

Abstract
Organoboron compounds are essential reagents in modern C−C coupling reactions. Their synthesis via catalytic C−H borylation by main group elements is emerging as a powerful tool alternative to transition metal based catalysis. Herein, a straightforward metal‐free synthesis of aryldifluoroboranes from BF3 and heteroarenes is reported. The reaction is assisted by sterically hindered amines and catalytic amounts of thioureas. According to computational studies the reaction proceeds via frustrated Lewis pair (FLP) mechanism. The obtained aryldifluoroboranes are further stabilized against destructive protodeborylation by converting them to the corresponding air stable tetramethylammonium organotrifluoroborates.
Keywords: boron trifluoride, borylation, C−H activation, frustrated Lewis pairs, protodeboronation
C−H activation: Indoles, pyrroles and indolenines undergo C−H borylation by boron trifluoride with assistance of base and thiourea additives.
Introduction
Efficient C−C and C−X couplings in contemporary organic synthesis including Suzuki–Miyaura coupling, Petasis reaction, Chan–Lam coupling etc., are performed by utilizing various organoboron compounds. [1] Consequently, a ready access to initial organoboron reagents is prerequisite in such cross‐coupling strategies. Among various organoboron compounds, organotrifluoroborates are widely used due to their increased stability under air and, especially, against protodeboronation. Generally, they are synthesized from boronic acids or their esters via fluorination with KHF2. [2] In turn, synthesis of organoboronic acids and organoboronates is widely studied including both transition metal [3] and main group catalysis. [4] However, direct synthesis of organotrifluoroborates by C−H borylation is still underdeveloped.
Borylation of C(sp2)−H bonds was traditionally performed by transition metal catalysts [5] until it was recently shown that geometrically arranged ansa‐aminoboranes, [6] usually used for hydrogen activation and reductions, are also capable of activating certain electron‐rich heteroarenes. [7] After that, substrate scope was notably expanded [6c] and several reports were published concerning the structure–activity correlation of such ansa‐systems. [8] Beside ansa‐aminoboranes, it is also possible to activate C(sp2)−H bonds by strong Lewis acids such as B(C6F5)3 [9] or BX3 (X=Cl, Br).[ 4c , 10 ] Depending on the conditions, reactions proceed either by direct electrophilic attack of the boron halide [10e] or by in situ generated borenium cations derived from BCl3,[ 4c , 10b , 11 ] BBr3,[ 10a , 10d , 10e , 11 ] or catecholborane. [12] Regardless of the mechanisms, such borylation reactions with BF3, the weakest among boron halides Lewis acid, has not been reported to the best of our knowledge.
Recently, we reported that combination of BF3⋅SMe2 with 1,2,2,6,6‐pentamethylpiperidine (PMP, 4 a) borylates terminal alkynes forming tri‐ and tetraalkynyl borates. [13] Such approach avoids usage of precious metals and utilizes one of inexpensive boron reagents along with the recoverable base. Also, we found that BF3⋅SMe2 adduct is very labile and can serve as the convenient alternative to reactive gaseous BF3. In continuation of our efforts, we focused on extending developed methodology to the borylation of more challenging C(sp2)−H bonds.
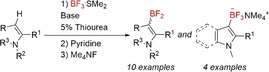
Herein, we present the direct C−H borylation of electron rich N‐heterocycles such as indoles, pyrroles, and indolenines by BF3⋅SMe2. These reactions are assisted by sterically hindered amines and promoted by catalytic amounts of various thioureas (Scheme 1). Borylation products, aryldifluoroboranes, are converted to organotrifluoroborates via complexation with pyridine and further fluorination by tetramethylammonium fluoride. Such procedure overcomes the problem of unwanted protodeboronation of organofluoroboranes and makes them practical starting materials for the C−C coupling reactions.
Scheme 1.
General pattern of N‐heteroarenes borylation by boron trifluoride.
