

Abstract
The reaction of the organometallic diarsene complex [Cp2Mo2(CO)4(μ,η2‐As2)] (B) (Cp = C5H5) with Ag[FAl{OC6F10(C6F5)}3] (Ag[FAl]) and Ag[Al{OC(CF3)3}4] (Ag[TEF]), respectively, yields three unprecedented supramolecular assemblies [(η2‐B)4Ag2][FAl]2 (4), [(μ,η1:η2‐B)3(η2‐B)2Ag3][TEF]3 (5) and [(μ,η1:η2‐B)4Ag3][TEF]3 (6). These products are only composed of the complexes B and AgI. Moreover, compounds 5 and 6 are the only supramolecular assemblies featuring B as a linking unit, and the first examples of [AgI]3 units stabilized by organometallic bichelating ligands. According to DFT calculations, complex B coordinates to metal centers through both the As lone pair and the As−As σ‐bond thus showing this unique feature of this diarsene ligand.
Keywords: argentophilicity, arsenic, self-assembly, silver, weakly coordinating anions
A linking moiety: The reaction of the diarsenic complex [(C5H5)2Mo2(CO)4(μ,η2‐As2)] (B) with AgI salts affords unprecedented assemblies in which B acts as a linking moiety stabilizing short Ag–Ag distances.
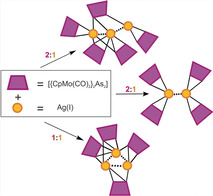
The interest in using metal‐directed self‐assembly for the design of well‐defined solid‐state structures has remarkably increased over the past decades. [1] Specifically, AgI complexes present an attractive research area because of their rich structural diversity and wide range of applications. [2] This diversity is due on the one hand to the flexible coordination sphere of the AgI ion which can adopt various coordination geometries (linear, trigonal planar, tetrahedral, square‐planar, trigonal bipyramidal, etc.) [3] and on the other hand to its ability to coordinate a variety of multitopic organic ligands bearing mainly N‐, O‐, S‐ or P‐ and, to a minor extent, Se, C, As or mixed‐donor atoms.[ 2 , 3 , 4 ] Besides organic molecules, very few examples of organometallic building blocks were used as linking moieties to AgI centers. [5] Due to the lack of such compounds, our group developed the concept of using organometallic polyphosphorus (Pn) ligand complexes with flexible coordination modes as connectors between metal ions. [6] This new approach allowed for the synthesis of a large variety of unprecedented supramolecular aggregates including 1D, 2D, and 3D coordination polymers (CPs), [7] inorganic nanospheres, [8] nanosized bowls [9] and capsules. [10] One of the simplest of such Pn compounds is the diphosphorus complex [Cp2Mo2(CO)4(μ,η2‐P2) (A) (Cp=η5‐C5H5). [11] Its reaction with a large number of AgI salts including those of the weakly coordinating anions [Al{OC(CF3)3}4]− ([TEF]−) and [FAl{OC6F10)(C6F5)}3]− ([FAl]−) allowed for the isolation of AgI dimers of the general formula [Ag2(η2‐A)2(μ,η1:η1‐A)2][X]2 ([X]−=[FAl]− (1), [TEF]− (2); Scheme 1). [7a] Notably, it is only possible to isolate these products selectively, if A is used in excess compared to the AgI salts. If, however, a stoichiometric reaction of for instance A and Ag[TEF] is conducted, the 1D polymer [Ag2(μ,η1:η1‐A)3]n[TEF]2 n (3) is formed instead. Interestingly, within the dimers 1 and 2, due to the weaker coordination of the terminal η2‐coordinated ligands A, as compared to the η1:η1‐coordinated ones, these can be easily substituted by for example, pyridyl functions upon the reaction of the AgI dimers with ditopic pyridine‐based organic molecules to form a new class of hybrid CPs in which both organometallic and organic units link AgI centers. [12]
Scheme 1.
