

Abstract
The year 2021 marks the 20th anniversary of the first publications reporting the discovery of the gene silencing mechanism, RNA interference (RNAi) in mammalian cells. Along with the many studies that delineated the proteins and substrates that form the RNAi pathway, this finding changed our understanding of the posttranscriptional regulation of mammalian gene expression. Furthermore, the development of methods that exploited the RNAi pathway began the technological revolution that eventually enabled the interrogation of mammalian gene function—from a single gene to the whole genome—in only a few days. The needs of the cancer research community have driven much of this progress. In this perspective, we highlight milestones in the development and application of RNAi‐based methods to study carcinogenesis. We discuss how RNAi‐based functional genetic analysis of exemplar tumor suppressors and oncogenes furthered our understanding of cancer initiation and progression and explore how such studies formed the basis of genome‐wide scale efforts to identify cancer or cancer‐type specific vulnerabilities, including studies conducted in vivo. Furthermore, we examine how RNAi technologies have revealed new cancer‐relevant molecular targets and the implications for cancer of the first RNAi‐based drugs. Finally, we discuss the future of functional genetic analysis, highlighting the increasing availability of complementary approaches to analyze cancer gene function.
Keywords: cancer dependencies, functional genomics, RNA interference, RNAi, shRNA, siRNA
Abbreviations
- ABC‐DLBCL
activated B‐cell‐diffuse large B‐cell lymphoma
- AHP
acute hepatic porphyria
- AML
acute myeloid leukemia
- ASCO
American Society of Clinical Oncology
- ATARiS
analytic technique for assessment of RNAi by similarity
- CCLE
Cancer Cell Encyclopedia
- CPT
camptothecin
- CRC
colorectal cancer
- CRISPR
clustered regularly interspaced short palindromic repeats
- CRISPRi
CRISPR interference
- CRISPRa
CRISPR activation
- CSA
common seed analysis
- DLBCL
diffuse large B‐cell lymphoma
- DOPC
dioleoyl‐phosphatidyl‐choline
- EWS
Ewing sarcoma
- GBC‐DLBCL
germinal B‐cell‐diffuse large B‐cell lymphoma
- dsRNA
double‐stranded RNA
- hATTR
hereditary transthyretin amyloidosis
- Hh
hedgehog
- HSC
hematopoietic stem cells
- OS
osteosarcoma
- LOF
loss‐of‐function
- miRNA
microRNA
- NSCLC
non‐small‐cell lung cancer
- RIGER
RNAi gene enrichment ranking
- RISC
RNA‐induced silencing complex
- RNAi
RNA interference
- RSA
redundant siRNA activity
- SCC
squamous cell carcinoma
- siRNA
small interfering RNA
- shRNA
short hairpin RNA
- TRC
the RNAi consortium
- TOP1
topoisomerase 1
- UTR
untranslated region
1. INTRODUCTION
Scientists worldwide routinely perform experiments in mammalian cells to assess gene function by inducing a gene‐specific reduction in expression followed by measurement of a biologically relevant endpoint, often in as little as 48 h. The development of the reagents and methods that induce this targeted decrease in gene expression began with two reports in the spring of 2001 of sequence‐specific silencing of gene expression in human and mouse cells. 1 , 2 These studies demonstrated that synthetic double‐stranded RNA (dsRNA) molecules of 21 or 22 nucleotides (nts) transfected into mammalian cells could induce a substantial decrease in the levels of a transcript containing a sequence complementary to one strand of the synthetic duplex RNA. Importantly, the reduction in messenger RNA (mRNA) levels resulted in reduced levels of the protein encoded by the targeted transcript, 1 and the synthetic double‐stranded RNA (dsRNA) did not trigger antiviral immune responses associated with exposure of mammalian cells to longer dsRNA molecules. 2 The rationale for testing whether dsRNA molecules of ~21 nts could target endogenous mRNAs in mammalian cells arose from previous studies conducted in model organisms. Specifically, studies performed in plants, fungi, Caenorhabditis elegans, and Drosophila melanogaster reporting a posttranscriptional gene silencing mechanism mediated by dsRNA that became referred to as RNA interference or RNAi. 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 Over the next few years, many groups defined the principal components—enzymes, substrates, and associated proteins—of the RNAi pathway. 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 Critical findings demonstrating RNAi's function in regulating endogenous gene expression soon emerged, building on earlier observations of the regulation of genes in C. elegans involved in development and differentiation by small RNAs. 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 The discovery of entire classes of regulatory noncoding RNAs, particularly microRNAs (miRNAs), that control gene expression via the RNAi pathway added additional levels of complexity to the regulation of the transcriptome, 43 , 44 , 45 , 46 , 47 leading to significant insights into how disruption of miRNA‐regulated gene expression contributes to disease states, including cancer. 48 , 49 , 50 , 51 , 52 , 53 , 54
Our ability to harness RNAi to study gene function represented a landmark achievement for the scientific community with immediate applicability across multiple biological contexts and disease phenotypes, especially cancer. Importantly, RNAi‐based technologies enabled researchers to conduct large‐scale loss‐of‐function (LOF) genomic screens that provided a means for unbiased discovery of drivers of carcinogenesis and drug development targets. Since 2001, hundreds of cancer‐based studies have reported the employment of RNAi‐based methods to study mammalian gene function, leading to the discovery of many genes with no previous links to cancer biology, and enhanced understanding of the functions of established oncogene and tumor suppressors. More recently, the extensive experience with RNAi and the global research efforts in optimizing screens and developing standardized pipelines for the analysis of LOF data across multiple cancer model systems have laid a foundation for the accelerated employment of CRISPR (clustered regularly interspaced short palindromic repeats)‐based methods for functional genetic analysis. In this perspective, we will discuss how the discovery of RNAi and the development of functional genomic approaches that utilize small RNA‐guided protein complexes have altered our understanding of carcinogenesis and furthered the identification of new cancer therapeutic targets (Figure 1). We will also consider the future impact of RNAi and other RNA‐based technologies on our understanding of tumor biology and cancer treatment.
Figure 1.
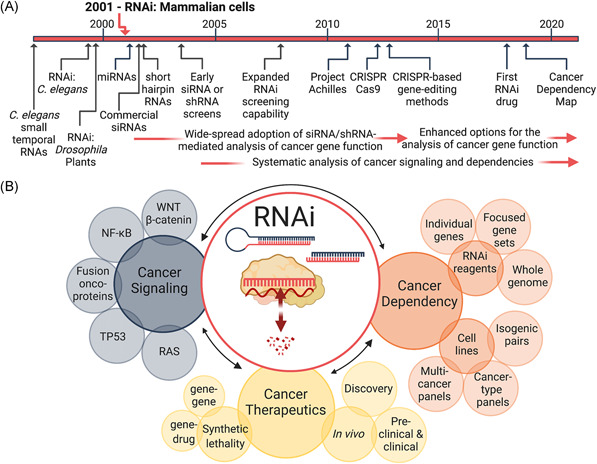
Cancer biology functional genomics. (A) Timeline highlighting the findings leading up to the identification of the RNAi pathway in mammalian cells and the subsequent development of RNAi‐based methods for the study of cancer biology. (B) A schematic representation of the topics discussed in this perspective. The ability to manipulate gene expression via the posttranscriptional gene silencing mechanism, RNAi, has enabled interrogation of cancer signaling pathways and cancer‐specific dependencies and further provided new avenues for the development of cancer therapeutics. C. elegans, Caenorhabditis elegans; CRISPR, clustered regularly interspaced short palindromic repeats; RNAi, RNA interference; shRNA, short hairpin RNA; siRNA, small interfering RNA
2. TURNING BIOLOGY INTO TECHNOLOGY
The initial descriptions of RNAi in mammalian cells led rapidly to the generation of the resources that enabled researchers to deplete protein expression with relative ease. Specifically, the field saw the development of the tools we still use today: expressed short hairpin RNAs (shRNAs) and synthetic small interfering RNAs (siRNAs) that mimic precursor or mature miRNAs 55 , 56 , 57 , 58 , 59 (Figure 2). However, the design of artificial mediators of RNAi came with challenges. In particular, the prioritization of sequences in a transcript that favor target mRNA cleavage and degradation required extensive optimization. Over time, studies that defined miRNA‐mediated gene regulation and iterative empirical studies that tested many different sequences corresponding to a specific transcript resulted in an enhanced understanding of the parameters that facilitate effective RNAi. 60 , 61 , 62 , 63 , 64 , 65 , 66 , 67 , 68 Based on these results, academic groups and commercial vendors developed computational models that could predict the sequence of active and specific siRNA or shRNA with some level of accuracy. Selection parameters included the nucleotide content at the 5ʹ ends of each strand of the dsRNA, with a high A/U nucleotide content at the 5ʹ end of one strand favoring its incorporation into the enzyme complex—the RNA‐induced silencing complex (RISC), responsible for the interaction with the target mRNA. 69 Our group contributed to these efforts by demonstrating the impact of targeting the coding region of protein‐coding genes versus regulatory 5ʹ or 3ʹ untranslated region (UTR) regions and sequence polymorphisms when selecting siRNA or shRNA sequences. 70 , 71
Figure 2.
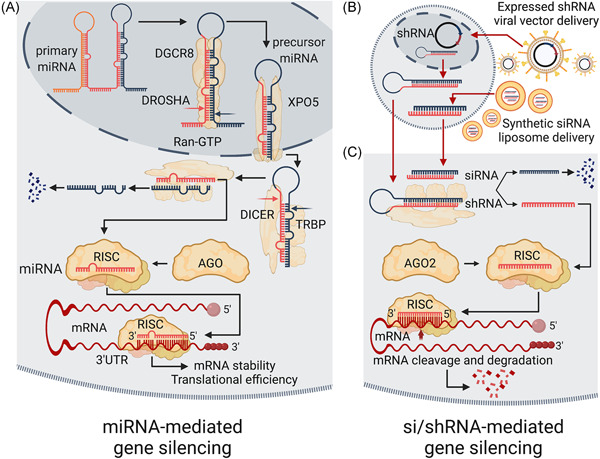
RNAi‐mediated regulation of gene expression. (A) Simplified schematic of miRNA biogenesis and their subsequent effect on gene expression. In brief, the ribonuclease III enzyme DROSHA, together with the DGCR8–microprocessor complex subunit, processes the pol II transcribed primary miRNA transcripts to release the precursor miRNA (pre‐miRNA). The translocation of the pre‐miRNA to the cytoplasm requires the RAN–GTP binding protein (Ran‐GTP)‐dependent export protein Exportin 5 (XPO5). In the cytoplasm, a second ribonuclease III enzyme, DICER further processes the pre‐miRNA and together with TRBP (TARBP2 subunit of RISC loading complex) facilitates the loading of the mature miRNA guide stranded into the RNA‐induced silencing complex or RISC, with a member of the argonaute (AGO) family of proteins at its core. The miRNA‐RISC binds one or more complementary mRNA sequences, predominantly in the 3ʹUTR of mRNAs with a tolerance of some nucleotide mismatches, altering mRNA stability and/or the efficiency of translation. (B) An outline of the principal methods used to deliver RNAi reagents (shRNAs or synthetic siRNAs) into cells. The delivery of shRNAs typically involves the cloning of the sequences corresponding to both strands of the duplex siRNA linked through a region that will form a stem‐loop into a viral vector system (usually lentiviral‐based) that will facilitate the expression of the shRNA in a mammalian cell. The delivery of chemically synthesized duplex RNA molecules, synthetic siRNAs, typically involves using a lipid‐based reagent that facilitates uptake into the cytoplasm. (C) A simplified schematic of shRNA and siRNA‐mediated targeting of a transcript for degradation via the RNAi pathway. Following the delivery or generation of the duplex RNA, the guide strand loads into RISC with argonaute RISC catalytic component 2 (AGO2) at its core. The RNA‐loaded AGO2‐RISC can interact with all regions of an mRNA, and through perfect or near‐perfect base‐pairing catalyzes the cleavage of the mRNA, resulting in transcript degradation. 3ʹUTR, 3ʹ untranslated region; miRNA, microRNA; mRNA, messenger RNA; RNAi, RNA interference; shRNA, short hairpin RNA; siRNA, small interfering RNA
Improved RNAi‐effector design tools lead to the generation of siRNA or shRNA libraries initially targeting subsets of genes, such as gene families or genes with similar functions. These libraries progressed to become genome‐wide in scale, paving the way for functional genomic studies previously impossible in mammalian cells. 72 , 73 The decision to use siRNA or shRNA screening platforms for functional genomic studies depended on many factors. However, in most cases, arrayed screens that involved the analysis of single or limited numbers of RNAi effectors per well of a multi‐well plate typically used siRNA resources, while expressed shRNA resources frequently utilized a pooled approach (Figure 3). Parameters used to select a screening approach included the biological question that the screen aimed to address, the model system, the assay endpoint, and access to suitable infrastructure. As the scale of experimental RNAi‐based workflows increased, so did the need to develop computational tools that facilitated assessment of data quality, the normalization of results across a screen, correction for sequence‐biases, the statistical classification of lead candidate genes for further analysis, and the need for appropriate experimental procedures to validate the results of large‐scale screens. 74
Figure 3.