Results and Discussion
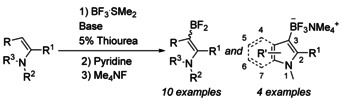
In our previous studies we showed that BF3 together with sterically hindered base, 4 a, can borylate phenylacetylene quantitatively within 15 minutes. [13] The BF3⋅SMe2/4 a pair was also proven efficient, yet full conversions were reached after 2 hours. When these Lewis pairs were applied to our model substrate, N‐methylindole 1 a, we achieved 62–65 % in situ yields of aryldifluoroborane 2 a and minor amounts of bis(indolyl) product 3 a (Scheme 2). Because of possibility of indole borylation to both 2nd and 3rd positions, regioselectivity of borylation was confirmed by X‐ray diffraction and 2D NMR spectra of the corresponding trifluoroborate (see Supporting Information for details).
Scheme 2.
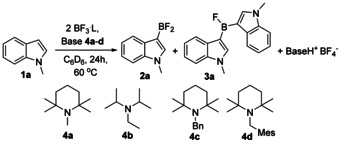
N‐Methylindole 1 a borylation with BF3⋅L and sterically hindered amines 4 a–d gives 3‐difluoroborane 2 a and minor amounts of 3 a.
A mixture of BF3⋅OEt2 and PMP was shown to borylate terminal acetylenes to a remarkable degree, [13] whereas only traces of 2 a were observed with this reagent combination (entry 3). Addition of SMe2 to the reaction mixture didn't enhance the reactivity of the BF3⋅OEt2/PMP system (entry 4). When the reaction temperature was raised to 120 °C, the in situ yield of 2 a raised to 30 % (entry 5). According to the previous studies, a higher energetic penalty required for dissociation of BF3⋅OEt2 results in the poor reactivity of this adduct that sharply contrasts with labile and reactive BF3⋅SMe2 and BF3⋅PMP (Table 1, column 4). [13]
Table 1.
In situ yields of the borylation product 2 a produced in the reactions of 1 a, BF3⋅L, and sterically hindered amines 4 a–d [a]
|
Entry |
Base |
L |
Strength of amine‐BF3 adduct, kcal mol−1[b] |
1 a, % |
2 a, %[c,d] |
|---|---|---|---|---|---|
|
1 |
4 a |
SMe2 |
−1.9 |
28 |
62 |
|
2 |
4 a |
4 a |
−1.9 |
20 |
65 |
|
3 |
4 a |
OEt2 |
−1.9 |
94 |
5 |
|
4[e] |
4 a |
OEt2 |
−1.9 |
83 |
3 |
|
5[f] |
4 a |
OEt2 |
−1.9 |
40 |
30 |
|
6 |
4 b |
SMe2 |
−7.8 |
55 |
22 |
|
7 |
4 b |
4 b |
−7.8 |
100 |
0 |
|
8 |
4 c |
SMe2 |
–[g] |
55 |
35 |
|
9 |
4 d |
SMe2 |
–[g] |
43 |
47 |
[a] Reactions were performed in the gas‐tight NMR tubes for 24 h at 60 °C in the argon atmosphere; Yields were determined by the 1H NMR spectroscopy against 1‐bromo‐3,5‐difluorobenzene as the internal standard. [b] Defined as ΔG of amine+BF3→amine‐BF3 as obtained from ref. [13]. [c] Characteristic signal of 2 a in 11B and 19F NMR spectra was observed at +24 ppm and −94 ppm, respectively. [d] In addition to 1 a and 2 a, some amount of unidentified product with indole pattern was observed in 1H NMR spectra (see Supporting Information for details). [e] 1 equiv. of SMe2 was added to the reaction. [f] Reaction temperature 120 °C. [g] No adduct with gaseous BF3 was detected.
We also explored the borylation of 1 a using other sterically hindered amines (Scheme 2, Table 1). Although 4 a is a sterically very demanding amine, it is capable of forming a weak Lewis adduct with BF3 (Entry 1). When sterically less hindered and hence more strongly interacting with BF3 N,N‐diisopropylethylamine (DIPEA, 4 b) was applied in combination with BF3⋅SMe2, the yield of 2 a reached only 22 % (entry 6). Moreover, when all the BF3 in the solution was in the form of adduct with 4 b, not even traces of borylation products were found (entry 7). Sterically more hindered than 4 a bases 4 c and 4 d did not form stable adducts even with gaseous BF3 (entries 8–9). However, borylation with 4 c–d was less effective as compared to 4 a, yet all the bases 4 a–d have similar Brønsted basicities. We hypothesized that 4 c and 4 d had stronger steric repulsive component in the borylation transitions states (vide infra) that conveyed into higher kinetic barriers in comparison to 4 a.
