

Abstract
Background and Aims
Itaconate, a metabolite of the tricarboxylic acid cycle, plays anti‐inflammatory roles in macrophages during endotoxemia. The mechanisms underlying its anti‐inflammatory roles have been shown to be mediated by the modulation of oxidative stress, an important mechanism of hepatic ischemia–reperfusion (I/R) injury. However, the role of itaconate in liver I/R injury is unknown.
Approach and Results
We found that deletion of immune‐responsive gene 1 (IRG1), encoding for the enzyme producing itaconate, exacerbated liver injury and systemic inflammation. Furthermore, bone marrow adoptive transfer experiments indicated that deletion of IRG1 in both hematopoietic and nonhematopoietic compartments contributes to the protection mediated by IRG1 after I/R. Interestingly, the expression of IRG1 was up‐regulated in hepatocytes after I/R and hypoxia/reoxygenation‐induced oxidative stress. Modulation of the IRG1 expression levels in hepatocytes regulated hepatocyte cell death. Importantly, addition of 4‐octyl itaconate significantly improved liver injury and hepatocyte cell death after I/R. Furthermore, our data indicated that nuclear factor erythroid 2–related factor 2 (Nrf2) is required for the protective effect of IRG1 on mouse and human hepatocytes against oxidative stress–induced injury. Our studies document the important role of IRG1 in the acute setting of sterile injury induced by I/R. Specifically, we provide evidence that the IRG1/itaconate pathway activates Nrf2‐mediated antioxidative response in hepatocytes to protect liver from I/R injury.
Conclusions
Our data expand on the importance of IRG1/itaconate in nonimmune cells and identify itaconate as a potential therapeutic strategy for this unfavorable postsurgical complication.
Abbreviations
- ALT
alanine aminotransferase
- DCFDA
2,7‐dichlorofluorescin diacetate
- DI
dimethyl itaconate
- ELISA
enzyme‐linked immunosorbent assay
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- H&E
hematoxylin and eosin
- 4‐HNE
4‐hydroxynonenal
- HO‐1
heme oxygenase‐1
- H/R
hypoxia/reoxygenation
- IL
interleukin
- I/R
ischemia–reperfusion
- IRG1
immune‐responsive gene 1
- Keap1
Kelch‐like ECH‐associated protein 1
- KO
knockout
- LC‐MS
liquid chromatography–mass spectrometry
- LDH
lactate dehydrogenase
- Ly6G
lymphocyte antigen 6G
- MCP1
monocyte chemoattractant protein 1
- MPO
myeloperoxidase
- NPC
nonparenchymal cell
- NQO1
reduced nicotinamide adenine dinucleotide phosphate: quinone oxidoreductase 1
- Nrf2
nuclear factor erythroid 2–related factor 2
- 4‐OI
4‐octyl itaconate
- PBS
phosphate‐buffered saline
- ROS
reactive oxygen species
- RT‐PCR
real‐time polymerase chain reaction
- TCA
tricarboxylic acid
- WT
wild type
Liver ischemia–reperfusion (I/R) injury remains a major contributor to liver dysfunction and failure after liver transplantation, liver resection, and hemorrhagic shock.1 Oxidative stress is a key factor in the mechanisms underlying liver I/R injury. Excessive production of reactive oxygen species (ROS) during I/R disturbs the cellular redox status, which directly leads to cellular injury.2 In response to oxidative stress, Kupffer cells and other cells produce inflammatory mediators that attract inflammatory cells into the liver and contribute to hepatocyte apoptosis.3 To maintain redox homeostasis, the liver has sophisticated antioxidant systems consisting of antioxidant proteins and enzymes that are regulated by redox‐sensitive transcription factors. Hence, the up‐regulation of antioxidant pathways represents an important protective mechanism to prevent liver injury during acute or chronic oxidative stress.4
Remodeling of the tricarboxylic acid (TCA) cycle is a metabolic adaptation accompanying inflammatory macrophage activation. This can result in the accumulation of metabolic intermediates that function as regulatory mediators of the inflammatory response.5 One of these regulatory metabolites is itaconate, which is synthesized by the decarboxylation of cis‐aconitic acid, a TCA cycle intermediate, by immune‐responsive gene 1 protein (IRG1).6 IRG1 is one of the most highly up‐regulated genes under proinflammatory conditions, such as during bacterial infections7, 8 and during the embryo implantation phase in the pregnant uterus.9, 10 The product of IRG1, itaconate, has recently become a focus of the field of immunometabolism due to its crucial role as an anti‐inflammatory metabolite that negatively regulates the inflammatory response and cytokine production.11, 12, 13, 14 Thus, the enzymatic function of IRG1 links cellular metabolism to immunity through the production of itaconate.
The nuclear factor erythroid 2–related factor 2 (Nrf2) is a ubiquitously expressed transcription factor that up‐regulates antioxidant defense pathways. Under homeostatic conditions, Nrf2 is maintained at a low level in the cytosol as an inactive complex by its inhibitor protein, Kelch‐like ECH‐associated protein 1 (Keap1), which degrades Nrf2 through a ubiquitin/proteosome pathway.15 During oxidative or electrophilic stress, cysteine residues in Keap1 are modified, resulting in stabilization of Nrf2 and its translocation from cytoplasm into the nucleus.16 Once in the nucleus, Nrf2 binds to the cis‐acting antioxidant response element within the gene promoters,17 and this leads to the transcriptional activation of genes that encode a series of antioxidant enzymes, including heme oxygenase‐1 (HO‐1) and reduced nicotinamide adenine dinucleotide phosphate:quinone oxidoreductase 1 (NQO1), as well as other cytoprotective pathways.18, 19 Previous studies have demonstrated that the Nrf2 pathway limits oxidant injury in models of I/R.18, 20 A link between IRG1 and Nrf2 was recently identified when itaconate was shown to directly alkylate cysteine residues in KEAP1, leading to an increase in Nrf2 in macrophages during endotoxemia.12
In this study, we hypothesized that the IRG1/itaconate pathway would be part of a protective response against oxidant injury in the liver, possibly through the up‐regulation of Nrf2. Using IRG1−/− mice, we found that IRG1 suppresses injury and inflammation during liver I/R and protects cultured hepatocytes from apoptosis following hypoxia/reoxygenation (H/R). Both liver I/R in vivo and hepatocyte H/R in vitro led to the up‐regulation of IRG1. The cell‐permeable itaconate analogue 4‐octyl itaconate (4‐OI) protected against liver I/R injury and rescued hepatocytes from H/R‐induced apoptosis through a mechanism that required Nrf2. These findings reveal an unrecognized role of the IRG1/itaconate/Nrf2 pathway in antioxidant defenses in hepatocytes during liver I/R.
Materials and Methods
Animals
IRG1 knockout (KO; IRG1−/−) mice (The Jackson Laboratory, no. 029340) and Nrf2 KO (Nrf2−/−) mice (The Jackson Laboratory, no. 017009) were bred and housed under specific pathogen‐free conditions in our facility. C57BL/6NJ (B6N) (no. 005304) and C57BL/6J (B6J) (no. 000664) controls were purchased from The Jackson Laboratory. IRG1−/− animals were congenic to the C57BL/6NJ background, and Nrf2−/− animals were congenic to the C57BL/6J background. Male mice aged 8‐12 weeks, weighing 21‐30 g, were used in our experiments. All experimental protocols were approved by the Institutional Animal Use and Care Committee of the University of Pittsburgh. The experiments were performed in accordance with National Institutes of Health guidelines for the use of laboratory animals.
Mouse Liver I/R Injury Model and Animal Treatment
A nonlethal model of segmental (70%) hepatic warm I/R was used, as described.21 Briefly, all structures in the portal triad (hepatic artery, portal vein, bile duct) to the left and median liver lobes were occluded with a microvascular clamp for 60 minutes, and then reperfusion was initiated by clamp removal. Ambient temperature of the mice during ischemia was maintained at 33°C using an incubator chamber. Sham‐operated animals underwent anesthesia, laparotomy, and exposure of the portal triad without hepatic ischemia. Animals were sacrificed at predetermined time points after reperfusion. For 4‐OI (Cayman Chemical) administration, mice were treated intraperitoneally with 4‐OI (25 mg/kg/dose) in (2‐hydroxypropyl)‐β‐cyclodextrin in phosphate‐buffered saline (PBS) or vehicle control 2 hours before hepatic ischemia and at the time of reperfusion.
