelial cells in vitro, thereby potentially facilitating migration toward the liver. In vivo, TGF‐β positively regulated liver metastases in a tumor cell/CAF co‐injection model, possibly by enhancing co‐travelling of tumor cells and CAFs through the vasculature. 92 Interestingly, increased adhesion to microenvironmental cells by TGF‐β was also observed in other tumor models. TGF‐β treatment of non‐invasive uveal melanoma cells increased adhesion to liver endothelial cells, promoting trans‐endothelial migration. 93 However, this was not observed for highly invasive uveal melanoma cell lines. TGF‐β treatment showed no effect on adhesion proteins in either cell line, so the mechanism behind this specific adhesion remains unclear. Similar positive effects on transendothelial migration by TGF‐β have been observed in hepatocellular carcinoma (HCC). 94 TGF‐β can thus promote migration towards the liver by promoting interactions with CAFs and endothelial cells.
4.3. TGF‐β signaling in tumor cells can increase liver metastasis formation
Deletions in genes encoding for TGF‐β components are associated with liver metastasis and poor prognosis. 68 Moreover, canonical TGF‐β signaling in tumor cells of CRC patients suppressed liver metastasis formation. 95 , 96 This suggests that TGF‐β acts as a tumor suppressor in liver metastasis. In contrast, TGF‐β might act as a promoter of liver metastasis as well, as high TGF‐β signaling is correlated with poor prognosis. 68 Indeed, pretreatment of tumor cells with TGF‐β before experimental liver metastasis formation increased the metastatic burden of a variety of cancer types. 61 , 97 , 98 In another study, TGF‐β pretreatment resulted in angiogenesis, as well as tumor cells with an enhanced mesenchymal and migratory character in vitro and tumor cells showing increased proliferation in vivo. This suggests that pretreatment with TGF‐β induced changes in both tumor cells and tumor microenvironment. 61 Moreover, knockdown of SMAD4 in CT26 CRC cells increased migration in vitro and liver metastasis in vivo through activation of, for example, TGF‐β‐induced SMAD4‐independent ERK signaling. 88 , 89 , 96 These results suggest that in these cells, TGF‐β has pro‐tumorigenic effects, and TGF‐β signaling inhibition might lead to anticancer effects. Indeed, the TβRI/II small molecule kinase inhibitor galunisertib was found to block ERK‐mediated non‐canonical TGF‐β signaling in KRAS‐mutated CRC cells, inhibiting the increased migratory phenotype in vitro and liver metastasis in vivo. 62
Besides inducing migration and EMT in tumor cells, TGF‐β can also affect the stem cell population. For example, pretreatment of orthotopically injected MDA‐MB‐231 breast cancer cells increased liver metastasis while the size of primary tumors was insignificantly affected. 99 TGF‐β pretreatment was found to increase stem cell markers in vitro and slightly increase the CD44+CD24− cancer stem cell population in liver metastases. Moreover, CD44+CD24+ cells were unable to form liver metastasis, in contrast to CD44+CD24− cells. This suggests that a CD44+CD24− cell population induced by TGF‐β could be responsible for increased outgrowth in the liver. 99 Similarly, in HCC, blocking TGF‐β signaling decreased expression of CD44 and stemness features in vitro and in vivo. Ex vivo treatment of HCC patient samples showed reduced CD44 gene expression amongst responding patients. 100
Combined, this suggests that based on cellular context TGF‐β signaling can have metastasis promoting or suppressive effects. The effect of TGF‐β signaling in most advanced tumor lesions is shifted towards migration, EMT or stemness, thereby promoting colonization of the liver
4.4. Angiogenesis can be promoted by TGF‐β signaling within the liver metastasis
Recruitment of new blood vessels is essential for the outgrowth of micrometastases. Both non‐canonical and canonical TGF‐β signaling can influence expression of angiogenic factors, as shown in Figure 5. TGF‐β is crucial in activating HSCs, upon which they secrete various angiogenic factors like VEGF and Jagged in the surrounding matrix. 56 , 57 , 101 This directly contributes to angiogenesis. 56 , 101 Combined, this suggests that TGF‐β can induce angiogenesis by activating HSCs, although it remains to be proven in a metastatic setting.
FIGURE 5.
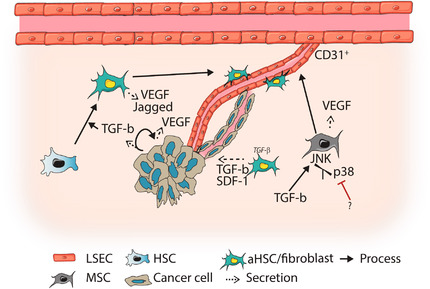
TGF‐β signaling influences angiogenic processes. Transforming growth factor (TGF)‐β signaling can elicit the secretion of angiogenic proteins, such as vascular endothelial growth factor (VEGF) and Jagged, by different cell types of the liver metastasis. This promotes (CD31+) vessel formation. TGF‐β and stromal derived factor (SDF)‐1 secreted by activated hepatic stellate cells (aHSCs) could promote vascular mimicry in tumor cells. Mesenchymal stem cells (MSCs) could promote angiogenesis through shifting TGF‐β responses towards c‐Jun N‐terminal kinases (JNK) signaling. This results in differentiation of MSCs into endothelial cells, and additional secretion of angiogenic factors. Abbreviation: LSEC, liver sinusoid endothelial cell
TGF‐β‐mediated regulation of angiogenesis is also studied in a cancer context. TGF‐β treatment increased VEGF levels in pancreatic tumor cells, and reduced tumor cell killing by peripheral blood mononuclear leukocytes. TGF‐β pretreatment also enhanced experimental liver metastasis formation. This could be due to increased angiogenesis, but remains to be proven. 102
A new mechanism of TGF‐β‐induced angiogenesis in mesenchymal stem cells was proposed by Battle and colleages. 103 They proposed that, upon activation of non‐canonical TGF‐β signaling, the balance between activation of P38 and JNK MAP kinases determined mesenchymal cell differentiation toward endothelial cells. 103 P38 MAPK was found to repress angiogenic programs in mesenchymal stem cells (MSCs), while JNK MAPK signaling promoted VEGF secretion, angiogenesis, and MSC differentiation toward endothelial cells. Indeed, in an inflammation‐induced CRC model in p38αΔFSP1 mice, increased CD31+ vessels and tumor growth were observed. 103 Expression of these proteins are also influenced by the surrounding niche, suggesting that TGF‐β in interplay with other environmental signals can promote MSC differentiation to promote angiogenesis.
Vascular mimicry can be an alternative approach to angiogenesis, were tumor cells form channel‐like structures to mediate oxygen and nutrient influx. 104 Interestingly, TGF‐β and SDF‐1 secretion by CAFs resulted in VM in HCC both in vitro and in subcutaneous tumors. This was mediated by TGF‐β‐induced ECM remodeling and expression of endothelial marker vascular endothelial VE‐cadherin by tumor cells. 104 Likewise, increased VE‐cadherin expression by HCC cells upon TGF‐β1 treatment was also observed by Zhang et al. 105 This resulted in VM and was dependent on TGF‐β induced EMT and Rho/ROCK2 signaling.
In conclusion, TGF‐β is able to promote angiogenesis by modulating the expression of angiogenic factors and endothelial proteins in multiple cell types, shown in Figure 5, orchestrating the formation of new blood vessels.
4.5. Tumor‐supportive role of TGF‐β‐activated stromal cells
In CRC patients, high TGF‐β levels are associated with relapse. Importantly, TGF‐β signaling as measured by nuclear p‐SMAD3 and TGF‐β transcriptional signatures were identified in every microenvironmental cell, and high TGF‐β signatures in CAFs predicted poor prognosis. 59 , 86 TGF‐β‐activated fibroblasts in another CRC model were found to secrete IL‐11, triggering GP130/STAT3 signaling in tumor cells. This repressed apoptosis, and thus enhanced colonization of the liver. 59 This suggests that stromal TGF‐β signaling could be key in overcoming the bottleneck of metastasis.