To gain mechanistic insight, computational studies of the borylation reaction of 1 a with BF3⋅SMe2/PMP pair were carried out. The mechanistic scenarios depicted in Scheme 3 were investigated via Density Functional Theory (DFT) calculations. [14] Despite the high nucleophilicity of indole, the 1 a‐BF3 adduct could not be identified computationally. [15] Therefore, the electrophilic substitution (SEAr) pathway can be readily excluded as a viable mechanism. For the borenium mediated pathway, the formation of the borenium/BF4 − species (4 a‐BF2 +/BF4 − ion pair) is thermodynamically uphill, but the relatively high barrier (34.7 kcal mol−1) represented by the subsequent BF2 + transfer transition state (TSbor) renders this pathway unlikely as well.
Scheme 3.
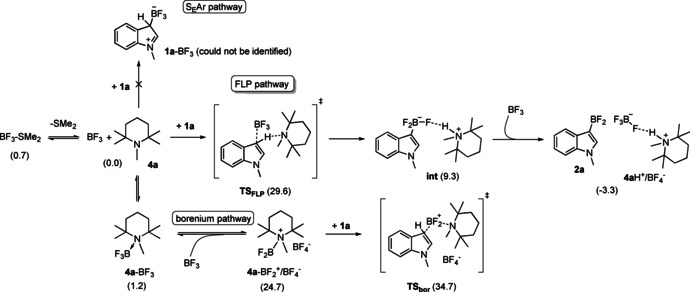
Computationally examined mechanistic pathways of 1 a borylation by a PMP/BF3 Lewis pair. Free energy data (in kcal mol−1) given in parentheses are with respect to the 1 a+4 a+BF3 reactant state.
Hence our attention turned towards the FLP‐type C−H activation mechanism, which has been described previously for ansa‐aminoboranes.[ 7 , 8 , 9 ] This mechanism assumes the cooperative action of Lewis acid/base centers, which was confirmed computationally (see Figures 1 and 2). [16] The reactants (1 a, 4 a and BF3) form a transient weakly bound complex (4 a⋅1 a⋅BF3), from which the C−H activation takes place concertedly via TSFLP. The obtained free energy barrier (29.6 kcal mol−1) is consistent with the reaction rate measured at elevated temperature. The concerted C−H activation leads to intermediate int, which is stabilized by N−H⋅⋅⋅F hydrogen bonding interactions. This species can favorably interact with an additional BF3 molecule, and the (int‐BF3) adduct can easily furnish the borylated product 2 a via a fluoride transfer (TSFtrans). The dissociation of the weakly bound 2 a⋅4 aH+/BF4 − species yield a more stable product state (2 a+4 aH+/BF4 −).
Figure 1.
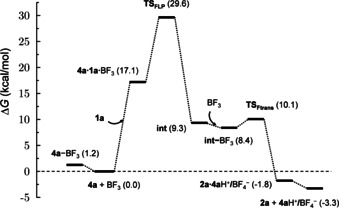
Free energy profile computed for the FLP‐type C−H activation pathway for 1 a borylation by a PMP/BF3 Lewis pair. Free energy data are given relative to the 1 a+4 a+BF3 reactant state.
Figure 2.
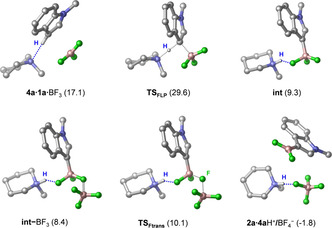
Structures of reaction intermediates and transition states identified computationally for the FLP‐type C−H activation pathway for 1 a borylation by PMP⋅BF3. H atoms (except that involved in the reaction) and the four methyl groups of PMP are omitted for clarity. Forming and breaking bonds are shown as black dotted lines in TS structures; H‐bonds are highlighted by blue dotted lines. Free energy data are given relative to the 1 a+4 a+BF3 reactant state (in kcal mol−1).