Reagents
The following reagents were used: mouse IRG1 enzyme‐linked immunosorbent assay (ELISA) Kit (no. MBS9319749) from MyBioSource; cleaved caspase‐3 antibody (no. 9664S), caspase‐3 antibody (no. 9662S), cleaved caspase‐7 antibody (no. 9491S), caspase‐7 antibody (no. 9491T), Nrf2 antibody (no. 12721S), HO‐1 antibody (no. 82206S), Myc‐Tag antibody (no. 2276S), β‐actin antibody (no. 8457S), α‐tubulin antibody (no. 2144S), and lamin B2 antibody (no. 13823) from Cell Signaling Technologies; NQO1 antibody (no. sc‐32793) from Santa Cruz Biotechnology; and Nrf2 antibody (no. ab137550) and HO‐1 antibody (no. ab13248) from Abcam.
Chimeric Mice
Chimeric mice were produced by adoptive transfer of donor bone marrow cells into irradiated recipient animals using combinations of wild‐type (WT; C57BL/6NJ) and KO (IRG1−/−) mice in the following recipient/donor combinations: WT/WT, WT/KO, KO/WT, KO/KO. Recipient mice (6‐8 weeks) were injected intraperitoneally with 200 μL of liposome‐encapsulated clodronate (CLD‐8909; Encapsula NanoSciences) 24 hours prior to irradiation. Recipient mice were then exposed to an otherwise lethal 1,000 cGy from a cesium source (Nordion International) 6 hours before receiving 5 × 106 bone marrow cells by tail vein injection. The bone marrow cells were prepared in a sterile manner from the tibia and femur bones of the donor mice. All animals were monitored 3 times weekly for the first 2 weeks to ensure successful bone marrow engraftment. The chimeric mice underwent hepatic I/R after 8‐12 weeks to ensure stable engraftment.
Hepatocyte and Nonparenchymal Cell Isolation and Treatment
Mice were perfused by an in situ collagenase (type VI; Sigma) technique. Hepatocytes were separated from nonparenchymal cells (NPCs) by two steps of differential centrifugation. Hepatocyte purity exceeded 99% as measured by light microscopy. Cell viability was typically > 95% as measured by trypan blue exclusion. NPCs were isolated, subjected to differential centrifugation, and purified without hepatocytes as determined by light microscopy. Hepatocytes were plated on gelatin‐coated culture plates in Williams’ E medium with 10% calf serum, 15 mM 4‐(2‐hydroxyethyl)‐1‐piperazine ethanesulfonic acid (HEPES), 1 μM insulin, 2 mM l‐glutamine, 100 U/mL penicillin, and 100 U/mL streptomycin. Cells were allowed to attach overnight. Medium was replaced with fresh medium before stimulation. To simulate ischemia, hepatocytes were cultured under the hypoxic condition (1% oxygen) in an atmosphere‐controlled chamber (MIC‐101, patent no. 5352414) containing a gas mixture composed of 94% N2, 5% CO2, and 1% O2. Cells were then reoxygenated under normoxic conditions in the standard cell culture incubator.
Isolation of Human Hepatocytes
Human hepatocytes, provided by David A. Geller (Department of Surgery, University of Pittsburgh), were isolated from histologically normal liver according to a protocol approved by the institutional review board. Human hepatocytes were prepared by a three‐step collagenase perfusion technique, as described.22 Isolated human hepatocytes were cultured in Williams’ E medium supplemented with 5% calf serum, 100 U/mL penicillin, 100 U/mL streptomycin, 2 mM l‐glutamine, and 15 mM HEPES.
Alanine Aminotransferase and Cytokine Abundance Assessment
Alanine aminotransferase (ALT) levels were determined by the DRI‐CHEM 4000 Chemistry Analyzer System (Heska). Cytokine abundance was analyzed by ELISA specific for interleukin 6 (IL‐6; R&D Systems) in the plasma according to the manufacturer’s instructions.
Myeloperoxidase Assay
A part of the ischemic liver lobe was homogenized, and myeloperoxidase (MPO) was quantified using a mouse MPO ELISA kit (Hycult Biotechnology) according to the manufacturer’s instructions.
Histological Analysis
Tissue samples were harvested and processed for hematoxylin and eosin (H&E) as described.23 Samples were imaged and scored using the Suzuki methodology by three independent members of the Center for Biologic Imaging (University of Pittsburgh). The tissues were assessed for the amount (percentage) of inflammation (sinusoidal congestion, cytoplasmic vacuolization, infiltrating inflammatory cells) and necrosis for characterizing liver damage after I/R.
Immunofluorescence
Liver samples were processed as described.24 In brief, sections of 6 μm were permeabilized with 0.1% Triton X‐100 for 20 minutes and rehydrated with five washes of PBS + 0.5% bovine serum albumin (PBB), and then the tissue sections underwent a blocking step of 45 minutes with PBS + 20% normal goat serum. Samples were incubated with primary antibodies (anti‐4‐hydroxynonenal [4‐HNE] at 1:100, Millipore no. 393207; anti‐lymphocyte antigen 6G [Ly6G] at 1:100, BD Biosciences no. 560599; anti‐F4/80 at 1:250, BD Biosciences no. 552958) in PBB + 0.1 Triton X‐100 for 12 hours or TMR red (1:1,000, Roche no. 12156792910) for 1 hour at room temperature. All immunofluorescent experiments staining sets involved staining β‐actin and nuclear stain for use in quantitation. Imaging conditions were maintained at identical settings within each antibody‐labeling experiment, with original gating performed using the primary depletion control. Large‐area images equivalent to nine unique fields were taken in X and Y with a Nikon A1 confocal microscope (purchased with 1S10OD019973‐01 awarded to Dr. Simon C. Watkins). Quantification was performed using NIS Elements (Nikon, Melville, NY).
Western Blot
Samples were lysed in lysis buffer (Cell Signaling Technology) with protease inhibitors and centrifuged at 16,000g for 10 minutes, and supernatant was collected. Protein concentrations of the supernatants were determined with a bicinchoninic acid assay kit (Thermo Fisher Scientific). Denatured protein samples were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred onto a polyvinylidene difluoride membrane at 250 mA for 2 hours. The membrane was blocked in 5% milk (Bio‐Rad) for 1 hour and then incubated overnight with primary antibody in 1% milk. Membranes were washed 3 times in trishydroxymethylaminomethane‐buffered saline Tween (TBS‐T) for 10 minutes, incubated with horseradish peroxidase–conjugated secondary antibody for 1 hour, and then washed 3 times for 10 minutes in TBS‐T before being developed for chemiluminescence (Bio‐Rad). Western images were quantified by densitometry using ImageJ software.
Real‐Time Polymerase Chain Reaction
Total RNA was extracted with the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. From 1 μg of RNA and with Reverse Transcription Supermix (Bio‐Rad no. 1708841), cDNA was generated and used for real‐time polymerase chain reaction (RT‐PCR) analysis. Universal SYBR Green Supermix (Bio‐Rad no. 1725121) and primer pairs specific for IRG1, IL‐6, monocyte chemoattractant protein 1 (MCP‐1), Ly6G, or glyceraldehyde 3‐phosphate dehydrogenase (GAPDH; Qiagen) were used to prepare the RT‐PCR mixes. All samples were assayed in duplicate, and results were normalized by GAPDH abundance.
Lactate Dehydrogenase Assay
Lytic cell death following H/R treatment was assessed by measurement of lactate dehydrogenase (LDH) released into the medium. At the end of treatment, 50 μL of medium solution was transferred to a 96‐well plate, and the LDH reaction was performed using the Pierce LDH Cytotoxicity Assay Kit (Thermo Scientific) according to the manufacturer’s instructions and analyzed by spectrophotometer (Biotech).
Cellular ROS Assay
Cellular ROS detection was performed using a 2′,7′‐dichlorofluorescin diacetate (DCFDA) Cellular Reactive Oxygen Species Detection Assay Kit (Abcam) based on fluorescent microplate measurement. Briefly, hepatocytes (2.5 × 104 cells/well) were seeded on a dark‐bottomed, 96‐well microplate and allowed to attach overnight. Cells were stained with 25 µM DCFDA 45 minutes before the end of H/R stimulation. Fluorescence signals were measured by microplate reader at excitation/emission of 485/535 nm. Changes were determined as percentage of control (normoxia) after background subtraction.