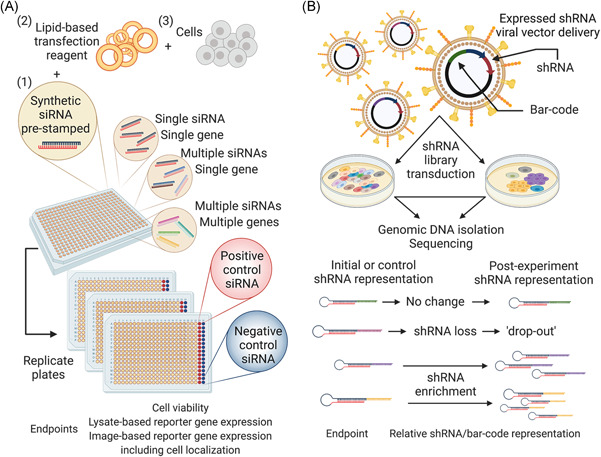
RNAi screening strategies. (A) A diagrammatic representation of plate‐based arrayed siRNA screening strategies compatible with endpoints such as cell viability, and luminescence or fluorescence reporter assays. Synthetic siRNAs are pre‐stamped into each well of a multi‐well plate (1), and the siRNA–liposome complex formed by adding a pre‐optimized amount of a lipid‐based transfection reagent (2). Following the addition of cells (3) to the siRNA–liposome complex, the transfected cells grow for 24–96 h under standard cell culture conditions before assessing a relevant endpoint. (B) A simplified outline of bar‐coded shRNA‐based pooled screening using a lentiviral vector delivery system. Cell populations transduced using pooled libraries that retain a normalized representation of each shRNA form the basis of different experimental groups, for example, different time‐points, Day 0 and Day 21, or two treatment conditions, vehicle or active compound. Following a selection period, the isolation of genomic DNA from each cell population allows for the assessment of the relative representation of each shRNA. RNAi, RNA interference; shRNA, short hairpin RNA; siRNA, small interfering RNA
As we recognize the successes that turned RNAi biology into RNAi technology, it is also critical to acknowledge the problems of data reproducibility and inconsistency that emerged as RNAi‐based studies became ubiquitous. The development of second‐ and even third‐generation RNAi reagents helped address some of the data reproducibility issues associated with early studies. However, it is vital to consider the RNAi mechanism's intrinsic features that generate variation, including differences in the efficacy of even highly optimized reagents and that RNAi reduces gene expression; it does not eliminate it. Over time, the increased publication of results that emphasize consistency in the phenotypic effects induced by siRNAs or shRNAs targeting the same gene has helped reduce the likelihood of false‐positive results. Nevertheless, many RNAi‐independent factors also need attention when assessing the results of any functional genomic study. For example, the number of cell lines used to define a tumor‐type or mutation‐specific effect. The signal‐to‐noise ratio of the endpoint assay used to identify candidate genes of interest can also influence the interpretation of a study's results. Finally, the failure to include appropriate secondary assays or orthogonal approaches to validate initial findings can contribute to the pursuit of nonspecific targets. 75 However, no technology is perfect, and it is essential to remember that the advent of RNAi‐based protocols for the manipulation of gene expression initiated whole new ways of examining mammalian biology, including the study of carcinogenesis.
3. UNRAVELING THE MISWIRING OF CANCER SIGNALING
Most tumor cells exhibit alterations in signaling pathways that regulate the many processes required for carcinogenesis, including cell proliferation or differentiation, and environmental responses. Thus, it is not surprising that the cancer community quickly adopted RNAi‐based functional genetic studies, including unbiased screens to study the genetic and epigenetic basis of cancer, and the functional consequences of alterations in specific genes and pathways. Here, we discuss selected RNAi‐mediated functional genetic studies of representative cancer‐associated genes and pathways.
3.1. WNT–β‐catenin signaling and other developmental pathways
Several subtypes of cancer exhibit disruption of signaling pathways involved in embryonic development and cell differentiation. Because of the crucial roles that developmental pathways, including Hedgehog (Hh) and WNT–β‐catenin signaling, play in carcinogenesis, their characterization in healthy or tumor cells became the primary focus of many RNAi‐based studies; an effort that continues using complementary functional genetic tools. Many of these RNAi studies used a reporter assay to measure the transcriptional output of a signaling pathway. For example, one of the first genome‐wide RNAi screens conducted in 2003 used a Drosophila cell‐based luciferase reporter assay of Hh signaling. The screen identified known components (e.g., Frizzled‐1 and Frizzled‐2 and GSK3‐beta) and new regulators of the Hh pathway, including a link to WNT signaling through the activity of CK1α. 76 Another siRNA screening study performed in mouse cells expressing a similar reporter assay of the activation of the Hh pathway also resulted in further investigation of the links between the Hh and WNT pathways, highlighting the function of STK11 as a regulator of both pathways. 77
Colorectal tumors frequently exhibit disruption of the WNT–β‐catenin signaling that involves secreted WNT glycoproteins acting as activators of specific receptors, stimulating additional cellular responses, including gene expression changes regulated by β‐catenin and members of the TCF/LEF family of transcription factors. To discover proteins that regulate the WNT‐pathway, DasGupta et al. 78 used a Drosophila‐based TCF‐luciferase reporter assay of β‐catenin transcriptional activity and identified over 200 putative regulators of WNT–β‐catenin signaling. Critically, follow‐up studies in zebrafish and mammalian cells confirmed the Drosophila‐based results, including regulatory functions for the kinases LATS1, LATS2, and CDC2/CDK1. In mammalian cells, a 2008 publication also employed a TCF‐luciferase reporter assay in a genome‐wide siRNA screen conducted in non‐CRC cells, but that resulted in a focus on the function of the TCF/LEF family member, TCF7L2, in colorectal cancer (CRC). 79 A study published in the same year reported a genome‐wide siRNA screen that also used a β‐catenin–responsive luciferase assay, but this study used DLD1, colon adenocarcinoma cells, that harbor inactivating mutations in the tumor suppressor APC. 80 Follow‐up analysis highlighted the function of AGGF1 that the authors linked to chromatin‐remodeling, though to date, this finding's significance remains unclear. A third study published in 2008 reported the results of shRNA screens (targeting ~1000 genes) conducted in human colon cancer cells that assessed either cell viability or a readout of β‐catenin‐dependent transcription. Of the nine genes highlighted by these screens, one–CDK8–maps to a region of chromosome 13 frequently amplified in CRC. Subsequent studies showed that the function of CDK8 as part of the mediator complex contributes to the transcriptional activity of β‐catenin. 81 We contributed to another study that used RNAi to study candidate genes found within the region of chromosome 13 amplified in CRC. Interestingly, we found that silencing LNX2 in CRC cell lines results in the downregulation of NOTCH‐regulated genes and TCF7L2‐target genes. Furthermore, the examination of tumor‐derived expression profiles found an LNX2‐TCF7L2 gene expression module in CRC samples. 82
A further study of β‐catenin transcriptional activity involved the correlation of data from a series of shRNA screens conducted as part of Project Achilles (discussed in further detail below in Section 4.4) with a cancer cell line's relative β‐catenin activity. 83 This analysis and follow‐up studies demonstrated the dependency of cells that exhibit high β‐catenin on the function of the transcriptional coactivator YAP1. 83 Building on these RNAi studies, more recent CRISPR–Cas9 screens have revealed further vulnerabilities in cancer cells that depend on WNT‐signaling. For example, a CRISPR‐screen conducted in a pancreatic ductal adenocarcinoma cell line that demonstrated the importance of the Frizzled receptor FZD5 in cells harboring a mutation in RNF43. 84 The same group also recently described using a positive‐selection assay to identify genes needed for β‐catenin activity in cells harboring APC mutations. 85 The screen identified BCL9L, a known co‐transcriptional activator of β‐catenin, and IPO11, a protein not previously linked to β‐catenin. Follow‐up studies demonstrated that IPO11 regulates the nuclear transport of β‐catenin in APC mutant cells that the activity of IPO11 could represent a mutation‐specific vulnerability.
3.2. NF‐κB signaling
The transcription factor complex NF‐κB regulates various biological processes, including B‐cell development and inflammation, and so alterations in proteins that form part of the NF‐κB signaling cascade contribute to the initiation and progression of several tumor types. An early focused shRNA screen used a cell‐based reporter of tumor necrosis factor (TNF) activation, an endpoint of NF‐κB signaling, to identify negative regulators of NF‐κB signaling by targeting members of the deubiquitinating family of proteins. 86 This study identified CYLD (CYLD lysine 63 deubiquitinase) as a negative regulator of NF‐κB that removes ubiquitin from the TNF receptor‐associated factor TRAF2; loss of CYLD drives a proliferative signal via the core NF‐κB regulator, the IKK complex. Interestingly, individuals with cylindromatosis harbor mutations in CYLD that results in multi‐focal skin lesions, and so this RNAi‐based study prompted the initiation of a clinical trial of the IKKβ signaling inhibitor salicylic acid. 87 Overall, treatment with salicylic acid resulted in the complete remission of 2 out of 12 cylindromatosis tumors, with some response in 8 other tumors.
In a hematologic tumor‐type, several RNAi screens performed by Staudt and co‐workers have probed the NF‐κB signaling pathway in a subtype of Diffuse Large B‐cell Lymphoma (DLBCL). In an initial study using a pooled shRNA screening strategy targeting 2500 genes performed in cell lines representing the two subgroups of DLBCL‐activated B‐cell (ABC) and germinal B‐cell (GBC) and an IKKβ reporter assay, revealed the dependence of the ABC‐DLBCL on NF‐κB signaling. 88 Specifically, the screens identified CARD11 and two interacting proteins, MALT1 and BCL10, as critical regulators of the ABC‐DLBCL. A follow‐up shRNA screen in cells representing the ABC‐subtype of DLBCL identified Myd88 as a consistent vulnerability across multiple ABC‐DLBCL cell lines. 89 This same screening project also identified IRAK1 as an essential gene for the viability of these cancer cells. More recently, the same group has used CRISPR–Cas9 functional genomic screens to identify a multi‐protein supercomplex in the DLBCL‐ABC subtype that drives JAK‐STAT3 signaling via NF‐κB regulated IL‐10 and MyD88. 90
3.3. Fusion oncogenes
Cancer genomes frequently show the presence of structural rearrangements. In some cases, genomic rearrangements result in the generation of a fusion oncogene that is the principal driver of tumorigenesis. One class of fusion oncogenes consist of a 5ʹ regulatory region and part of the protein‐coding sequences of one gene and a portion of the protein‐coding and 3ʹ regulatory sequences of a second gene. This class of fusion oncogenes includes those that encode the fusion kinase BCR‐ABL and the fusion transcription factor EWS‐FLI1, the principal oncogenic drivers of chronic myelogenous leukemia and Ewing sarcoma, respectively. The controlled depletion of fusion oncogene expression using RNAi has proven indispensable for assessing their function in carcinogenesis. Targeting a fusion transcript using an shRNA or siRNA can facilitate the systematic comparison of markers of cell state (e.g., the cell growth characteristics or differences in the transcriptome or the proteome) in the presence and absence of the oncogenic driver responsible for the disease. Here, as an example, we will discuss some of the RNAi‐focused studies of the EWS‐FLI1 fusion oncoprotein that is the primary oncogenic driver in most cases of the soft‐tissue and bone tumor Ewing sarcoma (EWS).
The early studies that used RNAi to probe the biology of EWS examined the dependency of EWS cells on the expression of the fusion oncoprotein EWS‐FLI1, which functions as an aberrant transcription factor. Some studies examined the effect of silencing the fusion transcript on the growth of EWS cells in vitro or in vivo, 91 , 92 , 93 , 94 , 95 , 96 while other early studies assessed the function of the EWS‐FLI1 protein. 97 Several RNAi‐based studies analyzed the function of EWS cell lines modified to express an shRNA targeting the EWS‐FLI1 fusion transcript in the presence of doxycycline. 98 , 99 Using the shRNA‐inducible system and increasingly sophisticated 'Omic‐scale methods to profile the transcriptome and epigenome of EWS cells following the silencing of EWS‐FLI1 enabled the identification of many of the cell signaling and metabolism pathway that EWS‐FLI1 deregulates as part of the tumorigenic process and the function of EWS‐FLI1 as an aberrant transcription factor. 98 , 99 , 100 , 101 , 102 , 103 , 104 , 105 , 106 In complementary studies, our work used RNAi screens and follow‐up gene‐specific RNAi analysis to discover and validate the sensitivity of EWS cells to the loss of proteins that process the EWS‐FLI1 fusion pre‐mRNA. 107 One aspect of this and subsequent studies highlighted the dependency of about one‐third of Ewing sarcomas that harbor chromosome 22 translocations in intron 8 of the 5ʹ partner gene, EWSR1. Specifically, our work demonstrated that EWS cells harboring EWSR1‐intron 8 breakpoints depend on the RNA binding protein HNRNPH1 to generate the in‐frame protein‐coding fusion mRNA in these tumor cells. 107 , 108
3.4. The TP53 tumor suppressor gene
The transcription factor TP53 is the most frequently mutated gene in cancer. 109 , 110 Significant consequences of changes in TP53 function include disruption of the cellular response to DNA damage and the maintenance of genome stability. The importance of understanding the complexity of TP53 biology made it a prime target for early RNAi‐based functional genetic studies. One of the earliest large‐scale shRNA screens employed a ~24,000 shRNA library targeting ~7900 human genes (combined with RNAi depletion of p16INK4A) and a modified human fibroblast line that enabled the identification of genes that resulted in a bypass of TP53‐dependent cell arrest. 72 This screen identified five candidate genes of interest that follow‐up experimentation linked to the regulation of p21 (CDKN1A) expression. 72 The same study also introduced the concept of molecular bar‐coding of shRNA libraries to ease the assessment of the relative enrichment and depletion, initially using arrays, and more recently, sequencing to quantify the relative levels of an shRNA sequence in the genomic DNA extracted from different cell populations (Figure 3B). The group then performed a TP53‐related shRNA screen using a bar‐coded shRNA library that studied the MCF7 breast cancer cell line's response to nutlin‐3, an MDM2 inhibitor. 111 MDM2 can bind TP53 and target it for proteasomal degradation. The inhibition of MDM2, therefore, stabilizes the TP53 protein, potentially activating the TP53 pathway. A comparison of the relative abundance of the shRNAs present in control and nutlin‐3 treated MCF7 cell populations following 14 days of culture identified, as expected, significant enrichment of shRNAs targeting TP53, and shRNAs corresponding to TP53BP1, among others. Subsequent experiments showed that the function of TP53BP1 in the DNA damage response and the alterations in this response in some tumor cells explained its identification in the RNAi screen.