We carried out kinetic studies of the 1 a borylation in the presence of varying amounts of BF3⋅SMe2 and 4 a. Analysis of initial rates revealed that the reaction is first order in 1 a and a positive order in BF3⋅SMe2. At the same time, addition of overstoichiometric 0.5 equivalents of 4 a resulted in a pronounced inhibiting effect (Figure 3). This effect was attributed to the zero kinetic order of the reaction in 4 a along with the positive order in free BF3. Additional 4 a decreased the concentration of BF3 and hence the reaction rate. Notwithstanding this explanation contrasted with the computationally found endergonicity of 4 a‐BF3 dissociation (Figure 1) it was known from our previous work [13] that this Lewis adduct was better stabilized in a more polar solvent, DCM. In the present work, the free energies were computed for benzene that was used as a solvent for borylation reaction. However, this might not be a completely adequate description of the real reaction mixture provided the presence of polar reactants (Me2S‐BF3, 4 a‐BF3) and products ([4 a‐H+][BF4 ‐]) in high concentrations.
Figure 3.
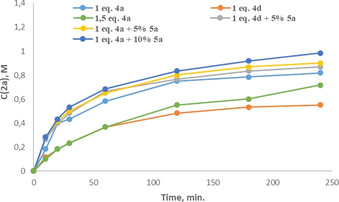
Kinetic profiles of 1 a borylation with BF3⋅SMe2, 4 a/4 d and various amounts of 5 a.
The ultimate picture of the kinetic processes seemed rather complex: apparently 4 a‐BF3 is generated in situ upon mixing of the reagents that discharged some amount of free Me2S. In addition to that, more Me2S was generated during the borylation, suppressing dissociation of the starting BF3 adducts. In order to derive the quantitative model of the observed kinetics a numerical simulation was carried out. Two most likely models were compared, one that followed the k[1 a][BF3] rate law (model A) and the model B with the k[1 a][BF3]2 law. Both of them described high percentage of the total observed variability but statistically were quite comparable (see Supporting Information for details). We concluded that there is more than one mechanism in action including also the FLP mechanism that is expected to follow the k[1 a][BF3][PMP] kinetic law. Yet such a kinetic model alone would poorly agree with the collected kinetic data, it might in combination with the models A or B.
Although 4 a together with BF3 gives the highest in situ yield in the borylation of 1 a among the bases 4 a–d, parallel decomposition of amine by Lewis acidic BF3 is an unwanted side reaction. [13] This drawback can be overcome by using the most sterically hindered base 4 d, where formation of its BF3 adduct is sterically unfavorable. However, borylation with 4 d proceeds much slower (Figure 3) and gives the lower yield of 2 a (Table 1). This inspired us to look for additives, which could promote the borylation reaction further. Considering that formation of borocations in benzene cannot be completely excluded (Scheme 3), we turned out attention to tetramethylthiourea (TMTU) that proved assisting the autoionization of BF3. [17] Accordingly, we found that the addition of small (5 mol %) amounts TMTU (5 a) to both BF3⋅SMe2/4 a and BF3⋅SMe2/4 d pairs increases the rate of 2 a formation (Figure 3). When stoichiometric amounts of 5 a were used in the absence of 4 a or 4 d, only 1 a, BF3⋅5 a adduct, some unidentified products but no traces of 2 a were detected by 1H or 11B NMR analysis.
Our computational analysis indicates that the formation of the borenium/BF4 − species with TMTU (5 a‐BF2 +/BF4 − ion pair) as well the subsequent BF2 + transfer process is notably more favored as compared to the analogous borenium formation process with PMP (see Scheme 4). [18] The barrier computed for the formation of borenium 1 a‐BF2 + is 31.0 kcal mol−1, which is comparable to that of the FLP pathway with PMP (29.6 kcal mol−1). Furthermore, the deprotonation of 1 a‐BF2 + by TMTU takes place via a similar barrier (29.9 kcal mol−1) and the proton is then delivered to the more basic PMP. Based on these results and considering the uncertainty in the computed barriers (a few kcal mol−1 at least), one possible explanation for the beneficial effect of TMTU is that it opens an alternative borylation pathway to the PMP assisted FLP mechanism. In this latter mechanism, TMTU acts as catalyst facilitating the formation and the deprotonation of the 1 a‐BF2 + borenium intermediate. We have also explored the FLP pathway with TMTU acting as a base and we obtained a barrier of 31.7 kcal mol−1, which is slightly higher than that of the TMTU promoted borenium pathway (31.0 kcal mol−1), but still comparable to the most favored PMP assisted FLP pathway (29.6 kcal mol−1). So an alternative explanation for the improved yields with TMTU is that it acts as a base in the FLP mechanism, which becomes important and beneficial at higher conversion rates, when most of the PMP is consumed.
Scheme 4.
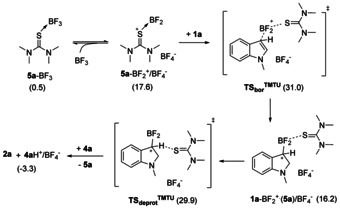
TMTU assisted borenium pathway examined computationally. Free energy data (in kcal mol−1) given in parentheses are with respect to the 1 a+5 a+BF3 reactant state.
To study the role of TMTU in more details, we introduced borylation of 1 a with other thioureas 5 b–e (Scheme 5). Among the series, cyclic thioureas 5 b and 5 d give the highest in situ yield of 2 a. Interestingly, in case of 5 d minor amounts of diarylfluoroborane 3 a were also found (Table 2).
Scheme 5.
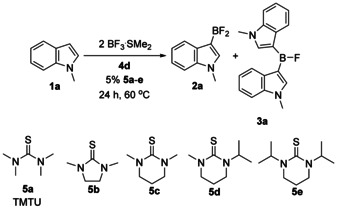
Screening of various thioureas as catalysts for borylation of 1 a.
Table 2.
In situ yields of products from the borylation of 1 a in the presence of thioureas 5 a–5 e.[a]
|
Thiourea |
1 a |
2 a |
3 a |
|---|---|---|---|
|
– |
43 |
47 |
– |
|
5 a [b] |
30 |
69 |
– |
|
5 b [b] |
22 |
77 |
– |
|
5 c [b] |
34 |
65 |
– |
|
5 d [b] |
19 |
79 |
2 |
|
5 e [b] |
32 |
68 |
– |
[a] Reactions were performed in the gas‐tight NMR tubes for 24 h at 60 °C in the argon atmosphere; Yields were determined by the 1H NMR spectroscopy against 1‐bromo‐3,5‐difluorobenzene as the internal standard; see the Supporting Information for details; [b] 5 mol % of thiourea 5 a–e was used.
We investigated the substrate scope of borylation with 5 d. In general, electron rich N‐alkylindoles and ‐pyrroles were borylated with high to moderate yields (Scheme 6, examples 2 a–d). Moreover, N‐allyl group remained untouched in the reaction conditions (entry 2 e). Incorporation of halogens atoms as electron withdrawing groups into the indole ring decreases the conversion (examples 2 f–h). Interestingly, terminal double bond in indolenine ring (entry 2 i,j) can also be borylated with a high yield. In general, electron‐rich alkyl‐ and halosubstituted N‐heteroarenes we found to be the best substrates in the developed reaction conditions. Attempted borylation of other substrates was less successful: 5‐Methoxy substituted indole 1 k produced complex mixture with no trace of borylation product. Set of signals around +15 ppm in the 11B NMR spectrum indicated possible ether cleavage. Attempt to use conventional enamines 1 l–m led only to their BF3 adducts whereas 2‐methylbenzothiophene (1 n) remained completely intact. Interestingly, 3‐ethynylthiophene 1 o gave a complex product mixture, however, characteristic for hetarene‐BF2 peaks at 22.95 ppm (11B NMR) and −84.96 (19F NMR) were detected. Importantly, no evidence of terminal alkyne CH bond activation was found even though this substrate is known to form trialkynylborane SMe2 adduct upon treatment with BF3⋅SMe2/4 a system. [19] Allyltrimethylsilane 1 p gave traces of borylation products according to 11B and 19F spectra. At the same time, the characteristic peak for trimethylsilylfluoride multiplet found in the 19F NMR spectrum at −157 ppm indicated TMS group cleavage.