Metabolite Analyses
Polar metabolites were extracted from the liver tissues by homogenizing in ice‐cold 80% methanol with 0.1% formic acid at a ratio of 15 µL/mg of tissue. Deuterated (D4)‐taurine and (D3)‐lactate (Sigma‐Aldrich) were added as an internal standard. Metabolite concentrations were quantified using liquid chromatography–mass spectrometry (LC‐MS) analysis. Briefly, samples were injected by Thermo Fisher Vanquish ultra‐high‐performance liquid chromatography and separated over a reversed‐phase Thermo Fisher Hypercarb porous graphitic column maintained at 50°C. The mobile phase consisted of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). The LC gradient was as follows: 0.0‐12.0 minutes 5%‐1,000% B, 12.0‐15.0 minutes 100% B, 15.0‐15.1 minutes 100%‐5% B, 15.1‐20.0 minutes 5% B. Spectra was acquired on a Thermo Fisher ID‐X Tribrid mass spectrometer. Calibration was performed prior to analysis using the Pierce Positive and Negative Ion Calibration Solutions (Thermo Fisher). Integrated peak areas were extracted manually using 2.7 Quan Browser (Thermo Fisher).
Mouse IRG1 Plasmid Transfections
pCMV6‐Myc‐IRG1 overexpressing plasmid (5 μg, mouse IRG1 transfection‐ready DNA; OriGene) was transfected into 1 × 106 hepatocytes using Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. The DNA–lipid complex was added to cells and incubated for 36 hours.
Nuclear Protein Extraction
Nuclear extraction was performed using an NE‐PER Nuclear Cytoplasmic Extraction Reagent kit (Thermo Fisher Scientific) according to the manufacturer’s instruction.
Statistics
Results are shown as mean ± SD from at least three independent experiments. Data were analyzed by GraphPad Prism 7 software. Comparisons between two experimental groups were performed by two‐tailed Student t test. Analysis of three or more independent groups was performed by one‐way analysis of variance. P < 0.05 was considered statistically significant.
Results
Deletion of IRG1 Worsens Liver Injury After I/R
To study the role of IRG1 and itaconate in hepatic I/R injury, IRG1−/− mice and control WT mice (C57BL/6NJ) were subjected to warm liver I/R with 1 hour of ischemia followed by 0, 6, or 24 hours of reperfusion (Fig. 1A‐C). Notably, IRG1−/− mice that are defective at producing itaconate showed exacerbated liver injury at both 6 and 24 hours after reperfusion, as demonstrated by significantly higher levels of serum ALT than those of control mice (Fig. 1B,C). Histological analysis showed significantly larger necrotic areas in liver sections of IRG1−/− mice after 6 and 24 hours of reperfusion (Fig. 1D,E), and this was associated with higher Suzuki’s liver injury scores (Fig. 1F). However, there was no significant difference in hepatic damage between WT and IRG1−/− mice after 1 hour of ischemia but no reperfusion (Fig. 1A,D,E), indicating that IRG1 mainly protects the liver from the injury that occurs during the reperfusion phase of hepatic I/R. We also confirmed that IRG1 mRNA expression was up‐regulated in the livers of WT mice subjected to I/R (Fig. 1G), and as expected, we observed significantly higher levels of itaconate in the livers of mice after I/R (Fig. 1H). Together, these indicate that IRG1 is up‐regulated in the setting of noninfectious inflammation in the liver.
Figure 1.
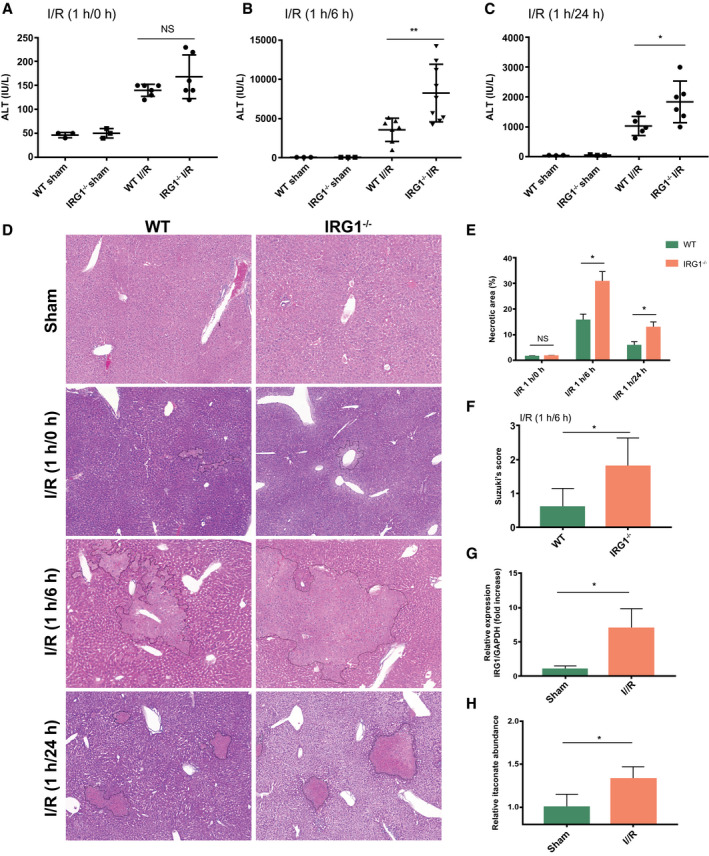
Deletion of IRG1 exacerbates liver injury after I/R. WT (C57BL/6NJ) and IRG1 KO (IRG1−/−) mice were subjected to liver ischemia for 1 hour and then reperfusion for 0, 6, and 24 hours (I/R 1/0, 1/6, 1 hour/24 hours) and sham surgery (sham) as control. (A) Serum ALT levels in WT and IRG1−/− mice after sham surgery or 1‐hour ischemia and 0‐hour reperfusion; n = 3 in each sham group, n = 6 in each I/R group. (B) Serum ALT levels in WT and IRG1−/− mice after sham surgery or 1‐hour ischemia and 6‐hour reperfusion; n = 3 in each sham group, n = 7‐9 in each I/R group. (C) Serum ALT levels in WT and IRG1−/− mice after sham surgery or 1‐hour ischemia and 24‐hour reperfusion; n = 3 in each sham group, n = 5‐6 in each I/R group. (D) Representative liver H&E staining (original magnification ×20) of WT and IRG1−/− mice after sham surgery or 1‐hour ischemia and 0‐, 6‐, or 24‐hour reperfusion. (E) Dotted lines in liver H&E staining indicate measured areas of necrosis, quantified in bar graph; n = 4 for each group. (F) Liver damages were evaluated by Suzuki’s histological score (I/R 1 hour/6 hours), quantified in bar graph; n = 5 for each group. (G) RT‐PCR for IRG1 mRNA expression in livers from WT mice after sham surgery or 1‐hour ischemia and 6‐hour reperfusion; n = 3 for sham group, n = 4 for I/R group. (H) LC‐MS analysis of itaconate abundance in livers from WT mice after sham surgery or 1‐hour ischemia and 6‐hour reperfusion; n = 4 for sham, n = 5 for I/R group. Data are presented as means ± SD. *P < 0.05, **P < 0.01. Abbreviation: NS, not significant.
Increased Systemic Inflammation and Hepatic Infiltration of Inflammatory Cells in IRG1−/− Mice After I/R
The pathogenesis of liver I/R injury includes both direct cellular damage as a result of the oxidative stress as well as injury resulting from activation of inflammatory pathways.25 Studies have shown that itaconate plays anti‐inflammatory roles in inflammatory macrophages during endotoxemia.12 Interestingly, we found that deletion of IRG1 led to increased systemic and local inflammation after hepatic I/R, as assessed by higher levels of both plasma IL‐6 and hepatic IL‐6 mRNA expression in IRG1−/− mice compared to control WT mice (C57BL/6NJ) (Fig. 2A,B). Levels of MCP‐1 and Ly6G mRNA as well as MPO protein levels were significantly higher in the livers of IRG1−/− mice than control mice after I/R (Fig. 2C‐E), indicating increased chemokine gene expression and neutrophil infiltration in the livers of IRG1−/− mice. Indeed, immunofluorescent staining confirmed the greater accumulation of Ly6G‐positive neutrophils in livers of IRG1−/− mice after I/R compared to WT mice (Fig. 2F,G). The hepatic accumulation of F4/80‐positive macrophages was also higher in livers of IRG1−/− mice than those of WT mice after I/R (Supporting Fig. S1A). Taken together, our results indicate that IRG1 suppresses both injury and inflammation in response to liver I/R injury in vivo.