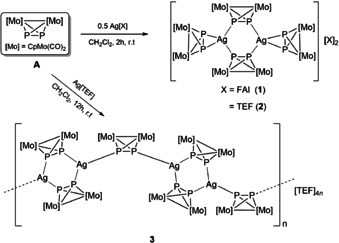
Reaction of A with Ag[FAl{OC(C6F5)(C6F10)}3] (AgFAl) and Ag[Al{OC(CF3)3}4] (AgTEF). Synthesis of the dimers 1 and 2 and the 1D CP 3.
Just as Pn complexes, arsenic‐based organometallic complexes have also been known for decades. [13] However, their coordination chemistry has so far been only very little investigated[ 14a , 14b , 14c , 14d ] and their use as linkers in supramolecular chemistry is rare. Moreover, coordination compounds of any polyarsenic linker and silver ions are extremely scarce.[ 14a , 14c , 14d , 14e ] Accordingly, we were keen to expand this research area by studying the supramolecular chemistry of polyarsenic Asn complexes and comparing it to that of their phosphorus analogues. In fact, because of the hindered accessibility of the lone pair on the heavier arsenic atoms, such Asn complexes were expected to have different coordination behaviors compared to their Pn analogues. Furthermore, due to the flexible coordination sphere of the AgI ion and its tendency to form Ag⋅⋅⋅Ag interactions,[ 3c , 15 ] the question arose whether it is possible to stabilize short Ag–Ag distances by using a certain combination of the Asn complexes and AgI ions. Herein, we report that the reaction of the diarsene complex [Cp2Mo2(CO)4(μ,η2‐As2)] (B) with Ag[FAl] and Ag[TEF] using various ratios of starting materials allowed for the isolation of the first homoleptic coordination compounds of B and silver; [(η2‐B)4Ag2][FAl]2 (4), [(μ,η1:η2‐B)3(η2‐B)2Ag3][TEF]3 (5) and [(μ,η1:η2‐B)4Ag3][TEF]3 (6). Moreover, the assemblies 5 and 6 are the first supramolecular compounds featuring complex B as a connecter between metal ions and, to the best of our knowledge, the first examples of trinuclear [AgI]3 units stabilized by organometallic bichelating ligands.
In order to evaluate the bonding situation in complex B towards unsaturated AgI centers, DFT calculations were performed at the B3LYP/def2TZVP level of theory. The results show that the lone pairs of the As atoms in B are lower in energy compared to those of the P atoms in A (Figure 1). Additionally, the energy of the As−As σ bond is higher compared to the P−P σ bond, which allows for a more effective overlap of these orbitals with the unoccupied orbitals of Ag instead of those of a lone pair. As for the As2 complex B, the As−As σ bond can therefore be assumed to be involved in the bonding with unsaturated transition metal fragments, rather than the lone pair. Moreover, the energy difference between the lone pairs and the E−E σ bond is considerably higher for the arsenic derivative B than for the phosphorus derivative A (0.55 eV and 2.85 eV for A and B, respectively). This indicates that A can easily participate in the bonding to transition metals with both orbitals (lone pair and E−E σ bond), while for B a considerably higher preference for the coordination via the As−As σ bond is expected. This preference is in line with the experimental results (vide infra).
Figure 1.
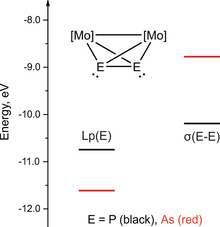
Frontier orbital energy diagram of [Cp2Mo2(CO)4(μ,η2‐E2)] (E=P, As), calculated at the B3LYP/def2‐TZVP level of theory.