Scheme 6.
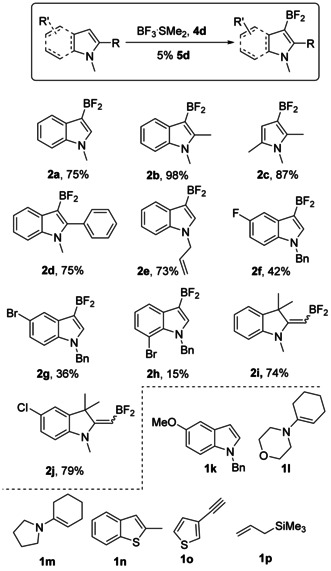
Substrate scope of the reaction and NMR yields of the N‐heteroarenes borylation products.
Aryldifluoroboranes 2 a–j were generated and partially characterized by the NMR spectroscopy in solution, however, attempted isolation of these compounds appeared to be unsuccessful due to high reactivity and reversibility of the borylation reaction (Figure 1). In line with the recent reports [15] we found protodeboronation to be the major decomposition pathway. Although only few 3‐BF3‐substituted indoles, all stabilized by the strongly electron withdrawing groups on nitrogen, were known in the literature, [1g] we attempted conversion of in situ produced 2 a to the corresponding indolyltrifluoroborate (6 a) (Scheme 7).
Scheme 7.
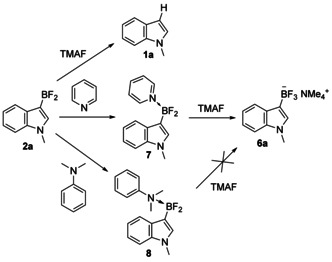
Aryldifluoroboranes are accessible with high reactivity and protodeboronation of 3‐subsituted indole 2 a takes place in the presence of TMAF. However, 2 a forms pyridine (7) and N,N‐dimethylaniline (8) adducts. The former can be further converted to trifluoroborate 11 a with TMAF.
We reported previously that trialkynylboranes are converted to trialkynylfluoroborates by the treatment with tetramethylammonium fluoride (TMAF). [13] However, all attempts to perform direct fluorination of 2 a to 6 a with TMAF led to the complete protodeborylation, and starting material 1 a was observed instead. The reason for instability of 2 a in the presence of TMAF is not clear, but to overcome this issue, we stabilized 2 a in the form of its pyridine or N,N‐dimethylaniline adducts. Addition of any of these amines to the 2 a containing reaction mixture resulted in instantaneous formation of the corresponding adducts 7 and 8. All further attempts to isolate them led to the decomposition to 1 a, but, to our delight, 7 produced trifluoroborate 6 a without noticeable protodeboronation upon treatment with TMAF. In contrast to 7, reaction of adduct 8 with TMAF led to a complex mixture of unidentified products. To demonstrate generality of the developed two‐step method, we applied it to other substrates and isolated the corresponding trifluoroborates with the overall yields equal or slightly lower than those measured in situ during the previous borylation experiments (Scheme 8). Although 5 d was found to be the most active additive for borylation in general, we found that commercially available 5 a gives similar yields with electron rich arenes (Scheme 8, substrates 6 a–c). Borylation of less active 1‐methyl‐2‐phenylindole was successfully performed with bulky thiourea 5d, giving corresponding trifluoroborate 6 d in good yield.
Scheme 8.