TGF‐β‐activated stromal cells might enhance liver metastasis by the induction of a fibrotic environment. 28 For example, in gastric cancer, TGF‐β‐activated HSCs secrete lysyl oxidase (LOX), which contributes to the fibrotic niche by crosslinking collagen fibers and increasing tumor cell proliferation through an AKT‐p70S6K‐hypoxia‐inducible factor(HIF)1‐α pathway. 106 Besides, TGF‐β‐induced expression of miRNA‐181a in stellate cells in vitro was shown to promote fibrosis through inhibition of Augmenter of Liver Regeneration. 107 The importance of a fibrotic environment for outgrowth of metastatic cells was shown by Barkan et al. 54 TGF‐β1‐induced lung fibrosis could reactivate dormant breast tumor cells through collagen 1 and integrin β1 signaling, resulting in phosphorylation of Src, focal adhesion kinase, and ERK that led to proliferation. 54
Stromal cell activation by TGF‐β might be enhanced by TGF‐β receptor recycling or degradation. Indeed, these processes were found altered in HSCs of CRC metastases. For example, the actin‐associated protein vasodilator‐stimulated phosphoprotein VASP mediates TβRII recycling to the cell membrane through rab11, thereby sensitizing HSCs to further TGF‐β stimulation. 108 In addition, TGF‐β signaling in HSCs is negatively regulated by RAS GTPase activating‐like protein IQGAP1, which, by binding the TβRII, mediated SMURF‐1‐dependent ubiquitination and degradation of the receptor. In CRC patients, reduced levels of IQGAP1 were found in stroma of liver metastasis. 51 Thus, TGF‐β receptor levels in HSCs are modulated in metastases to enhance signaling.
The source of TGF‐β that activates HSCs is often the tumor cell. For example, SDF‐1 secretion by aHSCs was found to bind C‐X‐C chemokine receptor type 4 (CXCR4), promoting TGF‐β signaling and secretion by CRC tumor cells. 50 Blocking CXCR4 or TGF‐β in vivo decreased the metastatic potential of CRC cell lines and reduced HSC activation. 50 Calon and colleagues tested the effect of TGF‐β secretion by TGF‐β‐deficient CRC cells, inducing stromal TGF‐β signaling, during liver metastasis formation in mice. 59 Secretion of TGF‐β was found to promote both tumor initiation and metastatic potential. Interestingly, high secretion of TGF‐β1 by the CRC cells, and thus stromal TGF‐β signaling, was essential at the initial phase of liver metastasis, since a 24 hour induction of TGF‐β secretion directly after splenic injection was sufficient to increase liver colonization. 59 Additionally, isolated CRC stem cells from patients were found to express higher levels of TGF‐β. 59 Thus, a feed forward loop can exist between aHSCs and tumors cells, whereby the HSCs stimulate tumor cells to secrete TGF‐β, which can activate HSCs, ultimately leading to enhanced liver metastasis formation.
At last, hepatocytes are also source of liver fibrosis next to HSCs. Lineage tracing in a CCL4 liver fibrosis model showed that hepatocytes contribute to fibroblast‐specific protein (FSP)1+ fibroblasts in fibrotic tissue. 109 This was mediated by TGF‐β‐promoted EMT in hepatocytes, a process that could be antagonized by TGF‐β family member Bone Morphogenetic Protein (BMP) 7. 109
In conclusion, during metastasis formation TGF‐β plays a significant role in promoting metastatic outgrowth by activating stromal cells and hepatocytes and promoting matrix remodeling and fibrosis. An overview of these processes is shown in Figure 6.
FIGURE 6.
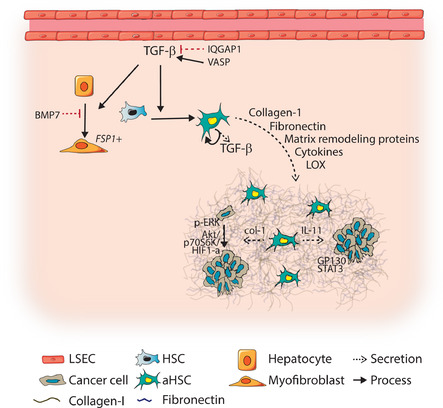
TGF‐β signaling in liver fibroblasts stimulates fibrotic environment. Transforming growth factor (TGF)‐β signaling in tumor stroma is essential for metastatic outgrowth in the liver. TGF‐β in the liver metastasis, secreted by various cell types in the metastases, promotes transdifferentiation of hepatocytes and hepatic stellate cells(HSCs) toward myofibroblasts and activated HSCs (aHSCs). In HSCs, vasodilator‐stimulated phosphoprotein (VASP) can sensitize HSCs toward TGF‐β signaling, while IQ motif containing GTPase activating protein 1(IQGAP1) represses TGF‐β signaling in HSCs. Activation of HSCs results in secretion of collagen‐1, fibronectin, matrix remodeling proteins, cytokines, and lysyl oxidase (LOX), triggering the formation of the fibrotic niche. Tumor cells in a fibrotic niche gain survival advantages through collagen‐1 induced extracellular signal‐regulated kinases (ERK) phosphorylation, which promotes proliferation. Moreover, LOX‐mediated crosslinking of collagen‐1 also increases proliferation through AKT‐p70S6K‐hypoxia‐inducible factor(HIF)1‐α signaling. Secretion of interleukin (IL)‐11 by aHSCs promote glycoprotein (GP)130/ signal transducer and activator of transcription (STAT3) signaling in tumor cells, thereby repressing apoptosis and promoting metastatic outgrowth. Abbreviations: HC, hepatocyte; LSEC, liver sinusoid endothelial cell
4.6. TGF‐β plays central role in regulating an immune‐suppressive metastatic niche
TGF‐β has a pronounced immune suppressive effect affecting multiple immune cell populations, which is depicted in Figure 7. 75 Therefore, TGF‐β is proposed to create immune permissive (metastatic) niches. In vivo studies of TGF‐β influencing the liver metastatic niche and outgrowth are limited due to the bias toward the use of immune suppressed or deficient mouse models. In light of immunotherapy, in vivo experiments that address the roles of TGF‐β will most likely move toward (humanized) immune competent models.
FIGURE 7.
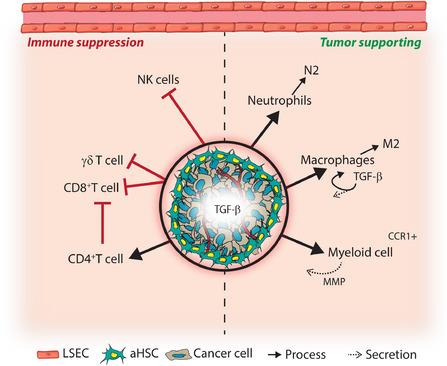
TGF‐β secretion and signaling in liver metastases can provide protection to innate and adoptive immune responses. Transforming growth factor (TGF)‐β secretion of tumor cells and fibroblasts, and the resulting fibrotic niche, provides a physical barrier against immune cells and triggers immune suppressive effects in immune cells. Neutrophils and macrophages could differentiate toward tumor supportive N2 and M2 type cells. Moreover, TGF‐β secretion by macrophages further enhance immune suppression. Anti‐tumor effects of Natural Killer (NK) cells can be inhibited by TGF‐β. Tumor supporting myeloid cells derived from the bone marrow support metastasis formation and are recruited to metastases through e.g. binding of secreted C‐C Motif Chemokine Ligand 15 (CCL15) to the C‐C chemokine receptor type 1 (CCR1) receptor. These cells support metastasis by contributing to immune suppression and secreting matrix modulating proteins such as matrix metalloproteases (MMPs). During liver metastasis formation, the effects of cytotoxic CD8+ and γδT cells are inhibited by TGF‐β, through e.g. reduction of T cell infiltration or proliferation, reduced T cell activation and inhibition of cytotoxic effector proteins, while immune suppressive CD4+ regulatory T cells are stimulated
The first line of defense in the liver are innate immune cells, including NK cells, neutrophils, and macrophages. In an intravenously injected B16 melanoma metastasis model liver metastases were only observed after depletion of NK cells. TGF‐β is known to suppress NK cells at multiple levels, suggesting that TGF‐β mediated NK cell suppression could positively affect liver metastasis formation. 75 Like metastases at other sites, metastases in the liver TGF‐β potentially induce differentiation of neutrophils and macrophages toward tumor promoting phenotypes N2 and M2, respectively. 75 , 77 At last, M2 macrophages produce TGF‐β, further contributing to immune suppression via a feed‐forward loop. 75 Thus, induced TGF‐β signaling in innate immune cells can promote an immune suppressed environment.
Creating an immune suppressive niche is essential for metastatic outgrowth. Recruitment of tumor promoting BMDCs contributes to this process. In addition, BMDCs are important in promoting invasion and metastasis through MMP secretion. 110 In experimental metastasis models of SMAD4‐deficient compared to SMAD4‐proficient CRC, BMDCs were recruited to liver metastases through increased secretion of C‐C motif chemokine ligand (CCL)9 (mouse ortholog of CCL15), the ligand for the C‐C chemokine receptor type 1 (CCR1) receptor on myeloid cells. In vitro, it was found that SMAD4 binds to the promotor of CCL15 to inhibit CCL15 expression. 111 The correlation between CCL15+ tumor cells and CCR1+ BDMCs was also observed in CRC liver metastasis of patients. 110 Thus, loss of SMAD4 during liver metastasis promotes secretion of CCL15 and recruitment of immune suppressive BMDCs.
Evading adaptive immune responses is another essential characteristic for successful metastasis formation, and exclusion of T cells is observed in growing metastases. 3 , 53 Also in C26 CRC liver metastases compared to wildtype livers, a decrease in CD8+ T cells was observed livers, while the population of T regulatory cells, suppressing immune responses, was increased. 112 This shift was accompanied by increased expression of IL‐10 and TGF‐β in liver tissue containing metastases. Similarly, in HCC, decreased numbers of γδ T cells were observed intratumorally compared to peritumorally, as well as reduced IFN‐γ and granzymes secretion exerting tumor cytotoxicity. 113 Ex vivo, this was dependent on CD4+ regulatory T cells, and IL‐10 and TGF‐β secretion. 113 In pancreatic cancer, ICAM‐1 cell surface levels were reduced after TGF‐β treatment in vitro. ICAM‐1, expressed by T cells, serves as a ligand for integrin β2 and results in T cell activation. 114 These studies suggest that TGF‐β mediates tumor cell evasion of the adaptive immune response, and that blocking TGF‐β would result in tumor cell killing. Indeed, Medina‐Echeverz and colleagues reported increased T cell‐mediated killing and IFN‐γ production in MC38 CRC and metastasizing melanoma mouse models upon treatment with high density lipoproteins containing the TGF‐β blocking peptide P144. 115
More recently, Tauriello et al. used an elegant genetically engineered CRC model to demonstrate the strong immune evasive effect of TGF‐β during liver metastasis. 60 Using a quadruple mutant (including TβRII) CRC organoid model, liver metastases were formed after implanting mouse tumor organoids in the caecum. These liver metastases were characterized by high stromal TGF‐β signaling and low T cell infiltration, reflecting a poor prognosis subtype of CRC. Blocking TGF‐β signaling in CAFs and T cells through galunisertib significantly reduced metastasis formation, mainly by enhancing T cell‐mediated tumor cell killing in early metastatic stages. 60 However, in established metastatic disease galunisertib resulted in mild effects. This was attributed to acquired resistance to T cell‐mediated killing due to increased expression of PD1/PDL‐1. 60 Indeed, combinational treatment of galunisertib and anti‐PD‐L1 antibodies significantly increased efficiency of the therapy.
In sum, studies in the liver suggest that, like in other organs, TGF‐β plays an important role in immune suppression at the innate and adaptive immune response level, as shown in Figure 7. This provides promise to TGF‐β inhibitors to treat metastatic liver disease. 116 This avenue of research could greatly improve response to immunotherapy and metastatic cancer patient survival.
5. IMPROVING CURRENT CLINICAL STRATEGIES
TGF‐β is important in creating the fibrotic niche and angiogenesis, and it supports immune evasion and tumor outgrowth in different phases of liver colonization, in both in TGF‐β wild‐type and deficient tumors. Orchestrating metastasis promoting TGF‐β signaling responses in the various liver niche cell types during the different phases of metastasis contributes to successful outgrowth. Thus, TGFβ poses an interesting potential therapeutic target.
The main treatment aiming for long‐term survival for patients with liver metastasis is surgery. Unfortunately, few patients qualify for surgery and additionally, surgery triggers fibrotic processes that could enhance the chance of recurrence. 25 Considering the effect of TGF‐β in all essential processes in cancer and metastasis formation, TGF‐β targeting therapies could pose a treatment target as monotherapy or in combinational therapy. A mouse model engineered to secrete a soluble TGF‐β antagonist showed protection against metastasis in, among others, the liver. 117 Several TGF‐β targeted therapies including receptor kinase inhibitors, small molecules, neutralizing antibodies and TGF‐β ligand traps, have been developed and are tested in (pre)clinical studies, as monotherapy or combinatorial treatment. 118
TGF‐β has pleiotropic effects, and as such, targeting TGF‐β might potentially lead to on‐target side effects. Indeed, the main adverse effects observed with TGF‐β targeting therapies are related to cardiovascular toxicity, and hyperproliferation due to blocking of the antiproliferative function of TGF‐β during tissue homeostasis. 12 Clinical trials on TGF‐β targeted drugs should carefully examine these on‐target side effects. Moreover, careful considerations on treatment schemes and doses (eg, drug holidays) are warranted. Importantly, the limited toxicity and side‐effects seen in a clinical trial with Galunisertib holds great promise for TGF‐β inhibition therapy. 119
5.1. TGF‐β inhibitors in (pre)clinical studies
TGFβRI/II kinase inhibitor Galunisertib is currently studied in phase II clinical trials. This drug showed moderate benefit for unresectable pancreatic cancer patients in combination with Gemcitabine, while also showing limited toxicity. 119 In contrast, a phase II clinical trials in glioblastoma patients did not find beneficial effects of galunisertib as monotherapy or in combination with Lomustine. 121 In HCC, galunisertib was shown to decrease stemness of invasive HCC cells in vitro, as well as in ex vivo samples of HCC patients responding to Galunisertib. 100 TGF‐β also plays an important role during fibrosis, therefore Hammad et al. studied the use of galunisertib in liver fibrosis. 122 In Abcb4Ko mice modeling chronic liver fibrosis, treatment with galunisertib did not reduce fibrosis, but did reduce the mRNA levels of fibrotic genes and levels of laminin‐332, and β‐catenin. Similar effects of galunisertib were seen in a human ex vivo liver fibrosis model. 123 These results indicate that inhibiting TGF‐β might not reverse fibrosis, but could remodel the matrix and potentially dysregulate stroma crosstalk in a metastatic setting. 122 Overall, galunisertib seems safe and shows some beneficial response as a monotherapy to treat metastases.
The monoclonal antibody fresolimumab neutralizing TGF‐β1, 2, and 3 showed modest effects in patients in a small phase 1 clinical trial of patients with malignant melanoma or renal carcinoma. 124 In metastatic breast cancer, the combination of fresolimumab with focal radiation was explored. Radiation evokes a damage response in tissues triggering, for example, TGF‐β signaling, therefore, combinatorial treatment with TGF‐β inhibitors could potentially be beneficial. Combination of radiation with higher doses of fresolimumab increased median overall survival compared to lower dosing, although with moderate response. 125 Interestingly, higher dose of fresolimumab showed increased levels of e.g. memory CD8+ T cells. 125
Thus, many TGF‐β inhibitory strategies show promising results in preclinical studies, however, the use of TGF‐β targeted therapy in the clinic has only shown moderate to small effects on the metastatic burden and proves to be challenging. As such, combinatorial treatment is more likely to be effective.
5.2. Combining TGF‐β inhibitors and immunotherapy
Immunotherapy is an extremely promising therapy showing great benefit for responding patients. Currently, the main immunotherapeutic drugs are the immune checkpoint inhibitors anti‐cytotoxic T lymphocyte antigen 4 antibody ipilimumab, and antibodies targeting programmed cell death 1 (PD1), pembrolizumab, and nivolumab. 126 Despite the long‐lasting, and sometimes curative responses in some patients, the percentage of responders is low. A meta‐analysis by Li et al. showed that patients with liver metastases of various cancer types treated with anti‐PD1/PD‐L1 had a better prognosis compared to conventional treatment. However, CD8+ T cell counts in the liver were lower compared to non‐liver metastatic sites, which might contribute to the higher immune tolerance of the liver. 127 , 128 Therefore, combinational therapies targeting the immune system might be of added value specifically in the liver. As TGF‐β shows a clear immune suppressive role, inhibition of TGF‐β might elevate this suppression, and combining this inhibition with immunotherapy might improve the response rate of immunotherapy. Additionally, TGF‐β‐induced EMT correlated with increased PD‐L1 levels through microRNA‐200/ZEB1 signaling, giving an additional reason for tumors to be more responsive to anti‐PD‐L1 treatments. 129
The potential of combining TGF‐β inhibition with immunotherapy targeting PD‐L1 was shown in studies performed by Tauriello and by Mariathasan. 60 , 116 , 130 Looking at a large phase 2 trial of metastatic urothelial cancer treated with PD‐L1 inhibitors, non‐responders were characterized by increased TGF‐β signaling in fibroblasts, mediating T cell exclusion. Combinatorial treatment induced stromal remodeling, decreased TGF‐β signaling and increased influx of CD8+ T cells. 130 Similarly, galunisertib treatment increased sensitivity to anti‐PD‐L1 treatment in a CRC mouse tumor organoid model, as TGF‐β targeting treatment increased stromal PD‐L1 and T cell programmed cell death (PD)‐1 expression. 60 Dual TGF‐β and PD‐L1 inhibition showed great synergistic effects on tumor regression in these models. 60 , 130 Similarly, TβRI inhibition together with anti‐CTLA‐4 treatment resulted in synergistic effects on tumor growth in a melanoma mouse model. 131 Interestingly, in this model, TGF‐β inhibition led to increased CAF proliferation and to reduced PD‐L1 surface expression on tumor cells through MMP9‐mediated cleavage, suggesting a resistance mechanism for anti‐PD1 therapy. 131 This could be circumvented by inhibiting TβRI 3 weeks after the start of PD‐1 inhibition, showing that there is room to explore optimal combination treatment conditions. 131
New studies of combinatorial treatment with TGF‐β inhibitors and immunotherapy are now emerging. 131 , 132 , 133 , 134 Bintrafusp alfa, a PD‐L1 IgG fused to two extracellular domains of TβRII molecules connected by peptide linkers was developed by Lan and colleagues. 120 This peptide traps TGF−β molecules and targets PD‐L1. In multiple syngeneic mouse models, this drug showed enhanced response compared to treatment with anti‐PD‐L or TGF‐β trap alone. Moreover, bintrafusp alfa reduced the development of (lung) metastasis in spontaneously metastasizing orthotopic breast cancer models. 120 , 135 Compared to anti‐PD‐L1 treatment alone, monotreatment with bintrafusp alfa increased the tumor density and activity of CD8+ T cells, NK cells, neutrophils, and tumor associated dendritic cell and M1 macrophages. 120 , 135 Interestingly, myofibroblasts also seemed to be affected by bintrafusp α, since a decrease in α‐SMA was seen. When bintrafusp alfa was combined with additional CXCR1/2‐IL8 inhibition, EMT related resistance mechanisms to checkpoint inhibitors were overcome, leading to an additional synergistic effect. 136 The first phase I clinical trials with bintrafusp alfa have been completed, showing tolerance and initial therapeutic benefit in various cancer types. 135 However, larger clinical trials are necessary to draw firm conclusions. Mouse models have shown the added benefit of bintrafusp alfa with radiation and chemotherapy, providing directions for potential use for bintrafusp alfa in the clinic. 120
Studies combining TGF‐β targeting therapies with immunotherapies are still in early stages, but aforementioned results clearly show the promises of this approach. 137
6. CONCLUSION AND FUTURE PERSPECTIVE
As an abundant cytokine with pleiotropic functions, TGF‐β has been shown to play important roles in multiple aspects of liver metastasis formation, as illustrated in Figure 3. Creating optimal growth conditions and adaptation at distant sites is essential for metastatic outgrowth. Hitting multiple aspects of this process therefore poses as a promising strategy for eliminating liver metastasis formation and preventing cancer‐related deaths. This review has illustrated that TGF‐β can be involved in all these individual processes. Tumor cells modulate TGF‐β signaling to their advantage, promoting EMT, motility, and therapy resistance mechanisms. Within the liver stroma, TGF‐β signaling in multiple cell types orchestrates a tumor supportive environment which is necessary to survive, grow, attract blood vessels, and shield metastases. Additionally, the importance of the immune suppressive function of TGF‐β in metastasis is becoming clear. Even though TGF‐β will most likely not be the sole regulator of any of these processes, the multiplicity of targets makes TGF‐β inhibition treatment an extremely attractive approach for many different cancer types. In patients, TGF‐β inhibitor responses might be heterogeneous due to mutational signatures (in TGF‐β components) and crosstalk with non‐canonical TGF‐β signaling. However, even if tumor cells remain unresponsive to TGF‐β, effects on the microenvironment still remain. Moreover, the strength of anti‐TGF‐β treatment will probably lay in combination therapy, since TGF‐β inhibitor treatment alone showed moderate responses. By combining anti‐TGF‐β treatment with immunotherapy, not only will the effect of immunotherapy be enhanced, potential mechanisms of resistance, fibrosis, and angiogenesis might also be affected, intensifying the anti‐cancer effect together with response rates of these drugs. Similarly, damage‐induced fibrosis due to chemotherapy or surgery might be dampened by this approach. Combinatorial therapy approaches will most likely be a focus of future research, to target the complexity and dynamics of metastases. One needs to keep in mind the pleiotropic nature of TGF‐β. On‐target side effects can be expected and are reported, so dosing (eg, through drug holidays) and toxicity should be carefully studied in order to safely apply these approaches in the clinic. Specific drugs delivered to for example aHSCs could help reduce the toxicity of TGF‐β targeted drugs. 138 Moreover, since the liver has essential roles in metabolism and waste removal, it is possible that TGF‐β targeting drugs accumulate in the liver, allowing for the use of lower systemic drug concentrations. This could make the liver specifically suitable for TGF‐β targeted therapies.
Current studies investigating the role of TGF‐β signaling in liver metastasis only provide a snapshot into metastases. However, cancer cells are not static but instead display dynamics and plasticity of processes, such as epithelial to mesenchymal plasticity. Considering the pleiotropic and dynamic nature of TGF‐β signaling, future studies should focus on unraveling these dynamics to gain better understanding on how and when to target TGF‐β signaling. Intravital imaging approaches in (immunocompetent) mice could advance this field of study. 139 , 140
AUTHOR CONTRIBUTIONS
DLM and LR initiated and designed the review. DLM wrote the manuscript. LR supervised the study. LR, PtD, and RH corrected the manuscript. RH performed a feasibility study and identified key references. All authors approved the final version.
DECLARATIONS
This work is part of the research program 016.176.081 (Veni, L. Ritsma), which is financed by the Dutch Organization for Scientific Research (NWO). Also, we would like to thank the financial support of the LUMC Gisela Their Fellowship grant, and a subsidy from the Leids Universiteits fonds (LUF, CWB 7204, L. Ritsma), and subsidies from the Cancer Genomics consortium (L. Ritsma and P. ten Dijke).
AUTHORS’ INFORMATION
D.L. Marvin is a PhD student under the supervision of Dr. L. Ritsma and Prof. P. ten Dijke. All three have an interest in studying the dynamics of TGF‐β signaling during metastatic colonization, particularly in the liver. Prof. ten Dijke is an expert in TGF‐β signaling. Dr. Ritsma is an expert in advanced microscopy techniques. R. Heijboer wrote her Masters’ thesis on the subject of TGF‐β signaling in liver metastasis under the guidance of D.L. Marvin.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
We acknowledge all investigators whose studies were not cited due to space limitations, for their contributions to this field of research. We thank Marten Hornsveld for his helpful comments on the manuscript. The cartoons used in Figure 1, 4, 5, 6, and 7 were created using Servier Medical Art (http://servier.com/Powerpoint-image-bank). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License.
Marvin DL, Heijboer R, ten Dijke P, Ritsma L. TGF‐β signaling in liver metastasis. Clin Transl Med. 2020;10:e160 10.1002/ctm2.160
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Dillekås H, Rogers MS, Straume O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019;8(12):5574‐5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. 2017;168(4):670‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luzzi KJ, MacDonald IC, Schmidt EE, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153(3):865‐873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ewing J. Neoplastic diseases: a treatise on tumours. Br J Surg. 1928;16(61):174‐175. [Google Scholar]
- 6. Hoshino A, Costa‐Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hess KR, Varadhachary GR, Taylor SH, et al. Metastatic patterns in adenocarcinoma. Cancer. 2006;106(7):1624‐1633. [DOI] [PubMed] [Google Scholar]
- 8. Mielgo A, Schmid MC. Liver tropism in cancer: the hepatic metastatic niche. Cold Spring Harb Perspect Med. 2020;10(3):a037259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Costa‐Silva B, Aiello NM, Ocean AJ, et al. Pancreatic cancer exosomes initiate pre‐metastatic niche formation in the liver. Nat Cell Biol. 2015;17(6):816‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vidal‐Vanaclocha F. The prometastatic microenvironment of the liver. Cancer Microenviron. 2008;1(1):113‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hao Y, Baker D, ten Dijke P. TGF‐β‐mediated epithelial‐mesenchymal transition and cancer metastasis. Int J Mol Sci. 2019;20(11):2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Colak S, ten Dijke P. Targeting TGF‐β signaling in cancer. Trends Cancer. 2017;3(1):56‐71. [DOI] [PubMed] [Google Scholar]
- 13. Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Bleser PJ, Niki T, Rogiers V, Geerts A. Transforming growth factor‐β gene expression in normal and fibrotic rat liver. J Hepatol. 1997;26(4):886‐893. [DOI] [PubMed] [Google Scholar]
- 15. Fabregat I, Moreno‐Càceres J, Sánchez A, et al. TGF‐β signalling and liver disease. FEBS J. 2016;283(12):2219‐2232. [DOI] [PubMed] [Google Scholar]
- 16. Budczies J, von Winterfeld M, Klauschen F, et al. The landscape of metastatic progression patterns across major human cancers. Oncotarget. 2015;6(1):570‐583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ma R, Feng Y, Lin S, et al. Mechanisms involved in breast cancer liver metastasis. J Transl Med. 2015;13(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jaques DP, Coit DG, Casper ES, Brennan MF. Hepatic metastases from soft‐tissue sarcoma. Ann Surg. 1995;221(4):392‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lorigan JG, Wallace S, Mavligit GM. The prevalence and location of metastases from ocular melanoma: imaging study in 110 patients. Am J Roentgenol. 1991;157(6):1279‐1281. [DOI] [PubMed] [Google Scholar]
- 20. Kanas GP, Taylor A, Primrose JN, et al. Survival after liver resection in metastatic colorectal cancer: review and meta‐analysis of prognostic factors. Clin Epidemiol. 2012;4(1):283‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Agarwala SS, Eggermont AMM, O'Day S, Zager JS. Metastatic melanoma to the liver: a contemporary and comprehensive review of surgical, systemic, and regional therapeutic options. Cancer. 2014;120(6):781‐789. [DOI] [PubMed] [Google Scholar]
- 22. Kow AWC. Hepatic metastasis from colorectal cancer. J Gastrointest Oncol. 2019;10(6):1274‐1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carvajal RD, Schwartz GK, Tezel T, Marr B, Francis JH, Nathan PD. Metastatic disease from uveal melanoma: treatment options and future prospects. Br J Ophthalmol. 2017;101(1):38‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chow FCL, Chok KSH. Colorectal liver metastases: an update on multidisciplinary approach. World J Hepatol. 2019;11(2):150‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paschos KA, Bird NC. Liver regeneration and its impact on post‐hepatectomy metastatic tumour recurrence. Anticancer Res. 2010;30(6):2161‐2170. [PubMed] [Google Scholar]
- 26. Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol. 2017;27(21):R1147‐R1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vollmar B, Menger MD. The hepatic microcirculation: mechanistic contributions and therapeutic targets in liver injury and repair. Physiol Rev. 2009;89(4):1269‐1339. [DOI] [PubMed] [Google Scholar]
- 28. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397‐411. [DOI] [PubMed] [Google Scholar]
- 29. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Poisson J, Lemoinne S, Boulanger C, et al. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J Hepatol. 2017;66(1):212‐227. [DOI] [PubMed] [Google Scholar]
- 31. Smedsrød B, De Bleser PJ, Braet F, et al. Cell biology of liver endothelial and Kupffer cells. Gut. 1994;35(11):1509‐1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brodt P. Role of the microenvironment in liver metastasis: from pre‐ to prometastatic niches. Clin Cancer Res. 2016;22(24):5971‐5982. [DOI] [PubMed] [Google Scholar]
- 33. Zeng Z, Li Y, Pan Y, et al. Cancer‐derived exosomal miR‐25‐3p promotes pre‐metastatic niche formation by inducing vascular permeability and angiogenesis. Nat Commun. 2018;9(1):5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee JW, Stone ML, Porrett PM, et al. Hepatocytes direct the formation of a pro‐metastatic niche in the liver. Nature. 2019;567(7747):249‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin Q, Ren L, Jian M, et al. The mechanism of the premetastatic niche facilitating colorectal cancer liver metastasis generated from myeloid‐derived suppressor cells induced by the S1PR1–STAT3 signaling pathway. Cell Death Dis. 2019;10(10):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ji Q, Zhou L, Sui H, et al. Primary tumors release ITGBL1‐rich extracellular vesicles to promote distal metastatic tumor growth through fibroblast‐niche formation. Nat Commun. 2020;11(1):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bouwens L, Jacobs R, Remels L, Wisse E. Natural cytotoxicity of rat hepatic natural killer cells and macrophages against a syngeneic colon adenocarcinoma. Cancer Immunol Immunother. 1988;27(2):137‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luo D, Vermijlen D, Kuppen PJK, Wisse E. MHC class I expression protects rat colon carcinoma cells from hepatic natural killer cell‐mediated apoptosis and cytolysis, by blocking the perforin/granzyme pathway. Comp Hepatol. 2002;1(2):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bayon LG, Izquierdo MA, Sirovich I, van rooijen N, Beelen RH, Meijer S. Role of Kupffer cells in arresting circulating tumor cells and controlling metastatic growth in the liver. Hepatology. 1996;23(5):1224‐1231. [DOI] [PubMed] [Google Scholar]
- 40. Ou J, Peng Y, Deng J, et al. Endothelial cell‐derived fibronectin extra domain A promotes colorectal cancer metastasis via inducing epithelial‐mesenchymal transition. Carcinogenesis. 2014;35(7):1661‐1670. [DOI] [PubMed] [Google Scholar]
- 41. Hu C‐T, Guo L‐L, Feng N, et al. MIF, secreted by human hepatic sinusoidal endothelial cells, promotes chemotaxis and outgrowth of colorectal cancer in liver prometastasis. Oncotarget. 2015;6(26):22410‐22423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scherbarth S, Orr FW. Intravital videomicroscopic evidence for regulation of metastasis by the hepatic microvasculature: effects of interleukin‐1α on metastasis and the location of B16F1 melanoma cell arrest. Cancer Res. 1997;57(18):4105‐4110. [PubMed] [Google Scholar]
- 43. Khatib AM, Kontogiannea M, Fallavollita L, Jamison B, Meterissian S, Brodt P. Rapid induction of cytokine and E‐selectin expression in the liver in response to metastatic tumor cells. Cancer Res. 1999;15(59):1356‐1361. [PubMed] [Google Scholar]
- 44. Khatib AM, Auguste P, Fallavollita L, et al. Characterization of the host proinflammatory response to tumor cells during the initial stages of liver metastasis. Am J Pathol. 2005;167(3):749‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mendoza L, Valcárcel M, Carrascal T, et al. Inhibition of cytokine‐induced microvascular arrest of tumor cells by recombinant endostatin prevents experimental hepatic melanoma metastasis. Cancer Res. 2004;64(1):304‐310. [DOI] [PubMed] [Google Scholar]
- 46. Huang C, Li N, Li Z, et al. Tumour‐derived interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nat Commun. 2017;8:14035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Benedicto A, Herrero A, Romayor I, et al. Liver sinusoidal endothelial cell ICAM‐1 mediated tumor/endothelial crosstalk drives the development of liver metastasis by initiating inflammatory and angiogenic responses. Sci Rep. 2019;9(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Olaso E, Santisteban A, Bidaurrazaga J, Gressner AM, Rosenbaum J, Vidal‐Vanaclocha F. Tumor‐dependent activation of rodent hepatic stellate cells during experimental melanoma metastasis. Hepatology. 1997;26(3):634‐642. [DOI] [PubMed] [Google Scholar]
- 49. Shimizu S, Yamada N, Sawada T, et al. In vivo and in vitro interactions between human colon carcinoma cells and hepatic stellate cells. Jpn J Cancer Res. 2000;91:1285‐1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tan H‐XX, Gong W‐ZZ, Zhou K, et al. CXCR4/TGF‐β1 mediated hepatic stellate cells differentiation into carcinoma‐associated fibroblasts and promoted liver metastasis of colon cancer. Cancer Biol Ther. 2020;21(3):258‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu C, Billadeau DD, Abdelhakim H, et al. IQGAP1 suppresses TβRII‐mediated myofibroblastic activation and metastatic growth in liver. J Clin Invest. 2013;123(3):1138‐1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burnier J V, Wang N, Michel RP, et al. Type IV collagen‐initiated signals provide survival and growth cues required for liver metastasis. Oncogene. 2011;30(35):3766‐3783. [DOI] [PubMed] [Google Scholar]
- 53. Quaranta V, Rainer C, Nielsen SR, et al. Macrophage‐derived granulin drives resistance to immune checkpoint inhibition in metastatic pancreatic cancer. Cancer Res. 2018;78(15):4253‐4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Barkan D, El Touny LH, Michalowski AM, et al. Metastatic growth from dormant cells induced by a Col‐I‐enriched fibrotic environment. Cancer Res. 2010;70(14):5706‐5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82(1):4‐7. [DOI] [PubMed] [Google Scholar]
- 56. Olaso E, Salado C, Egilegor E, et al. Proangiogenic role of tumor‐activated hepatic stellate cells in experimental melanoma metastasis. Hepatology. 2003;37(3):674‐685. [DOI] [PubMed] [Google Scholar]
- 57. Wirz W, Antoine M, Tag CG, et al. Hepatic stellate cells display a functional vascular smooth muscle cell phenotype in a three‐dimensional co‐culture model with endothelial cells. Differentiation. 2008;76(7):784‐794. [DOI] [PubMed] [Google Scholar]
- 58. Gordon‐Weeks AN, Lim SY, Yuzhalin AE, et al. Neutrophils promote hepatic metastasis growth through fibroblast growth factor 2–dependent angiogenesis in mice. Hepatology. 2017;65(6):1920‐1935. [DOI] [PubMed] [Google Scholar]
- 59. Calon A, Espinet E, Palomo‐Ponce S, et al. Dependency of colorectal cancer on a TGF‐β‐driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22(5):571‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tauriello DVF, Palomo‐Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538‐543. [DOI] [PubMed] [Google Scholar]
- 61. Zubeldia IG, Bleau AM, Redrado M, etal. Epithelial to mesenchymal transition and cancer stem cell phenotypes leading to liver metastasis are abrogated by the novel TGFβ1‐targeting peptides P17 and P144. Exp Cell Res. 2013;319(3):12‐22. [DOI] [PubMed] [Google Scholar]
- 62. Zhang B, Halder SK, Zhang S, Datta PK. Targeting transforming growth factor‐β signaling in liver metastasis of colon cancer. Cancer Lett. 2009;277(1):114‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Derynck R, Budi EH. Specificity, versatility, and control of TGF‐b family signaling. Sci Signal. 2019;12(570):eaav5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang YE. Non‐Smad signaling pathways of the TGF‐β family. Cold Spring Harb Perspect Biol. 2017;9(2):a022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Frey RS, Mulder KM. Involvement of extracellular signal‐regulated kinase 2 and stress‐activated protein kinase/Jun N‐terminal kinase activation by transforming growth factor β in the negative growth control of breast cancer cells. Cancer Res. 1997;57(4):628‐633. [PubMed] [Google Scholar]
- 66. Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates smad‐independent activation of JNK and p38 by TGF‐β. Mol Cell. 2008;31(6):918‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mao R, Fan Y, Mou Y, Zhang H, Fu S, Yang J. TAK1 lysine 158 is required for TGF‐β‐induced TRAF6‐mediated Smad‐independent IKK/NF‐κB and JNK/AP‐1 activation. Cell Signal. 2011;23(1):222‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Korkut A, Zaidi S, Kanchi RS, et al. A pan‐cancer analysis reveals high‐frequency genetic alterations in mediators of signaling by the TGF‐β superfamily. Cell Syst. 2018;7(4):422‐437. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ivanović V, Demajo M, Krtolica K, et al. Elevated plasma TGF‐β 1 levels correlate with decreased survival of metastatic breast cancer patients. Clin Chim Acta. 2006;371(1‐2):191‐193. [DOI] [PubMed] [Google Scholar]
- 70. Picon A, Gold LI, Wang J, Cohen A, Friedman E. A subset of metastatic human colon cancers expresses elevated levels of transforming growth factor beta1. Cancer Epidemiol Prev Biomarkers. 1998;7(6):497‐504. [PubMed] [Google Scholar]
- 71. Friess H, Yamanaka Y, Büchler M, et al. Enhanced expression of transforming growth factor β isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology. 1993;105(6):1846‐1856. [DOI] [PubMed] [Google Scholar]
- 72. Antognelli C, Cecchetti R, Riuzzi F, Peirce MJ, Talesa VN. Glyoxalase 1 sustains the metastatic phenotype of prostate cancer cells via EMT control. J Cell Mol Med. 2018;22(5):2865‐2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang J, Antin P, Berx G, et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2020;21(6):341‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor‐β1 gene results in multifocal inflammatory disease [14]. Nature. 1992;359(6397):693‐699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Batlle E, Massagué J. Transforming growth factor‐b signaling in immunity and cancer. Immunity. 2019;50(4):924‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fionda C, Stabile H, Cerboni C, et al. Hitting more birds with a stone: impact of TGF‐β on ILC activity in cancer. J Clin Med. 2020;9(1):143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang F, Wang H, Wang X, et al. TGF‐β induces M2‐like macrophage polarization via SNAILmediated suppression of a pro‐inflammatory phenotype. Oncotarget. 2016;7(32):52294‐52306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Oh SH, Swiderska‐Syn M, Jewell ML, Premont RT, Diehl AM. Liver regeneration requires Yap1‐TGFβ‐dependent epithelial‐mesenchymal transition in hepatocytes. J Hepatol. 2018;69(2):359‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bird TG, Müller M, Boulter L, et al. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci Transl Med. 2018;10(454):eaan1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mekala S, Tulimilli SRV, Geesala R, Manupati K, Dhoke NR, Das A. Cellular crosstalk mediated by platelet‐derived growth factor BB and transforming growth factor β during hepatic injury activates hepatic stellate cells. Can J Physiol Pharmacol. 2018;96(8):728‐741. [DOI] [PubMed] [Google Scholar]
- 81. Cai X, Li Z, Zhang Q, et al. CXCL6‐EGFR‐induced kupffer cells secrete TGF‐β1 promoting hepatic stellate cell activation via the SMAD2/BRD4/C‐MYC/ EZH2 pathway in liver fibrosis. J Cell Mol Med. 2018;22(10):5050‐5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fabregat I, Caballero‐Díaz D. Transforming growth factor‐β‐induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front Oncol. 2018;8:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hu X, Marietta A, Dai W‐X, et al. Prediction of hepatic metastasis and relapse in colorectal cancers based on concordance analyses with liver fibrosis scores. Clin Transl Med. 2020;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cox TR, Erler JT. Molecular pathways: connecting fibrosis and solid tumor metastasis. Clin Cancer Res. 2014;20(14):3637‐3643. [DOI] [PubMed] [Google Scholar]
- 85. Cox TR, Bird D, Baker AM, et al. LOX‐mediated collagen crosslinking is responsible for fibrosis‐enhanced metastasis. Cancer Res. 2013;73(6):1721‐1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Calon A, Lonardo E, Berenguer‐Llergo A, et al. Stromal gene expression defines poor‐prognosis subtypes in colorectal cancer. Nat Genet. 2015;47(4):320‐329. [DOI] [PubMed] [Google Scholar]
- 87. Voorneveld PW, Jacobs RJ, De Miranda NFCC, et al. Evaluation of the prognostic value of pSMAD immunohistochemistry in colorectal cancer. Eur J Cancer Prev. 2013;22(5):420‐424. [DOI] [PubMed] [Google Scholar]
- 88. Zhang B, Halder SK, Kashikar ND, et al. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology. 2010;138(3):969‐980. e1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tsushima H, Ito N, Tamura S, et al. Circulating transforming growth factor β1 as a predictor of liver metastasis after resection in colorectal cancer. Clin Cancer Res. 2001;7(5):1258‐1262. [PubMed] [Google Scholar]
- 90. Erin N, Ogan N, Yerlikaya A. Secretomes reveal several novel proteins as well as TGF‐β1 as the top upstream regulator of metastatic process in breast cancer. Breast Cancer Res Treat. 2018;170(2):235‐250. [DOI] [PubMed] [Google Scholar]
- 91. Hsu YL, Huang MS, Hung JY, et al. Bone‐marrow‐derived cell‐released extracellular vesicle miR‐92a regulates hepatic pre‐metastatic niche in lung cancer. Oncogene. 2020;39(4):739‐753. [DOI] [PubMed] [Google Scholar]
- 92. Gonzalez‐Zubeldia I, Dotor J, Redrado M, et al. Co‐migration of colon cancer cells and CAFs induced by TGFβ1 enhances liver metastasis. Cell Tissue Res. 2015;359(3):829‐839. [DOI] [PubMed] [Google Scholar]
- 93. Woodward JKL, Rennie IG, Burn JL, Sisley K. A potential role for TGFβ in the regulation of uveal melanoma adhesive interactions with the hepatic endothelium. Investig Ophthalmol Vis Sci. 2005;46(10):3473‐3477. [DOI] [PubMed] [Google Scholar]
- 94. Koudelkova P, Costina V, Weber G, et al. Transforming growth factor‐β drives the transendothelial migration of hepatocellular carcinoma cells. Int J Mol Sci. 2017;18(10):2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Are C, Simms N, Rajupt A, Brattain M. The role of transforming growth factor‐β in suppression of hepatic metastasis from colon cancer. HPB. 2010;12(7):498‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ai X, Wu Y, Zhang W, et al. Targeting the ERK pathway reduces liver metastasis of Smad4‐inactivated colorectal cancer. Cancer Biol Ther. 2013;14(11):1059‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Teraoka H, Sawada T, Yamashita Y, etal. TGF‐β1 promotes liver metastasis of pancreatic cancer by modulating the capacity of cellular invasion. Int J Oncol. 2001;19(4):709‐715. [DOI] [PubMed] [Google Scholar]
- 98. Reed JA, McNutt NS, Prieto VG, Albino AP. Expression of transforming growth factor‐β2 in malignant melanoma correlates with the depth of tumor invasion: implications for tumor progression. Am J Pathol. 1994;145(1):97‐104. [PMC free article] [PubMed] [Google Scholar]
- 99. Zhang C, Gao H, Li C, et al. TGFβ1 promotes breast cancer local invasion and liver metastasis by increasing the CD44high/CD24‐ subpopulation. Technol Cancer Res Treat. 2018;17:1533033818764497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rani B, Malfettone A, Dituri F, et al. Galunisertib suppresses the staminal phenotype in hepatocellular carcinoma by modulating CD44 expression. Cell Death Dis. 2018;9(3):373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Jin X, Aimaiti Y, Chen Z, Wang W, Li D. Hepatic stellate cells promote angiogenesis via the TGF‐β1‐Jagged1/VEGFA axis. Exp Cell Res. 2018;373(1‐2):34‐43. [DOI] [PubMed] [Google Scholar]
- 102. Teraoka H, Sawada T, Nishihara T, et al. Enhanced VEGF production and decreased immunogenicity induced by TGF‐β 1 promote liver metastasis of pancreatic cancer. Br J Cancer. 2001;85(4):612‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Batlle R, Andrés E, Gonzalez L, et al. Regulation of tumor angiogenesis and mesenchymal–endothelial transition by p38α through TGF‐β and JNK signaling. Nat Commun. 2019;10(1):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yang J, Lu Y, Lin YY, et al. Vascular mimicry formation is promoted by paracrine TGF‐β and SDF1 of cancer‐associated fibroblasts and inhibited by miR‐101 in hepatocellular carcinoma. Cancer Lett. 2016;383(1):18‐27. [DOI] [PubMed] [Google Scholar]
- 105. Zhang X, Zhang J, Zhou H, Liu G, Li Q. Rho kinase mediates transforming growth factor‐β1‐induced vasculogenic mimicry formation: involvement of the epithelial–mesenchymal transition and cancer stemness activity. Acta Biochim Biophys Sin (Shanghai). 2020;52(4):411‐420. [DOI] [PubMed] [Google Scholar]
- 106. Li Q, Zhu CC, Ni B, et al. Lysyl oxidase promotes liver metastasis of gastric cancer via facilitating the reciprocal interactions between tumor cells and cancer associated fibroblasts. EBioMedicine. 2019;49:157‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gupta P, Sata TN, Yadav AK, et al. TGF‐β induces liver fibrosis via miRNA‐181amediated down regulation of augmenter of liver regeneration in hepatic stellate cells. PLoS One. 2019;14(6):e0214534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Tu K, Li J, Verma VK, etal. Vasodilator‐stimulated phosphoprotein promotes activation of hepatic stellate cells by regulating Rab11‐dependent plasma membrane targeting of transforming growth factor β receptors. Hepatology. 2015;61(1):361‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zeisberg M, Yang C, Martino M, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282(32):23337‐23347. [DOI] [PubMed] [Google Scholar]
- 110. Kitamura T, Kometani K, Hashida H, et al. SMAD4‐deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat Genet. 2007;39(4):467‐475. [DOI] [PubMed] [Google Scholar]
- 111. Itatani Y, Kawada K, Fujishita T, et al. Loss of SMAD4 from colorectal cancer cells promotes CCL15 expression to recruit CCR1+ myeloid cells and facilitate liver metastasis. Gastroenterology. 2013;145(5):1064‐1075. [DOI] [PubMed] [Google Scholar]
- 112. Huang X, Zou Y, Lian L, et al. Changes of T cells and cytokines TGF‐β1 and IL‐10 in mice during liver metastasis of colon carcinoma: implications for liver anti‐tumor immunity. J Gastrointest Surg. 2013;17(7):1283‐1291. [DOI] [PubMed] [Google Scholar]
- 113. Yi Y, He H‐W, Wang J‐X, et al. The functional impairment of HCC‐infiltrating γδ T cells, partially mediated by regulatory T cells in a TGFβ‐ and IL‐10‐dependent manner. J Hepatol. 2013;58(5):977‐983. [DOI] [PubMed] [Google Scholar]
- 114. Sawada T, Kimura K, Nishihara T, etal. TGF‐β1 down‐regulates ICAM‐1 expression and enhances liver metastasis of pancreatic cancer. Adv Med Sci. 2006;51:60‐65. [PubMed] [Google Scholar]
- 115. Medina‐Echeverz J, Fioravanti J, Diáz‐Valdés N, et al. Harnessing high density lipoproteins to block transforming growth factor beta and to inhibit the growth of liver tumor metastasis. PLoS One. 2014;9(5):e96799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ganesh K, Massagué J. TGF‐β inhibition and immunotherapy: checkmate. Immunity. 2018;48(4):626‐628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yang Y, Dukhanina O, Tang B, et al. Lifetime exposure to a soluble TGF‐β antagonist protects mice against metastasis without adverse side effects. J Clin Invest. 2002;109(12):1607‐1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Huynh LK, Hipolito CJ, ten Dijke P. A perspective on the development of TGF‐β inhibitors for cancer treatment. Biomolecules. 2019;9(11):743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Melisi D, Garcia‐Carbonero R, Macarulla T, et al. Galunisertib plus gemcitabine vs. gemcitabine for first‐line treatment of patients with unresectable pancreatic cancer. Br J Cancer. 2018;119(10):1208‐1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lan Y, Zhang D, Xu C, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD‐L1 and TGF‐. Sci Transl Med. 2018;10(424):eaan5488. [DOI] [PubMed] [Google Scholar]
- 121. Brandes AA, Carpentier AF, Kesari S, et al. A phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 2016;18(8):1146‐1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hammad S, Cavalcanti E, Werle J, et al. Galunisertib modifies the liver fibrotic composition in the Abcb4Ko mouse model. Arch Toxicol. 2018;92(7):2297‐2309. [DOI] [PubMed] [Google Scholar]
- 123. Luangmonkong T, Suriguga S, Bigaeva E, et al. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br J Pharmacol. 2017;174(18):3107‐3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Morris JC, Tan AR, Olencki TE, etal. Phase I study of GC1008 (Fresolimumab): a human anti‐transforming growth factor‐β (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One. 2014;9(3):e90353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Formenti SC, Lee P, Adams S, et al. Focal irradiation and systemic TGFb blockade in metastatic breast cancer. Clin Cancer Res. 2018;24(11):2493‐2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Li S, Sun S, Xiang H, Yang J, Peng M, Gao Q. Liver metastases and the efficacy of the PD‐1 or PD‐L1 inhibitors in cancer: a meta‐analysis of randomized controlled trials. Oncoimmunology. 2020;9(1):1746113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Tumeh PC, Hellmann MD, Hamid O, et al. Liver metastasis and treatment outcome with anti‐PD‐1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res. 2017;5(5):417‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Chen L, Gibbons DL, Goswami S, et al. Metastasis is regulated via microRNA‐200/ZEB1 axis control of tumour cell PD‐L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Zhao F, Evans K, Xiao C, etal. Stromal fibroblasts mediate anti–PD‐1 resistance via MMP‐9 and dictate TGFβ inhibitor sequencing in melanoma. Cancer Immunol Res. 2018;6(12):1459‐1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Groeneveldt C, van Hall T, van der Burg SH, ten Dijke P, van Montfoort N. Immunotherapeutic potential of TGF‐β inhibition and oncolytic viruses. Trends Immunol. 2020;41(5):406‐420. [DOI] [PubMed] [Google Scholar]
- 133. Penafuerte C, Bautista‐Lopez N, Bouchentouf M, Birman E, Forner K, Galipeau J. Novel TGF‐β antagonist inhibits tumor growth and angiogenesis by inducing IL‐2 receptor‐driven STAT1 activation. J Immunol. 2011;186(12):6933‐6944. [DOI] [PubMed] [Google Scholar]
- 134. Ng S, Deng J, Chinnadurai R, Yuan S, Pennati A, Galipeau J. Stimulation of natural killer cell‐mediated tumor immunity by an IL15/TGFβ‐neutralizing fusion protein. Cancer Res. 2016;76(19):5683‐5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Lind H, Gameiro SR, Jochems C, et al. Dual targeting of TGF‐β and PD‐L1 via a bifunctional anti‐PD‐L1/TGF‐ΒRII agent: status of preclinical and clinical advances. J Immunother Cancer. 2020;8:e000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Horn LA, Riskin J, Hempel HA, et al. Simultaneous inhibition of CXCR1/2, TGF‐β, and PD‐L1 remodels the tumor and its microenvironment to drive antitumor immunity. J Immunother cancer. 2020;8(1):e000326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Holmgaard RB, Schaer DA, Li Y, et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti‐tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer. 2018;6(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. van Beuge MM, Prakash J, Lacombe M, et al. Enhanced effectivity of an ALK5‐inhibitor after cell‐specific delivery to hepatic stellate cells in mice with liver injury. PLoS One. 2013;8(2):e56442‐e56442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Beerling E, Ritsma L, Vrisekoop N, Derksen PWB, Van Rheenen J. Intravital microscopy: new insights into metastasis of tumors. J Cell Sci. 2011;124(3):299‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Ritsma L, Steller EJA, Beerling E, et al. Intravital microscopy through an abdominal imaging window reveals a pre‐micrometastasis stage during liver metastasis. Sci Transl Med. 2012;4(158):158ra145‐158ra145. [DOI] [PubMed] [Google Scholar]