Inspired by these calculations, complex B [13a] was reacted with the AgI salt Ag[FAl]. This reaction was conducted using a 2:1 ratio of B:Ag[FAl] in CH2Cl2 at room temperature (Scheme 2). This specific ratio of the reactants was studied in order to compare the formed product to that obtained from a similar reaction of the P‐donor analog A affording the AgI dimer 1 (Scheme 1). From this reaction, however, compound 4 was isolated as red prisms in 36 % yield suitable for X‐ray structure analysis. In the solid state, 4 is air‐ and light‐stable for several hours while it decomposes gradually after one hour in solvents such as CH3CN or CH2Cl2 under air. Compound 4 crystallizes in the orthorhombic space group Pccn. Its molecular structure (Figure 2 a) reveals a unique AgI dimer stabilized by four As2 ligands B. The entire molecular complex lies on the twofold axis along the z direction and is additionally disordered over two positions lying closely together with occupancies of 0.75 and 0.25, respectively. As regards the interpretation of the structure, this type of disorder is ambiguous and allows for three possible individual cores for 4, with two of them, core 4 a and core 4 b, possessing twofold rotational symmetry and core 4 c being asymmetric (Figure 2 b; for further details see the Supporting Information). The said disorder implies that the crystal structure of 4 is always a mixture of complexes with different cores. If the cores 4 a and 4 b co‐crystallize, they should form the mixture of 75 % of 4 a and 25 % of 4 b. The cores 4 c and 4 a can co‐exist in a 1:1 ratio. In principle, any mixture of all three complexes 4 a–4 c is possible with a ratio that does not contradict the crystallographic occupancies of the atoms, for example, the mixture of 4 a, 4 b, and 4 c in a ratio of 0.25:0.25:0.5. Thus, the question as to which of these alternatives do really exist cannot be answered by means of the X‐ray structural data.
Scheme 2.
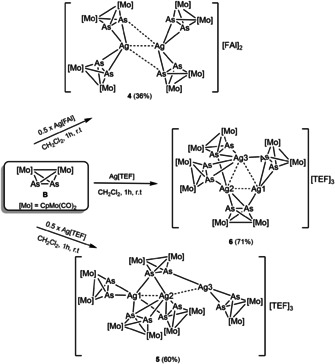
Reaction of B with Ag[FAl{OC(C6F5)(C6F10)}3] (Ag[FAl]) and Ag[Al{OC(CF3)3}4] (Ag[TEF]). Synthesis of the supramolecular compounds 4–6. Yields are shown in parentheses.
Figure 2.
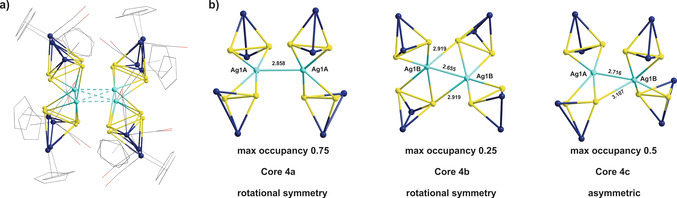
a) The disordered complex 4 (2z axis is directed vertically to the plane of the picture). b) Possible individual cores of 4 in the disordered structure.
In order to elucidate which of the above‐mentioned cores represents an energy minimum in the gas phase, we performed DFT calculations using the range‐separated hybrid functional ωB97XD, [15] which also incorporates dispersion corrections together with the def2SVP basis set. Starting from the experimental geometry of the core 4 b, the geometry optimization in the gas phase leads to a geometry that is very similar to that of the core 4 a. The Ag⋅⋅⋅Ag distance in the optimized geometry is with 3.188 Å longer than the one found experimentally for core 4 a (Figure 3, left). Interestingly, the geometry optimization of a [({CpMo(CO)2}2As2)2Ag]+ unit, starting from the experimental coordinates of half a core of 4 a, leads to a more symmetric geometry containing a distorted tetrahedrally coordinated AgI center (Figure 3, right structure), which indicates that attraction forces should be present between the two [({CpMo(CO)2}2As2)2Ag]+ units in the solid state. This is also reflected by the gas phase “dimerization” energy of two [({CpMo(CO)2}2As2)2Ag]+ units to the gas‐phase‐optimized geometry of 4 of −9.90 kJ mol−1 (for further details see ESI).
Figure 3.
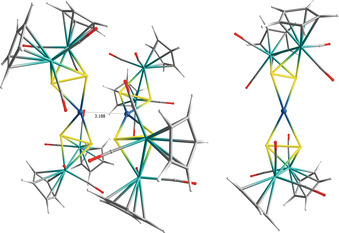
Gas‐phase‐optimized geometry of 4 at the ωB97XD/def2SVP level of theory.
Irrespective of which structures are adopted by 4 in the solid state, its composition (a AgI dimer stabilized by four As2 ligands B) is only slightly related to the AgI dimer 2, obtained from a similar reaction with the phosphorus analog A (Scheme 1). Still, two main differences are perceived between the two dimers 2 and 4. First, although two of the E2 units (E=P, As) in both dimers possess an η2‐coordination mode each, the remaining ones each possess a bridging μ,η1:η1‐coordination in 2 and a bridging μ,η1:η2‐coordination or an η2‐coordination in 4. Additionally, the distances between the metal centers in 2 [d(Ag⋅⋅⋅Ag)>4.85 Å] are much larger than those in 4 [2.65 Å> d(Ag⋅⋅⋅Ag)>2.86 Å]. Therefore, there is no argentophilic interaction in 2, while there is a possible metal–metal interaction in 4 (the sum of the van der Waals radii for silver (3.44 Å)). [16] The As−As (2.331(1)‐2.414(2) Å) bond lengths in 4 are slightly elongated compared to those in the non‐coordinated ligand complex B (As−As=2.312(3) Å). [13a] The As−Ag bond lengths are in the range of 2.613(1)–2.919(6) Å. As expected, these lengths are longer than the P−Ag bond lengths (2.442(5)–2.688(5) Å) found in the AgI dimer based on the lighter analog [Cp2Mo2(CO)4(μ,η2‐P2)]. [7a]
The crystallographic features of 4, including the flexible coordination mode of the As2 ligand complex B and the short Ag⋅⋅⋅Ag contacts, prompted us to further study the effect of the change in the stoichiometry of the reactants and the used counteranion on the outcome of the reaction. Obviously, a higher amount of AgI salts would lead to a higher number of AgI ions with a possible metal‐metal interaction in the formed products in the solid‐state. Thus, the reaction of B with the AgI salt Ag[Al{OC(CF3)3}4] (Ag[TEF]) was studied, due to the very high solubility of the [TEF] salts. In this case, two B:Ag[TEF] ratios (2:1 and 1:1) were used to be able to compare the outcome of these reactions to similar ones based on the diphosphorus analogue A (Scheme 1). These reactions were performed in CH2Cl2 and subsequently layered with n‐pentane. The 2:1 ratio reaction afforded compound 5 and the 1:1 reaction produced compound 6 in yields of 60 and 71 %, respectively. Compounds 5 and 6 were selectively isolated from their corresponding crude reaction mixtures as red crystals, showing air and light stability in the solid state. Their single‐crystal X‐ray structure analysis reveals composition ratios of 5:3 (for 5) and 4:3 (for 6) of B:Ag[TEF]. In contrast, such reactions with the complex A yielded the dimer 2 and the one‐dimensional coordination polymer (3). Both compounds, 5 and 6, represent unprecedented AgI trimers with the formulas [Ag3(μ,η2‐B)2(μ,η1:η2‐B)3][TEF]3 and [Ag3(μ,η1:η2‐B)4][TEF]3, respectively.
Compounds 5 and 6 crystallize in the monoclinic space groups P21/n and P21/c, respectively. The central structural motif of 5 consists of a bent trinuclear Ag3 chain while it shows an almost equilateral Ag3 triangle in 6 (Figure 4). In 5, these AgI ions are stabilized by five Mo2As2 ligands B with two of them showing an η2‐coordination mode and three others a μ,η2:η1‐coordination. Interestingly, one of these bridging ligands B connects all the three AgI ions, Ag1, Ag2 and Ag3, while the other two ligands B connect each only the Ag1 and Ag2 ions. Additionally, the intermetallic Ag⋅⋅⋅Ag distances in 5 (2.8376(3)–2.9053(3) Å) are significantly shorter than the sum of the van der Waals radii for two silver atoms (3.44 Å), indicating the possible existence of argentophilic interactions. [16] As a consequence, all the AgI ions in 5 show different coordination environments: Ag1 is hexacoordinated to five As atoms and one AgI ion, Ag2 is heptacoordinated to five As atoms and two AgI ions and Ag3 is tetracoordinated to three As atoms and one AgI ion. The Ag3 core in 6 is stabilized by four bridging Mo2As2 ligands B, each showing an η2:η1‐coordination. All AgI ions in 6 show different coordination spheres: Ag3 is heptacoordinated to five As atoms and two AgI ions, Ag2 is hexacoordinated to four As atoms and two AgI ions and Ag1 is pentacoordinated to three As atoms and two AgI ions. The intermetallic Ag⋅⋅⋅Ag distances in 6 range between 2.858(2) and 2.980(1) Å and are also within the range of argentophilic interactions. [16] The As−As bond lengths in 5 (2.321(1)–2.458(3) Å) and 6 (2.378(5)–2.409(5) Å) are elongated compared to those in the non‐coordinated complex B (2.312(3) Å). [13a] The Ag−As bond lengths are in the range of (2.438(1)–3.123(1) Å) and (2.573(8)–2.989(8) Å), respectively.
Figure 4.
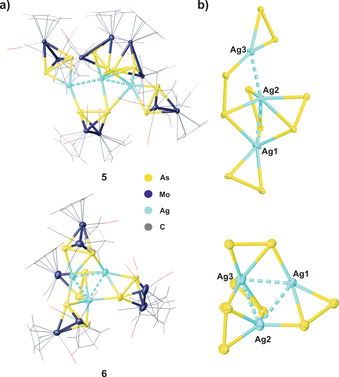
a) Molecular structures of the supramolecular assemblies 5 and 6 in the solid state. Counter anions are omitted for clarity. b) Structures of the cationic fragments in 5 and 6 showing the Ag−As cores.
Compounds 4–6 are well soluble in common organic solvents such as CH2Cl2 and CH3CN, little soluble in THF and insoluble in n‐pentane. Their 1H and 13C{1H} NMR spectra in CD3CN at room temperature show signals typical for Cp and CO ligands. In the ESI mass spectra in CH3CN, peaks for the cations [Ag(B)2]+ and [Ag(B)(CH3CN)]+ are mainly detected in the positive ion mode and a peak for the [TEF] or the [FAl] anions in the negative ion mode. These data indicate that only a partial dissociation of the assemblies 4–6 occurs in solutions of CH3CN. The solid state IR spectra of 4 show each three strong broad absorptions between 1921 and 2048 cm−1, while those of 5 and 6 show each two absorptions between 1942 and 1980 cm−1, attributable to the stretching vibrations of the CO ligands in the coordinated ligand units B. These vibrations appear at lower energies as compared to those reported for the free complex B (1900 and 1949 cm−1). [13a]
In summary, we synthesized the first homoleptic complexes (4–6) of the tetrahedral diarsene complex Mo2As2 (B) and AgI ions. In so doing, the potential of B as a connector in supramolecular chemistry stabilizing short Ag⋅⋅⋅Ag distances was demonstrated for the first time. By using various stoichiometric ratios of the starting materials and changing the counteranion, a variety of solid‐state AgI coordination compounds stabilized by four or five of these ligand complexes is selectively accessible. The solid‐state structures of these products allow for a comparison to corresponding P‐containing derivatives obtained from similar reactions using the lighter analogue P2 complex A as a building block. The 2:1 stoichiometric ratio reactions of the Mo2P2 ligand complex (A) and Ag[FAl] or Ag[TEF] afforded the AgI dimers 1 and 2, whereas a 1:1 reaction with Ag[TEF] gave the 1D polymer 3. Similar reactions of the Mo2As2 ligand complex (B) using similar ratios afforded products with entirely different structures (4–6). According to DFT calculations, the reactivity difference of the complexes A and B towards AgI salts originates from the difference in the donor nature of both complexes. Specifically, the As−As σ bond is better accessible for coordination to metal centers than the P−P σ bond. This σ‐donation towards AgI offers the As2 units more flexibility and promotes the formation of unprecedented dimers (4) and trimers as cycle (6) or catena (5) compounds showing remarkable Ag⋅⋅⋅Ag interactions. Current investigations in this field focus on three‐component reactions of the complex B with AgI salts and N‐donor organic molecules to build unprecedented supramolecular architectures with (As,N) mixed‐donor ligands.
Experimental Section
Crystallographic data:
Deposition numbers 1985242, 1985244, and 1985245 (4, 5, and 6) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the European Research Council (Grant ERC‐2013‐AdG 339072). Open access funding enabled and organized by Projekt DEAL.
M. E. Moussa, J. Schiller, E. Peresypkina, M. Seidl, G. Balázs, P. Shelyganov, M. Scheer, Chem. Eur. J. 2020, 26, 14315.
Dedicated to Professor Christoph Janiak on the occasion of his 60th birthday
References
- 1.
- 1a. Mako T. L., Racicot J. M., Levine M., Chem. Rev. 2019, 119, 322–477; [DOI] [PubMed] [Google Scholar]
- 1b. Gan M.-M., Liu J.-Q., Zhang L., Wang Y.-Y., Hahn F. E., Han Y.-F., Chem. Rev. 2018, 118, 9587–9641; [DOI] [PubMed] [Google Scholar]
- 1c. Lu Y., Zhang H.-N., Jin G.-X., Acc. Chem. Res. 2018, 51, 2148–2158; [DOI] [PubMed] [Google Scholar]
- 1d. Lescop C., Acc. Chem. Res. 2017, 50, 885–894; [DOI] [PubMed] [Google Scholar]
- 1e. Cook T. R., Stang P., Chem. Rev. 2015, 115, 7001–7045; [DOI] [PubMed] [Google Scholar]
- 1f. Han M., Engelhard D. M., Clever G. H., Chem. Soc. Rev. 2014, 43, 1848–1860; [DOI] [PubMed] [Google Scholar]
- 1g. Harris K., Fujita D., Fujita M., Chem. Commun. 2013, 49, 6703–6712; [DOI] [PubMed] [Google Scholar]
- 1h. Smulders M. M. J., Riddell I. A., Browne C., Nitschke J. R., Chem. Soc. Rev. 2013, 42, 1728–1754. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Rogovoy M. I., Samsonenko D. G., Rakhmanova M. I., Artem'ev A. V., Inorg. Chim. Acta 2019, 489, 19–26; [Google Scholar]
- 2b. Rocha C. S., Filho L. F. O. B., De Souza A. E., Diniz R., Denadai A. M. L., Beraldo H., Teixeira L. R., Polyhedron 2019, 170, 723–730; [Google Scholar]
- 2c. Dosen M., Kawada Y., Shibata S., Tsuge K., Sasaki Y., Kobayashi A., Kato M., Ishizaka S., Kitamura N., Inorg. Chem. 2019, 58, 8419–8431; [DOI] [PubMed] [Google Scholar]
- 2d. Alderson J. M., Corbin J. R., Schomaker J. M., Acc. Chem. Res. 2017, 50, 2147–2158; [DOI] [PubMed] [Google Scholar]
- 2e. Medici S., Peana M., Crisponi G., Nurchi V. M., Lachowicz J. I., Remelli M., Zoroddu M. A., Coord. Chem. Rev. 2016, 327, 349–359; [Google Scholar]
- 2f. Zhang T., Huang H.-Q., Mei H.-X., Wang D.-F., Wang X.-X., Huang R.-B., Zheng L.-S., J. Mol. Struct. 2015, 1100, 237–244; [Google Scholar]
- 2g. Bai H.-Y., Yang J., Liu B., Ma J.-F., Kan W.-Q., Liu Y.-Y., Liu Y.-Y., CrystEngComm CrystEngComm. 2011, 13, 5877–5884; [Google Scholar]
- 2h. Lin R., Yip J. H. K., Inorg. Chem. 2006, 45, 4423–4430. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Young A. G., Hanton L. R., Coord. Chem. Rev. 2008, 252, 1346–1386; [Google Scholar]
- 3b. Low F. H., Klausmeyer K. K., Inorg. Chim. Acta 2008, 361, 1298–1310; [Google Scholar]
- 3c. Khlobystov A. N., Blake A. J., Champness N. R., Lemenovskii D. A., Majouga A. G., Zyk N. V., Schröder M., Coord. Chem. Rev. 2001, 222, 155–192. [Google Scholar]
- 4.