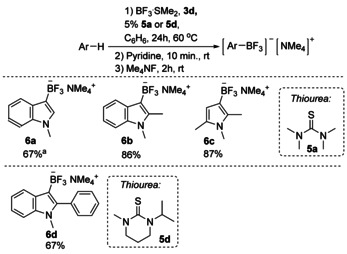
A one‐pot thiourea‐assisted synthesis of aryltrifluoroborates from heteroarenes. [a] compound 11 a was isolated along with 15 % of tetramethyl‐ammonium diindolyldifluoroborate (see Supporting Information for details).
Conclusions
In conclusion, we developed a straightforward method of N‐heteroarenes C−H borylation by boron trifluoride and sterically hindered amines. Addition of thioureas as additives had a beneficial effect on the yield of borylation products. Indoles were selectively borylated into the 3‐position and indolenines to the terminal position of the double bond. Borylation intermediates, aryldifluoroboranes, are very reactive and prone to protodeboronation in presence of Me4NF. This problem was overcame by intermediate formation of their pyridine adducts prior to treatment with Me4NF that furnished 3‐substituted indolyl‐ and pyrrolyltrifluoroborates with high overall yields. Expansion of the reactivity of boron trifluoride towards other C−H bonds as well as studies towards avoiding protodeboronation in other organoboron compounds are subjects of ongoing studies in our groups.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was funded by Academy of Finland (decision 316207) and by NKFIH (grant K‐115660). We thank the CSC‐IT Center for Science, Finland, for providing computational resources (CSC project 2000358).
V. Iashin, D. Berta, K. Chernichenko, M. Nieger, K. Moslova, I. Pápai, T. Repo, Chem. Eur. J. 2020, 26, 13873.
Contributor Information
Imre Pápai, Email: papai.imre@ttk.mta.hu.
Timo Repo, Email: timo.repo@helsinki.fi.
References
- 1.
- 1a. Vieira A. S., Fiorante P. F., Hough T. L. S., Ferreira F. P., Lüdtke D. S., Stefani H. A., Org. Lett. 2008, 10, 5215–5218; [DOI] [PubMed] [Google Scholar]
- 1b. Mitchell T. A., Bode J. W., J. Am. Chem. Soc. 2009, 131, 18057–18059; [DOI] [PubMed] [Google Scholar]
- 1c. Vo C.-V. T., Mitchell T. A., Bode J. W., J. Am. Chem. Soc. 2011, 133, 14082–14089; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Zeng J., Vedachalam S., Xiang S., Liu X.-W., Org. Lett. 2011, 13, 42–45; [DOI] [PubMed] [Google Scholar]
- 1e. Roscales S., Csaky A. G., Chem. Commun. 2014, 50, 454–456; [DOI] [PubMed] [Google Scholar]
- 1f. Brady P. B., Carreira E. M., Org. Lett. 2015, 17, 3350–3353; [DOI] [PubMed] [Google Scholar]
- 1g. Shih J.-L., Nguyen T. S., May J. A., Angew. Chem. Int. Ed. 2015, 54, 9931–9935; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10069–10073; [Google Scholar]
- 1h. Wrigstedt P., Iashin V., Lagerblom K., Keskiväli J., Chernichenko K., Repo T., Eur. J. Org. Chem. 2017, 880–891. [Google Scholar]
- 2.
- 2a. Farooq O., J. Fluor. Chem. 1995, 70, 225–227; [Google Scholar]
- 2b. Vedejs E., Chapman R. W., Fields S. C., Lin S., Schrimpf M. R., J. Org. Chem. 1995, 60, 3020–3027; [Google Scholar]
- 2c. Frohn H. J., Franke H., Fritzen P., Bardin V. V., J. Organomet. Chem. 2000, 598, 127–135; [Google Scholar]
- 2d. Lennox A. J. J., Lloyd-Jones G. C., Angew. Chem. Int. Ed. 2012, 51, 9385–9388; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9519–9522. [Google Scholar]
- 3.
- 3a. Kallepalli V. A., Shi F., Paul S., Onyeozili E. N., Maleczka R. E., Smith M. R., J. Org. Chem. 2009, 74, 9199–9201; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Stahl T., Müther K., Ohki Y., Tatsumi K., Oestreich M., J. Am. Chem. Soc. 2013, 135, 10978–10981; [DOI] [PubMed] [Google Scholar]
- 3c. Seechurn C. C. C. J., Sivakumar V., Satoskar D., Colacot T. J., Organometallics 2014, 33, 3514–3522; [Google Scholar]
- 3d. Takaya J., Ito S., Nomoto H., Saito N., Kirai N., Iwasawa N., Chem. Commun. 2015, 51, 17662–17665; [DOI] [PubMed] [Google Scholar]
- 3e. Wang G., Xu L., Li P., J. Am. Chem. Soc. 2015, 137, 8058–8061. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Stephan D. W., Acc. Chem. Res. 2015, 48, 306–316; [DOI] [PubMed] [Google Scholar]
- 4b. Fontaine F.-G., Rochette É., Acc. Chem. Res. 2018, 51, 454–464; [DOI] [PubMed] [Google Scholar]
- 4c. Bagutski V., Del Grosso A., Carrillo J. A., Cade I. A., Helm M. D., Lawson J. R., Singleton P. J., Solomon S. A., Marcelli T., Ingleson M. J., J. Am. Chem. Soc. 2013, 135, 474–487. [DOI] [PubMed] [Google Scholar]
- 5. Gandeepan P., Müller T., Zell D., Cera G., Warratz S., Ackermann L., Chem. Rev. 2019, 119, 2192–2452. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Chernichenko K., Kótai B., Nieger M., Heikkinen S., Pápai I., Repo T., Dalton Trans. 2017, 46, 2263–2269; [DOI] [PubMed] [Google Scholar]
- 6b. Zhivonitko V. V., Sorochkina K., Chernichenko K., Kótai B., Földes T., Pápai I., Telkki V.-V., Repo T., Koptyug I., Phys. Chem. Chem. Phys. 2016, 18, 27784–27795; [DOI] [PubMed] [Google Scholar]
- 6c. Chernichenko K., Lindqvist M., Kótai B., Nieger M., Sorochkina K., Pápai I., Repo T., J. Am. Chem. Soc. 2016, 138, 4860–4868; [DOI] [PubMed] [Google Scholar]
- 6d. Lindqvist M., Borre K., Axenov K., Kótai B., Nieger M., Leskelä M., Pápai I., Repo T., J. Am. Chem. Soc. 2015, 137, 4038–4041; [DOI] [PubMed] [Google Scholar]
- 6e. Chernichenko K., Kótai B., Pápai I., Zhivonitko V., Nieger M., Leskelä M., Repo T., Angew. Chem. Int. Ed. 2015, 54, 1749–1753; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1769–1773; [Google Scholar]
- 6f. Chernichenko K., Madarász Á., Pápai I., Nieger M., Leskelä M., Repo T., Nat. Chem. 2013, 5, 718. [DOI] [PubMed] [Google Scholar]
- 7. Légaré M.-A., Courtemanche M.-A., Rochette É., Fontaine F.-G., Science 2015, 349, 513. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Légaré M.-A., Rochette É., Légaré Lavergne J., Bouchard N., Fontaine F.-G., Chem. Commun. 2016, 52, 5387–5390; [DOI] [PubMed] [Google Scholar]
- 8b. Légaré Lavergne J., Jayaraman A., Misal Castro L. C., Rochette É., Fontaine F.-G., J. Am. Chem. Soc. 2017, 139, 14714–14723; [DOI] [PubMed] [Google Scholar]
- 8c. Shao Y., Zhang J., Li Y., Liu Y., Ke Z., Org. Lett. 2018, 20, 1102–1105. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Focante F., Camurati I., Nanni D., Leardini R., Resconi L., Organometallics 2004, 23, 5135–5141; [Google Scholar]
- 9b. Liu Y.-L., Kehr G., Daniliuc C. G., Erker G., Chem. Eur. J. 2017, 23, 12141–12144. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Ishida N., Moriya T., Goya T., Murakami M., J. Org. Chem. 2010, 75, 8709–8712; [DOI] [PubMed] [Google Scholar]
- 10b. Grosso A. D., Helm M. D., