

Abstract
Acute lymphoblastic leukemia (ALL) is the most common childhood cancer with high cure rates leading to rising numbers of long-term survivors. Adult survivors of childhood ALL are at increased risk of obesity, cardiovascular disease, and other chronic illnesses. We hypothesize that ALL therapy is associated with long-term gut microbiome alterations that contribute to predisposition to chronic medical conditions. We conducted a pilot study to test whether differences can be detected between stool microbiota of pediatric ALL survivors and their siblings. Stool samples were collected from 38 individuals under age 19 who were at least 1 year after completion of therapy for ALL. Stool samples collected from 16 healthy siblings served as controls. 16S ribosomal RNA gene sequencing was performed on the stool samples. Comparing microbiota of survivors to sibling controls, no statistically significant differences were found in alpha or beta diversity. However, among the top 10 operational taxonomic units (OTUs) from component 1 in sparse partial least squares discriminant analysis (sPLS-DA) with different relative abundance in survivors versus siblings, OTUs mapping to the genus Faecalibacterium were depleted in survivors. Differences in gut microbial composition were found between pediatric survivors of childhood ALL and their siblings. Specifically, the protective Faecalibacterium is depleted in survivors, which is reminiscent of gut microbiota alteration found in adult survivors of childhood ALL and reported in obesity, suggesting that microbiota alterations in pediatric ALL survivors start in childhood and may play a role in predisposition to chronic illness in later years of survivorship.
Keywords: Acute lymphoblastic leukemia, ALL survivors, Faecalibacterium, microbiome, stool microbiota
Introduction
Approximately 3,000 children in the United States are diagnosed with acute lymphoblastic leukemia (ALL) each year. With 5-year survival rates >90%, the number of individuals living in the US who were treated for childhood ALL has been rising and is estimated currently to be >50,000.1–3 Long-term survivors of ALL have higher rates of obesity and metabolic syndrome compared to their siblings and the general population, as well as several-fold increase in cardiovascular disease morbidity and mortality.4–13
While cranial irradiation and transplant were identified as risk factors for obesity in the past, contemporary ALL treatment has largely eliminated radiation, yet obesity continues to be highly prevalent. Multiple studies, including those from the Childhood Cancer Survivor Study (CCSS), the St. Jude Lifetime Cohort Study (SJLIFE), and the French Leucémie de l’Enfant et de l’Adolescent L.E.A.program, have reported obesity rates of 31.7–42.8% in ALL survivors compared to approximately 20% in controls and relative risk of metabolic syndrome of 1.43–2.0 among survivors.6,8,10–12 The recent comprehensive SJLIFE analysis of ALL survivors treated over 4 decades showed not only continued prevalence of treatment-related sequelae but also doubling of the number of late health conditions per survivor compared to controls.14 While the success of ALL treatment continues to improve, the long-term morbidity in the survivors remains a challenge.15
The gut microbiome has emerged as an orchestrator of gastrointestinal and systemic functions through various immune, metabolic, and inflammatory mechanisms in conjunction with the host.16,17 Dysbiosis is seen in many disease states and can contribute to disease pathophysiology.18–20 Microbiota analyses have shown obesity to be associated with low microbiota diversity, fewer taxa, and altered Firmicutes to Bacteroidetes ratio compared to leanness, in adults as well as in children.21–27 Furthermore, changes in gut microbiome have been shown to precede the development of obesity, and fecal transplants from obese mice or humans into lean mice resulted in lean mice becoming obese, and vice versa, suggesting a causal role for microbes in body phenotype and associated health risks.28,29
In cancer, gut microbiome has been implicated in cancer treatment toxicities in studies done largely in adults, but studies in pediatric cancer have been limited.30–32 During cancer treatment, chemotherapy and antibiotics decrease both diversity and abundance of intestinal flora, conferring immediate toxicities, allowing pathogenic bacteria to predominate, leading to potential far-reaching systemic effects.33–37 A significant drop in microbiota abundance is observed immediately after chemotherapy treatment.38 Profound antibiotic-related effects on the microbiota have been identified, with certain components not recovering even after years.39–41 A few published gut microbiota studies in pediatric leukemia patients show that AML treatment resulted in 10,000-fold decrease in protective anaerobic bacteria and 100-fold increase in pathogenic aerobic enterococci.42,43 In pediatric B-cell ALL, a stool microbiota study of 28 patients compared to 23 sibling controls revealed that microbiota diversity was already lower at leukemia diagnosis.44 A larger study of serial stool samples in pediatric ALL patients found that changes in the relative abundance of certain bacterial taxa predicted subsequent infection risks.45 These studies show dysbiosis at diagnosis and during therapy, but whether dysbiosis persists after treatment is largely unexplored.
Chemotherapy and antibiotics are indispensable for ALL treatment. Just as efforts have been made to reduce the immediate toxicities of chemotherapy, it may be possible to reduce the long-term health effects of chemotherapy and antibiotics post treatment. Manipulations of microbiota to restore and maintain diversity are novel interventions being tested. These include noninvasive approaches such as diet manipulations, oral administration of pre-biotics, pro-biotics, or post-biotics, and more direct techniques such as fecal microbiota transplantation (FMT) being tried in the stem cell transplant setting. A Malaysian study published in 2017 by Chua et al. found that adults who were long-term survivors of childhood ALL had decreased stool microbiota diversity that correlated with markers of immune activation.52 Studies such as this aimed at understanding the microbiome of cancer survivors may reveal opportunities for novel strategies to improve long-term health outcomes.
We hypothesize that ALL treatment may be associated with long-term alterations in gut microbiome that can be a potential factor in the predisposition to chronic medical conditions in survivors of childhood ALL. In a pilot study, we investigated whether microbiota alterations can be detected in stool samples of pediatric ALL survivors compared to those of their siblings.
Methods
Recruitment
ALL survivors were identified through the roster of the Pediatric Specialists of Virginia Long Term Follow Up clinic. Survivors of childhood ALL who were at least 1 year off therapy and <19 years of age were recruited for study enrollment by informed consent. Survivors who came to clinic during the recruitment period were approached in person. Survivors who were not scheduled to come to clinic were recruited by telephone. Studies have shown that fecal microbiomes are similar among family members, therefore, healthy full siblings were recruited as controls when available.28,46 Stools were collected on stool cards (Hemoccult II SENSA cards® (Beckman Coulter, CA)) by subjects or their parents. Stool cards were either mailed back or brought back to clinic. Samples and clinical data were de-identified and assigned subject ID numbers. Subjects with siblings were tracked as families. The study was approved by the Inova Hospital IRB and signed consents were obtained for all participants.
Clinical database
A clinical database was created for this study and included the following fields: date of birth, age, gender, date of ALL diagnosis, treatment protocol, end of treatment date, stool sample collection date, prophylactic and treatment antibiotic courses received during therapy, radiation, diet, probiotics usage, weight, height, BMI percentiles and z-scores at diagnosis and at time of stool collection. For siblings, only anthropometric data, diet, antibiotic and probiotics usage were collected.
Sample preparation and sequencing
Stool cards were stored at −80° prior to processing. DNA was extracted from stool cards as previously described.47 Prepped samples were loaded on the EZ1 Advanced (Qiagen, Valencia, CA) using the EZ1 Tissue Kit and the Bacterial DNA Extraction protocol card. Samples were cleaned and concentrated using the DNeasy PowerClean Cleanup kit (Qiagen, Valencia, CA). Sequencing libraries were prepared using a Nextera XT kit (Illumina, San Diego, CA) using a modified Illumina 16S Metagenomics Sequencing Library Preparation protocol for analysis of hypervariable region V4. Each sample was sequenced on the Illumina MiSeq with paired-end reads of 301 bp. Sequencing of negative controls of lysis buffer and positive controls of Staphylococcus aureus (Strain NCTC 8532, ATCC, VA) and Escherichia coli (Strain NCTC 9001, ATCC, VA) were included. The sequence data are being deposited in NCBI Sequence Read Archive (SRA).
16S rRNA data preprocessing
QIIME 1.9 was used for the 16S rRNA data preprocessing to generate the OTU (operational taxonomic unit) table and the phylogenetic tree.48 Fastq files of the demultiplexed sequencing reads from each sample were treated as single-ended reads and quality filtered using default QIIME settings.49 Open-reference OTU picking against the Greengenes database was performed on the filtered sequences.50 The OTU table and the phylogenetic tree were imported into R Bioconductor package Phyloseq (Version 1.25.2), where a single rarefaction at 4,897 was performed and OTUs that were not observed more than 3 times in at least 20% of the samples were removed (Supplemental Figure S2).
Data analysis
All data analysis in this manuscript was performed using R Version 3.5.0 with the Phyloseq package unless otherwise noted. Four alpha diversity measures were calculated, including number of OTUs, Fisher, Shannon, and Simpson. Beta diversities were calculated by unweighted unifrac, weighted unifrac, dpcoa, jsd, and bray. PERMANOVA was performed using the adonis function in R package Vegan 2.5 to compare the bray distance between survivors and siblings.
The OTUs were further filtered by removing those with less than 100 raw counts. The related abundances at OTU and each taxa level (Phylum, Class, Order, Family and Genus) were calculated using normalized counts for each sample. Two sample Wilcoxon Test was used to compare the groups at Phylum and genus levels, as well as at the OTU level.
Sparse partial least squares discriminant analysis (sPLS-DA) was performed using the R mixOmics Package (Version 6.3.2) for selecting the most discriminative OTUs on the first and second principal components that best discriminate survivor and sibling groups.51 The abundances of the selected OTUs were shown on clustering heatmaps.
Results
Patient characteristics
One hundred thirty-four ALL survivors on our Long Term Follow Up roster met eligibility criteria, which included prior treatment for either B-cell or T-cell ALL, at least 1 year off therapy, and age younger than 19 years. Of these 134 eligible survivors, 38 (28%) were lost to follow-up and could not be contacted, 34 (25%) declined participation, 60 (45%) verbally consented, but 6 did not return signed consent forms. 54 (40%) survivors were given stool cards; 39 stool cards were returned, for a stool card recovery rate of 72%. One survivor stool sample was excluded, because the survivor relapsed shortly after submitting the stool card. Of 28 siblings who consented, 18 returned stool samples, 2 of which were excluded due to low number of sequencing reads. Thus, the analysis included 38 survivors, or 28% of all eligible survivors, and 16 full siblings of 12 survivors (Supplemental Figure S1).
Participant characteristics are shown in Table 1. Of the 38 survivors, 34 had B-cell ALL, including 27 standard risk and 7 high risk, and 4 had T-cell ALL. The male:female ratio was 16:22 among survivors and 9:7 among siblings. The ages of survivors ranged from 7–18 years (median 12 years), the age at diagnosis ranged from 1 to 12 years (median 4 years), and the number of years off therapy ranged from 1.3 to 11.9 years (median 4.4 years). The ages of siblings ranged from 4 to 15 years (median 10.5 years). Siblings were significantly younger than survivors (p = 0.017). The BMI percentile and z-scores of survivors tended to be higher than those of siblings, but the differences were not statistically significant. In the 6 months prior to stool collection, 8/38 (21.1%) survivors and 4/16 (25%) siblings reported being treated with antibiotics. Reasons listed for antibiotics in survivors included Strep pharyngitis, bronchitis, chest infection, urinary tract infection, upper airways infection, sinus infection, and wisdom teeth infection. Siblings took antibiotics for ear infection, Strep pharyngitis, skin infection, sinus infection. Probiotic usage was reported in 20/38 (52.6%) survivors and 10/16 (62.5%) siblings. Probiotics included mostly yogurt (30/54), with 2 mentions of Kids Culturelle, 1 Bio-Kult, 1 Animal Parade AcidophiKidz supplement. There were no statistical differences in antibiotic or probiotic usage in the 6 months prior to stool collection between the two groups.
Table 1.
Participant characteristics.
| Survivors | Siblings | p value | |
|---|---|---|---|
| Number | N = 38 | N = 16 | |
| ALL type | 34 pre-B cell | N/A | |
| 27 standard risk | |||
| 7 high risk | |||
| 4 T-cell | N/A | ||
| Male (%) | 16 (42) | 9 (56) | 0.514 |
| Age (yr) at diagnosis, range (median) | 1–12 (4) | N/A | |
| Age (yr) at EOT*, range (median) | 3–14 (6) | N/A | |
| Yrs from EOT to stool collection, range (median) | 1.3–11.9 (4.4) | N/A | |
| Age at stool collection, range (median) | 7–18 (12) | 4–15 (10.5) | 0.017 |
| BMI %ile at stool collection, mean (sd) | 59.2 (32.7) | 55.11 (35.92) | 0.698 |
| BMI z-score at stool collection, mean (sd) | 0.74 (2.52) | 0.06 (1.39) | 0.215 |
| Number parenteral antibiotic courses, range (median) | 1–15 (7) | N/A | |
| Antibiotics in last 6 months, true (%) | 8 (21.1) | 4 (25%) | 1 |
| Probiotics in last 6 months, true (%) | 20 (52.6) | 10 (62.5) | 0.714 |
Alpha diversity
Alpha diversity evaluates the richness, or the number of species within samples, and their relative evenness. While the means of alpha diversity measured by 4 different methods were all decreased in the survivors compared to siblings, the differences did not reach statistical significance by Wilcoxon rank sum test (number of OTUs, p = 0.91; Fisher, p = 0.91; Shannon, p = 0.57; Simpson, p = 0.56) (Figure 1). Comparing survivors with siblings in the same family, alpha diversity of survivors was lower or the same as siblings in 10 out of 12 families; however, in 2 families the survivors demonstrated much higher alpha diversity than their siblings, for unclear reasons.
Figure 1.
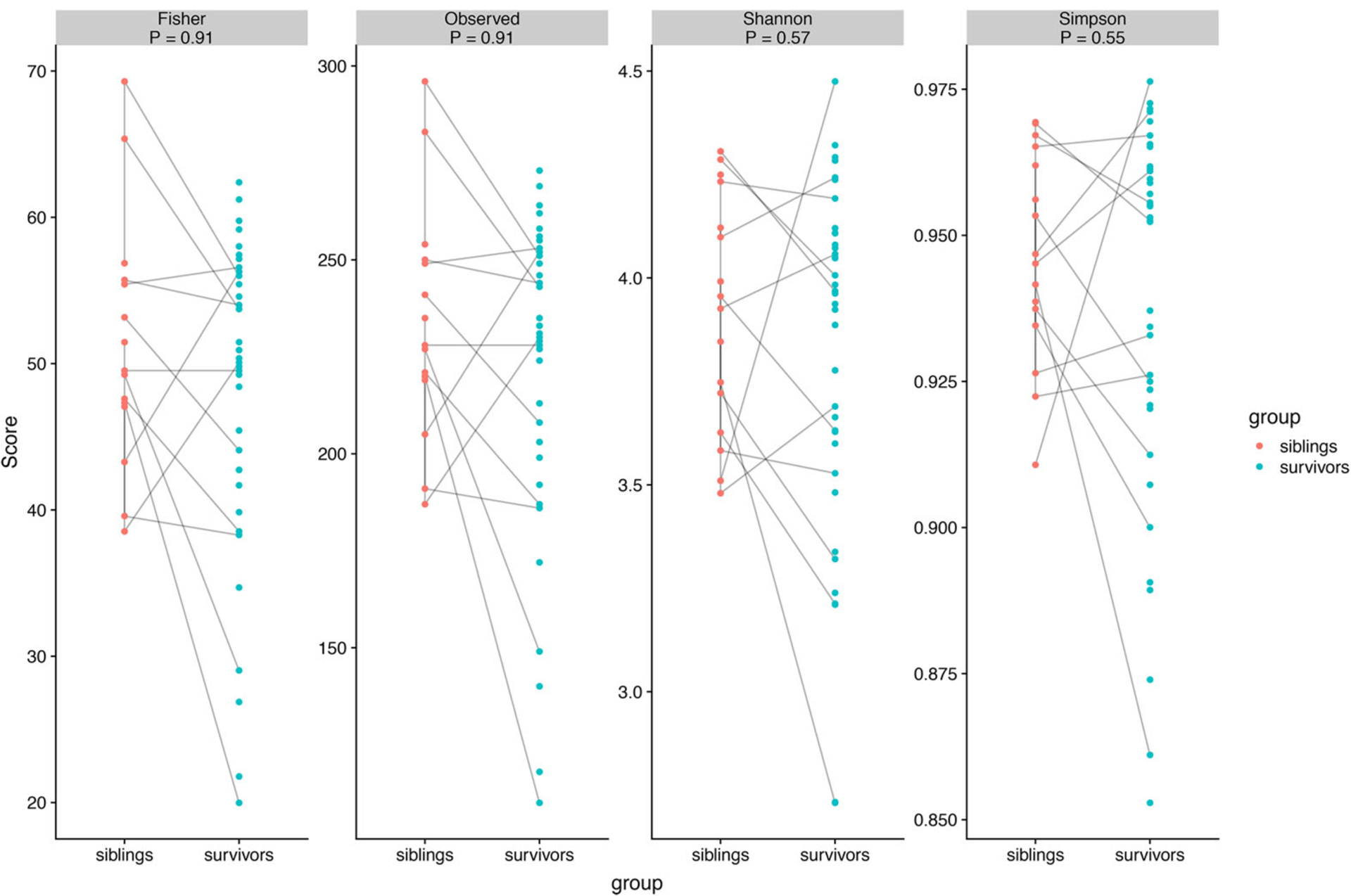
Alpha diversity of gut microbiota of pediatric ALL survivors and siblings with p-values, measured by Fisher, Number of OTUs (observed), Shannon and Simpson methods.
Beta diversity
To compare microbial composition between samples, beta diversities were calculated based on the differences in shared species and their abundance. Principal coordinate analysis (PCoA) based on Bray-Curtis distances using all filtered OTUs did not reveal a separation between ALL survivors and their healthy siblings (p = 0.23, Adonis Test with 9999 permutations) (Figure 2). In this unsupervised analysis, survivors were not more similar to each other than to siblings.
Figure 2.
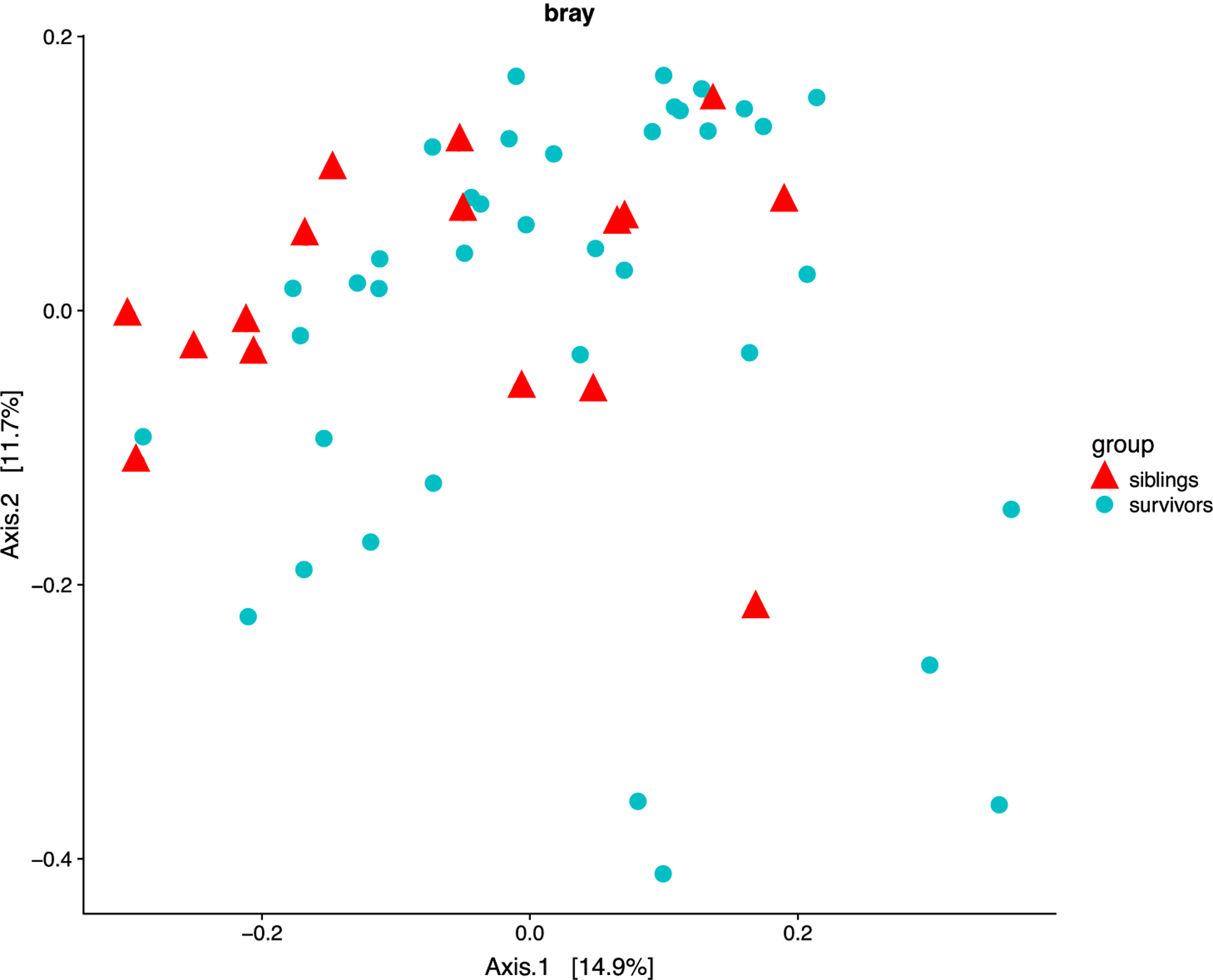
Beta diversity of gut microbiota of pediatric ALL survivors and siblings by Bray distance. Axis 1 and 2 are principal components 1 and 2.
Relative abundance
The relative abundance of bacteria in the survivors compared to siblings was not statistically different at the phylum level (p > 0.05 for all) (Supplemental Figure S3). However, using the top 10 OTUs in the sparse partial least squares discriminant analysis (sPLS-DA), survivors and siblings separated into 2 clusters (Figure 3A,B). Members of the Lachnospiraceae and Ruminococcaceae families, including Faecalibacterium, were depleted in survivors compared to siblings (Figure 3C). The most depleted OTU was in the Lachnospiraceae family, but the species could not be identified from the available 16S rRNA sequences. The relative abundance of 3 OTUs mapping to Faecalibacterium were notably lower in survivors than controls. Statistically significant correlation was not found between Faecalibacterium abundance and length of survivorship in this study with limited sample size. Thus, although a difference in overall gut microbiota diversity between survivors and siblings was not demonstrated in this pilot study, the relative abundance of at least 10 specific microbial taxa were significantly different between the 2 groups. Gut microbial composition in childhood ALL survivors is not the same as in their siblings, with lower abundance of members of Ruminococcaceae and Lachnospiraceae families, notably Faecalibacterium.
Figure 3.
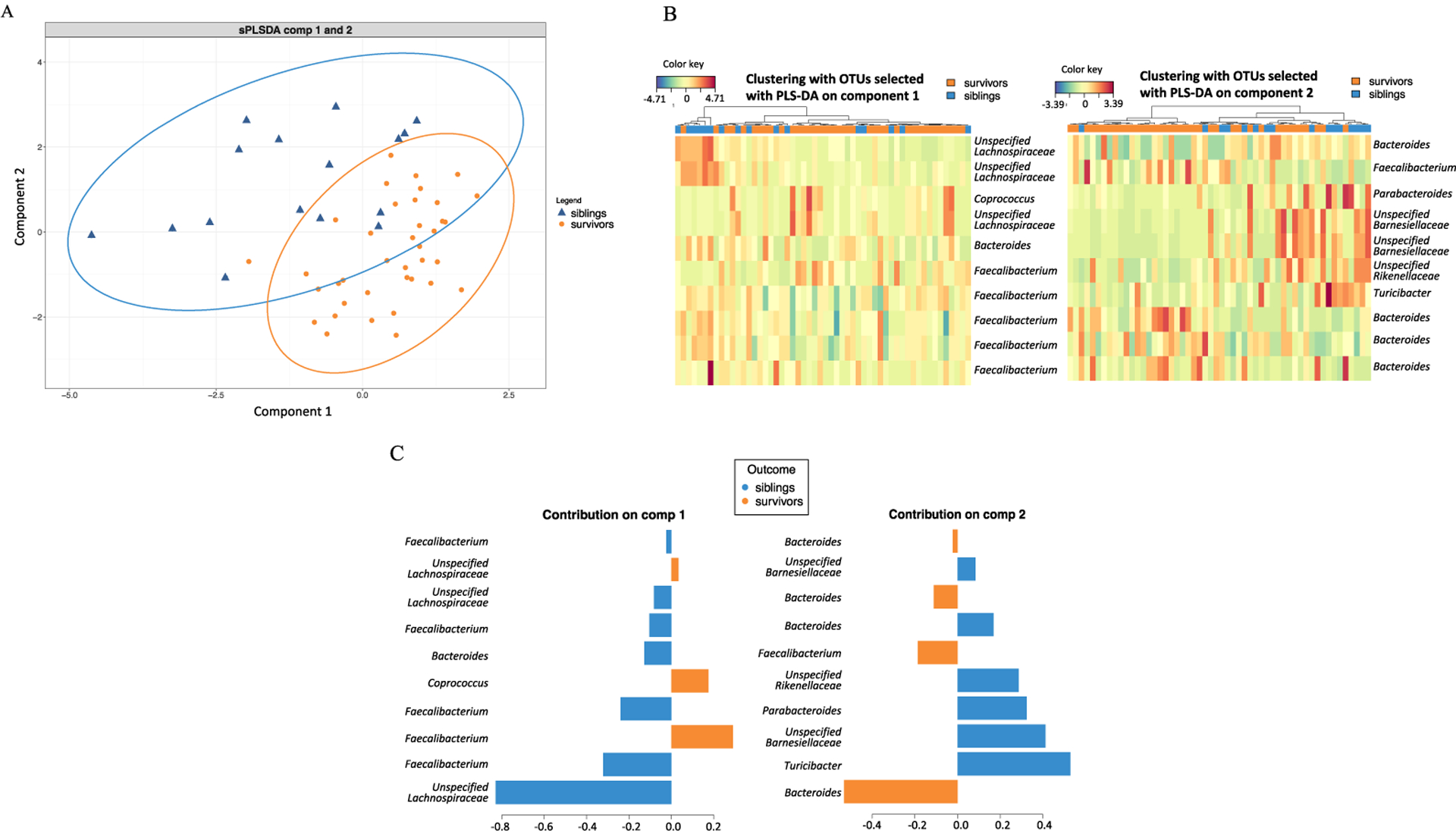
A. sPLS-DA analysis of the 2 groups, survivors and siblings, based on the top 10 OTUs most different between survivors and siblings. B. Clustering heatmaps of 10 selected OTUs from sPLS-DA plot component 1 and 2, labeled with genus name when known, or unspecified family name when genus could not be identified. C. Contribution plot showing the contribution of the 10 OTUs from component 1 and 2 of survivors and siblings.
Further analysis of relative abundance of taxa against BMI, probiotic usage, high versus standard risk ALL, and courses of antibiotics received during therapy did not reveal statistically significant correlations.
Discussion
This pilot study was designed to see if any difference in stool microbiota could be detected between pediatric survivors of ALL and their siblings. Differential relative abundances of certain bacterial taxa were detected. Children off therapy for ALL displayed depletion of OTUs in Ruminococcaceae and Lachnospiraceae families of bacteria. Specifically, the decrease in Ruminococcaceae Faecalibacterium seen here has been reported by Chua et al. in survivors of childhood ALL when they have reached adulthood.52 Our study suggests that this alteration in microbiota starts in childhood. Faecalibacterium is decreased in obesity, diabetes, ulcerative colitis, Crohn’s disease, and after hematopoietic stem cell transplant.53,54 Faecalibacterium prausnitzii ferments dietary fiber, is a major producer of the beneficial short chain fatty acid butyrate, and has anti-inflammatory properties that ameliorate colitis, thus is a “protective” bacterium that is integral to intestinal health.53,55,56 Depletion of this beneficial bacterium, Faecalibacterium, as well as other microbial species, in ALL survivors compared to their siblings found in this study supports the proposition that gut microbiota dysbiosis could be a factor in predisposition to obesity and chronic illness as childhood ALL survivors grow into adulthood.
While decreased microbial diversity is associated with obesity, metabolic syndrome, and other disease states,57,58 studies suggest that in some settings, such as in cancer treatment and after antibiotic therapy, overall diversity is unchanged but microbial composition is altered due to dominance or depletion of selected taxa.41,43,45,54 Even short-term changes during critical periods of gut microbiome development can have long-term health consequences.59 Repeated courses of chemotherapy and antibiotics commonly administered in leukemia treatment may cause changes in the microbiota that never recover and contribute to chronic illness predisposition. We found changes in microbial composition in survivors who were many years off therapy, which further supports the long-lasting effects of treatment on the gut microbiome. The study by Chua et al. found significantly lower alpha diversity among 73 adult survivors of childhood ALL compared to 61 healthy controls,52 but in our smaller study of pediatric survivors of ALL, we did not find statistically significant decrease in alpha diversity in survivors relative to their siblings, which could be due to small sample size or perhaps not enough years have lapsed since treatment. However, we found that the relative abundances of at least 10 different OTUs were significantly different between the survivors and siblings, indicating altered microbial composition in ALL survivors.
The difference in microbial composition was found in survivors 1.3 to 11.9 years off therapy (median 4.4 years), suggesting that microbiota alterations likely present at the conclusion of therapy can persist for years. Depletion of Faecalibacterium in both adult and childhood ALL survivors further supports that dysbiosis associated with cancer treatment can be long-lasting. This pilot study is too small to correlate the extent of dysbiosis to length of time off therapy. No correlation was found between microbial composition change and BMI of survivors, possibly because the times off therapy for survivors in this study were too short for the development of obesity commonly reported in adult survivors of childhood ALL.
Antibiotics can have profound effects on gut microbiota.39–41 All patients treated for ALL receive pneumocystis prophylaxis with Bactrim or pentamidine throughout the 2–3 years of therapy and for 3 months afterward. Broad-spectrum intravenous (IV) antibiotic is the mainstay of fever and neutropenia management during leukemia treatment. The survivors in this study received between 1 and 15 courses of IV antibiotics during therapy (median 7 courses), yet alpha diversity did not correlate with the number of antibiotic courses. A published microbiome study of 16 pediatric ALL patients during the first 6 months of treatment stratified by the presence or absence of infectious complications found significant differences in phylogenetic diversity between the two groups, and notably Faecalibacterium was the only species that was depleted in those with infectious complications.60 Whether the reduction in Faecalibacterium was due to antibiotic usage or the infection itself could not be discerned. A larger study correlating stool microbiota to infection in 199 children with ALL did not attribute special significance to Faecalibacterium.45 In the pilot data reported here, depletion of Faecalibacterium could not be correlated to the number of IV antibiotic courses received.
Chemotherapy also alters the gut microbiome. Childhood ALL therapy is very protocol-driven and the backbone of standard risk ALL protocols has been fairly constant over the years, with high-risk groups receiving more intense chemotherapy. The St. Jude ALL study found significant decreases in diversity after intensive induction and re-induction chemotherapy in pediatric ALL patients,45 similar to studies of adult leukemia/lymphoma and stem cell transplant patients receiving chemotherapy and immunosuppressive therapy. It is impossible to discern whether the differential microbial composition in the ALL survivors reported here, or in the above studies, is due to chemotherapy, antibiotics, infections, leukemia itself, or a combination of all of these factors.
A major limitation of this pilot study is small sample size of both the survivors and siblings. Only 28% of eligible survivors donated stool samples, and even fewer siblings participated. The high rate of declination to participate in this study was unexpected (34 out of 96 survivors contacted, or 35% declined), as was a similarly high rate of failure to return the stool card after consent was signed (21 of 60 consented, or 35% did not return sample), despite repeated reminders and concerted efforts of study staff. A higher participation rate would be more reassuring regarding possible selection bias of subjects. However, using siblings as controls rather than unrelated controls strengthens this study. Future studies may consider multi-institutional recruitment to increase the number of eligible subjects.
Potential confounding factors in this study were antibiotic and probiotic usage in the months prior to stool collection. Up to 21% of survivors and 25% of siblings received antibiotics, which could have masked potential differences between survivors and siblings. However, given the small sample size to begin with, we did not exclude participants who took antibiotics. In future studies with larger numbers, recent antibiotic use could be either accounted for or excluded from analysis. Consumption of yogurt was common and was similar between survivors and siblings, while other probiotic supplements were rare. Thus, probiotic usage was less likely to have significantly affected the results.
Our pilot study used 16S rRNA gene sequencing of stool bacteria. 16S rRNA exists in all bacteria and has species-specific variable regions. Sequencing of 16S rRNA variable regions provides overviews of microbiota composition, but often lacks resolution to identify species or strains. We encountered a number of “unspecified” OTUs, meaning the identification beyond phylum or family level could not be assigned. This challenge could be overcome by shotgun metagenomic analysis, designed to sequence all genes of all microbes in a sample without restriction, which enables more precise taxonomy and identification of specific species and strains.61 In addition to the presence of specific microbes, the function of the microbiome can be examined through metabolomics. Future studies may employ both metagenomic sequencing and metabolomics to gain deeper insight into the roles the species play in the microbiomes of ALL survivors.
Another limitation of this study is that only stool samples during survivorship were collected. We observed decreased relative abundance of Ruminococcaceae and Lachnospiraceae in long-term survivors, but we are unable to determine when or how the changes occurred since stool was sampled only at one point in time. Future studies need to include repeated longitudinal sampling from the same individual at the time of diagnosis, during therapy, at the completion of therapy, and during off therapy followup, to track microbiota alterations as they occur. Longitudinal sampling will more likely enable correlation of microbiota profiles to specific clinical events. Such longitudinal data will also potentially reveal cause and effect relationships and address whether microbiota alterations during ALL therapy persist long term. In addition, several studies have indicated that microbiota profiles, such as species dominance, are predictive of subsequent infections.43,45,60 With more precise knowledge of the timing of relative species changes, interventions based on microbiota profiles may be designed to avert impending infections. There are now at least three published reports of longitudinal stool sampling during the initial part of childhood ALL treatment,44,45,62 but these did not extend to the end of therapy and beyond, to speak to whether or how microbiota alterations might recover. Such studies are difficult and time-consuming, given that current childhood ALL therapy lasts 2.5 to 3 years.
In conclusion, the pilot study reported here showed that differences in gut microbial composition could be detected between pediatric survivors of ALL and their siblings. The preliminary data can serve as a basis for larger longitudinal microbiome studies to confirm these findings and to extend knowledge of the microbiota in survivorship, which could lead to potential microbial-targeted interventions to improve the long-term health of ALL survivors.
Supplementary Material
Acknowledgements
We thank the PSV Long Term Follow Up team for access to patient roster and are grateful to the off therapy ALL patients and their families for donating samples to this research project. We gratefully acknowledge the support of research activities by Inova Children’s Hospital and the Pediatric Specialists of Virginia Philanthropy Fund, which generously provided gift cards for the study participants.
Funding
This project was supported by a Pilot Project Grant from the Inova Translational Medicine Institute (Yang) and a Research Seed Grant from the Inova Research Council (Yang). Research reported in this publication was also supported in part by the National Institute of Child Health and Human Development under Award Number K23HD099240 (Hourigan). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- ALL
acute lymphoblastic leukemia
- OTU
operational taxonomic unit
- sPLS-DA
sparse partial least squares discriminant analysis
- PCoA
principal coordinate analysis
Footnotes
Supplemental data for this article can be accessed at https://doi.org/10.1080/08880018.2020.1759740.
Conflict of interest
The authors have no conflicts of interest to disclose.
References
- 1.Tai EW, Ward KC, Bonaventure A, Siegel DA, Coleman MP. Survival among children diagnosed with acute lymphoblastic leukemia in the United States, by race and age, 2001 to 2009: findings from the CONCORD-2 study. Cancer. 2017;123(Suppl 24):5178–5189. doi: 10.1002/cncr.30899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noone AM, Howlader N, Krapcho M, et al. (eds). SEER Cancer Statistics Review, 1975–2015. Bethesda, MD: National Cancer Institute. [Google Scholar]
- 3.Mariotto AB, Rowland JH, Yabroff KR, et al. Long-term survivors of childhood cancers in the United States. Cancer Epidemiol Biomarkers Prev. 2009;18(4):1033–1040. doi: 10.1158/1055-9965.EPI-08-0988. [DOI] [PubMed] [Google Scholar]
- 4.Robison LL, Hudson MM. Survivors of childhood and adolescent cancer: life-long risks and responsibilities. Nat Rev Cancer. 2014;14(1):61–70. doi: 10.1038/nrc3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hudson MM, Oeffinger KC, Jones K, et al. Age-dependent changes in health status in the Childhood Cancer Survivor cohort. J Clin Oncol. 2015;33(5):479–491. doi: 10.1200/JCO.2014.57.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Essig S, Li Q, Chen Y, et al. Risk of late effects of treatment in children newly diagnosed with standard-risk acute lymphoblastic leukaemia: a report from the Childhood Cancer Survivor Study cohort. Lancet Oncol. 2014;15(8):841–851. doi: 10.1016/S1470-2045(14)70265-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown AL, Lupo PJ, Danysh HE, et al. Prevalence and predictors of overweight and obesity among a multiethnic population of pediatric acute lymphoblastic leukemia survivors: a cross-sectional assessment. J Pediatr Hematol Oncol. 2016;38(6):429–436. doi: 10.1097/MPH.0000000000000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson CL, Liu W, Yang JJ, et al. Genetic and clinical factors associated with obesity among adult survivors of childhood cancer: a report from the St. Jude lifetime cohort. Cancer. 2015;121(13):2262–2270. doi: 10.1002/cncr.29153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang FF, Kelly MJ, Saltzman E, Must A, Roberts SB, Parsons SK. Obesity in pediatric ALL survivors: a meta-analysis. Pediatrics. 2014;133(3):e704–15. doi: 10.1542/peds.2013-3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saultier P, Auquier P, Bertrand Y, et al. Metabolic syndrome in long-term survivors of childhood acute leukemia treated without hematopoietic stem cell transplantation: An L.E.A. study. Haematologica. 2016;101(12):1603–1610. doi: 10.3324/haematol.2016.148908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnea D, Raghunathan N, Friedman DN, Tonorezos ES. Obesity and metabolic disease after childhood cancer. Oncology. 2015; 29(11):849–855. [PMC free article] [PubMed] [Google Scholar]
- 12.Nottage KA, Ness KK, Li C, Srivastava D, Robison LL, Hudson MM. Metabolic syndrome and cardiovascular risk among long-term survivors of acute lymphoblastic leukaemia - from the St. Jude lifetime cohort. Br J Haematol. 2014;165(3):364–374. doi: 10.1111/bjh.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen MH, Colan SD, Diller L. Cardiovascular disease: cause of morbidity and mortality in adult survivors of childhood cancers. Circ Res. 2011;108(5):619–628. doi: 10.1161/CIRCRESAHA.110.224519. [DOI] [PubMed] [Google Scholar]
- 14.Mulrooney DA, Hyun G, Ness KK, et al. The changing burden of late health outcomes in adult survivors of childhood acute lymphoblastic leukaemia: a report from the St Jude Lifetime Cohort Study. Lancet Haematol. 2019;6(6):e306–e316. doi: 10.1016/S2352-3026(19)30050-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ariffin H Challenges in surviving childhood leukaemia. Lancet Haematol. 2019;6(6): e288–e289. doi: 10.1016/S2352-3026(19)30047-X. [DOI] [PubMed] [Google Scholar]
- 16.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014; 157(1):121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kho ZY, Lai SK. The human gut microbiome - a potential controller of wellness and disease. Frontiers Microbiol. 2018;9:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ihekweazu FD, Versalovic J. Development of the pediatric gut microbiome: impact on health and disease. Am J Med Sci. 2018;356(5):413–423. doi: 10.1016/j.amjms.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis. 2015;26:26191. doi: 10.3402/mehd.v26.26191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Chatelier E, Nielsen J, Qin E, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500(7464):541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 21.Backhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101(44):15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis CD. The gut microbiome and its role in obesity. Nutr Today. 2016;51(4):167–174. doi: 10.1097/NT.0000000000000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102 (31):11070–11075. doi: 10.1073/pnas.0504978102. Erratum in: Cell Metab. 2016 Mar 8;23(3):564–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ussar S, Griffin NW, Bezy O, et al. Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab. 2015; 22(3):526–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riva A, Borgo F, Lassandro C, et al. Pediatric obesity is associated with an altered gut microbiota and discordant shifts in firmicutes populations. Environ Microbiol. 2017;19(1): 95–105. doi: 10.1111/1462-2920.13463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalliomäki M, Collado MC, Salminen S, Isolauri E. Early differences in fecal microbiota composition in children may predict overweight. Am J Clin Nutr. 2008;87(3):534–538. doi: 10.1093/ajcn/87.3.534. [DOI] [PubMed] [Google Scholar]
- 27.Gao X, Jia R, Xie L, et al. Obesity in school-aged children and its correlation with gut E.coli and bifidobacteria: a case- control study. BMC Pediatr. 2015;15(1):64. doi: 10.1186/s12887-015-0384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ridaura VK, Faith JJ, Rey FE, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341(6150):1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy S, Trinchieri G. Microbiota: a key orchestrator of cancer therapy. Nat Rev Cancer. 2017;17(5):271–285. doi: 10.1038/nrc.2017.13. [DOI] [PubMed] [Google Scholar]
- 31.Bhuta R, Nieder M, Jubelirer T, Ladas EJ. The gut microbiome and pediatric cancer: current research and gaps in knowledge. J Natl Cancer Inst Monogr. 2019;2019(54):169–173. doi: 10.1093/jncimonographs/lgz026. [DOI] [PubMed] [Google Scholar]
- 32.Wen Y, Jin R, Chen H. Interactions between gut microbiota and acute childhood leukemia. Front Microbiol. 2019;10:1300. doi: 10.3389/fmicb.2019.01300. eCollection 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fijlstra M, Ferdous M, Koning AM, Rings E, Harmsen HJM, Tissing WJE. Substantial decreases in the number and diversity of microbiota during chemotherapy-induced gastrointestinal mucositis in a rat model. Support Care Cancer. 2015;23(6):1513–1522. doi: 10.1007/s00520-014-2487-6. [DOI] [PubMed] [Google Scholar]
- 34.Montassier E, Gastinne T, Vangay P, et al. Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment Pharmacol Ther. 2015;42(5):515–528. doi: 10.1111/apt.13302. [DOI] [PubMed] [Google Scholar]
- 35.Abeles SR, Jones MB, Santiago-Rodriguez TM, et al. Microbial diversity in individuals and their household contacts following typical antibiotic courses. Microbiome. 2016;4(1):39. doi: 10.1186/s40168-016-0187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khosravi A, Yáñez A, Price JG, et al. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe. 2014;15(3):374–381. doi: 10.1016/j.chom.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bai L, Zhou P, Li D, Ju X. Changes in the gastrointestinal microbiota of children with acute lymphoblastic leukaemia and its association with antibiotics in the short term. J Med Microbiol. 2017. doi: 10.1099/jmm.0.000568. [DOI] [PubMed] [Google Scholar]
- 38.Zwielehner J, Lassl C, Hippe B, et al. Changes in human fecal microbiota due to chemotherapy analyzed by TaqMan-PCR, 454 sequencing and PCR-DGGE fingerprinting. PLoS One. 2011;6(12):e28654. doi: 10.1371/journal.pone.0028654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by Deep16S rRNA sequencing. PLoS Biol. 2008;6(11): 2383–2400. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA. 2011; 108(Supplement_1):4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jernberg C, Löfmark S, Edlund C, Jansson JK. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. Isme J. 2007;1(1):56–66. doi: 10.1038/ismej.2007.3. [DOI] [PubMed] [Google Scholar]
- 42.Galloway-Peña JR, Smith DP, Sahasrabhojane P, et al. The role of the gastrointestinal microbiome in infectious complications during induction chemotherapy for acute myeloid leukemia. Cancer. 2016;122(14):2186–2196. doi: 10.1002/cncr.30039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Vliet MJ, Tissing WJE, Dun CAJ, et al. Chemotherapy treatment in pediatric patients with acute myeloid leukemia receiving antimicrobial prophylaxis leads to a relative increase of colonization with potentially pathogenic bacteria in the gut. Clin Infect Dis. 2009;49(2): 262–270. doi: 10.1086/599346. [DOI] [PubMed] [Google Scholar]
- 44.Rajagopala SV, Yooseph S, Harkins DM, et al. Gastrointestinal microbial populations can distinguish pediatric and adolescent acute lymphoblastic leukemia (ALL) at the time of disease diagnosis. BMC Genomics. 2016;17(1):635. doi: 10.1186/s12864-016-2965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hakim H, Dallas R, Wolf J, et al. Gut microbiome composition predicts infection risk during chemotherapy in children with acute lymphoblastic leukemia. Clin Infect Dis. 2018; 67(4):541–548. doi: 10.1093/cid/ciy153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong WSW, Clemency N, Klein E, et al. Collection of non-meconium stool on fecal occult blood cards is an effective method for fecal microbiota studies in infants. Microbiome. 2017;5(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bokulich NA, Subramanian S, Faith JJ, et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon. Nat Methods. 2013;10(1):57–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McDonald D, Price MN, Goodrich J, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012; 6(3):610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lê Cao KA, Boitard S, Besse P. Sparse PLS discriminant analysis: biologically relevant feature selection and graphical displays for multiclass problems. BMC Bioinf. 2011;12(1):253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chua LL, Rajasuriar R, Azanan MS, et al. Reduced microbial diversity in adult survivors of childhood acute lymphoblastic leukemia and microbial associations with increased immune activation. Microbiome. 2017;5(1):35. doi: 10.1186/s40168-017-0250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lopez-Siles M, Duncan SH, Garcia-Gil LJ, Martinez-Medina M. Faecalibacterium prausnitzii: from microbiology to diagnostics ad prognostics. ISME J. 2017;11(4):841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biagi E, Zama D, Nastasi C, et al. Gut microbiota trajectory in pediatric patients undergoing hematopoietic SCT. Bone Marrow Transplant. 2015;50(7):992–998. doi: 10.1038/bmt.2015.16. [DOI] [PubMed] [Google Scholar]
- 55.Miquel S, Martín R, Rossi O, et al. Faecalibacterium prausnitzii and human intestinal health. Curr Opin Microbiol. 2013;16(3):255–261. doi: 10.1016/j.mib.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 56.Zhou L, Zhang M, Wang Y, et al. Faecalibacterium prausnitzii produces butyrate to maintain Th17/Treg balance and to ameliorate colorectal colitis by inhibiting histone deacetylase 1. Inflamm Bowel Dis. 2018. [DOI] [PubMed] [Google Scholar]
- 57.Chen X, Devaraj S. Gut microbiome in obesity, metabolic syndrome, and diabetes. Curr Diab Rep. 2018;18(12):129. doi: 10.1007/s11892-018-1104-3. [DOI] [PubMed] [Google Scholar]
- 58.Mazidi M, Rezaie P, Kengne AP, Mobarhan MG, Ferns GA. Gut microbiome and metabolic syndrome. Diabetes Metab Syndr. 2016;10(2):S150–S137. doi: 10.1016/j.dsx.2016.01.024. [DOI] [PubMed] [Google Scholar]
- 59.Cox LM, Yamanishi S, Sohn J, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014;158(4):705–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nearing JT, Connors J, Whitehouse S, et al. Infectious complications are associated with alterations in the gut microbiome in pediatric patients with acute lymphoblastic leukemia. Front Cell Infect Microbiol. 2019;9:28. doi: 10.3389/fcimb.2019.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hilton SK, Castro-Nallar E, Pérez-Losada M, et al. Metataxonomic and metagenomic approaches vs. culture-based techniques for clinical pathology. Front Microbiol. 2016;7:484. doi: 10.3389/fmicb.2016.00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rajagopala SV, Singh H, Yu Y, et al. Persistent gut microbial dysbiosis in children with acute lymphoblastic leukemia (ALL) during chemotherapy. Microb Ecol. 2019;79:1034–1043. doi: 10.1007/s00248-019-01448-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.