- 4a. Weis P., Hettich C., Kratzert D., Krossing I., Eur. J. Inorg. Chem. 2019, 1657–1668; [Google Scholar]
- 4b. Hamze R., Shi S., Kapper S. C., Ravinson D. S. M., Estergreen L., Jung M.-C., Tadle A. C., Haiges R., Djurovich P. I., Peltier J. L., Jazzar R., Bertrand G., Bradforth S. E., Thompson M. E., J. Am. Chem. Soc. 2019, 141, 8616–8626; [DOI] [PubMed] [Google Scholar]
- 4c. Ma L.-L., Sun Y.-Y., Wang Y.-Y., Hahn F. E., Han Y.-F., Angew. Chem. Int. Ed. 2019, 58, 3986–3991; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4026–4031; [Google Scholar]
- 4d. Nahra F., Hecke K. V., Kennedy A. R., Nelson D. J., Dalton Trans. 2018, 47, 10671–10684; [DOI] [PubMed] [Google Scholar]
- 4e. Perras J. H., Mezibroski S. M. J., Wiebe M. A., Ritch J. S., Dalton Trans. 2018, 47, 1471–1478. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Škoch K., Císařová I., Schulz J., Siemeling U., Štěpnička P., Dalton Trans. 2017, 46, 10339–10354; [DOI] [PubMed] [Google Scholar]
- 5b. Škoch K., Císařová I., Štĕpnička P., Inorg. Chem. Commun. 2017, 84, 234–236; [Google Scholar]
- 5c. Škoch K., Uhlík F., Císařová I., Štĕpnička P., Dalton Trans. 2016, 45, 10655–10671. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Whitmire K. H., Coord. Chem. Rev. 2018, 376, 114–195; [Google Scholar]
- 6b. Scheer M., Dalton Trans. 2008, 4372–4386. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Moussa M. E., Fleischmann M., Peresypkina E. V., Dütsch L., Seidl M., Balázs G., Scheer M., Eur. J. Inorg. Chem. 2017, 3222–3226; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Heindl C., Peresypkina E., Lüdeker D., Brunklaus G., Virovets A. V., Scheer M., Chem. Eur. J. 2016, 22, 2599–2604; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Fleischmann M., Welsch S., Peresypkina E. V., Virovets A. V., Scheer M., Chem. Eur. J. 2015, 21, 14332–14336; [DOI] [PubMed] [Google Scholar]
- 7d. Bai J., Virovets A. V., Scheer M., Angew. Chem. Int. Ed. 2002, 41, 1737–1740; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1808–1811. [Google Scholar]
- 8.
- 8a. Peresypkina E., Heindl C., Virovets A., Brake H., Mädl E., Scheer M., Chem. Eur. J. 2018, 24, 2503–2508; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Heindl C., Peresypkina E., Virovets A. V., Bushmarinov I. S., Medvedev M. G., Krämer B., Dittrich B., Scheer M., Angew. Chem. Int. Ed. 2017, 56, 13237–13243; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13420–13426; [Google Scholar]
- 8c. Peresypkina E., Heindl C., Virovets A., Scheer M., in Clusters–Contemporary Insight in Structure and Bonding, Structure and Bonding (Ed.: Dehnen S.), Springer, Berlin, 2016, pp. 321–373; [Google Scholar]
- 8d. Heindl C., Peresypkina E. V., Virovets A. V., Kremer W., Scheer M., J. Am. Chem. Soc. 2015, 137, 10938–10941; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. Dielmann F., Heindl C., Hastreiter F., Peresypkina E. V., Virovets A. V., Gschwind R. M., Scheer M., Angew. Chem. Int. Ed. 2014, 53, 13605–13608; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13823–13827; [Google Scholar]
- 8f. Scheer M., Schindler A., Merkle R., Johnson B. P., Linseis M., Winter R., Anson C. E., Virovets A. V., J. Am. Chem. Soc. 2007, 129, 13386–13387; [DOI] [PubMed] [Google Scholar]
- 8g. Bai J., Virovets A. V., Scheer M., Science 2003, 300, 781–783. [DOI] [PubMed] [Google Scholar]
- 9. Brake H., Peresypkina E., Heindl C., Virovets A. V., Kremer W., Scheer M., Chem. Sci. 2019, 10, 2940–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Welsch S., Gröger C., Sierka M., Scheer M., Angew. Chem. Int. Ed. 2011, 50, 1435–1438; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1471–1474. [Google Scholar]
- 11.
- 11a. Scherer O. J., Schwalb J., Sitzmann H., Inorg. Synth. 1990, 27, 224–227; [Google Scholar]
- 11b. Scherer O. J., Sitzmann H., Wolmershäuser G., J. Organomet. Chem. 1984, 268, C9–C12. [Google Scholar]
- 12.
- 12a. Moussa M. E., Peresypkina E., Virovets A. V., Venus D., Balázs G., Scheer M., CrystEngComm 2018, 20, 7417–7422; [Google Scholar]
- 12b. Attenberger B., Welsch S., Zabel M., Peresypkina E., Scheer M., Angew. Chem. Int. Ed. 2011, 50, 11516–11519; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11718–11722. [Google Scholar]
- 13.
- 13a. Sullivan P. J., Rheingold A. L., Organometallics 1982, 1, 1547–1549; [Google Scholar]
- 13b. Blechschmitt K., Pfisterer H., Zahn T., Ziegler M., Angew. Chem. Int. Ed. Engl. 1985, 24, 66–67; [Google Scholar]; Angew. Chem. 1985, 97, 73–74. [Google Scholar]
- 14.
- 14a. Schwarzmaier C., Sierka M., Scheer M., Angew. Chem. Int. Ed. 2013, 52, 858–861; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 891–894; [Google Scholar]
- 14b. Krauss H., Balázs G., Bodensteiner M., Scheer M., Chem. Sci. 2010, 1, 337–342; [Google Scholar]
- 14c. Scheer M., Gregoriades L. J., Virovets A. V., Kunz W., Neueder R., Krossing I., Angew. Chem. Int. Ed. 2006, 45, 5689–5693; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5818–5822; [Google Scholar]
- 14d. Gregoriades L. J., Krauss H., Wachter J., Virovets A. V., Sierka M., Scheer M., Angew. Chem. Int. Ed. 2006, 45, 4189–4192; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 4295–4298; [Google Scholar]
- 14e. Fenske D., Simon F., Z. Anorg. Allg. Chem. 1996, 622, 45–52. [Google Scholar]
- 15. Meijboom R., Bowen R. J., Berners-Price S. J., Coord. Chem. Rev. 2009, 253, 325–342. [Google Scholar]
- 16.
- 16a. Chai J.-D., Head-Gordon M., Phys. Chem. Chem. Phys. 2008, 10, 6615–6620; [DOI] [PubMed] [Google Scholar]
- 16b. Schmidbaur H., Schier A., Angew. Chem. Int. Ed. 2015, 54, 746–784; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 756–797. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary