

Abstract
Objective
Anti–topoisomerase I (anti–topo I) autoantibodies in systemic sclerosis (SSc) are associated with diffuse skin involvement and interstitial lung fibrosis. Thus far, however, the relationship between anti–topo I antibody response and disease course has not yet been fully evaluated. This study was undertaken to gain insight into the association between characteristics of the anti–topo I antibody response and clinical disease course in SSc patients positive for anti–topo I antibodies.
Methods
Levels of anti–topo I IgG, anti–topo I IgM, and anti–topo I IgA were assessed in consecutive serum samples obtained from patients at baseline who were positive for anti–topo I IgG in the Leiden Combined Care In Systemic Sclerosis (CCISS) cohort. One‐year disease progression was defined by a relevant increase in modified Rodnan skin thickness score (MRSS), decline in pulmonary function, development of digital ulcers, renal crisis, and pulmonary hypertension, and/or mortality. Validation was performed in SSc patients who were positive for anti–topo I from the Oslo University Hospital and University Hospital Zurich.
Results
Of the 103 patients with anti–topo I IgG in the CCISS cohort, clinical data were available to assess 1‐year disease progression in 81 patients. Of these 81 patients, 23 (28%) had disease progression. At baseline, patients with disease progression were significantly more often anti–topo I IgM–positive than those who did not experience disease progression (21 [91%] of 23 versus 33 [57%] of 58; P < 0.01). This finding was confirmed in the independent validation samples.
Conclusion
In SSc patients who were anti–topo I IgG–positive, presence of anti–topo I IgM, which might be considered as a surrogate for an ongoing autoreactive B cell immune response, is associated with disease progression.
INTRODUCTION
Anti–topoisomerase I (anti–topo I) antibodies are highly specific for systemic sclerosis (SSc) (1). Individuals with isolated Raynaud’s phenomenon have an increased risk of developing SSc when positive for anti–topo I antibodies (2), indicating the potential importance of the presence of anti–topo I antibodies in a preclinical phase. In established SSc, anti–topo I antibodies are associated with diffuse cutaneous SSc (dcSSc) and severe interstitial lung disease (ILD), and their presence indicates an unfavorable prognosis (3, 4, 5, 6, 7). This association with a typical clinical phenotype suggests that the immune response involved in anti–topo I antibody production may play a role in disease pathophysiology. The exact pathogenicity of anti–topo I antibodies, however, has not yet been elucidated.
In daily clinical practice, anti–topo I antibody–positive SSc is heterogeneous. Not all patients with anti–topo I antibodies demonstrate a severe disease course, and some patients experience only moderate skin and lung fibrosis (6, 8). Based on the hypothesis that topo I represents a candidate autoantigen in the pathogenesis of SSc, different groups have studied immunization with topo I in mouse models. These studies demonstrated that a specific antibody response can be induced, resulting in varying extents of fibrosis in the skin and lungs of immunized mice (9, 10).
Anti–topo I antibodies can be classified according to their immunoglobulin class or isotype as IgG, IgA, or IgM. In clinical practice, anti–topo I positivity is commonly based on the presence of anti–topo I antibodies of the IgG isotype. Previous small studies in SSc have shown that the levels of anti–topo I antibodies of either the IgG and IgA isotype correlated with the severity of skin disease (11, 12, 13). Loss of the anti–topo I antibody response has been associated with a favorable disease course in a small patient group (14). However, the relationship between anti–topo I isotype profile and anti–topo I isotype levels and disease course has not yet been fully evaluated in larger SSc cohorts. By taking advantage of our well‐described SSc cohort from whom comprehensive clinical data are collected annually, we investigated the association between the presence and levels of anti–topo I antibodies of the IgG, IgM, and IgA isotypes and disease course in anti–topo I IgG–positive SSc.
PATIENTS AND METHODS
Patient population
The Combined Care in Systemic Sclerosis (CCISS) cohort Leiden is a prospective cohort that started in April 2009 and includes all consecutive SSc patients evaluated at the Leiden University Medical Center (15).
As described previously (15), all patients in the cohort underwent annual extensive screening during a 1–2‐day health care program, including detailed physical examination, modified Rodnan skin thickness score (MRSS) assessment (16), laboratory testing (with autoantibody screening performed at baseline), pulmonary function test and, optionally, echocardiography (mandatory at baseline), Holter evaluation (mandatory at baseline), cardiopulmonary exercise tests (CPETs), and high‐resolution computed tomography (HRCT) (mandatory at baseline).
Patients were requested to complete the following questionnaires at every visit: the Scleroderma Health Assessment Questionnaire (SHAQ) (17), Short Form 36 (SF‐36) (18, 19), Mouth Handicap in Systemic Sclerosis (MHISS) scale (20, 21), EuroQol 5‐domain (EQ‐5D) (22, 23, 24), and Scleroderma Clinical Trial Consortium Gastrointestinal Tract Instrument 2.0 (SCTC GIT 2.0) (25, 26). Additionally, at every visit, serum samples were collected and stored in the Leiden Scleroderma Biobank. All patients who entered the cohort before September 24, 2016, and who were anti–topo I IgG antibody–positive were selected for the present study. Only patients who had a clinical SSc diagnosis at inclusion and fulfilled the 2013 American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) classification criteria for SSc (27) at any point during their disease course were evaluated.
Ethics approval for data collection was obtained from the local ethics committee (CME no. B16.037). Research was done without patient involvement, and all participants provided written informed consent. Data are available upon request from the corresponding author.
Disease progression
Clinical data were collected, with censoring on January 1, 2018. Progression of skin disease was defined as a ≥5‐point and ≥25% increase on the MRSS (28). Worsening of lung involvement was defined as follows: 1) a relative decline in the forced vital capacity (FVC) of ≥10%, with an FVC % predicted (FVC%) of <80% at follow‐up, or 2) a relative decline in the FVC ranging from ≥5% to <10%, together with either a ≥15% relative decline in the diffusing capacity for carbon monoxide (DLco), with a DLco % predicted (DLco%) of <80% at follow‐up, or an increase in lung involvement of >20% (as determined by HRCT) (29). Patients were categorized as either “disease progressors” or “disease nonprogressors” based on the presence or absence of any of the following features: progression of skin and/or lung disease, incident digital ulcers, and newly diagnosed myocardial involvement, scleroderma renal crisis, and/or pulmonary hypertension. In addition, patients were categorized as “disease progressors” if death had occurred during follow‐up. Use of aggressive immunosuppression in both disease progressors and disease nonprogressors was assessed, including hematopoietic stem cell transplantation (HSCT), cyclophosphamide, and mycophenolate mofetil.
Anti–topo I assay and measurements
Total anti–topo I antibody levels of the IgG, IgM, and IgA isotypes were measured in consecutive serum samples collected before January 1, 2017 and obtained from patients at baseline or during follow‐up by fluorescence enzyme‐linked immunosorbent assay (FELISA), using a Phadia250 system (ThermoFisher Scientific). If necessary, sera were diluted to obtain a reliable anti–topo I antibody isotype–specific level. For determination of anti–topo I IgG isotype positivity, a cutoff value of 7 AU/ml was specified by the manufacturer. For determination of anti–topo I IgA and anti–topo I IgM isotype positivity, no cutoff values were available from the manufacturer, and therefore we measured these anti–topo I isotypes in the serum of 51 controls without rheumatic disease, and determined the cutoff value to be the mean + 2 SD above the values in controls (432 AU/ml for anti–topo I IgM and 77 AU/ml for anti–topo I IgA). To evaluate the specificity of the assay, we measured the levels of anti–topo I IgG, IgM, and IgA isotypes in the serum of 5 SSc patients who were positive for antinuclear antibodies (ANAs) who lacked SSc‐specific antibodies and the serum of 5 SSc patients who were positive for anticentromere antibodies (ACAs). None of these patients were positive for any of the isotypes in the anti–topo I antibody assay.
Data validation
For validation of the main findings, baseline serum samples from anti–topo I IgG–positive patients from the Oslo University Hospital (30) and from the University Hospital Zurich (31) were tested for the presence and levels of anti–topo I antibodies of the IgG, IgM, and IgA isotypes using the same methodology described earlier. Baseline and follow‐up clinical data were also collected. At both centers, longitudinal data on SSc patients have been collected according to European Scleroderma Trials and Research (EUSTAR) recommendations (32). Details of these cohorts can be found elsewhere (30, 31). Collection and analysis of biomaterial and their clinical associations was approved by the Cantonal Ethics Committee in Switzerland (PB_2016‐02014 and BASEC‐Nr. 2018‐01873) and by the Data Protection Authority in Norway (no. 2006/119).
Statistical analysis
Descriptive statistics were used to clinically characterize the study population clinically. Contingency tables were evaluated by Fisher’s exact test, chi‐square test, or Mann‐Whitney test as appropriate. Correlations between isotype levels were assessed using Spearman’s correlation test. Disease progression over time was summarized with Kaplan‐Meier survival curves. To exclude potential bias conferred by the presence of anti–topo I IgM and/or anti–topo I IgA in patients negative for anti–topo I IgG using a cutoff value of mean + 4 SD above the value in controls, a sensitivity analysis was performed (Supplementary Figure 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41403/abstract). Additionally, robustness of the data was evaluated in a separate analysis also by using a different cutoff value for anti–topo I IgM positivity. Statistical analysis was performed using SPSS version 23.0 and GraphPad Prism 7. P values less than 0.05 were considered significant.
RESULTS
Baseline characteristics and anti–topo I isotype expression in the study population
In total, 103 patients who were anti–topo I IgG–positive patients from the CCISS cohort were included. Of these patients, a total of 333 samples were available (range 1–8 samples per patient). The cohort consisted of 70 female patients (68%), with a mean age of 53 years, and 48% had dcSSc (Table 1). At baseline, median disease duration since onset of first non–Raynaud’s symptom was 2.8 years. Patients were followed up clinically for a median of 3.4 years (ranging up to 8.4 years). All but 1 patient evaluated in the cohort were anti–topo I IgA antibody–positive at baseline. This patient also had a low level of anti–topo I IgG (24 AU/ml). At baseline, 65% of patients (67 of 103) were anti–topo I IgM–positive. Anti–topo I isotype levels at baseline were weakly correlated with each other (rs = 0.25 for anti–topo I IgG and anti–topo I IgM, rs = 0.30 for anti–topo I IgG and anti–topo I IgA, and rs = 0.45 for anti–topo I IgA and anti–topo I IgM [each P = <0.01]). None of the anti–topo I isotype levels were correlated with disease duration (Supplementary Figures 2 and 3, http://onlinelibrary.wiley.com/doi/10.1002/art.41403/abstract). Associations between baseline anti–topo I isotype levels and skin scores are presented in Figure 1; levels of anti–topo I IgG correlated with skin scores (rs = 0.41; P < 0.01), but other isotypes did not correlate with skin scores.
Table 1.
Baseline characteristics of all SSc patients in the study population who were positive for anti–topoisomerase I IgG*
|
All patients (n = 103) |
|
|---|---|
| Demographic characteristics | |
| Female sex | 70 (68) |
| Age, mean ± SD years | 53.0 ± 14.8 |
| Smoking (ever) | 50 (49) |
| Disease duration | |
| Since onset of first Raynaud’s symptom, median (IQR) years | 5.8 (2.1–13.4) |
| Since onset of first non‐Raynaud’s symptom, median (IQR) years | 2.8 (0.8–9.3) |
| Organ involvement | |
| Diffuse cutaneous SSc | 49 (48) |
| Modified Rodnan skin thickness score, median (IQR) | 6 (2–12) |
| FVC%, mean ± SD | 87 ± 27 |
| DLco%, mean ± SD | 63 ± 17 |
| History of renal crisis | 3 (3) |
| Digital ulcers | 14 (14) |
| Pulmonary hypertension | 5 (5) |
| History of immunosuppressive treatment† | |
| HSCT | 7 (7) |
| CYC (ever) | 24 (23) |
| MMF (ever) | 1 (1) |
Except where indicated otherwise, values are the number (%) of patients. SSc = systemic sclerosis; IQR = interquartile range; FVC% = forced vital capacity % predicted; DLco% = diffusing capacity for carbon monoxide % predicted.
Immunosuppressive treatment includes the use of hematopoietic stem cell transplantation (HSCT), cyclophosphamide (CYC), or mycophenolate mofetil (MMF).
Figure 1.
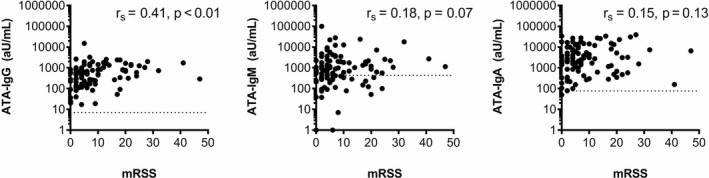
Correlation between baseline levels of anti–topoisomerase I antibody (ATA) IgG, IgM, and IgA and modified Rodnan skin thickness score (MRSS) in patients from the Leiden Combined Care in Systemic Sclerosis cohort (n = 103). Spearman’s correlation analyses indicated that only anti–topoisomerase I IgG levels were significantly correlated with MRSS scores.
During follow‐up, 12 patients died, with causes of death listed as the following: combined pulmonary and cardiac failure (n = 6), cardiac ischemia (n = 1), sepsis during preparation for HSCT (n = 1), gastrointestinal ischemia (n = 1), influenza (n = 1), multi‐organ failure during acute myeloid leukemia treatment (n = 1), and unclear causes (n = 1).
Loss and gain of anti–topo I IgG, IgA, and IgM isotypes over time
Change of isotype profile over time was assessed in 75 of the 103 patients (28 patients did not have follow‐up samples available) (Table 2 and Supplementary Figure 4, http://onlinelibrary.wiley.com/doi/10.1002/art.41403/abstract). Of these patients, 4 (5%) showed loss of anti–topo I IgG; all 4 patients were negative for anti–topo I IgM at baseline. Two of these patients were treated with intravenous cyclophosphamide prior to baseline sampling, 1 was treated with HSCT prior to baseline sampling, and 1 was treated with HSCT 3 months following baseline sampling. Three of these 4 patients were anti–topo I IgA–positive at baseline, and 2 of them also showed loss of anti–topo I IgA. In total, there were 4 patients (5%) who lost anti–topo I IgA over time, of whom 1 also lost anti–topo I IgM, but remained positive for anti–topo I IgG. During follow‐up, more changes were observed in the expression of anti–topo I IgM as compared to anti–topo I IgG and anti–topo IgA. Among the 45 patients who were positive for anti–topo I IgM at baseline, 14 (31%) lost positivity over follow‐up, and 3 (10%) of the 29 patients who were negative for anti–topo I IgM at baseline gained an anti–topo I IgM response over follow‐up.
Table 2.
Changes in the presence of anti–topo I isotypes in paired samples from 75 systemic sclerosis patients positive for anti–topo I IgG*
| Anti–topo I isotype status at baseline/last follow‐up visit† | ||||
|---|---|---|---|---|
| +/+ | +/− | −/− | −/+ | |
| Anti–topo I IgG | 71 | 4 | – | – |
| Anti–topo I IgM | 31 | 14 | 27 | 3 |
| Anti–topo I IgA | 70 | 4 | 1 | 0 |
Values are the number of patients. Anti–topo I = anti–topoisomerase I autoantibody.
Status of first available serum sample/status of last available serum sample.
Frequent disease progression in anti–topo I IgG–positive SSc patients who are also positive for anti–topo I IgM
To assess the association between anti–topo I isotype profile and disease progression, we used data from 81 patients with 1‐year clinical follow‐up data available. During the first year starting from sampling, none of these patients received HSCT, 16 patients were treated with cyclophosphamide, and 7 received mycophenolate mofetil.
In total, 23 patients showed disease progression according to predefined criteria, which consisted of the following: death (n = 4, which included combined pulmonary and cardiac failure in 3 patients and multi‐organ failure during acute myeloid leukemia treatment in 1 patient), progression of skin disease (n = 12), progression of lung disease (n = 4), and digital ulcers (n = 5). None of the patients developed clinically meaningful myocardial involvement or renal crisis. Correlations between anti–topo I isotype levels at baseline and 1‐year change in MRSS, FVC%, and DLco% are shown in Supplementary Figure 5, http://onlinelibrary.wiley.com/doi/10.1002/art.41403/abstract. Baseline levels of anti–topo I IgM and anti–topo I IgA correlated with a decrease in FVC%, and anti–topo I IgM isotype level also correlated with a decrease in DLco%. Baseline levels of anti–topo I IgG, IgM, and IgA were not correlated with 1‐year change in MRSS.
In total, 23 patients (28%) showed disease progression according to prespecified criteria during the first year. Clinical characteristics and anti–topo I isotype profiles at baseline stratified for disease progression are presented in Table 3. At baseline, there were no differences in clinical characteristics between patients in the absence or presence of disease progression. Treatment strategy was also comparable between patients in the absence or presence of disease progression. Strikingly, while the clinical characteristics were similar, anti–topo I isotype levels at baseline were significantly higher, and anti–topo I IgM positivity was significantly more frequent in patients who had disease progression (91% versus 57%; P < 0.01). Kaplan‐Meier analysis underlined the prognostic value of anti–topo I IgM positivity (P = 0.02 by log rank test and Mantel‐Cox test) (Figure 2). Sensitivity analysis yielded similar results (Supplementary Figure 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41403/abstract).
Table 3.
Baseline characteristics of SSc patients positive for anti–topo I IgG, stratified according to presence or absence of disease progression over 1 year of follow‐up*
|
Disease progressors (n = 23) |
Disease nonprogressors (n = 58) |
P | |
|---|---|---|---|
| Demographic characteristics | |||
| Female sex | 14 (61) | 39 (67) | 0.59 |
| Age, mean ± SD years | 55.3 ± 16.3 | 51.9 ± 13.9 | 0.21 |
| Smoking (ever) | 12 (52) | 30 (52) | 0.95 |
| Disease duration | |||
| Since onset of first Raynaud’s symptom, median (IQR) years | 3.8 (1.3–8.4) | 5.6 (2.1–12.9) | 0.21 |
| Since onset of first non‐Raynaud’s symptom, median (IQR) years | 1.9 (0.6–4.5) | 3.5 (0.7–11.4) | 0.07 |
| Organ involvement | |||
| Diffuse cutaneous SSc | 12 (52) | 28 (48) | 1.00 |
| Modified Rodnan skin thickness score, median (IQR) | 6 (2–19) | 6 (3–13) | 0.86 |
| FVC%, mean ± SD | 89 ± 26 | 89 ± 28 | 0.92 |
| DLco%, mean ± SD | 62 ± 18 | 64 ± 16 | 0.83 |
| History of renal crisis | 0 (0) | 2 (4) | 1.00 |
| Digital ulcers | 0 (0) | 5 (9) | 0.31 |
| Pulmonary hypertension | 2 (9) | 2 (4) | 0.59 |
| History of immunosuppressive treatment† | |||
| HSCT | 0 (0) | 7 (12) | 0.18 |
| CYC (ever) | 4 (17) | 16 (28) | 0.34 |
| MMF (ever) | 1 (4) | 0 (0) | 0.28 |
| Use of immunosuppressive treatment during 1‐year follow‐up† | |||
| HSCT | 0 (0) | 0 (0) | – |
| CYC | 11 (19) | 5 (26) | 0.52 |
| MMF | 1 (5) | 6 (10) | 0.67 |
| Anti–topo I antibody characteristics | |||
| IgG level, median (IQR) AU/ml | 813 (542–1,263) | 396 (115–832) | <0.01 |
| IgA positivity | 23 (100) | 57 (98) | 1.00 |
| IgA level, median (IQR) AU/ml | 9,898 (2,743–16,656) | 2,045 (462–5,314) | <0.01 |
| IgM positivity | 21 (91) | 33 (57) | 0.04 |
| IgM level, median (IQR) AU/ml | 1065 (869–3,853) | 588 (223–1,610) | 0.01 |
In 22 individuals, clinical follow‐up data were not available and they could not be classified as either disease progressors or disease nonprogressors. Except where indicated otherwise, values are the number (%) of patients. SSc = systemic sclerosis; anti–topo I = anti–topoisomerase I; IQR = interquartile range; FVC% = forced vital capacity % predicted; DLco% = diffusing capacity for carbon monoxide % predicted.
Immunosuppressive treatment includes the use of hematopoietic stem cell transplantation (HSCT), cyclophosphamide (CYC), or mycophenolate mofetil (MMF).
Figure 2.
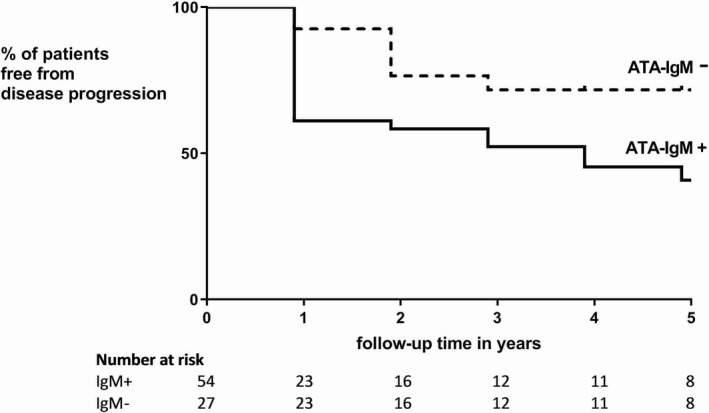
Percentage of patients with systemic sclerosis (among 81 with ≥1 year of follow‐up data available) who did not experience disease progression over time, according to the presence or absence of anti–topoisomerase I antibodies (ATAs) of the IgM isotype. Disease progression occurred more often in patients who were positive for anti–topoisomerase I IgM (P = 0.02, by log rank test and Mantel‐Cox test).
Validation in other cohorts
To confirm our results, we also performed anti–topo I isotype level measurements in 90 SSc patients who were positive for anti–topo I IgG (60 patients from University Hospital Zurich and 30 patients from Oslo University Hospital). Baseline characteristics of these patients are presented in Supplementary Table 1 (http://onlinelibrary.wiley.com/doi/10.1002/art.41403/abstract). Cross‐sectional analysis confirmed the correlation between anti–topo I IgG levels and skin scores at baseline (rs = 0.37; P < 0.01). In addition, this sample set, anti–topo I IgG isotype levels were correlated with the FVC (rs = −0.30; P < 0.01) and with the DLco (rs = −0.24; P = 0.03).
In validation samples, clinical follow‐up data at 1 year were available for 63 patients. During this year, 5 patients died, progression of skin disease was observed in 6 patients, progression of lung disease was observed in 7 patients, incident renal crisis developed in 1 patient, and digital ulcers developed in 5 patients. In total, 24 patients from the validation sample set experienced disease progression. Again, there were no clinical differences between disease progressors and disease nonprogressors at baseline, but disease progressors more often expressed anti–topo I IgM (96% versus 71%; P = 0.04) (Supplementary Table 1, http://onlinelibrary.wiley.com/doi/10.1002/art.41403/abstract). Thus, these data confirm that anti–topo I IgG–positive SSc patients who are also positive for anti–topo I IgM have a higher risk for disease progression compared to anti–topo I IgG–positive patients not expressing anti–topo I IgM.
DISCUSSION
This study shows that anti–topo I IgG–positive SSc patients who are also positive for anti–topo I IgM more often experience progression of disease compared to anti–topo I IgG–positive patients who are negative for anti–topo I IgM. Importantly, disease progressors could not be identified based on baseline clinical parameters. Additionally, our study shows that anti–topo I IgG–positive patients are almost always positive for anti–topo I IgA. Alteration from a positive to negative response (or vice versa) for anti–topo I IgG and anti–topo I IgA isotypes is relatively rare, while loss and gain of the anti–topo I IgM response occurs frequently. Over one‐third of patients who were positive for anti–topo I IgM at baseline eventually became negative for anti–topo I IgM during follow‐up.
Our observations that the majority of anti–topo I IgG–positive SSc patients express high levels of anti–topo I IgA while only some of these patients also expressed anti–topo I IgM are consistent with previous study findings from the early 1990s (33, 34). The sustained anti–topo I IgG response observed in SSc patients, with little or no fluctuations in disease activity and no seroreverting (in some cases, even after high‐dose cyclophosphamide treatment in the context of HSCT therapy), suggests that this response is long‐lived and that its generation depends on T cell help. Hence, it is conceivable that long‐lived plasma cells secreting anti–topo I IgG without the need for antigenic triggering may be responsible for a large fraction of the anti–topo I IgG levels measured in serum. However, we consider it possible that there is also a short‐lived, more dynamic part of the anti–topo I antibody response, triggered due to the continuous presence of autoantigens and, potentially, additional/external (yet unknown) triggers such as Toll‐like receptor ligands. Such triggers would be able to recruit naive B cells from the repertoire and would explain why IgM‐secreting plasma cells arise that, due to their short lifespan (i.e., the lack of a long‐lived memory compartment) and the short half‐life of IgM, more closely reflect disease‐relevant processes, with possible clinical consequences in the near future.
Anti–topo I IgG levels have previously been described as being correlated with skin scores (11, 12, 13). In a study by Kuwana and colleagues, it was reported that in 28 SSc patients, 21% of anti–topo I IgG–positive patients lost their anti–topo I IgG response over time, which was associated with a favorable disease course (14). Notably, although the results were not significant, none of these patients who lost positivity for anti–topo I IgG were anti–topo I IgM–positive at baseline, whereas one‐third of patients who remained positive for anti–topo I IgG over time were also positive for anti–topo I IgM at baseline. In our cohort, loss of anti–topo I IgM response over time was less common (5%).
The discrepancy between the study by Kuwana et al and our present study might be explained by methodologic differences. In their ELISAs, Kuwana and colleagues used a cutoff value for antibody positivity that was 3 times the SD of values in healthy controls (35). We used a cutoff value for anti–topo I IgG positivity prespecified by the manufacturer and used in clinical routine, which corresponds to the mean + 8 SD of the values in healthy controls (data not shown). Consequently, Kuwana et al might have included patients with already lower anti–topo I IgG levels at baseline. Additionally, in another study that included 21 patients, decreasing levels of anti–topo I IgG were accompanied by atrophic changes of the skin, while increasing levels were associated with new onset or worsening of organ involvement. Thus, our work as well as the work from others shows that the anti–topo I antibody response is related to disease course. Nonetheless, the frequency of a positive anti–topo I IgM response in patients not experiencing disease progression implies that anti–topo I IgM status alone is not sufficient to function as a biomarker in everyday clinical practice, but may be useful for clinical trial enrichment. As disease progression is highly unlikely in patients negative for anti–topo I IgM (<10%), anti–topo I IgM status might be useful in the decision to refrain from aggressive treatment like HSCT.
Our hypothesis that anti–topo I antibodies or the underlying immune dysregulation in anti–topo I–positive SSc is (at least partly) responsible for clinical heterogeneity might not seem to correspond with the heterogeneity observed among patients who are positive for both anti–topo I IgG and anti–topo I IgM at the first measurement and onwards. A pathophysiologic explanation for the clinical heterogeneity observed in these patients might be found in the presence of additional triggers responsible for transforming anti–topo I antibodies into pathogenic factors. For example, it has been speculated that anti–topo I antibodies trigger adhesion and activation of monocytes by binding to DNA–topoisomerase I expressed on fibroblasts. This could potentially lead to amplification of the fibrogenetic cascade (36, 37, 38).
Consistent with these findings, it is tempting to speculate that the presence of anti–topo I antibodies may only be pathogenic in an environment in which there is insufficient clearance of apoptotic bodies of endothelial cells containing DNA–topoisomerase I. Consequently, the production of anti–topo I IgG might be an ongoing process in all patients positive for anti–topo I antibodies; however, if not accompanied by the presence of extracellular DNA–topoisomerase I, the ability of anti–topo I antibodies to trigger fibrosis is lost. Clinically, this might result in different anti–topo I antibody–positive subsets of patients depending on the level of endothelial cell apoptosis. This could also fit with the observation that more severe capillary loss is associated with more severe organ involvement independent of autoantibody subtype (39). Alternatively, other characteristics of anti–topo I antibodies or their underlying immune response, such as epitope recognition patterns, extent of T cell and/or B cell activation, or interaction with cytokines, could be important for pathogenicity.
The present study had some limitations. As we included only patients who were positive for anti–topo I IgG at baseline, we cannot exclude the possibility that some SSc patients solely express anti–topo I IgM and/or IgA and are not included in our analyses. However, based on our sensitivity analysis, we conclude that in SSc patients, continuous expression of anti–topo I IgM without switching to anti–topo I IgG is unlikely. Also, as data were derived from a cohort study, treatment was uncontrolled. However, significant differences in treatment between disease progressors and disease nonprogressors were not observed. In addition, because of the exploratory character of the study, we deliberately did not correct for multiple testing as this would lead to increased chances of false‐negative findings (40), which cannot be easily justified in an explorative study. Instead, we validated our main findings in an independent cohort.
Finally, we used a composite of several individually validated scores for different organs to define overall disease progression, including all‐cause mortality. We acknowledge that a precise determination of cause of death is often difficult, leading to weak data quality. To address this, recorded causes of death are described in the Results. Using a composite end point for disease progression is common in SSc studies (41, 42), as the heterogeneous nature of the disease, which involves multiple organs, implicates the use of composite indices. Availability of a validated composite index for disease progression could have substantiated our findings. However, although it lacks validation, our composite index has face validity and, most importantly, as our data have been validated in an independent second cohort, our analyses are robust.
In conclusion, our results indicate that the anti–topo I antibody response is relevant to the disease course of SSc. Further research with regard to the anti–topo I antibody response in patients with SSc, focusing on specific epitopes and other antibody characteristics, such as Fc glycosylation, could help clarify their role in disease pathogenesis. Most importantly, our data indicate that expression of anti–topo I IgM is associated with an unfavorable disease course—a finding that we validated in other cohorts. Whether the presence of the IgM isotype of other SSc‐specific autoantibodies is of equal importance as that of anti–topo I IgM in the progression of the disease remains to be elucidated.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Boonstra had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Boonstra, Huizinga, Toes, Scherer, Vries‐Bouwstra.
Acquisition of data
Boonstra, Bakker, Grummels, Ninaber, Marsan, Huizinga, Jordan, Hoffman‐Vold, Distler, Toes, Scherer, Vries‐Bouwstra.
Analysis and interpretation of data
Boonstra, Bakker, Ninaber, Marsan, Wortel, Huizinga, Jordan, Hoffman‐Vold, Distler, Toes, Scherer, Vries‐Bouwstra.
Supporting information
Supplementary Material
No potential conflicts of interest relevant to this article were reported.
Contributor Information
Maaike Boonstra, Email: m.boonstra@lumc.nl.
Jeska K. de Vries‐Bouwstra, Email: j.k.de_vries-bouwstra@lumc.nl.
References
- 1. Reveille JD, Solomon DH, for the American College of Rheumatology Ad Hoc Committee of Immunologic Testing Guidelines . Evidence‐based guidelines for the use of immunologic tests: anticentromere, Scl‐70, and nucleolar antibodies. Arthritis Rheum 2003;49:399–412. [DOI] [PubMed] [Google Scholar]
- 2. Koenig M, Joyal F, Fritzler MJ, Roussin A, Abrahamowicz M, Boire G, et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud's phenomenon to systemic sclerosis: a twenty‐year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum 2008;58:3902–12. [DOI] [PubMed] [Google Scholar]
- 3. Patterson KA, Roberts‐Thomson PJ, Lester S, Tan JA, Hakendorf P, Rischmueller M, et al. Interpretation of an extended autoantibody profile in a well‐characterized Australian Systemic Sclerosis (scleroderma) Cohort using principal components analysis. Arthritis Rheumatol 2015;67:3234–44. [DOI] [PubMed] [Google Scholar]
- 4. Mierau R, Moinzadeh P, Riemekasten G, Melchers I, Meurer M, Reichenberger F, et al. Frequency of disease‐associated and other nuclear autoantibodies in patients of the German Network for Systemic Scleroderma: correlation with characteristic clinical features. Arthritis Res Ther 2011;13:R172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vanthuyne M, Smith V, de Langhe E, van Praet J, Arat S, Depresseux G, et al. The Belgian Systemic Sclerosis Cohort: correlations between disease severity scores, cutaneous subsets, and autoantibody profile. J Rheumatol 2012;39:2127–33. [DOI] [PubMed] [Google Scholar]
- 6. Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum 2005;35:35–42. [DOI] [PubMed] [Google Scholar]
- 7. Khanna D, Denton CP. Evidence‐based management of rapidly progressing systemic sclerosis. Best Pract Res Clin Rheumatol 2010;24:387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boonstra M, Ninaber MK, Marsan NA, Huizinga TW, Scherer HU, de Vries‐Bouwstra JK. Prognostic properties of antitopoisomerase antibodies in patients identified by the ACR/EULAR 2013 systemic sclerosis criteria [letter]. Rheumatology (Oxford) 2019;58:730–2. [DOI] [PubMed] [Google Scholar]
- 9. Yoshizaki A, Yanaba K, Ogawa A, Asano Y, Kadono T, Sato S. Immunization with DNA topoisomerase I and Freund's complete adjuvant induces skin and lung fibrosis and autoimmunity via interleukin‐6 signaling. Arthritis Rheum 2011;63:3575–85. [DOI] [PubMed] [Google Scholar]
- 10. Mehta H, Goulet PO, Nguyen V, Pérez G, Koenig M, Senécal JL, et al. Topoisomerase I peptide‐loaded dendritic cells induce autoantibody response as well as skin and lung fibrosis. Autoimmunity 2016;49:503–13. [DOI] [PubMed] [Google Scholar]
- 11. Hu PQ, Fertig N, Medsger TA Jr, Wright TM. Correlation of serum anti‐DNA topoisomerase I antibody levels with disease severity and activity in systemic sclerosis. Arthritis Rheum 2003;48:1363–73. [DOI] [PubMed] [Google Scholar]
- 12. Perera A, Fertig N, Lucas M, Rodriguez‐Reyna TS, Hu P, Steen VD, et al. Clinical subsets, skin thickness progression rate, and serum antibody levels in systemic sclerosis patients with anti–topoisomerase I antibody. Arthritis Rheum 2007;56:2740–6. [DOI] [PubMed] [Google Scholar]
- 13. Hasegawa M, Imura‐Kumada S, Matsushita T, Hamaguchi Y, Fujimoto M, Takehara K. Anti–topoisomerase I antibody levels as serum markers of skin sclerosis in systemic sclerosis. J Dermatol 2013;40:89–93. [DOI] [PubMed] [Google Scholar]
- 14. Kuwana M, Kaburaki J, Mimori T, Kawakami Y, Tojo T. Longitudinal analysis of autoantibody response to topoisomerase I in systemic sclerosis. Arthritis Rheum 2000;43:1074–84. [DOI] [PubMed] [Google Scholar]
- 15. Meijs J, Schouffoer AA, Marsan NA, Kroft LJ, Stijnen T, Ninaber MK, et al. Therapeutic and diagnostic outcomes of a standardised, comprehensive care pathway for patients with systemic sclerosis. RMD Open 2016;2:e000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clements P, Lachenbruch P, Siebold J, White B, Weiner S, Martin R, et al. Inter and intraobserver variability of total skin thickness score (modified Rodnan TSS) in systemic sclerosis. J Rheumatol 1995;22:1281–5. [PubMed] [Google Scholar]
- 17. Steen VD, Medsger TA Jr. The value of the Health Assessment Questionnaire and special patient‐generated scales to demonstrate change in systemic sclerosis patients over time. Arthritis Rheum 1997;40:1984–91. [DOI] [PubMed] [Google Scholar]
- 18. Ware JE, Kosinski M, Keller S. SF‐36 physical and mental health summary scales: a user's manual. Boston: Health Assessment Lab, New England Medical Center; 1994. [Google Scholar]
- 19. Newnham EA, Harwood KE, Page AC. Evaluating the clinical significance of responses by psychiatric inpatients to the mental health subscales of the SF‐36. J Affect Disord 2007;98:91–7. [DOI] [PubMed] [Google Scholar]
- 20. Mouthon L, Rannou F, Bérezné A, Pagnoux C, Arène JP, Foïs E, et al. Development and validation of a scale for mouth handicap in systemic sclerosis: the Mouth Handicap in Systemic Sclerosis scale. Ann Rheum Dis 2007;66:1651–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schouffoer A, Strijbos E, Schuerwegh M, Mouthon L, Vlieland MV. Translation, cross‐cultural adaptation, and validation of the Mouth Handicap in Systemic Sclerosis questionnaire (MHISS) into the Dutch language. Clin Rheumatol 2013;32:1649. [DOI] [PubMed] [Google Scholar]
- 22. Clements PJ, Wong WK, Hurwitz EL, Furst DE, Mayes M, White B, et al. The Disability Index of the Health Assessment Questionnaire is a predictor and correlate of outcome in the high‐dose versus low‐dose penicillamine in systemic sclerosis trial. Arthritis Rheum 2001;44:653–61. [DOI] [PubMed] [Google Scholar]
- 23. Dolan P. Modeling valuations for EuroQol health states. Med Care 1997;35:1095–108. [DOI] [PubMed] [Google Scholar]
- 24. Lamers L, Stalmeier P, McDonnell J, Krabbe P, van Busschbach J. Kwaliteit van leven meten in economische evaluaties: het Nederlands EQ‐5D-tarief. Ned Tijdschr Geneeskd 2005;149:1574–8. [PubMed] [Google Scholar]
- 25. Meijs J, Pors D, Vliet VT, Huizinga T, Schouffoer A. Translation, cross‐cultural adaptation, and validation of the UCLA Scleroderma Clinical Trial Consortium Gastrointestinal Tract Instrument (SCTC GIT) 2.0 into Dutch. Clin Exp Rheumatol 2013;32 Suppl 86:S41–8. [PubMed] [Google Scholar]
- 26. Khanna D, Hays RD, Park GS, Braun‐Moscovici Y, Mayes MD, McNearney TA, et al. Development of a preliminary scleroderma gastrointestinal tract 1.0 quality of life instrument. Arthritis Rheum 2007;57:1280–6. [DOI] [PubMed] [Google Scholar]
- 27. Van Den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum 2013;65:2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khanna D, Furst DE, Clements PJ, Allanore Y, Baron M, Czirjak L, et al. Standardization of the modified Rodnan skin score for use in clinical trials of systemic sclerosis. J Scleroderma Relat Disord 2017;2:11–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khanna D, Mittoo S, Aggarwal R, Proudman SM, Dalbeth N, Matteson EL, et al. Connective Tissue Disease‐associated Interstitial Lung Diseases (CTD‐ILD): report from OMERACT CTD‐ILD Working Group [review]. J Rheumatol 2015;42:2168–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoffmann‐Vold AM, Midtvedt O, Molberg O, Garen T, Gran JT. Prevalence of systemic sclerosis in Southeast Norway. Rheumatology (Oxford) 2012;51:1600–5. [DOI] [PubMed] [Google Scholar]
- 31. Frauenfelder T, Winklehner A, Nguyen TD, Dobrota R, Baumueller S, Maurer B, et al. Screening for interstitial lung disease in systemic sclerosis: performance of high‐resolution CT with limited number of slices: a prospective study. Ann Rheum Dis 2014;73:2069–73. [DOI] [PubMed] [Google Scholar]
- 32. Jordan S, Maurer B, Toniolo M, Michel B, Distler O. Performance of the new ACR/EULAR classification criteria for systemic sclerosis in clinical practice. Rheumatology (Oxford) 2015;54:1454–8. [DOI] [PubMed] [Google Scholar]
- 33. Verheijen R, de Jong BA, van Venrooij WJ. A recombinant topoisomerase I ELISA: screening for IgG, IgM and IgA anti–topo I autoantibodies in human sera. Clin Exp Immunol 1992;89:456–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hildebrandt S, Jackh G, Weber S, Peter HH. A long‐term longitudinal isotypic study of anti‐topoisomerase I autoantibodies. Rheumatol Int 1993;12:231–4. [DOI] [PubMed] [Google Scholar]
- 35. Kuwana M, Medsger TA Jr, Wright TM. Detection of anti‐DNA topoisomerase I antibody by an enzyme‐linked immunosorbent assay using overlapping recombinant polypeptides: part 1. Clin Immunol Immunopathol 1995;76:266–78. [DOI] [PubMed] [Google Scholar]
- 36. Henault J, Robitaille G, Senecal JL, Raymond Y. DNA topoisomerase I binding to fibroblasts induces monocyte adhesion and activation in the presence of anti–topoisomerase I autoantibodies from systemic sclerosis patients. Arthritis Rheum 2006;54:963–73. [DOI] [PubMed] [Google Scholar]
- 37. Senecal J, Hénault J, Raymond Y. The pathogenic role of autoantibodies to nuclear autoantigens in systemic sclerosis (scleroderma). J Rheumatol 2005;32:1643. [PubMed] [Google Scholar]
- 38. Henault J, Tremblay M, Clement I, Raymond Y, Senecal JL. Direct binding of anti‐DNA topoisomerase I autoantibodies to the cell surface of fibroblasts in patients with systemic sclerosis. Arthritis Rheum 2004;50:3265–74. [DOI] [PubMed] [Google Scholar]
- 39. Markusse IM, Meijs J, de Boer B, Bakker JA, Schippers HPC, Schouffoer AA, et al. Predicting cardiopulmonary involvement in patients with systemic sclerosis: complementary value of nailfold videocapillaroscopy patterns and disease‐specific autoantibodies. Rheumatology (Oxford) 2017;56:1081–8. [DOI] [PubMed] [Google Scholar]
- 40. Feise RJ. Do multiple outcome measures require p‐value adjustment? BMC Med Res Methodol 2002;2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sullivan KM, Goldmuntz EA, Keyes‐Elstein L, McSweeney PA, Pinckney A, Welch B, et al. Myeloablative autologous stem‐cell transplantation for severe scleroderma. N Engl J Med 2018;378:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Avouac J, Walker UA, Hachulla E, Riemekasten G, Cuomo G, Carreira PE, et al. Joint and tendon involvement predict disease progression in systemic sclerosis: a EUSTAR prospective study. Ann Rheum Dis 2016;75:103–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material