Figure 2.

Increased systemic inflammation and hepatic neutrophil infiltration in IRG1−/− mice after I/R. (A) ELISA for plasma IL‐6 levels of WT (C57BL/6NJ) and IRG1−/− mice after sham surgery or 1‐hour ischemia and 6‐hour reperfusion; n = 3 in each sham group, n = 6 in each I/R group. (B‐D) RT‐PCR for mRNA expression of IL‐6, MCP‐1, and Ly6G in livers from WT and IRG1−/− mice after sham surgery or 1‐hour ischemia and 6‐hour reperfusion; n = 3 for sham group, n = 5 for I/R group. (E) MPO levels in livers of WT and IRG1−/− mice after sham surgery or 1‐hour ischemia and 6‐hour reperfusion; n = 3 for sham group, n = 5 for I/R group. (F) Immunofluorescence images of livers from WT and IRG1−/− mice after 1‐hour ischemia and 6‐hour reperfusion; liver tissues were stained with Ly6G (white), Hoecsht nuclear stain (blue), and β‐actin (green); images were taken using confocal microscopy (scale bar, 50 µm). (G) Number of infiltrating neutrophils was determined by Ly6G (no.)/total nuclei (no.) and is represented in bar graph; n = 5 for each group. Images are representative of data from multiple mice per experimental group. Data are presented as means ± SD. *P < 0.05. Abbreviation: DAPI, 4´,6‐diamidino‐2‐phenylindole.
Deletion of IRG1 in Either Hematopoietic or Nonhematopoietic Compartments Increases Liver Injury
IRG1 expression is well characterized in myeloid cells.7, 26 However, multiple cell types participate in the adaptive and maladaptive responses in the liver to I/R. Surprisingly, we found that IRG1 expression was up‐regulated in both hepatocytes and NPCs from WT mice subjected to I/R (Fig. 3A). To determine if bone marrow–derived cells and/or non–bone marrow–derived cells were involved in the IRG1‐mediated protection from hepatic I/R injury, we generated bone marrow chimeras by adoptive transfer of donor bone marrow cells into irradiated recipient mice using the following combinations of WT (C57BL/6NJ) and IRG1−/− (KO) mice (recipient/donor): WT/WT, WT/KO, KO/WT, KO/KO. Chimeric mice underwent hepatic I/R 8‐12 weeks after adoptive transfer. Liver injury was assessed by measuring serum ALT and area of necrosis in liver sections stained with H&E. Surprisingly, WT recipients receiving IRG1−/− bone marrow cells (WT/KO) as well as IRG1−/− mice receiving WT bone marrow cells (KO/WT) had higher ALT levels and worse necrosis than WT/WT mice (Fig. 3B‐D). Importantly, KO/KO mice exhibited the most severe liver injury after I/R among the chimeric groups (Fig. 3B‐D), indicating that deletion of IRG1 in both hematopoietic and nonhematopoietic compartments contributes to the protection mediated by IRG1 after I/R. We also confirmed significantly lower itaconate levels in the livers of WT/KO, KO/WT, and KO/KO chimeras after I/R than those of WT/WT mice (Fig. 3E). Taken together, these observations reveal the regulatory role of IRG1 in immune cells in the setting of liver I/R as well as an unrecognized role of IRG1 in hepatocytes.
Figure 3.

Deletion of IRG1 in either hematopoietic or nonhematopoietic compartments increases liver injury. (A) RT‐PCR for IRG1 mRNA expression in hepatocytes and NPCs isolated from WT (C57BL/6NJ) mice after sham surgery (sham) or 1‐hour ischemia and 6‐hour reperfusion (I/R); n = 4 for sham group, n = 5 for I/R group. Chimeric mice were constructed by adoptive transfer of donor bone marrow cells into irradiated recipient animals using the following recipient/donor combinations of WT (C57BL/6NJ) and IRG1−/− (KO) mice: WT/WT, WT/KO, KO/WT, KO/KO. All chimeric mice were monitored 3 times weekly for the first 2 weeks to ensure successful bone marrow engraftment. Then, the chimeric mice underwent hepatic I/R (1 hour/6 hour) after 8‐12 weeks to ensure stable engraftment. (B) Serum ALT levels in each group of chimeric mice after 1‐hour ischemia and 6‐hour reperfusion; n = 5‐6 in each group. (C) Representative liver H&E staining (original magnification ×20) from each group of chimeric mice after 1‐hour ischemia and 6‐hour reperfusion. (D) Dotted lines in liver H&E staining indicate measured areas of necrosis, quantified in bar graph; n = 4 in each group. (E) LC‐MS analysis of itaconate levels in livers from each group of chimeric mice after 1‐hour ischemia and 6‐hour reperfusion; n = 5 for each group. Images are representative of data from multiple mice per experimental group. Data are presented as means ± SD. *P < 0.05, **P < 0.01.
IRG1 Protects Hepatocytes From Oxidative Cell Death In Vivo and In Vitro
To further explore the protective role of IRG1 during oxidative stress in liver cells, we first assessed levels of apoptosis in livers and hepatocytes from WT (C57BL/6NJ) and IRG1−/− mice. We observed more TMR red–positive cells in the livers of IRG1−/− mice after hepatic I/R than in those of WT mice (Fig. 4A,B). This was associated with markedly increased levels of cleaved caspase‐3 and cleaved caspase‐7 in livers of IRG1−/− mice compared to those of WT mice after I/R (Fig. 4C), indicating a higher level of apoptosis in the IRG1‐deficient livers after I/R. To understand the role of IRG1 in hepatocytes under oxidative conditions, we mimicked the I/R process by subjecting mouse primary hepatocytes to 10‐hour hypoxia followed by reoxygenation (H/R) for 0, 4, or 10 hours. Notably, IRG1 expression in hepatocytes was up‐regulated in response to H/R (Fig. 4D,E). LDH release was significantly higher in IRG1−/− hepatocytes after H/R than in WT hepatocytes (Fig. 4F), and this was associated with increased cleaved caspase‐3 and cleaved caspase‐7 assessed by western blot (Fig. 4G). Importantly, restoring IRG1 expression in IRG1−/− hepatocytes by transfection with Myc‐tagged IRG1 plasmids rescued the IRG1−/− hepatocytes from oxidative damage, as determined by lower levels of cleaved caspase‐3 and LDH release under both normoxic and H/R conditions, compared to cells transfected with the empty vector (Fig. 4H‐J). The expression of Myc was used to confirm effective transfection (Fig. 4H). These findings suggest that IRG1 deficiency renders hepatocytes more susceptible to oxidative stress both in vivo and in vitro.
Figure 4.

IRG1 protects hepatocytes from oxidative injury in vivo and in vitro. (A) Confocal microscopic images of TMR (red), Hoecsht nuclear stain (blue), and β‐actin (green) in liver sections from WT (C57BL/6NJ) and IRG1−/− mice subjected to sham surgery or liver ischemia for 1 hour and reperfusion for 6 hours (scale bar, 50 µm), (B) Percentage of TMR red–positive cells was quantified and is represented in bar graph; n = 4 per group. (C) Western blot for apoptosis markers (cleaved caspase‐3, cleaved caspase‐7, etc.) in whole‐liver lysates from WT and IRG1−/− mice subjected to sham surgery or liver I/R (1 hour/6 hours). (D) RT‐PCR for IRG1 mRNA expression in WT hepatocytes subjected to normoxia (control) or 10‐hour hypoxia and 0‐hour, 4‐hour, or 10‐hour reoxygenation (10/0, 10/4, 10 hours/10 hours); n = 5 for each group. (E) IRG1 protein level in WT hepatocytes subjected to normoxia (control) or 10‐hour hypoxia and 0‐hour, 4‐hour, or 10‐hour reoxygenation (10/0, 10/4, 10 hours/10 hours); n = 6 for each group. (F) LDH release (lytic cell death) in WT (C57BL/6NJ) and IRG1−/− hepatocytes subjected to normoxia or 10‐hour hypoxia and 10‐hour reoxygenation (H/R); n = 8 for normoxia group, n = 24 for H/R group. (G) Western blot for apoptosis markers (cleaved caspase‐3, cleaved caspase‐7, etc.) in whole‐cell lysates from WT and IRG1−/− hepatocytes after normoxia or 10‐hour hypoxia and 10‐hour reoxygenation (H/R). (H,I) Western blot for Myc‐Tag, cleaved caspase‐3, and caspase‐3 in whole‐cell lysates from IRG1−/− hepatocytes transfected with IRG1‐expressing plasmid (IRG1) or empty vector followed by normoxia or H/R (10 hour/10 hour). (J) LDH release in IRG1−/− hepatocytes transfected with IRG1 plasmid or empty vector followed by H/R (10 hour/10 hour); n = 8 in each group. Images are representative of data from multiple mice per experimental group. Data are presented as means ± SD. *P < 0.05, **P < 0.01. Abbreviations: cl, cleaved; NS, not significant.
Itaconate Ameliorates Hepatic I/R Injury and Rescues Hepatocytes From Injury Resulting From H/R
Having established that IRG1 is part of a protective response to I/R injury, we then sought to determine if the enzymatic product of IRG1, itaconate, also protected the liver from I/R. To do this, we used 4‐OI, a recently described synthesized cell‐permeable itaconate derivative in our in vivo and in vitro models.12 Administration of 4‐OI in the I/R model significantly reduced hepatic injury in both control (C57BL/6NJ) and IRG1−/− mice compared with mice subjected to I/R and receiving vehicle treatment. Both the circulating ALT levels and histologic examination of liver necrosis were lower in mice receiving 4‐OI at 6 hours of reperfusion (Fig. 5A‐C). Similarly, 4‐OI treatment reduced the apoptotic cell death in livers after I/R compared with vehicle treatment, as determined by lower numbers of TMR red–positive cells observed in livers of 4‐OI‐treated mice (Supporting Fig. S1B). Consistent with these in vivo observations, 4‐OI significantly reduced LDH release by cultured WT hepatocytes after H/R (Fig. 5D) in association with less cleaved caspase‐3 and caspase‐7, as assessed by western blot (Fig. 5E,F). This hepatocellular protective effect of 4‐OI could also be observed in IRG1−/− hepatocytes subjected to H/R (Fig. 5G,H). We also found that IRG1‐deficient hepatocytes up‐regulated caspase‐3 activation to a greater degree after tumor necrosis factor alpha exposure in vitro than WT hepatocytes, suggesting that IRG1 may play a protective role in other settings of liver injury (Supporting Fig. S2A). Together, these data show that itaconate can protect hepatocytes from oxidative cell death both in vivo and in vitro.
Figure 5.
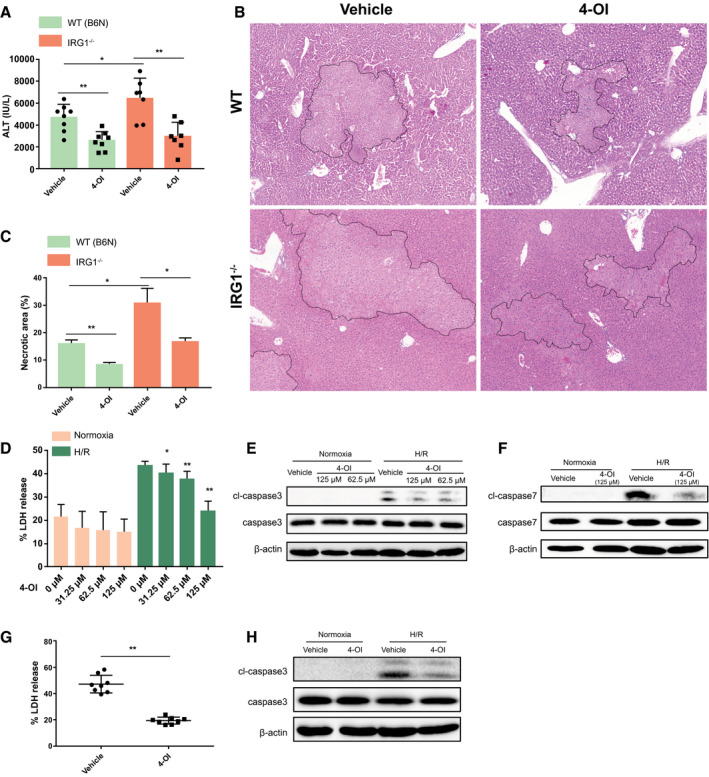
Itaconate ameliorates hepatic I/R injury and rescues hepatocytes from H/R. WT (C57BL/6NJ, B6N) and IRG1−/− mice were subjected to ischemia for 1 hour and then reperfusion for 6 hours. 4‐OI or vehicle control (25 mg/kg body weight) was injected intraperitoneally 2 hours before hepatic I/R and at the time of reperfusion. (A) Serum ALT levels in each group of mice after 1‐hour ischemia and 6‐hour reperfusion; n = 7‐8 in each group. (B) Representative liver H&E staining (original magnification ×20) from each group of mice after 1‐hour ischemia and 6‐hour reperfusion. (C) Dotted lines in liver H&E staining indicate measured areas of necrosis, quantified in bar graph; n = 3 in each group. Primary hepatocytes isolated from WT mice were pretreated with 4‐OI or vehicle control 1 hour prior to normoxia or 10‐hour hypoxia and 10‐hour reoxygenation (H/R). (D) Cell death was assessed by LDH release from hepatocytes after normoxia or H/R; n = 8 for each group. (E,F) Western blot for apoptosis markers (cleaved caspase‐3, cleaved caspase‐7, etc.) in whole‐cell lysates from hepatocytes after normoxia or H/R. Primary hepatocytes isolated from IRG1−/− mice were pretreated with 4‐OI (125 µM) or vehicle control 1 hour prior to normoxia or 10‐hour hypoxia and 10‐hour reoxygenation (H/R). (G) Cell death was accessed by LDH release from hepatocytes after H/R; n = 8 for each group. (H) Western blot for cleaved caspase‐3 and caspase‐3 in whole‐cell lysates from hepatocytes after normoxia or H/R. Images are representative of data from multiple mice per experimental group. Data are presented as means ± SD. *P < 0.05, **P < 0.01. Abbreviation: cl, cleaved.
Deficiency of IRG1 Suppressed Nrf2 Pathway Activation in Hepatocytes After Oxidative Stress
Nrf2, a master cellular regulator of redox homeostasis, has been shown to protect against liver I/R injury.19, 20 A recent study suggested that Nrf2 is involved in the anti‐inflammatory actions of itaconate in macrophages during endotoxemia.12 Given these findings, we hypothesized that IRG1‐mediated hepatoprotection in liver I/R occurs by up‐regulation of Nrf2–antioxidant pathways in hepatocytes in the setting of oxidative stress. To test this hypothesis, we first assessed the Nrf2 expression in livers of mice subjected to hepatic I/R. As expected, Nrf2 expression increased in livers of WT (C57BL/6NJ) mice after I/R. In contrast, Nrf2 expression was not increased in livers of IRG1−/− mice subjected to I/R (Fig. 6A). In addition, levels of HO‐1 and NQO1, both downstream of Nrf2, were lower in livers of IRG1−/− mice compared with WT mice following I/R (Fig. 6B). Importantly, we observed more lipid peroxidation in the livers of IRG1−/− mice after hepatic I/R than in those of WT mice, as measured by 4‐HNE staining (Fig. 6C). In vitro, we also found lower levels of Nrf2 in hepatocytes isolated from IRG1−/− mice compared with those from WT mice after H/R (Fig. 6D). Importantly, the nuclear translocation of Nrf2 was attenuated in IRG1−/− hepatocytes after H/R (Fig. 6E). The expression of HO‐1 and NQO1 in hepatocytes from IRG1−/− mice was significantly lower when compared with WT hepatocytes following H/R (Fig. 6F). Consistent with the in vivo results, ROS generation in IRG1−/− hepatocytes was found to be significantly higher after H/R compared with WT hepatocytes (Fig. 6G). These data indicate an important role for IRG1 in regulating antioxidant responses in hepatocytes during I/R.
Figure 6.
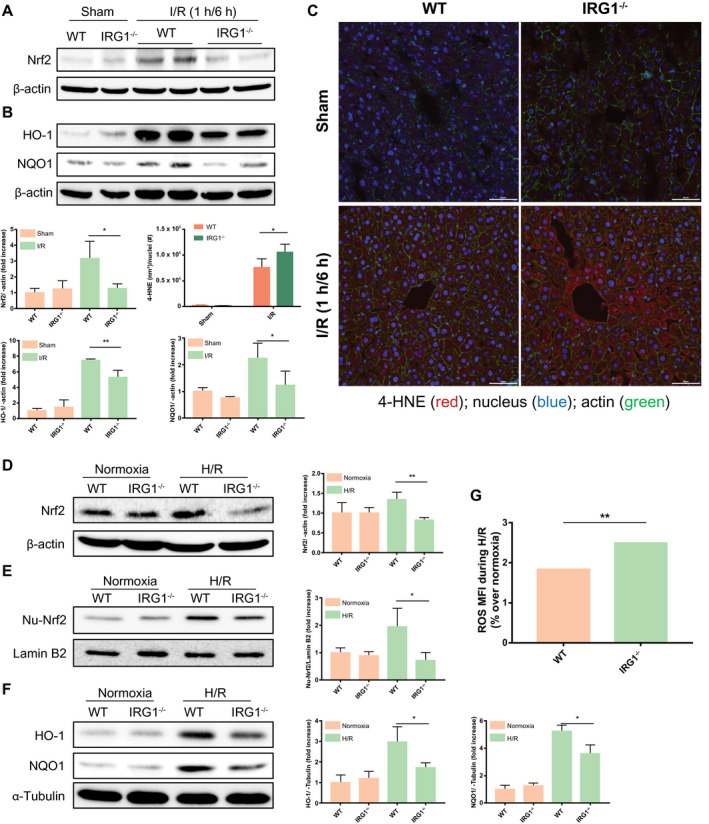
Deficiency of IRG1 suppressed Nrf2 pathway activation in hepatocytes after oxidative stress. (A,B) Western blot for Nrf2 and its downstream mediators (HO‐1 and NQO1) in whole‐liver lysates from WT (C57BL/6NJ) and IRG1−/− mice subjected to sham surgery or liver I/R (1 hour/6 hours). Quantitation of band density was performed across at least three separate blots and is presented in bar graph. (C) Confocal microscopic images of 4‐HNE (red), Hoecsht nuclear stain (blue), and β‐actin (green) in liver sections from WT and IRG1−/− mice subjected to sham surgery or liver ischemia for 1 hour and reperfusion for 6 hours (scale bar, 50 µm). Area of 4‐HNE staining was quantified by total nuclei number and is represented in bar graph; n = 3 for sham group, n = 4 for I/R group. (D) Western blot for Nrf2 protein in whole‐cell lysates from WT (C57BL/6NJ) and IRG1−/− hepatocytes subjected to normoxia or 10‐hour hypoxia and 10‐hour reoxygenation (H/R). (E) Total nuclear protein was extracted from WT and IRG1−/− hepatocytes after normoxia or H/R (10 hours/10 hours); nuclear Nrf2 was assessed by western blot. (F) Western blot for HO‐1 and NQO1 in whole‐cell lysates from WT and IRG1−/− hepatocytes subjected to normoxia or 10‐hour hypoxia and 10‐hour reoxygenation. Quantitation of band density above was performed across at least three separate blots and is presented in bar graph. (G) Cellular ROS was measured by DCFDA mean fluorescence intensity. Results were presented as percentage change of ROS (over respective normoxic controls) in hepatocytes subjected to H/R (10 hours/10 hours); n = 8 for each group. Images are representative of data from multiple mice per experimental group. Data are presented as means ± SD. *P < 0.05, **P < 0.01. Abbreviations: MFI, mean fluorescence intensity; Nu, nuclear.
Itaconate Protects Hepatocytes Against Oxidative Stress Damage in an Nrf2‐Dependent Manner
We next assessed whether treatment with exogenous itaconate induces Nrf2 activation in hepatocytes during H/R. Pretreatment with 4‐OI increased the expression of Nrf2 and its downstream target genes (HO‐1 and NQO1) in WT hepatocytes in a dose‐dependent manner both under normoxic conditions and after H/R (Fig. 7A,B; Supporting Fig. S2B‐D). Similarly, treatment with 4‐OI boosted the nuclear translocation of Nrf2 in hepatocytes before and after H/R (Fig. 7C; Supporting Fig. S2E). Furthermore, production of ROS in hepatocytes pretreated with 4‐OI was markedly reduced after H/R (Fig. 7D). To further determine whether 4‐OI is protective through a mechanism that is dependent on Nrf2, WT (C57BL/6J) and Nrf2−/− mice received either 4‐OI or vehicle control and then were subjected to I/R. As expected, Nrf2−/− mice exhibited more liver damage than WT mice in the vehicle‐treated groups, as assessed by a higher level of circulating ALT and greater liver necrosis after I/R (Fig. 7E‐G). Importantly, 4‐OI treatment significantly reduced hepatic injury in WT mice after I/R but had no effect on I/R‐induced liver injury in Nrf2−/− mice (Fig. 7E‐G). Furthermore, this phenomenon was also observed in cultured hepatocytes pretreated with 4‐OI in vitro, as assessed by LDH release by WT (C57BL/6J) and Nrf2−/− hepatocytes after H/R (Fig. 7H). In addition, 4‐OI treatment did not change the expression of Nrf2 and HO‐1 in Nrf2−/− hepatocytes as seen in WT hepatocytes (Fig. 7I; Supporting Fig. S2F,G), suggesting that the protective effect of 4‐OI against oxidative stress damage in hepatocytes is mediated by Nrf2. We also confirmed that 4‐OI increased the expression of Nrf2 and HO‐1 in primary human hepatocytes both under normoxic conditions and after H/R (Fig. 7J; Supporting Fig. S2H,I). Taken together, our results indicate that itaconate is an important inducer of Nrf2‐mediated antioxidative response in hepatocytes during hepatic I/R injury.
Figure 7.
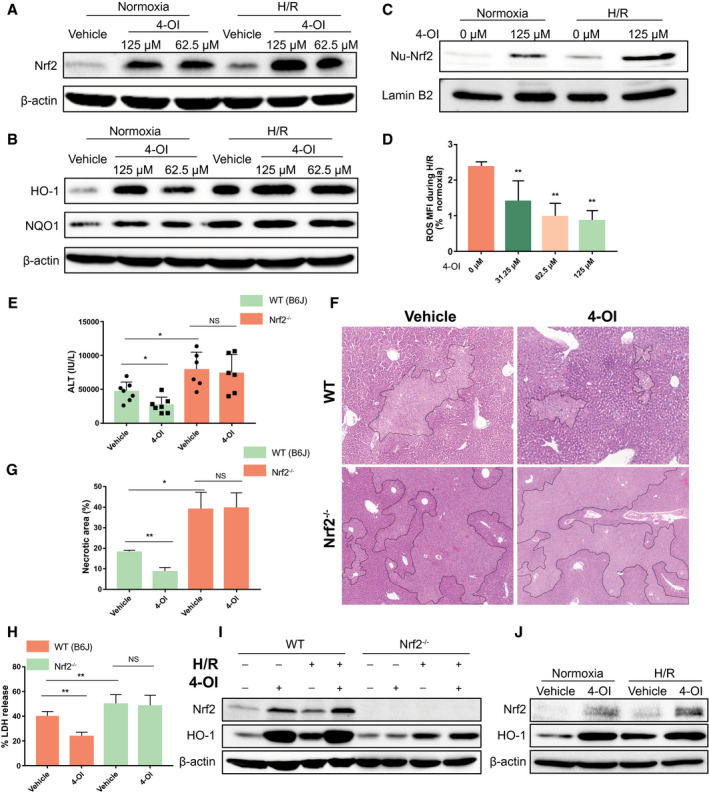
Itaconate protects hepatocytes against oxidative stress damage in an Nrf2‐dependent manner. Primary hepatocytes isolated from WT (C57BL/6J) mice were pretreated with 4‐OI or vehicle control 1 hour prior to normoxia or 10‐hour hypoxia and 10‐hour reoxygenation (H/R). (A,B) Western blot for Nrf2 and its downstream mediators (HO‐1 and NQO1) in whole‐cell lysates from hepatocytes subjected to normoxia or H/R. (C) Total nuclear protein was extracted from hepatocytes after normoxia or H/R. Nuclear Nrf2 was assessed by western blot. (D) Cellular ROS was measured by DCFDA mean fluorescence intensity. Results were presented as percentage change of ROS (over respective normoxic controls) in hepatocytes subjected to H/R; n = 8 for each group. WT (C57BL/6J, B6J) and Nrf2−/− mice were subjected to ischemia for 1 hour and then reperfusion for 6 hours (I/R). 4‐OI or vehicle control (25 mg/kg body weight) was injected intraperitoneally 2 hours before hepatic I/R and at the time of reperfusion. (E) Serum ALT levels in each group of mice after hepatic I/R; n = 6‐7 in each group. (F) Representative liver H&E staining (original magnification ×20) from each group of mice after hepatic I/R. (G) Dotted lines in liver H&E staining indicate measured areas of necrosis, quantified in bar graph; n = 3 in each group. Primary hepatocytes isolated from WT (C57BL/6J, B6J) and Nrf2−/− mice were pretreated with 4‐OI (125 µM) or vehicle control 1 hour prior to normoxia or H/R (10 hours/10 hours). (H) Cell death was assessed by LDH release from hepatocytes after normoxia or H/R; n = 8 for each group. (I) Western blot for Nrf2 and HO‐1 in whole‐cell lysates from hepatocytes after normoxia or H/R. (J) Western blot for Nrf2 and HO‐1 in whole‐cell lysates from primary human hepatocytes pretreated with 4‐OI (125 µM) or vehicle control 1 hour before normoxia or 10‐hour hypoxia and 10‐hour reoxygenation (H/R). Images are representative of data. Data are presented as means ± SD. *P < 0.05, **P < 0.01. Abbreviations: MFI, mean fluorescence intensity; NS, not significant; Nu, nuclear.