A seminal investigation, published in 2003, employed an RNAi‐based strategy to investigate the effect of alterations in gene expression on in vivo tumor development focused on TP53 and its function in lymphoma. 112 The study by Hemann et al. described three shRNAs targeting Tp53 that reduced protein expression each by a different amount relative to a control shRNA. The stable introduction of each of these shRNAs into hematopoietic stem cells (HSCs) isolated from the fetal livers of Eµ‐Myc mice (a B‐cell lymphoma model) generated a Tp53‐epi‐allelic panel of cells. Critically, the transplantation of the Tp53‐shRNA expressing cells into irradiated recipient mice showed a correlation between the levels of Tp53 and a reduction in survival. Specifically, those cells expressing the lowest level of Tp53 showed an accelerated decrease in survival compared with control mice, and histopathology consistent with that observed in mice bearing Tp53‐null tumors. In contrast, the lymphoma generated by the HSCs expressing an intermediate level of Tp53 showed a different phenotype including, a longer time to lethality and less pronounced systematic disease. In Section 5, we will discuss additional TP53‐related in vivo RNAi studies.
Since the publication of these early studies (pre‐2006), hundreds of reports have described application of RNAi‐based strategies to probe TP53 function. Here, we will highlight one further study, focused on the bone tumor, osteosarcoma (OS). 113 The genomes of OS tumor samples are complex, with almost no consistent genetic changes, except for mutations altering TP53 expression or function. 114 As part of an integrated study of OS, Perry and co‐workers conducted a pooled shRNA screen (40,000 shRNAs targeting ~8400 mouse genes) using tumor cells derived from a mouse model of OS (conditional deletion of Tp53 and Rb in pre‐osteoblasts). This screen identified Pik3ca as a lead candidate gene. Subsequent analysis demonstrated a dependence of mouse OS cells on the PI3K/mTOR pathway that non‐tumorigenic mouse bone cells did not exhibit. Furthermore, Perry and co‐workers reported that small‐molecule inhibition of PI3K and/or mTOR inhibited the growth of mouse and human OS lines, suggesting genetic alterations, including alterations in TP53 function, may sensitize OS cells to disruption of signaling through this pathway. To date, the rarity and genetic complexity of OS have meant clinical translation of this finding has proven a challenge, but this is a targetable pathway that, in the long‐term, may have applicability for the treatment of OS.
3.5. The Ras oncoprotein family
Activating mutations in KRAS, NRAS, and HRAS, which encode for members of the RAS family of proteins, occur at high prevalence in many cancer types, particularly subtypes of lung and pancreatic cancers. 115 One of the earliest shRNA screens first focused on identifying genes that may alter RAS activity in the absence of a mutation using modified primary fibroblasts, with enhanced potential to transform. The screen identified the transcription factor PITX1 as a putative suppressor of RAS activity, 116 a finding subsequent studies of hepatocarcinogenesis confirmed. 117 Another study conducted a genome‐wide RNAi screen in K‐Ras‐transformed NIH‐3T3 cells to investigate the suppression of a proapoptotic pathway by RAS proteins. 118 The screen implicated over 25 candidate genes in the regulation of FAS expression, a proapoptotic molecule. A follow‐up study by the same group described how several of these candidate genes form a RAS‐regulated gene network and suppresses FAS expression by altering DNA methyltransferase, DNMT1 activity. 119
An early attempt to identify proteins that mutant RAS cells are selectively dependent on for cell survival employed a 4000 gene siRNA library to screen the colon cancer cell line DLD‐1 that harbors one mutant and one wild‐type KRAS allele. 120 The screen identified 75 candidate genes that Sarthy et al. re‐screened using isogenic KRAS cell lines. From this follow‐up screen, they selected to examine the relative dependency of the KRAS‐mutant line on the function of the negative regulator of apoptosis BIRC5 (Survivin). Three independent shRNA‐based RNAi screening studies, published in 2009, used similar approaches to discover mutant RAS dependencies. 121 , 122 , 123 Each used slightly different model systems, isogenic cancer cell lines, multiple KRAS mutant and wild‐type cell lines, or cell lines exhibiting different dependencies on mutant KRAS activity, and different shRNAs libraries. Given these differences, it is unsurprising that while there was some limited overlap in these screens' results, each group focused on different lead candidate genes. Unfortunately, none of these leads proved to be a critical breakthrough, illustrating the difficulty of these types of large scale functional studies, 124 particularly when dealing with the analysis of an oncoprotein that influences many interconnected signaling pathways.
Besides the complexity RAS signaling, there is also the possible impact of functional redundancy, where, under certain circumstances, members of a family of proteins can substitute, at least in part, for each other's function. To determine if the depletion of more than one gene within the KRAS‐signaling pathway and its associated pathways could reveal KRAS‐co‐dependencies, Lee et al. 125 used previously validated siRNAs designed using a third‐generation sequence algorithm and a screening workflow that pooled siRNAs targeting multiple genes. 71 , 126 This study demonstrated that the combined targeting of RAF‐kinase signaling (BRAF and CRAF/RAF1) and autophagy (autophagy E1 ligase ATG7) induces G1‐arrest and apoptosis of KRAS‐mutant cells while mediating minimal effects on nonmutant cells. The study by Lee et al. 125 also showed evidence that RAC1 function inhibition could act as a co‐target of the RAF kinases. Interestingly, two studies also published in 2019 broadly supported the concept of parallel targeting RAF signaling and autophagy as an approach for the treatment of RAS‐mutant tumors, particularly RAS‐driven pancreatic adenocarcinoma. 127 , 128
Enhanced shRNA screening efforts conducted in large numbers of cancer cell lines (discussed in further detail in Section 4.4) have also generated additional potential modulators of KRAS signaling. 129 , 130 For example, McDonald et al. 130 reported the susceptibility of a subset of KRAS‐mutant lung cancer cell lines that lack dependency on KRAS. Instead, they found that cells harboring mutations in KEAP1 exhibit a dependency on NFE2L2, and cells that have alterations in the expression of SMARCA4 showed a dependency on SMARCA2 function. 130
The publication of these comprehensive shRNA screening efforts came at the same time as reports of the first wave of genome‐wide CRISPR–Cas9 screens designed to examine RAS biology that broadly used the same approaches as used for earlier RNAi studies. 131 , 132 For example, Wang et al. 131 highlighted five genes—RCE1 and ICMT that encode proteins required for the generation of the mature membrane‐associated form of RAS; two genes encoding proteins that function within the MAPK signaling cascade—RAF1 and SHOC2; and PREX1, encoding an RHO, small GTP‐binding protein that binds and activates RAC1 and thus Rac signaling. Interestingly, the dependency on PREX1 proved specific for AML cell lines, due to the lack of a paralog of this gene in these lines, versus other cancer cell lines. Martin et al. 132 used a similar screening strategy to find genes that altered the growth of KRAS‐mutant DLD1 cells and not KRAS‐wild‐type HCT‐116 cells and a follow‐up screen in LS513 cells, another KRAS‐mutant line. The results of these screens highlighted a new area of vulnerability in KRAS‐mutant cells, that of dependence on mitochondrial pathways. Additional studies, including the use of other KRAS‐mutant/wild‐type isogenic cell line pairs, demonstrated that KRAS‐mutant cells exhibit selective dependency on proteins involved in mitochondrial oxidative phosphorylation or translation. Importantly, KRAS‐mutant cells proved more sensitive to the use of inhibitors of these processes; however, it is still too early to know if any of these new putative mutant‐KRAS vulnerabilities are reproducible and will prove tractable as drug targets. These, and other large‐scale functional genomic screening studies conducted in isogenic, or cancer or mutation‐specific cell lines, highlight the importance of such approaches to find new cancer‐specific dependencies. In the next section, we will discuss the cancer research community's efforts to use RNAi and other LOF approaches to discover cancer dependencies systematically.
4. MAPPING CANCER DEPENDENCIES—MORE REALLY IS BETTER
As RNAi technology's capabilities improved, cancer researchers began to assess the feasibility of conducting systemic large screen studies across multiple cancer cell lines, initially representing a single tumor type, and then an increasingly diverse variety of cancers. The predominant rationale for developing these efforts was the need to conduct functional genetic screens in cell lines representing the genetic and histopathologic heterogeneity of human cancer. Crucially, the scale of these studies needed the statistical power to identify the effect of a perturbation in the context of another variable, for example, an oncogenic mutation. Most such studies used cell survival as an endpoint. Well‐based arrayed siRNA screens typically used the quantification of a metabolite as a surrogate for cell viability measured 48–96 h posttransfection (Figure 3A), while screens using pooled retro‐ or lentiviral shRNA libraries assessed the relative enrichment or depletion (or, “drop‐out”) of a cell harboring a given shRNA, usually 14–21 days post‐transduction (Figure 3B). If performed at scale, both approaches can identify: genes coding for proteins with essential functions; proteins that most cancer cells, irrespective of tumor type, depend on for cell growth—pan‐cancer dependency genes; and those proteins, that when depleted, alters the survival of a specific subtypes of cancers. A protein that a particular subtype of cancer depends on for survival may reflect differences in cell lineage or mutational state. Early studies typically involved the publication of large‐scale LOF data sets generated using screening approaches as supplementary information or deposition of results in data repositories (e.g., PubChem: https://pubchem.ncbi.nlm.nih.gov/; GenomeRNAi: http://genomernai.org 133 ). However, with time we have seen more structured efforts develop, including web‐based sites that enable the community to search standardized and regularly updated LOF data sets and retrieve gene‐specific or whole‐genome data.
4.1. Early siRNA screens
An early study that tested the hypothesis that RNAi screens conducted in a panel of cell lines could identify pan or mutation‐specific gene dependencies employed siRNAs targeting 700 kinases and kinase‐related genes. 134 Screens performed in breast, lung, and cervical cancer cell lines (five in total) demonstrated a cell line‐specific dependency on the expression of PIK3CA and the presence of a PIK3CA mutation. This study also identified correlations between protein expression, including the mitotic regulator WEE1, and sensitivity to its depletion by RNAi. Interestingly, a study published in the same year reported the results of siRNA screens conducted in the breast cancer cell lines MDA‐MB‐231, BT‐20, and HCC‐1937 also described the sensitivity of these lines to the loss of WEE1 function. 135 These screens and others employing siRNAs targeting subsets of human genes conducted in human cancer cell lines began to establish the infrastructure needed to study many cell lines in parallel, for example, siRNA screens performed in a panel of 34 breast cancer cell lines. 136 This study of over 30 breast cancer cell lines reported that over 90% exhibited dependence on the mitosis‐associated kinases PLK1, WEE1, and AURKA. This screening effort also highly ranked kinases previously linked to breast cancer, such as PI3KCA, ERBB2, and CSK1, and the examination of a subset of lines showed the relative dependency of PTEN‐deficient cells on the function of MPS1, a mitotic spindle checkpoint protein. The same study also revealed that the activity of the ADCK2 kinase influences estrogen receptor (ER) signaling and the proliferation of ER‐positive breast cancer cells. 136
4.2. Early shRNA screens
Complementary efforts, over the same period, focused on the employment of large‐scale shRNA libraries, with three of the earliest studies published in 2008 employing libraries of increasing complexity, ranging from ~3000 genes (8000 shRNAs) to 9500 genes (45,000 shRNAs), and a wider variety of cell lines, beginning with three cancer cell lines (two colon lines and one breast line) and one non‐transformed line, and by the third study, a screen of 12 cancer cell lines. 137 , 138 , 139 The late 2008 study published by Luo et al. 139 of 12 cancer cell lines screened using a subset of a genome‐wide shRNA library 140 , 141 ; the RNAi Consortium (TRC) enabled the authors to report that 268 genes had essential functions across all the cell lines studied. Overall, this finding reflected the activity of the proteins encoded by these genes in processes such as translation, mRNA biogenesis, including splicing, and protein degradation via the proteasome. The study by Luo and co‐workers also examined their data to identify genes that, when silenced, decreased the growth of cell lines representing one tumor‐type versus another, identifying 63 genes that non‐small‐cell lung cancer (NSCLC) exhibited greater dependency on for cell survival than other cell lines. Among cell‐line specific genetic vulnerabilities, this study also identified the sensitivity of the chronic myelogenous leukemia cell line K562 that harbors the BCR–ABL fusion gene to the shRNAs targeting the BCR or ABL genes. Luo and co‐workers also confirmed the utility of RNAi screens to identify oncogenes within amplified regions, such as the identifying CRKL as a putative oncogene in NSCLC, a finding supported by follow‐up studies by the same group. 142
4.3. Statistical analysis of RNAi screens
An essential part of developing the workflows for large‐scale RNAi screening efforts (siRNA and shRNA) was the need for robust statistical analysis of the data these efforts generated. Such data analysis requires consideration of many technical and biological variables, including, though not limited to, the following: (1) The definition, quantification, and application of basic quality control metrics. (2) A means for appropriately handling inter‐ and intra‐screen variation to facilitate valid comparisons within and between screens. (3) The consideration of RNAi‐specific variables, including differences in the efficacy with which an siRNA or shRNA silences the expression of a target transcript or miRNA‐like off‐target effects. (4) The method used for the normalization and ranking of screening results and the criterion used to define a candidate gene for further analysis. (5) The ability to integrate functional genomic data sets and other relevant large‐scale profiling results, including gene copy number and expression data.
Over time, several groups have reported statistical approaches designed to reduce the false‐negative or positive rates of RNAi screens. For example, a statistical analysis of siRNA data used the median and the median of absolute deviations (±3) coupled with assessing the normalized data in quartiles, to center and identify candidates of interest. 143 This approach proved more robust than the mean ± three standard deviations more typically used to analyze small molecule screening data. Other statistical methods incorporated consideration of the consistency of phenotype generated by siRNAs targeting the same gene. For example, if an RNAi library contains four to six siRNAs targeting each gene, then there is enough redundancy in the data set to determine if the clustering of the results of these siRNAs is significant (Redundant siRNA Activity—RSA score) and more likely to represent a high confidence data point. 144 The Luo study mentioned above 139 also used a ranking‐based approach called RIGER—“RNAi gene enrichment ranking” score method to identify lead candidate genes. Like the previous study, RIGER also considers the ranked position of the shRNAs targeting a given gene, but in this case, applied gene‐set enrichment methods and Kolmogorov–Smirnov statistical analysis to guide the selection of genes for follow‐up analysis. Other statistical methods assessed the consistency between the effects of siRNAs targeting each gene and considered the impact of miRNA‐like off‐target effects. For example, one study used a genome‐wide RNAi screening data set to determine the effect of siRNAs with identical seed sequences, developing a common seed analysis (CSA) score for each siRNA in the screen. Such analysis allows for the de‐prioritizations of siRNAs with a high CSA score due to a promiscuous seed sequence. 145 An extension of this study used the heptamer seed sequence at the 5ʹ end of the guide strand to identify previously unidentified candidate genes of interest within genome‐wide siRNAs screens. 146 Finally, with the increased availability of large‐scale RNAi screening data using the same reagents in different screens, it was possible to devise computational methods that encompassed several features of these earlier efforts, in an easier to apply fashion. One such example, ATARiS—Analytic Technique for Assessment of RNAi by Similarity—used the large data sets developed as part of Project Achilles (discussed below) to develop gene and RNAi reagent level scores. By comparing the phenotypic effects of individual reagents across multiple screens, ATARiS facilitated the removal of reagents with significant off‐target effects, while retaining RNAi effectors that mediated on‐target suppression of gene expression. 147 ATARiS proved to be a starting point for a series of subsequent algorithms used to analyze RNAi data, such as DEMETER 129 and DEMETER2 148 and the analysis of CRISPR–Cas9 gene editing screens conducted in cancer cell lines 149 that must take account of gene copy number effects. 150 , 151
4.4. Building a cancer dependency map
The profiling of the genome, epigenome, and transcriptome of human tumors has revolutionized our ability to identify causally relevant changes in cancer cells that can assist both the diagnosis and treatment of cancer. However, while these high‐resolution methods can detect molecular alterations in an individual tumor, we still lack an understanding of the impact of many of these changes on the affected gene's function. Enhancing the scale of cancer LOF screens and integrating these data sets with other data, including gene copy number, mutation status, and expression patterns, has the potential to address this deficit. In 2011, Cheung et al. 152 published the results of a systematic study of over 100 cancer cell lines employing an shRNA library targeting 11,194 human genes (54,020 shRNAs in total). This effort, part of a broader program called Project Achilles, aimed to identify cancer cell‐type‐specific genetic vulnerabilities across multiple cancer cell lines representing ovarian, colon, pancreatic, esophageal, and NSCLC, or glioblastoma. The initial study confirmed the dependency of colon cancer cell lines on the expression of KRAS, CTNNB1, and BRAF, and as discussed above, the differences in the sensitivity of wild‐type and mutant PIK3CA cell lines to the targeting of PIK3CA by RNAi. Next, to aid the discovery of new cancer‐type specific dependencies, Cheung et al. 152 integrated gene copy number and RNAi data, identifying PAX8 as a dependency in ovarian cancer cell lines that harbor amplification of the region containing this gene. Another study, published in 2012, employed an shRNA library targeting 16,000 genes to screen breast, pancreatic, and ovarian cancer cell lines (72 lines in total). 153 In this case, the integration of expression profiles that correlate with different subtypes of breast cancer and RNAi screening data identified potential breast cancer subtype‐specific vulnerabilities; findings supported and extended by a follow‐up study that included the results of shRNA screens performed in 77 breast cancer cell lines. 154 Most of these studies used statistical analysis to reduce the false‐negative and positive rates, but others also took the approach of increasing the number of RNAi effectors per gene. For example, a study by Hoffman et al. 155 showed that applying the RSA method 144 using data from ~17 shRNAs per gene allowed the identification of high confidence lead candidate genes. In this study, the shRNA library targeted genes coding for proteins involved in the regulation of chromatin state, and its employment in screens of 58 cancer cell lines identified the dependence of SMARCA4‐deficient cells on the function of another SWI/SNF complex member, SMARCA2. 155
By 2014, Project Achilles included a data set of shRNA screens targeting 11,000 genes (5 shRNAs per gene) performed in 216 cancer cell lines, 156 and by 2017 this resource had expanded to over 500 cell lines. 129 A complementary effort, also published in 2017, reported shRNA screens targeting 7837 genes (~20 shRNAs per gene) in 398 cancer cell lines. 130 Referred to as Project DRIVE, this study noted many of the previously defined dependencies, including cell lines harboring NRAS, BRAF, KRAS, or PIKSCA mutations. Integration with other data sets, such as gene expression profiles, revealed other vulnerabilities. For example, McDonald et al. 131 reported the dependence of several cancer cell lines on the expression of lineage‐specific genes, particularly transcription factors such as SOX10. Critically, these and other efforts supported the development of web interfaces that facilitate the ability of the research community to search or download data sets and associated software packages, now harmonized within a single portal—the DepMap portal—https://depmap.org/portal/. Today, the DepMap portal integrates the data from RNAi and several CRISPR–Cas9 LOF large‐scale screening projects encompassing over 700 cancer cell lines, along with many other data sets, including the multiparametric analysis of the cell lines that make up the Cancer Cell Encyclopedia (CCLE), 157 and drug sensitivity data. 158 Importantly, this represents an ongoing collaborative effort involving multiple institutions and funding sources that continues to employ pooled CRISPR–Cas9 gene‐editing screens to define cancer‐specific vulnerabilities. 149 , 159 , 160 The reader is encouraged to explore the DepMap portal interface and make use of the regularly updated results available at https://depmap.org/portal/download/. However, it is essential to remember that any hypotheses generated using these, and other large‐scale cancer cell line data, will always need extensive experimental investigation and validation using orthogonal approaches.
5. RNAi SCREENING IN THE MOUSE
The examination of gene function through the application of small RNA‐guided functional genetic tools has accelerated our ability to delineate the activity of complex transcriptional and signaling cascades and identify new candidate target proteins for the treatment of cancer and other diseases. However, most such studies have involved the use of cell lines established years or even decades ago and grown as monolayers on treated plastic. While informative, such experiments lack the biological context that a whole‐organism study can offer. For this reason, many groups sought to adapt RNAi and now CRISPR‐based technologies for application in vivo. C. elegans proved a remarkably straightforward organism for the study of gene function in vivo by RNAi because there is no need for a carrier molecule to introduce the dsRNA, 161 , 162 , 163 but mouse models have and remain the principal whole‐organism used for the study of cancer biology. As discussed above, an early in vivo RNAi study that investigated the function of TP53 in B‐cell lymphomagenesis used shRNAs targeting Tp53 to generate an epi‐allelic cell series and showed a correlation between the levels of Tp53 protein and disease state. 112 However, difficulties in predicting and controlling the exact level of silencing by a given RNAi effector that emerged over the next few years meant that few others adopted this approach. The study by Hemann et al. 112 did though, along with others (e.g., 164 , 165 , 166 ), spur the use of ex vivo manipulated cells that expressed a gene‐specific shRNA (constitutive or inducible expression) to study the consequence of targeting the candidate gene of interest on the initiation or maintenance of tumor growth, through the reintroduction of the modified cells to mice as transplants or xenografts. Here, we focus on the discovery potential of in vivo RNAi screens, typically conducted using pooled shRNA libraries. For an expanded discussion of the study of cancer in vivo using complementary gain‐ or loss‐of‐function functional genomic approaches, we direct the reader to a recent review by Weber et al. 167
One early in vivo RNAi screen interrogated ~300 mouse genes for potential tumor suppressor function using p53 −/−; Myc hepatocytes (a liver tumorigenesis model) transduced ex vivo and transplanted subcutaneously into immunocompromised mice. 168 By analyzing the relative depletion or enrichment of specific shRNAs assessed by analysis of genomic DNA harvested from the resulting xenografts, Zender et al. identified the known tumor suppressor PTEN and several previously unknown putative tumor suppressors, including XPO4, that follow‐up studies implicated in the regulation of proteins involved in TGF‐β signaling or translation initiation. Another study from the same group used an analogous RNAi screening approach, but targeting ~1000 genes and HSCs from Eµ‐Myc mice to identify B‐cell lymphoma‐associated tumor suppressors. 169 This screen identified several putative tumor suppressors, including a member of the DNA damage response machinery, Rad17, a finding corroborated by further studies, including a recent analysis of homozygous deletions in over 2000 primary tumors. 170 A complementary in vivo RNAi study discovered proteins involved in cell motility using shRNAs targeting about 1000 genes and the Eµ‐Myc B‐cell lymphoma model, focusing on proteins that modulated the growth of cells grown in vivo but not in cell culture. 171 Subsequently, the same group reported a genome‐scale shRNA screen designed to identify modulators of B‐cell leukemia progression, again comparing in vivo versus in vitro growth effects. This screen identified several genes where growth in vivo generated specific dependencies, including a dependency on the function of the transcriptional regulator Phf6. 172 As an example of RNAi in vivo studies focused on a solid tumor, one study, by Iorns et al. 173 used modified nontransformed human mammary epithelial cells transduced with an shRNA library ex vivo and grown as xenografts in the mammary fat pads of severely immunocompromised mice. The screen identified established tumor suppressors such as TP53, and several additional tumor suppressor candidate genes, including the leukemia inhibitory factor receptor, 173 that more recent studies have shown may influence the metastasis of breast cancer cells to bone. 174
Another highly informative series of in vivo RNAi studies used lentiviral packaged shRNAs injected into mouse embryos in utero. 175 , 176 , 177 This approach facilitates the genetic modification of the progenitor cells that will form the mouse epidermis and, depending on the embryo's genetic background, can enable examination of normal and oncogenic growth. Fuchs and co‐workers first assessed this method by analyzing the effects of silencing individual genes and then conducting large‐scale pooled shRNA screens using a similar strategy. One of the shRNA screens conducted in the context of a mutated HRAS demonstrated dependency of the mutant cells on the function of Ctnnb1 or Mllt6, findings the authors successfully replicated in cell line models of human squamous cell carcinoma (SCC). 176 A separate shRNA‐screen (347 mouse genes) also used an in utero approach, but this time using embryos derived from TGFβ‐Receptor‐II conditional knockout mice, the same group identified Myh9 as a putative tumor suppressor in SCC. 177 Further studies suggested that Myh9 acts as a regulator of Tp53 stability and cellular localization. 177
6. WHEN TWO (OR MORE) ARE BETTER THAN ONE
Combination chemotherapy remains the backbone of cancer care and is likely to remain so for many tumor‐types for years to come. For this reason, determining the optimal genetic background in which a drug acts or identifying more effective drug combinations was an early focus of RNAi studies and screens. Many of these studies built on the concept of synthetic lethality that had derived from results in fruit flies and yeast, which showed that in some cases, the function of one gene could compensate for the LOF of another, resulting in no discernable phenotype after genetic disruption of one or other of these genes. However, mutation of both genes results in organismal demise, or synthetic lethality. 178 , 179 , 180 , 181 In cancer, synthetic lethality typically refers to a gene that is nonessential when depleted in a nontransformed cell, but if depleted in a transformed cell harboring a tumor‐specific mutation or combination of mutations, the cancer cell dies. Several studies described above illustrate the use of RNAi screens to identify synthetic lethality interactions, and the cancer dependency map also aims to identify such interactions by integrating LOF phenotypic information with mutational status or gene copy number. Here, we will highlight two further examples of the identification of potential synthetic gene–gene interactions revealed by RNAi studies of MTAP and PRMT5, and ARID1A and ARID1B. We will also discuss another aspect of synthetic lethal interactions of interest to the cancer community, that of gene–drug interactions, focusing on genes that modulate chemotherapy agents.
6.1. MTAP and PRMT5
The protein 5‐methylthioadenosine phosphorylase (MTAP) catalyzes the breakdown of methylthioadenosine (MTA) to adenine and methionine. The proximity of the MTAP gene locus to CDKN2A (p16) results in the frequent loss of this gene in several cancer types. Three separate studies published in 2016 combined genetic information and the results of shRNA functional genetic studies to identify MTAP synthetic lethal interactions, all of which identified PRMT5 as a lead candidate. 182 , 183 , 184 Two groups used the shRNA screening data from the cancer cell lines that are part of the CCLE and identified the subset of genes, that when depleted, resulted in the loss of viability of MTAP‐deficient cell lines, 182 , 183 while the third performed shRNA screens in HCT116‐MTAP wild‐type and homozygous deleted isogenic cell lines. 184 In addition to identifying protein arginine methyltransferase 5 (PRMT5), the screens also identified several known PRMT5 cofactors or proteins that function within the same pathway, including MEP50/WD77 and MAT2A. Biochemical studies demonstrated that the accumulation of MTA due to loss of MTAP resulted in the structural reordering of the cofactor binding pocket of PRMT5, decreasing its catalytic activity; however, tumor cell lines remain viable but sensitive to further disruption of PRMT5 function. 182 , 183 , 184 These studies highlighted a potential therapeutic strategy in MTAP deficient tumors where molecules that competitively bind to and inhibit PRMT5 in the same manner as MTA would lead to selective lethality in tumor cells. Building on this concept and other evidence, 185 several PRMT5 inhibitors (GSK3326595, JNJ‐64619178, and PF‐06939999) are now the subjects of current early phase clinical trials in patients with different subtypes of leukemia or lymphoma or advanced solid tumors (NCT02783300, NCT03614728, NCT03573310, and NCT03854227).
6.2. ARIDIA and ARID1B
Another cancer‐relevant synthetic lethal interaction that emerged from an analysis of the results of Project Achilles screens involved the examination of the shRNA screens conducted in ARID1A‐mutant cell lines (18 lines) versus non‐ARID1A‐mutant lines (>100 lines). 186 Multiple cancers types exhibit mutations in ARID1A, a gene that encodes a member of the SWI/SNF chromatin‐remodeling complex. Analysis of Project Achilles shRNA screening data showed ARID1B expression as essential for the viability of cell lines harboring ARID1A inactivating mutations. Follow‐up studies demonstrated tumor cells harboring ARID1A mutations depend on the ARID1B, a close homolog of ARID1A, for the assembly of the SWI/SNF chromatin‐remodeling complexes. In the absence of both ARID1A and ARID1B, the disruption to the generation of SWI/SNF complexes results in cell death. 186 To date, there is no published ARID1B inhibitor; however, a recent analysis of inhibitors of other epigenetic regulators revealed that inhibition of EZH2, a methyltransferase member of the polycomb repressive complex 2, has efficacy in ARID1A‐mutant ovarian clear cell carcinoma cell lines in vitro and in vivo. 187 Clinical trials with EZH2 inhibitors in various cancer types are ongoing, including the recruitment of patients with tumors associated with mutations in members of the SWI/SNF complex, such as rhabdoid tumors and synovial sarcoma (NCT02601950 and NCT02601937).
6.3. Chemotherapeutic agents plus one
The applications of RNAi screening to find synthetic lethal gene–drug interactions relevant to cancer treatment began in 2006 with a siRNA screen targeting 20,000 genes conducted in HeLa cells. 188 This study compared the effects of each siRNA alone or combined with either the nucleoside analog pro‐drug, gemcitabine, the microtubule stabilizer, paclitaxel, or the DNA cross‐linker, cisplatin. 188 In this study, the silencing of genes coding for members of the BRCA family of proteins or the RAD6/RA7D18 DNA repair pathways significantly enhanced cisplatin activity. The authors, Bartz et al., attributed this finding to the silenced cells' inability to mitigate cisplatin‐induced DNA damage in the context of mutant TP53 and loss of BRCA1 or other members of the BRCA1/2 network. Their findings contributed to the understanding of BRCA1‐deficient and BRCA1‐like tumors and their response to DNA‐damaging agents. Subsequently, in 2007, Whitehurst et al. 189 performed siRNA screens in combination with paclitaxel, but in the NSCLC cell line NCI‐H1155. The study identified 87 candidate paclitaxel‐sensitizing genes that included multiple genes encoding components of the proteasome or the mitotic spindle apparatus. Our siRNA–chemotherapeutic drug combination studies included several examining the topoisomerase 1 (TOP1) inhibitor camptothecin (CPT). TOP1 inhibitors create drug‐stabilized cleavage complexes that result in DNA damage, leading to their use as part of chemotherapeutic regimens to treat ovarian, lung, and colon cancers. For example, the treatment of ovarian and colon cancers often involves administrating CPT's clinical analogs—topotecan and irinotecan, respectively. However, TOP1 inhibitors have not performed successfully in clinical trials testing their efficacy to treat advanced breast cancer patients. 190 To determine if combining TOP1 inhibition with the inhibition of a second target could broaden the employment of TOP1 inhibitors, we conducted several siRNA screens to find proteins, that when inhibited, increase the efficacy of the TOP1 inhibitor, CPT, using different breast cancer cell lines representing the hardest to treat subtype, so‐called triple‐negative breast cancer. 191 , 192 One screen identified ataxia telangiectasia and Rad3‐related protein (ATR) as a sensitizing target of CPT, 191 which prompted a follow‐up RNAi screen and follow‐up study of an ATR inhibitor. 193 Together, these studies contributed to the rationale of two multicenter clinical trials sponsored by the National Cancer Institute that are ongoing (NCT02487095 and NCT02595931) testing the combination of ATR inhibition and the TOP1 inhibitor topotecan. Recently, the results of a phase one clinical trial combining topotecan with the ATR inhibitor M6620 (previously VX‐970) demonstrated that the maximum dose of the combination was tolerable and showed promise in refractory small‐cell lung cancer. 194
7. INTO THE CLINIC
The previous section discussed how RNAi studies guided the development of clinical trials testing traditional small‐molecule drugs, but can we use the RNAi mechanism directly as a therapeutic approach? Groups began testing the potential of employing RNAi effector molecules to treat human diseases within a year of the initial studies reporting its presence in mammalian cells, targeting viral pathogens, including HIV, and mutant transcripts, including those associated with neurodegenerative diseases or cancer. 91 , 195 , 196 , 197 , 198 However, it took almost two decades for these initial efforts to bear fruit, with the approval of the first RNAi‐based therapy in 2018 for the treatment of hereditary transthyretin amyloidosis (hATTR) based on the completion of a successful Phase III trial of Patisiran, a siRNA targeting the TTR transcript. 199 In late 2019, another siRNA‐based drug, Givosiran—a siRNA targeting hepatic delta‐aminolevulinic acid synthase 1 (ALAS1)—received approval for the treatment of acute hepatic porphyria (AHP), in part, because of the results of a recently published Phase III trial. 200 Other RNAi therapies at an advanced stage of development include a siRNA targeting of PCSK9, the subject of several recent clinical trials 201 , 202 , 203 , 204 involving patients with heterozygous familial hypercholesterolemia or atherosclerotic cardiovascular disease. However, building on these successes for the treatment of cancer has proved a challenge, and to date, the effective delivery of RNAi effector molecules into enough tumor cells to mediate a clinically relevant response remains the most significant obstacle. Several recent reviews have discussed the development of nucleic acid‐based therapies and RNAi therapeutics, and for further details, we direct the reader to these. 205 , 206 , 207 , 208 , 209 Here, in brief, we will discuss two clinically focused efforts testing the use of siRNA‐mediated gene silencing to treat solid tumors—the targeting of mutant‐KRAS transcripts (siG12D‐LODER) and the silencing of the gene encoding the EPHA2 receptor tyrosine kinase.
As discussed in Section 3.4, mutations in KRAS are present in many tumors, and several early RNAi studies assessed the feasibility of selectively targeting the transcripts encoding mutant KRAS. 210 , 211 , 212 , 213 , 214 The last of these studies, authored by Zorde Khvalevsky et al., described an investigational agent named siG12D‐LODER™ that consists of a siRNA targeting the KRAS transcript present in tumors harboring the KRAS‐G12D mutations encapsulated within a biodegradable polymeric matrix. When implanted intratumorally siG12D‐LODER™, Zorde Khvalevsky et al. 214 observed reduced KRAS expression (measured by antibody staining) and decreased growth of orthotopic human pancreatic tumor xenografts in vivo. A subsequent open‐label Phase 1/2a trial of siG12D‐LODER™ assessed the safety of intratumoral insertion of the encapsulated siRNA in 15 patients with advanced pancreatic cancer and receiving standard chemotherapy (NCT01188785). An article reporting the findings from this trial stated that patients tolerated the combination of the siRNA‐based intervention and chemotherapy. 215 As of 2020, a multicenter Phase 2 study of siG12D‐LODER™ in patients with advanced pancreatic cancer, in combination chemotherapy, remains active (NCT01676259), with an abstract submitted to the 2020 ASCO Annual Meeting reporting completion of trial accrual by December 2020. 216 Another mRNA target that is the subject of an early‐stage clinical trial encodes the receptor tyrosine kinase EPHA2. A variety of solid tumors, including ovarian cancer, overexpress EPHA2, and several studies performed by Sood and co‐workers, have demonstrated the reduction of EPHA2 expression by RNAi (alone or in combination with another intervention) decreases the growth of several different xenograft tumor models. 217 , 218 , 219 , 220 , 221 , 222 An ongoing clinical trial is assessing the intravenous administration of a nanoliposome (DOPC)‐encapsulated EPHA2 siRNA (EPHARNA) in patients with solid tumors (NCT01591356). Hopefully, promising results from these two ongoing trials, and other similar efforts investigating the in vivo or ex vivo application of siRNAs (see Das et al. 223 for a recent summary), will accelerate the further assessment of RNAi as a treatment approach for cancer.
8. CONCLUSION
Twenty years on from the first descriptions of RNAi in mammalian cells and the rapid development of the functional genetic tools that could target every gene in the human genome, the cancer biology research community now has available a remarkable array of approaches that perturb gene expression in a controlled and accurate fashion. Building on both the successes and limitations of RNAi‐based technologies, the advent of approaches that make use of methods that employ CRISPR‐based resources has expanded our options tremendously. We are now able to effectively edit DNA directly through the application of sequence‐specific guide RNAs and the Cas endonucleases (e.g., Cas9 or Cas12 and modified variants), including the ability to make increasingly precise single‐nucleotide changes (please see the following recent review 224 ). Alternatively, we can perturb gene expression using either modified Cas9 (e.g., the catalytically inactivated version of Cas9 and dCas9) or an alternative enzyme such as Cas13 to inhibit gene expression (CRISPRi) or, through a further modification of dCas9, activate expression (CRISPRa). For further descriptions of CRISPR‐based functional genetic tools, we direct the reader to recent in‐depth reviews or commentaries and the citations and links therein. 225 , 226 , 227 , 228 , 229 , 230 , 231 We also see the emergence of systems that mediate the selective degradation of a protein. 232 The access to this diversity of functional genetic tools offers the cancer community the opportunity to address myriad questions. As a starting point, gene‐editing tools may enhance our ability to study the early steps of carcinogenesis by generating model systems that better mimic the accumulation of mutations observed in many tumors. It may also be possible to generate better metastasis models that require the concurrent activation of some genes and the inactivation of others. Furthermore, we now have the potential to examine the temporal effects of depleting genes over a broader time frame, from minutes using protein degradation tools through to hours or days using CRISPR‐ or RNAi‐based approaches and the ability to reverse this effect. Employment of sophisticated temporal control over gene expression may also allow us to mimic better the dynamic and heterogeneous patterns of gene expression present in a growing tumor and thus improve insights into how we can target a higher proportion of cells within a tumor.
Twenty years ago, few of us would have dreamed that the discovery of gene expression changes mediated by small RNAs transfected into mammalian cells would trigger the technological revolution that continues to impact how many of us conduct cancer research. It is clear that small RNAs have enabled us to dream big, and it will be exciting to see what the next 20 years of cancer‐focused functional genetic studies yields.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
ACKNOWLEDGMENTS
We apologize to those colleagues in the RNAi and cancer functional genomics research fields whose studies we could not include in this perspective. Figures created with Biorender. com. The Intramural Research Program of the National Cancer Institute, Center for Cancer Research, supported this study—Project Number ZIA BC 011704 (NJC). We thank other members of the Caplen laboratory for their constructive comments. Natasha J. Caplen thanks all past members of her laboratory, our current and former collaborators and colleagues, and leaders of the NCI and NIH intramural programs that contributed to or supported our functional genetic studies and trans‐NIH funded RNAi screening efforts.
Sundara Rajan S, Ludwig KR, Hall KL, Jones TL, Caplen NJ. Cancer biology functional genomics: From small RNAs to big dreams. Molecular Carcinogenesis. 2020;59:1343–1361. 10.1002/mc.23260
Soumya Sundara Rajan and Katelyn R. Ludwig contributed equally to this study.
REFERENCES
- 1. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21‐nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–498. [DOI] [PubMed] [Google Scholar]
- 2. Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double‐stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci USA. 2001;98(17):9742–9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double‐stranded RNA in Caenorhabditis elegans . Nature. 1998;391(6669):806–811. [DOI] [PubMed] [Google Scholar]
- 4. Montgomery MK, Xu S, Fire A. RNA as a target of double‐stranded RNA‐mediated genetic interference in Caenorhabditis elegans . Proc Natl Acad Sci USA. 1998;95(26):15502–15507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kennerdell JR, Carthew RW. Use of dsRNA‐mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell. 1998;95(7):1017–1026. [DOI] [PubMed] [Google Scholar]
- 6. Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 1999;286(5441):950–952. [DOI] [PubMed] [Google Scholar]
- 7. Tuschl T, Zamore PD, Lehmann R, Bartel DP, Sharp PA. Targeted mRNA degradation by double‐stranded RNA in vitro. Genes Dev. 1999;13(24):3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: double‐stranded RNA directs the ATP‐dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101(1):25–33. [DOI] [PubMed] [Google Scholar]
- 9. Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA‐directed nuclease mediates post‐transcriptional gene silencing in Drosophila cells. Nature. 2000;404(6775):293–296. [DOI] [PubMed] [Google Scholar]
- 10. Caplen NJ, Fleenor J, Fire A, Morgan RA. dsRNA‐mediated gene silencing in cultured Drosophila cells: a tissue culture model for the analysis of RNA interference. Gene. 2000;252(1‐2):95–105. [DOI] [PubMed] [Google Scholar]
- 11. Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409(6818):363–366. [DOI] [PubMed] [Google Scholar]
- 12. Nykanen A, Haley B, Zamore PD. ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell. 2001;107(3):309–321. [DOI] [PubMed] [Google Scholar]
- 13. Hutvagner G, Zamore PD. A microRNA in a multiple‐turnover RNAi enzyme complex. Science. 2002;297(5589):2056–2060. [DOI] [PubMed] [Google Scholar]
- 14. Schwarz DS, Hutvagner G, Haley B, Zamore PD. Evidence that siRNAs function as guides, not primers, in the Drosophila and human RNAi pathways. Mol Cell. 2002;10(3):537–548. [DOI] [PubMed] [Google Scholar]
- 15. Martinez J, Patkaniowska A, Urlaub H, Luhrmann R, Tuschl T. Single‐stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell. 2002;110(5):563–574. [DOI] [PubMed] [Google Scholar]
- 16. Schwarz DS, Hutvagner G, Du T, Xu ZS, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115(2):199–208. [DOI] [PubMed] [Google Scholar]
- 17. Lee Y, Ahn C, Han J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425(6956):415–419. [DOI] [PubMed] [Google Scholar]
- 18. Yi R, Qin Y, Macara IG, Cullen BR. Exportin‐5 mediates the nuclear export of pre‐microRNAs and short hairpin RNAs. Genes Dev. 2003;17(24):3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu J, Carmell MA, Rivas FV, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305(5689):1437–1441. [DOI] [PubMed] [Google Scholar]
- 20. Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118(1):57–68. [DOI] [PubMed] [Google Scholar]
- 21. Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303(5654):95–98. [DOI] [PubMed] [Google Scholar]
- 22. Zeng Y, Cullen BR. Structural requirements for pre‐microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res. 2004;32(16):4776–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell. 2004;15(2):185–197. [DOI] [PubMed] [Google Scholar]
- 24. Gregory RI, Yan K, Amuthan G, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432(7014):235–240. [DOI] [PubMed] [Google Scholar]
- 25. Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432(7014):231–235. [DOI] [PubMed] [Google Scholar]
- 26. Han JJ, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha‐DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18(24):3016–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Landthaler M, Yalcin A, Tuschl T. The human DiGeorge syndrome critical region gene 8 and its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol. 2004;14(23):2162–2167. [DOI] [PubMed] [Google Scholar]
- 28. Song JJ, Smith SK, Hannon GJ, Joshua‐Tor L. Crystal structure of Argonaute and its implications for RISC slicer activity. Science. 2004;305(5689):1434–1437. [DOI] [PubMed] [Google Scholar]
- 29. Matranga C, Tomari Y, Shin C, Bartel DP, Zamore PD. Passenger‐strand cleavage facilitates assembly of siRNA into Ago2‐containing RNAi enzyme complexes. Cell. 2005;123(4):607–620. [DOI] [PubMed] [Google Scholar]
- 30. Okada C, Yamashita E, Lee SJ, et al. A high‐resolution structure of the pre‐microRNA nuclear export machinery. Science. 2009;326(5957):1275–1279. [DOI] [PubMed] [Google Scholar]
- 31. Elkayam E, Kuhn CD, Tocilj A, et al. The structure of human argonaute‐2 in complex with miR‐20a. Cell. 2012;150(1):100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin‐4 encodes small RNAs with antisense complementarity to lin‐14. Cell. 1993;75(5):843–854. [DOI] [PubMed] [Google Scholar]
- 33. Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin‐14 by lin‐4 mediates temporal pattern formation in C. elegans . Cell. 1993;75(5):855–862. [DOI] [PubMed] [Google Scholar]
- 34. Pasquinelli AE, Reinhart BJ, Slack F, et al. Conservation of the sequence and temporal expression of let‐7 heterochronic regulatory RNA. Nature. 2000;408(6808):86–89. [DOI] [PubMed] [Google Scholar]
- 35. Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, Ruvkun G. The lin‐41 RBCC gene acts in the C. elegans heterochronic pathway between the let‐7 regulatory RNA and the LIN‐29 transcription factor. Mol Cell. 2000;5(4):659–669. [DOI] [PubMed] [Google Scholar]
- 36. Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA‐interference enzyme Dicer in the maturation of the let‐7 small temporal RNA. Science. 2001;293(5531):834–838. [DOI] [PubMed] [Google Scholar]
- 37. Grishok A, Pasquinelli AE, Conte D, et al. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell. 2001;106(1):23–34. [DOI] [PubMed] [Google Scholar]
- 38. Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans . Genes Dev. 2001;15(20):2654–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans . Science. 2001;294(5543):862–864. [DOI] [PubMed] [Google Scholar]
- 40. Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans . Science. 2001;294(5543):858–862. [DOI] [PubMed] [Google Scholar]
- 41. Lagos‐Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–858. [DOI] [PubMed] [Google Scholar]
- 42. Bernstein E, Kim SY, Carmell MA, et al. Dicer is essential for mouse development. Nature Genet. 2003;35(3):215–217. [DOI] [PubMed] [Google Scholar]
- 43. Yekta S, Shih IH, Bartel DP. MicroRNA‐directed cleavage of HOXB8 mRNA. Science. 2004;304(5670):594–596. [DOI] [PubMed] [Google Scholar]
- 44. Lim LP, Lau NC, Garrett‐Engele P, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433(7027):769–773. [DOI] [PubMed] [Google Scholar]
- 45. Sood P, Krek A, Zavolan M, Macino G, Rajewsky N. Cell‐type‐specific signatures of microRNAs on target mRNA expression. Proc Natl Acad Sci USA. 2006;103(8):2746–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grimson A, Farh KK, Johnston WK, Garrett‐Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gu S, Jin L, Zhang F, Sarnow P, Kay MA. Biological basis for restriction of microRNA targets to the 3ʹ untranslated region in mammalian mRNAs. Nat Struct Mol Biol. 2009;16(2):144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down‐regulation of micro‐RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99(24):15524–15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101(9):2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Calin GA, Ferracin M, Cimmino A, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353(17):1793–1801. [DOI] [PubMed] [Google Scholar]
- 51. Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let‐7 microRNA family. Cell. 2005;120(5):635–647. [DOI] [PubMed] [Google Scholar]
- 52. Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–838. [DOI] [PubMed] [Google Scholar]
- 53. Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9(3):189–198. [DOI] [PubMed] [Google Scholar]
- 54. Mackiewicz M, Huppi K, Pitt JJ, Dorsey TH, Ambs S, Caplen NJ. Identification of the receptor tyrosine kinase AXL in breast cancer as a target for the human miR‐34a microRNA. Breast Cancer Res Treat. 2011;130(2):663–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci. 2001;114(24):4557–4565. [DOI] [PubMed] [Google Scholar]
- 56. Elbashir SM, Harborth J, Weber K, Tuschl T. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods. 2002;26(2):199–213. [DOI] [PubMed] [Google Scholar]
- 57. Paddison PJ, Caudy AA, Hannon GJ. Stable suppression of gene expression by RNAi in mammalian cells. Proc Natl Acad Sci USA. 2002;99(3):1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296(5567):550–553. [DOI] [PubMed] [Google Scholar]
- 59. McManus MT, Petersen CP, Haines BB, Chen J, Sharp PA. Gene silencing using micro‐RNA designed hairpins. RNA. 2002;8(6):842–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Birmingham A, Anderson EM, Reynolds A, et al. 3ʹ UTR seed matches, but not overall identity, are associated with RNAi off‐targets. Nat Methods. 2006;3(3):199–204. [DOI] [PubMed] [Google Scholar]
- 61. Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Gene Dev. 2003;17(4):438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18(5):504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115(2):209–216. [DOI] [PubMed] [Google Scholar]
- 64. Jackson AL, Bartz SR, Schelter J, et al. Expression profiling reveals off‐target gene regulation by RNAi. Nat Biotechnol. 2003;21(6):635–637. [DOI] [PubMed] [Google Scholar]
- 65. Jackson AL, Burchard J, Leake D, et al. Position‐specific chemical modification of siRNAs reduces "off‐target" transcript silencing. RNA. 2006;12(7):1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. [DOI] [PubMed] [Google Scholar]
- 67. Semizarov D, Frost L, Sarthy A, Kroeger P, Halbert DN, Fesik SW. Specificity of short interfering RNA determined through gene expression signatures. Proc Natl Acad Sci USA. 2003;100(11):6347–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Scacheri PC, Rozenblatt‐Rosen O, Caplen NJ, et al. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci USA. 2004;101(7):1892–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22(3):326–330. [DOI] [PubMed] [Google Scholar]
- 70. Martin SE, Caplen NJ. Mismatched siRNAs downregulate mRNAs as a function of target site location. FEBS Lett. 2006;580(15):3694–3698. [DOI] [PubMed] [Google Scholar]
- 71. Martin SE, Jones TL, Thomas CL, et al. Multiplexing siRNAs to compress RNAi‐based screen size in human cells. Nucleic Acids Res. 2007;35(8):e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Berns K, Hijmans EM, Mullenders J, et al. A large‐scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428(6981):431–437. [DOI] [PubMed] [Google Scholar]
- 73. Paddison PJ, Silva JM, Conklin DS, et al. A resource for large‐scale RNA‐interference‐based screens in mammals. Nature. 2004;428(6981):427–431. [DOI] [PubMed] [Google Scholar]
- 74. Echeverri CJ, Beachy PA, Baum B, et al. Minimizing the risk of reporting false positives in large‐scale RNAi screens. Nat Methods. 2006;3(10):777–779. [DOI] [PubMed] [Google Scholar]
- 75. Lin A, Giuliano CJ, Palladino A, et al. Off‐target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med. 2019;11(509):eaaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lum L, Yao S, Mozer B, et al. Identification of Hedgehog pathway components by RNAi in Drosophila cultured cells. Science. 2003;299(5615):2039–2045. [DOI] [PubMed] [Google Scholar]
- 77. Jacob LS, Wu X, Dodge ME, et al. Genome‐wide RNAi screen reveals disease‐associated genes that are common to Hedgehog and Wnt signaling. Sci Signal. 2011;4(157):ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. DasGupta R, Kaykas A, Moon RT, Perrimon N. Functional genomic analysis of the Wnt‐wingless signaling pathway. Science. 2005;308(5723):826–833. [DOI] [PubMed] [Google Scholar]
- 79. Tang W, Dodge M, Gundapaneni D, Michnoff C, Roth M, Lum L. A genome‐wide RNAi screen for Wnt/beta‐catenin pathway components identifies unexpected roles for TCF transcription factors in cancer. Proc Natl Acad Sci USA. 2008;105(28):9697–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Major MB, Roberts BS, Berndt JD, et al. New regulators of Wnt/beta‐catenin signaling revealed by integrative molecular screening. Sci Signal. 2008;1(45):ra12. [DOI] [PubMed] [Google Scholar]
- 81. Firestein R, Bass AJ, Kim SY, et al. CDK8 is a colorectal cancer oncogene that regulates beta‐catenin activity. Nature. 2008;455(7212):547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Camps J, Pitt JJ, Emons G, et al. Genetic amplification of the NOTCH modulator LNX2 upregulates the WNT/beta‐catenin pathway in colorectal cancer. Cancer Res. 2013;73(6):2003–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rosenbluh J, Nijhawan D, Cox AG, et al. β‐Catenin‐driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151(7):1457–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Steinhart Z, Pavlovic Z, Chandrashekhar M, et al. Genome‐wide CRISPR screens reveal a Wnt‐FZD5 signaling circuit as a druggable vulnerability of RNF43‐mutant pancreatic tumors. Nat Med. 2017;23(1):60–68. [DOI] [PubMed] [Google Scholar]
- 85. Mis M, O'Brien S, Steinhart Z, et al. IPO11 mediates betacatenin nuclear import in a subset of colorectal cancers. J Cell Biol. 2020;219(2):e201903017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF‐kappaB. Nature. 2003;424(6950):797–801. [DOI] [PubMed] [Google Scholar]
- 87. Oosterkamp HM, Neering H, Nijman SMB, et al. An evaluation of the efficacy of topical application of salicylic acid for the treatment of familial cylindromatosis. Br J Dermatol. 2006;155(1):182–185. [DOI] [PubMed] [Google Scholar]
- 88. Ngo VN, Davis RE, Lamy L, et al. A loss‐of‐function RNA interference screen for molecular targets in cancer. Nature. 2006;441(7089):106–110. [DOI] [PubMed] [Google Scholar]
- 89. Ngo VN, Young RM, Schmitz R, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Phelan JD, Young RM, Webster DE, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature. 2018;560(7718):387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dohjima T, Lee NS, Li H, Ohno T, Rossi JJ. Small interfering RNAs expressed from a Pol III promoter suppress the EWS/Fli‐1 transcript in an Ewing sarcoma cell line. Mol Ther. 2003;7(6):811–816. [DOI] [PubMed] [Google Scholar]
- 92. Chansky HA, Barahmand‐Pour F, Mei Q, et al. Targeting of EWS/FLI‐1 by RNA interference attenuates the tumor phenotype of Ewing's sarcoma cells in vitro. J Orthop Res. 2004;22(4):910–917. [DOI] [PubMed] [Google Scholar]
- 93. Hu‐Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ. Sequence‐specific knockdown of EWS‐FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing's sarcoma. Cancer Res. 2005;65(19):8984–8992. [DOI] [PubMed] [Google Scholar]
- 94. Matsunobu T, Tanaka K, Nakamura T, et al. The possible role of EWS‐Fli1 in evasion of senescence in Ewing family tumors. Cancer Res. 2006;66(2):803–811. [DOI] [PubMed] [Google Scholar]
- 95. Takigami I, Ohno T, Kitade Y, et al. Synthetic siRNA targeting the breakpoint of EWS/Fli‐1 inhibits growth of Ewing sarcoma xenografts in a mouse model. Int J Cancer. 2011;128(1):216–226. [DOI] [PubMed] [Google Scholar]
- 96. Chaturvedi A, Hoffman LM, Welm AL, Lessnick SL, Beckerle MC. The EWS/FLI oncogene drives changes in cellular morphology, adhesion, and migration in Ewing sarcoma. Genes Cancer. 2012;3(2):102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Prieur A, Tirode F, Cohen P, Delattre O. EWS/FLI‐1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin‐like growth factor binding protein 3. Mol Cell Biol. 2004;24(16):7275–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Siligan C, Ban J, Bachmaier R, et al. EWS‐FLI1 target genes recovered from Ewing's sarcoma chromatin. Oncogene. 2005;24(15):2512–2524. [DOI] [PubMed] [Google Scholar]
- 99. Kinsey M, Smith R, Lessnick SL. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing's sarcoma. Mol Cancer Res. 2006;4(11):851–859. [DOI] [PubMed] [Google Scholar]
- 100. Owen LA, Lessnick SL. Identification of target genes in their native cellular context: an analysis of EWS/FLI in Ewing's sarcoma. Cell Cycle. 2006;5(18):2049–2053. [DOI] [PubMed] [Google Scholar]
- 101. Smith R, Owen LA, Trem DJ, et al. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing's sarcoma. Cancer Cell. 2006;9(5):405–416. [DOI] [PubMed] [Google Scholar]
- 102. Owen LA, Kowalewski AA, Lessnick SL. EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing's sarcoma. PLOS One. 2008;3(4):e1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Stoll G, Surdez D, Tirode F, et al. Systems biology of Ewing sarcoma: a network model of EWS‐FLI1 effect on proliferation and apoptosis. Nucleic Acids Res. 2013;41(19):8853–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tomazou EM, Sheffield NC, Schmidl C, et al. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS‐FLI1. Cell Rep. 2015;10(7):1082–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sen N, Cross AM, Lorenzi PL, et al. EWS‐FLI1 reprograms the metabolism of Ewing sarcoma cells via positive regulation of glutamine import and serine‐glycine biosynthesis. Mol Carcinog. 2018;57(10):1342–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Aynaud MM, Mirabeau O, Gruel N, et al. Transcriptional programs define intratumoral heterogeneity of Ewing sarcoma at single‐cell resolution. Cell Rep. 2020;30(6):1767–1779. [DOI] [PubMed] [Google Scholar]
- 107. Grohar PJ, Kim S, Rangel Rivera GO, et al. Functional genomic screening reveals splicing of the EWS‐FLI1 fusion transcript as a vulnerability in Ewing sarcoma. Cell Rep. 2016;14(3):598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Neckles C, Boer RE, Aboreden N, et al. HNRNPH1‐dependent splicing of a fusion oncogene reveals a targetable RNA G‐quadruplex interaction. RNA. 2019;25(12):1731–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Levine AJ. p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer. 2020;20(8):471–480. [DOI] [PubMed] [Google Scholar]
- 110. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Brummelkamp TR, Fabius AWM, Mullenders J, et al. An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat Chem Biol. 2006;2(4):202–206. [DOI] [PubMed] [Google Scholar]
- 112. Hemann MT, Fridman JS, Zilfou JT, et al. An epi‐allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet. 2003;33(3):396–400. [DOI] [PubMed] [Google Scholar]
- 113. Perry JA, Kiezun A, Tonzi P, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci USA. 2014;111(51):E5564–E5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chen X, Bahrami A, Pappo A, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7(1):104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS‐targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19:533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kolfschoten IGM, van Leeuwen B, Berns K, et al. A genetic screen identifies PITX1 as a suppressor of RAS activity and tumorigenicity. Cell. 2005;121(6):849–858. [DOI] [PubMed] [Google Scholar]
- 117. Calvisi DF, Ladu S, Conner EA, et al. Inactivation of Ras GTPase‐activating proteins promotes unrestrained activity of wild‐type Ras in human liver cancer. J Hepatol. 2011;54(2):311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR. An elaborate pathway required for Ras‐mediated epigenetic silencing. Nature. 2007;449(7165):1073–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wajapeyee N, Malonia SK, Palakurthy RK, Green MR. Oncogenic RAS directs silencing of tumor suppressor genes through ordered recruitment of transcriptional repressors. Genes Dev. 2013;27(20):2221–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sarthy AV, Morgan‐Lappe SE, Zakula D, et al. Survivin depletion preferentially reduces the survival of activated K‐Ras‐transformed cells. Mol Cancer Ther. 2007;6(1):269–276. [DOI] [PubMed] [Google Scholar]
- 121. Luo J, Emanuele MJ, Li D, et al. A genome‐wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Scholl C, Fröhling S, Dunn IF, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137(5):821–834. [DOI] [PubMed] [Google Scholar]
- 123. Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS‐driven cancers require TBK1. Nature. 2009;462(7269):108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kaelin WG, Jr. Molecular biology. Use and abuse of RNAi to study mammalian gene function. Science. 2012;337(6093):421–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Lee CS, Lee LC, Yuan TL, et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci USA. 2019;116(10):4508–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yuan TL, Fellmann C, Lee CS, et al. Development of siRNA payloads to target KRAS‐mutant cancer. Cancer Discov. 2014;4(10):1182–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kinsey CG, Camolotto SA, Boespflug AM, et al. Protective autophagy elicited by RAF‐‐>MEK‐‐>ERK inhibition suggests a treatment strategy for RAS‐driven cancers. Nat Med. 2019;25(4):620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Bryant KL, Stalnecker CA, Zeitouni D, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25(4):628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tsherniak A, Vazquez F, Montgomery PG, et al. Defining a cancer dependency map. Cell. 2017;170(3):564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. McDonald ER, de Weck A, Schlabach MR, et al. Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large‐scale, deep RNAi screening. Cell. 2017;170(3):577–592. [DOI] [PubMed] [Google Scholar]
- 131. Wang T, Yu H, Hughes NW, et al. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell. 2017;168(5):890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Martin TD, Cook DR, Choi MY, Li MZ, Haigis KM, Elledge SJ. A role for mitochondrial translation in promotion of viability in K‐Ras mutant cells. Cell Rep. 2017;20(2):427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Schmidt EE, Pelz O, Buhlmann S, Kerr G, Horn T, Boutros M. GenomeRNAi: a database for cell‐based and in vivo RNAi phenotypes, 2013 update. Nucleic Acids Res. 2013;41(Database issue):D1021–D1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Iorns E, Lord CJ, Grigoriadis A, et al. Integrated functional, gene expression and genomic analysis for the identification of cancer targets. PLOS One. 2009;4(4):e5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Murrow LM, Garimella SV, Jones TL, Caplen NJ, Lipkowitz S. Identification of WEE1 as a potential molecular target in cancer cells by RNAi screening of the human tyrosine kinome. Breast Cancer Res Treat. 2010;122(2):347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Brough R, Frankum JR, Sims D, et al. Functional viability profiles of breast cancer. Cancer Discov. 2011;1(3):260–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Schlabach MR, Luo J, Solimini NL, et al. Cancer proliferation gene discovery through functional genomics. Science. 2008;319(5863):620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Silva JM, Marran K, Parker JS, et al. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science. 2008;319(5863):617–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Luo B, Cheung HW, Subramanian A, et al. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci USA. 2008;105(51):20380–20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Moffat J, Grueneberg DA, Yang X, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high‐content screen. Cell. 2006;124(6):1283–1298. [DOI] [PubMed] [Google Scholar]
- 141. Root DE, Hacohen N, Hahn WC, Lander ES, Sabatini DM. Genome‐scale loss‐of‐function screening with a lentiviral RNAi library. Nat Methods. 2006;3(9):715–719. [DOI] [PubMed] [Google Scholar]
- 142. Cheung HW, Du J, Boehm JS, et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non‐small cell lung cancers. Cancer Discov. 2011;1(7):608–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Douglas Zhang X, Yang XC, Chung N, et al. Robust statistical methods for hit selection in RNA interference high‐throughput screening experiments. Pharmacogenomics. 2006;7(3):299–309. [DOI] [PubMed] [Google Scholar]
- 144. König R, Chiang C, Tu BP, et al. A probability‐based approach for the analysis of large‐scale RNAi screens. Nat Methods. 2007;4(10):847–849. [DOI] [PubMed] [Google Scholar]
- 145. Marine S, Bahl A, Ferrer M, Buehler E. Common seed analysis to identify off‐target effects in siRNA screens. J Biomol Screen. 2012;17(3):370–378. [DOI] [PubMed] [Google Scholar]
- 146. Buehler E, Khan AA, Marine S, et al. siRNA off‐target effects in genome‐wide screens identify signaling pathway members. Sci Rep. 2012;2:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Shao DD, Tsherniak A, Gopal S, et al. ATARiS: computational quantification of gene suppression phenotypes from multisample RNAi screens. Genome Res. 2013;23(4):665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. McFarland JM, Ho ZV, Kugener G, et al. Improved estimation of cancer dependencies from large‐scale RNAi screens using model‐based normalization and data integration. Nat Commun. 2018;9(1):4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Meyers RM, Bryan JG, McFarland JM, et al. Computational correction of copy number effect improves specificity of CRISPR‐Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49(12):1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Aguirre AJ, Meyers RM, Weir BA, et al. Genomic copy number dictates a gene‐independent cell response to CRISPR/Cas9 targeting. Cancer Discov. 2016;6(8):914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Munoz DM, Cassiani PJ, Li L, et al. CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false‐positive hits for highly amplified genomic regions. Cancer Discov. 2016;6(8):900–913. [DOI] [PubMed] [Google Scholar]
- 152. Cheung HW, Cowley GS, Weir BA, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage‐specific dependencies in ovarian cancer. Proc Natl Acad Sci USA. 2011;108(30):12372–12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Marcotte R, Brown KR, Suarez F, et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012;2(2):172–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Marcotte R, Sayad A, Brown KR, et al. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell. 2016;164(1–2):293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Hoffman GR, Rahal R, Buxton F, et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1‐deficient cancers. Proc Natl Acad Sci USA. 2014;111(8):3128–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Cowley GS, Weir BA, Vazquez F, et al. Parallel genome‐scale loss of function screens in 216 cancer cell lines for the identification of context‐specific genetic dependencies. Sci Data. 2014;1:140035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ghandi M, Huang FW, Jané‐Valbuena J, et al. Next‐generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569(7757):503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Corsello SM, Nagari RT, Spangler RD, et al. Discovering the anti‐cancer potential of non‐oncology drugs by systematic viability profiling. Nat Cancer. 2020;1(2):235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Dempster JM, Pacini C, Pantel S, et al. Agreement between two large pan‐cancer CRISPR‐Cas9 gene dependency data sets. Nat Commun. 2019;10(1):5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Pacini C, Dempster JM, Gonçalves E, et al. Integrated cross‐study datasets of genetic dependencies in cancer. bioRxiv. 2020:2020.2005.2022.110247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Timmons L, Fire A. Specific interference by ingested dsRNA. Nature. 1998;395(6705):854. [DOI] [PubMed] [Google Scholar]
- 162. Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans . Gene. 2001;263(1–2):103–112. [DOI] [PubMed] [Google Scholar]
- 163. Tabara H, Grishok A, Mello CC. RNAi in C. elegans: soaking in the genome sequence. Science. 1998;282(5388):430–431. [DOI] [PubMed] [Google Scholar]
- 164. Czauderna F. Functional studies of the PI(3)‐kinase signalling pathway employing synthetic and expressed siRNA. Nucleic Acids Res. 2003;31(2):670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Spankuch B, Matthess Y, Knecht R, Zimmer B, Kaufmann M, Strebhardt K. Cancer inhibition in nude mice after systemic application of U6 promoter‐driven short hairpin RNAs against PLK1. J Natl Cancer Inst. 2004;96(11):862–872. [DOI] [PubMed] [Google Scholar]
- 166. Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. CEACAM6 gene silencing impairs anoikis resistance and in vivo metastatic ability of pancreatic adenocarcinoma cells. Oncogene. 2004;23(2):465–473. [DOI] [PubMed] [Google Scholar]
- 167. Weber J, Braun CJ, Saur D, Rad R. In vivo functional screening for systems‐level integrative cancer genomics. Nat Rev Cancer. 2020;20:573–593. [DOI] [PubMed] [Google Scholar]
- 168. Zender L, Xue W, Zuber J, et al. An oncogenomics‐based in vivo RNAi screen identifies tumor suppressors in liver. Cancer Cell. 2008;135(5):852–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Bric A, Miething C, Bialucha CU, et al. Functional identification of tumor‐suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell. 2009;16(4):324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Cheng J, Demeulemeester J, Wedge DC, et al. Pan‐cancer analysis of homozygous deletions in primary tumours uncovers rare tumour suppressors. Nat Commun. 2017;8(1):1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Meacham CE, Ho EE, Dubrovsky E, Gertler FB, Hemann MT. In vivo RNAi screening identifies regulators of actin dynamics as key determinants of lymphoma progression. Nat Genet. 2009;41(10):1133–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Meacham CE, Lawton LN, Soto‐Feliciano YM, et al. A genome‐scale in vivo loss‐of‐function screen identifies Phf6 as a lineage‐specific regulator of leukemia cell growth. Genes Dev. 2015;29(5):483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Iorns E, Ward TM, Dean S, et al. Whole genome in vivo RNAi screening identifies the leukemia inhibitory factor receptor as a novel breast tumor suppressor. Breast Cancer Res Treat. 2012;135(1):79–91. [DOI] [PubMed] [Google Scholar]
- 174. Johnson RW, Finger EC, Olcina MM, et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat Cell Biol. 2016;18(10):1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Beronja S, Livshits G, Williams S, Fuchs E. Rapid functional dissection of genetic networks via tissue‐specific transduction and RNAi in mouse embryos. Nat Med. 2010;16(7):821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Beronja S, Janki P, Heller E, et al. RNAi screens in mice identify physiological regulators of oncogenic growth. Nature. 2013;501(7466):185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Schramek D, Sendoel A, Segal JP, et al. Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science. 2014;343(6168):309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Bender A, Pringle JR. Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae . Mol Cell Biol. 1991;11(3):1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Hennessy KM, Lee A, Chen E, Botstein D. A group of interacting yeast DNA replication genes. Genes Dev. 1991;5(6):958–969. [DOI] [PubMed] [Google Scholar]
- 180. Lucchesi JC. Synthetic lethality and semi‐lethality among functionally related mutants of Drosophila melanfgaster . Genetics. 1968;59(1):37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278(5340):1064–1068. [DOI] [PubMed] [Google Scholar]
- 182. Kryukov GV, Wilson FH, Ruth JR, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. 2016;351(6278):1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Mavrakis KJ, McDonald ER 3rd, Schlabach MR, et al. Disordered methionine metabolism in MTAP/CDKN2A‐deleted cancers leads to dependence on PRMT5. Science. 2016;351(6278):1208–1213. [DOI] [PubMed] [Google Scholar]
- 184. Marjon K, Cameron MJ, Quang P, et al. MTAP deletions in cancer create vulnerability to targeting of the MAT2A/PRMT5/RIOK1 axis. Cell Rep. 2016;15(3):574–587. [DOI] [PubMed] [Google Scholar]
- 185. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13(1):37–50. [DOI] [PubMed] [Google Scholar]
- 186. Helming KC, Wang X, Wilson BG, et al. ARID1B is a specific vulnerability in ARID1A‐mutant cancers. Nat Med. 2014;20(3):251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Bitler BG, Aird KM, Garipov A, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A‐mutated cancers. Nat Med. 2015;21(3):231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Bartz SR, Zhang Z, Burchard J, et al. Small interfering RNA screens reveal enhanced cisplatin cytotoxicity in tumor cells having both BRCA network and TP53 disruptions. Mol Cell Biol. 2006;26(24):9377–9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Whitehurst AW, Bodemann BO, Cardenas J, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446(7137):815–819. [DOI] [PubMed] [Google Scholar]
- 190. Levine EG, Cirrincione CT, Szatrowski TP, Canellos G, Norton L, Henderson IC. Phase II trial of topotecan in advanced breast cancer: a Cancer and Leukemia Group B study. Am J Clin Oncol. 1999;22(3):218–222. [DOI] [PubMed] [Google Scholar]
- 191. Zhang YW, Jones TL, Martin SE, Caplen NJ, Pommier Y. Implication of checkpoint kinase‐dependent up‐regulation of ribonucleotide reductase R2 in DNA damage response. J Biol Chem. 2009;284(27):18085–18095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. E. Martin S, Wu ZH, Gehlhaus K, et al. RNAi screening identifies TAK1 as a potential target for the enhanced efficacy of topoisomerase inhibitors. Curr Cancer Drug Targets. 2011;11(8):976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Josse R, Martin SE, Guha R, et al. ATR inhibitors VE‐821 and VX‐970 sensitize cancer cells to topoisomerase I inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res. 2014;74(23):6968–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Thomas A, Redon CE, Sciuto L, et al. Phase I study of ATR inhibitor M6620 in combination with topotecan in patients with advanced solid tumors. J Clin Oncol. 2018;36(16):1594–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Caplen NJ, Zheng Z, Falgout B, Morgan RA. Inhibition of viral gene expression and replication in mosquito cells by dsRNA‐triggered RNA interference. Mol Ther. 2002;6(2):243–251. [DOI] [PubMed] [Google Scholar]
- 196. Novina CD, Murray MF, Dykxhoorn DM, et al. siRNA‐directed inhibition of HIV‐1 infection. Nat Med. 2002;8(7):681–686. [DOI] [PubMed] [Google Scholar]
- 197. Caplen NJ, Taylor JP, Statham VS, Tanaka F, Fire A, Morgan RA. Rescue of polyglutamine‐mediated cytotoxicity by double‐stranded RNA‐mediated RNA interference. Hum Mol Genet. 2002;11(2):175–184. [DOI] [PubMed] [Google Scholar]
- 198. Koldehoff M, Steckel NK, Beelen DW, Elmaagacli AH. Therapeutic application of small interfering RNA directed against BCR‐ABL transcripts to a patient with Imatinib‐resistant chronic myeloid leukaemia. Clin Exp Med. 2007;7(2):47–55. [DOI] [PubMed] [Google Scholar]
- 199. Adams D, Gonzalez‐Duarte A, O'Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. [DOI] [PubMed] [Google Scholar]
- 200. Balwani M, Sardh E, Ventura P, et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N Engl J Med. 2020;382(24):2289–2301. [DOI] [PubMed] [Google Scholar]
- 201. Raal FJ, Kallend D, Ray KK, et al. Inclisiran for the treatment of heterozygous familial hypercholesterolemia. N Engl J Med. 2020;382(16):1520–1530. [DOI] [PubMed] [Google Scholar]
- 202. Ray KK, Wright RS, Kallend D, et al. Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N Engl J Med. 2020;382(16):1507–1519. [DOI] [PubMed] [Google Scholar]
- 203. Fitzgerald K, Frank‐Kamenetsky M, Shulga‐Morskaya S, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single‐blind, placebo‐controlled, phase 1 trial. Lancet. 2014;383(9911):60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Fitzgerald K, White S, Borodovsky A, et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N Engl J Med. 2017;376(1):41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Barata P, Sood AK, Hong DS. RNA‐targeted therapeutics in cancer clinical trials: current status and future directions. Cancer Treat Rev. 2016;50:35–47. [DOI] [PubMed] [Google Scholar]
- 206. Lieberman J. Tapping the RNA world for therapeutics. Nat Struct Mol Biol. 2018;25(5):357‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi‐based therapeutics. Nat Rev Drug Discov. 2019;18(6):421–446. [DOI] [PubMed] [Google Scholar]
- 208. Bajan S, Hutvagner G. RNA‐based therapeutics: from antisense oligonucleotides to miRNAs. Cells. 2020;9(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Crooke ST, Witztum JL, Bennett CF, Baker BF. RNA‐targeted therapeutics. Cell Metab. 2019;29(2):501. [DOI] [PubMed] [Google Scholar]
- 210. Fleming JB, Shen GL, Holloway SE, Davis M, Brekken RA. Molecular consequences of silencing mutant K‐ras in pancreatic cancer cells: justification for K‐ras‐directed therapy. Mol Cancer Res. 2005;3(7):413–423. [DOI] [PubMed] [Google Scholar]
- 211. Rejiba S, Wack S, Aprahamian M, Hajri A. K‐ras oncogene silencing strategy reduces tumor growth and enhances gemcitabine chemotherapy efficacy for pancreatic cancer treatment. Cancer Sci. 2007;98(7):1128–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Zhang YA, Nemunaitis J, Samuel SK, Chen P, Shen Y, Tong AW. Antitumor activity of an oncolytic adenovirus‐delivered oncogene small interfering RNA. Cancer Res. 2006;66(19):9736–9743. [DOI] [PubMed] [Google Scholar]
- 213. Sunaga N, Shames DS, Girard L, et al. Knockdown of oncogenic KRAS in non‐small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther. 2011;10(2):336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Zorde Khvalevsky E, Gabai R, Rachmut IH, et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc Natl Acad Sci USA. 2013;110(51):20723–20728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Golan T, Khvalevsky EZ, Hubert A, et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget. 2015;6(27):24560–24570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Varghese AM, Ang C, Dimaio CJ, et al. A phase II study of siG12D‐LODER in combination with chemotherapy in patients with locally advanced pancreatic cancer (PROTACT). J Clin Oncol. 2020;38(15_suppl):TPS4672. [Google Scholar]
- 217. Landen CN Jr., Chavez‐Reyes A, Bucana C, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65(15):6910–6918. [DOI] [PubMed] [Google Scholar]
- 218. Landen CN, Merritt WM, Mangala LS, et al. Intraperitoneal delivery of liposomal siRNA for therapy of advanced ovarian cancer. Cancer Biol Ther. 2006;5(12):1708–1713. [DOI] [PubMed] [Google Scholar]
- 219. Tanaka T, Mangala LS, Vivas‐Mejia PE, et al. Sustained small interfering RNA delivery by mesoporous silicon particles. Cancer Res. 2010;70(9):3687–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Shen H, Rodriguez‐Aguayo C, Xu R, et al. Enhancing chemotherapy response with sustained EphA2 silencing using multistage vector delivery. Clin Cancer Res. 2013;19(7):1806–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Nishimura M, Jung EJ, Shah MY, et al. Therapeutic synergy between microRNA and siRNA in ovarian cancer treatment. Cancer Discov. 2013;3(11):1302–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Wagner MJ, Mitra R, McArthur MJ, et al. Preclinical mammalian safety studies of EPHARNA (DOPC nanoliposomal EphA2‐targeted siRNA). Mol Cancer Ther. 2017;16(6):1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Das M, Musetti S, Huang L. RNA interference‐based cancer drugs: the roadblocks, and the "Delivery" of the promise. Nucleic Acid Ther. 2019;29(2):61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR‐Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020;38(7):824–844. [DOI] [PubMed] [Google Scholar]
- 225. Haley B, Roudnicky F. Functional genomics for cancer drug target discovery. Cancer Cell. 2020;38(1):31–43. [DOI] [PubMed] [Google Scholar]
- 226. Hanna RE, Doench JG. Design and analysis of CRISPR‐Cas experiments. Nat Biotechnol. 2020;38(7):813–823. [DOI] [PubMed] [Google Scholar]
- 227. Smargon AA, Shi YJ, Yeo GW. RNA‐targeting CRISPR systems from metagenomic discovery to transcriptomic engineering. Nat Cell Biol. 2020;22(2):143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Yin H, Xue W, Anderson DG. CRISPR‐Cas: a tool for cancer research and therapeutics. Nat Rev Clin Oncol. 2019;16(5):281–295. [DOI] [PubMed] [Google Scholar]
- 229. Zhan T, Rindtorff N, Betge J, Ebert MP, Boutros M. CRISPR/Cas9 for cancer research and therapy. Semin Cancer Biol. 2019;55:106–119. [DOI] [PubMed] [Google Scholar]
- 230. Huang CH, Lee KC, Doudna JA. Applications of CRISPR‐Cas enzymes in cancer therapeutics and detection. Trends Cancer. 2018;4(7):499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Fellmann C, Gowen BG, Lin PC, Doudna JA, Corn JE. Cornerstones of CRISPR‐Cas in drug discovery and therapy. Nat Rev Drug Discov. 2017;16(2):89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Verma R, Mohl D, Deshaies RJ. Harnessing the power of proteolysis for targeted protein inactivation. Mol Cell. 2020;77(3):446–460. [DOI] [PubMed] [Google Scholar]