Discussion
The finding that IRG1 catalyzes the synthesis of itaconate from cis‐aconitate identified a function for this protein known to be among the most highly induced in lipopolysaccharide‐treated macrophages.6 Functions ascribed to itaconate include the inhibition of succinate dehydrogenase and the up‐regulation of the antioxidant transcription factor Nrf2.11, 12 In experiments aimed at determining the role of the IRG1/itaconate pathway in liver I/R, we found that IRG1 is markedly up‐regulated during liver I/R and in hepatocytes exposed to oxidant stress and that IRG1 and itaconate protected hepatocytes from oxidant‐associated cell death. We confirmed that Nrf2 also protects the liver and hepatocytes from ischemic injury and linked the protective effects of IRG1 and itaconate to Nrf2 signaling. These findings show that IRG1 plays critical roles in antioxidant defenses in hepatocytes by Nrf2.
Although IRG1 and itaconate have been well described in macrophages,27, 28 the expression and functions of this pathway in nonimmune cells are largely unknown. IRG1 was found to be highly expressed in the uterine epithelium of mice undergoing early pregnancy9, 10 where it is important to embryonic implantation.9 IRG1 expression was also induced in alveolar epithelial cells after respiratory syncytial virus infection29 and in glioma cells.30 Here, we expand the cell types known to express IRG1 by showing that hepatocytes rapidly up‐regulate IRG1 in response to H/R. These results document the critical role of IRG1 in the acute setting of sterile injury induced by I/R. Whether inducible IRG1 expression is typical of epithelial cells is unknown.
IRG1 links cellular metabolism with immune defense by catalyzing itaconic acid production, and metabolic reprogramming is emerging as a key hallmark of innate immunity.6 In parallel with the enhanced glycolysis, a disrupted TCA cycle is an important feature of inflammatory macrophages where the accumulation of metabolic intermediates, such as citrate, succinate, and itaconate, functions as a regulator of inflammatory responses.31, 32 Isocitrate dehydrogenase expression is inhibited in inflammatory macrophages, and this leads to the accumulation of citrate, which contributes to lipogenesis and the production of nitric oxide. Citrate can also be redirected to itaconate by IRG1. Itaconate has been demonstrated to have anti‐inflammatory properties by inhibiting the activity of succinate dehydrogenase, an enzyme critical for driving the inflammatory response by oxidizing succinate.11, 33 Moreover, a recent study demonstrates that a cell‐permeable derivative of itaconate can dampen the sustained inflammatory response through activation of the transcription factor Nrf2.12 Because IRG1 expression by both bone marrow–derived and non–bone marrow–derived cells contributed to the IRG1‐mediated protection in liver I/R, it is likely that the described pathways for IRG1 observed in macrophages contributed to the protection. However, it would seem that an additional important mechanism of protection is through the induction of Nrf2‐dependent antioxidant pathways in hepatocytes.
Dimethyl itaconate (DI), a cell membrane–permeable itaconate derivative, has been used as an exogenous itaconate surrogate in studies exploring the role of IRG1.11, 13 However, a recent finding revealed that DI is not metabolized into itaconate within cells.34 To overcome the limitations of DI, a cell‐permeable itaconate derivative, 4‐OI, was developed, making it a suitable cell‐permeable itaconate surrogate with which to probe the physiological function of itaconate.12 In our study, we demonstrated that treatment with 4‐OI ameliorated hepatic I/R injury in vivo and rescued hepatocytes from injury resulting from H/R in vitro; thus, it may provide a basis for the development of 4‐OI for the prevention of hepatic I/R injury.
Nrf2 is a transcription factor associated with intracellular signaling that protects organs against oxidative stress.35, 36, 37 A number of studies indicate that depletion of Nrf2 increases susceptibility to toxin‐induced liver injury,38, 39, 40 all of which provide strong evidence for Nrf2 as a hepatoprotective pathway. Additionally, it has been shown that Nrf2 activation protects the liver from I/R injury in mice.18, 20, 41, 42, 43, 44 Specifically, the effect of cell‐specific Nrf2 induction was also explored, and activation of the Nrf2–antioxidant response element pathway in either hepatocytes18, 20, 43 or myeloid cells44 decreases hepatocellular damage, apoptosis, inflammation, and oxidative stress after hepatic I/R injury. Our work provides support for the recent findings that itaconate can activate Nrf2‐driven signaling.12 Interestingly, we demonstrate that treatment with 4‐OI increases the expression and nuclear translocation of Nrf2 as well as up‐regulation of its downstream protective pathways (HO‐1 and NQO1) not only in mouse hepatocytes but also in primary human hepatocytes, both under basic conditions and after H/R.
In summary, our study reveals a function for IRG1 and its enzymatic product itaconate in the regulation of the protective anti‐inflammatory and antioxidative responses to liver I/R. Importantly, our study reveals an unappreciated hepatoprotective role of hepatocyte‐specific IRG1 during liver I/R injury by activating Nrf2 antioxidative response in hepatocytes. Our study also provides evidence that administration of 4‐OI can reduce hepatic I/R injury, and this may be a potential pharmacological strategy to prevent this complication in the clinical setting.
Author Contributions
M.D., F.H. and T.R.B. designed the study; Z.Y., G.F., P.A.L., S.L., P.S. conducted experiments; Z.Y., Z.L., C.Y., W.L., H.X. analyzed data; M.D., M.J.S., F.H. and T.R.B. interpreted results of experiments; ZY generated figures and drafted manuscript; M.D., M.J.S. and T.R.B. edited and revised manuscript; All authors reviewed and approved final version of manuscript.
Supporting information
Acknowledgment
We thank Stacy Gelhaus Wendell, Hong Liao, Danielle Reiser, and Richard A. Shapiro for technical assistance with in vivo and in vitro experiments.
Potential conflict of interest: Nothing to report.
Contributor Information
Feizhou Huang, Email: huangfeizhou@csu.edu.cn.
Timothy R. Billiar, Email: billiartr@upmc.edu.
REFERENCES
Author names in bold designate shared co‐first authorship.
- 1. Busuttil R. Liver ischaemia and reperfusion injury. Br J Surg 2010;94:787‐788. [DOI] [PubMed] [Google Scholar]
- 2. Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: present concepts. J Gastroenterol Hepatol 2011;26:173‐179. [DOI] [PubMed] [Google Scholar]
- 3. Hoek JB, Pastorino JG. Ethanol, oxidative stress, and cytokine‐induced liver cell injury. Alcohol 2002;27:63‐68. [DOI] [PubMed] [Google Scholar]
- 4. Jadeja RN, Upadhyay KK, Devkar RV, Khurana S. Naturally occurring Nrf2 activators: potential in treatment of liver injury. Oxid Med Cell Longev 2016;2016:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol 2017;38:395‐406. [DOI] [PubMed] [Google Scholar]
- 6. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune‐responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci USA 2013;110:7820‐7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Degrandi D, Hoffmann R, Beuter‐Gunia C, Pfeffer K. The proinflammatory cytokine‐induced IRG1 protein associates with mitochondria. J Interferon Cytokine Res 2009;29:55‐68. [DOI] [PubMed] [Google Scholar]
- 8. Basler T, Jeckstadt S, Valentin‐Weigand P, Goethe R. Mycobacterium paratuberculosis, Mycobacterium smegmatis, and lipopolysaccharide induce different transcriptional and post‐transcriptional regulation of the IRG1 gene in murine macrophages. J Leukoc Biol 2006;79:628‐638. [DOI] [PubMed] [Google Scholar]
- 9. Cheon Y‐P, Xu X, Bagchi MK, Bagchi IC. Immune‐responsive gene 1 is a novel target of progesterone receptor and plays a critical role during implantation in the mouse. Endocrinology 2003;144:5623‐5630. [DOI] [PubMed] [Google Scholar]
- 10. Chen B, Zhang D, Pollard JW. Progesterone regulation of the mammalian ortholog of methylcitrate dehydratase (immune response gene 1) in the uterine epithelium during implantation through the protein kinase C pathway. Mol Endocrinol 2003;17:2340‐2354. [DOI] [PubMed] [Google Scholar]
- 11. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab 2016;24:158‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti‐inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018;556:113‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ–ATF3 inflammatory axis. Nature 2018;556:501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nair S, Huynh JP, Lampropoulou V, Loginicheva E, Esaulova E, Gounder AP, et al. Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. J Exp Med 2018;215:1035‐1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Itoh K, Mimura J, Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal 2010;13:1665‐1678. [DOI] [PubMed] [Google Scholar]
- 16. Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1–Nrf2 pathway. Trends Pharmacol Sci 2013;34:340‐346. [DOI] [PubMed] [Google Scholar]
- 17. Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke–induced emphysema in mice. J Clin Invest 2004;114:1248‐1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ke B, Shen X‐D, Zhang Y, Ji H, Gao F, Yue S, et al. KEAP1–NRF2 complex in ischemia‐induced hepatocellular damage of mouse liver transplants. J Hepatol 2013;59:1200‐1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shin SM, Yang JH, Ki SH. Role of the Nrf2–ARE pathway in liver diseases. Oxid Med Cell Longev 2013;2013:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ge M, Yao W, Yuan D, Zhou S, Chen X, Zhang Y, et al. Brg1‐mediated Nrf2/HO‐1 pathway activation alleviates hepatic ischemia–reperfusion injury. Cell Death Dis 2017;8:e2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia–reperfusion. J Exp Med 2005;201:1135‐1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deng M, Loughran PA, Zhang L, Scott MJ, Billiar TR. Shedding of the tumor necrosis factor (TNF) receptor from the surface of hepatocytes during sepsis limits inflammation through cGMP signaling. Sci Signal 2015;8:ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lei Z, Deng M, Yi Z, Sun Q, Shapiro RA, Xu H, et al. cGAS‐mediated autophagy protects the liver from ischemia–reperfusion injury independently of STING. Am J Physiol Gastrointest Liver Physiol 2018;314:G655‐G667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun Q, Gao W, Loughran P, Shapiro R, Fan J, Billiar TR, et al. Caspase 1 activation is protective against hepatocyte cell death by up‐regulating beclin 1 protein and mitochondrial autophagy in the setting of redox stress. J Biol Chem 2013;288:15947‐15958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klune JR, Tsung A. Molecular biology of liver ischemia/reperfusion injury: established mechanisms and recent advancements. Surg Clin North Am 2010;90:665‐677. [DOI] [PubMed] [Google Scholar]
- 26. Li Y, Zhang P, Wang C, Han C, Meng J, Liu X, et al. Immune responsive gene 1 (IRG1) promotes endotoxin tolerance by increasing A20 expression in macrophages through reactive oxygen species. J Biol Chem 2013;288:16225‐16234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rodríguez N, Mages J, Dietrich H, Wantia N, Wagner H, Lang R, et al. MyD88‐dependent changes in the pulmonary transcriptome after infection with Chlamydia pneumoniae . Physiol Genomics 2007;30:134‐145. [DOI] [PubMed] [Google Scholar]
- 28. Preusse M, Tantawy MA, Klawonn F, Schughart K, Pessler F. Infection‐ and procedure‐dependent effects on pulmonary gene expression in the early phase of influenza A virus infection in mice. BMC Microbiol 2013;13:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ren K, Lv Y, Zhuo Y, Chen C, Shi H, Guo L, et al. Suppression of IRG‐1 reduces inflammatory cell infiltration and lung injury in respiratory syncytial virus infection by reducing production of reactive oxygen species. J Virol 2016;90:7313‐7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pan J, Zhao X, Lin C, Xu H, Yin Z, Liu T, et al. Immune responsive gene 1, a novel oncogene, increases the growth and tumorigenicity of glioma. Oncol Rep 2014;32:1957‐1966. [DOI] [PubMed] [Google Scholar]
- 31. Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O’Neill LA, et al. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat Metab 2019;1:16‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murphy MP, O’Neill LA. Krebs cycle reimagined: the emerging roles of succinate and itaconate as signal transducers. Cell 2018;174:780‐784. [DOI] [PubMed] [Google Scholar]
- 33. Tannahill G, Curtis A, Adamik J, Palsson‐McDermott E, McGettrick A, Goel G, et al. Succinate is an inflammatory signal that induces IL‐1β through HIF‐1α. Nature 2013;496:238‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. ElAzzouny M, Tom CT, Evans CR, Olson LL, Tanga MJ, Gallagher KA, et al. Dimethyl itaconate is not metabolized into itaconate intracellularly. J Biol Chem 2017;292:4766‐4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest 2016;116:984‐995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wei Y, Gong J, Yoshida T, Eberhart CG, Xu Z, Kombairaju P, et al. Nrf2 has a protective role against neuronal and capillary degeneration in retinal ischemia–reperfusion injury. Free Radic Biol Med 2011;51:216‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tanaka N, Ikeda Y, Ohta Y, Deguchi K, Tian F, Shang J, et al. Expression of Keap1–Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res 2011;1370:246‐253. [DOI] [PubMed] [Google Scholar]
- 38. Enomoto A, Itoh K, Nagayoshi E, Haruta J, Kimura T, O’Connor T, et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE‐regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci 2001;59:169‐177. [DOI] [PubMed] [Google Scholar]
- 39. Zhang Y‐KJ, Yeager RL, Tanaka Y, Klaassen CD. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine‐ and choline‐deficient diet. Toxicol Appl Pharmacol 2010;245:326‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morito N, Yoh K, Itoh K, Hirayama A, Koyama A, Yamamoto M, et al. Nrf2 regulates the sensitivity of death receptor signals by affecting intracellular glutathione levels. Oncogene 2003;22:9275‐9281. [DOI] [PubMed] [Google Scholar]
- 41. Kudoh K, Uchinami H, Yoshioka M, Seki E, Yamamoto Y. Nrf2 activation protects the liver from ischemia/reperfusion injury in mice. Ann Surg 2014;260:118‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu D, Chen L, Chen X, Wen Y, Yu C, Yao J, et al. The triterpenoid CDDO‐imidazolide ameliorates mouse liver ischemia–reperfusion injury through activating the Nrf2/HO‐1 pathway enhanced autophagy. Cell Death Dis 2017;8:e2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee L‐Y, Harberg C, Matkowskyj KA, Cook S, Roenneburg D, Werner S, et al. Over‐activation of the Nrf2–antioxidant response element pathway in hepatocytes decreases hepatic ischemia reperfusion injury in mice. Liver Transpl 2016;22:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee LY, Harberg C, Matkowskyj KA, Cook S, Roenneburg D, Werner S, et al. Cell‐specific overactivation of nuclear erythroid 2 p45‐related factor 2–mediated gene expression in myeloid cells decreases hepatic ischemia/reperfusion injury. Liver Transpl 2016;22:1115‐1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials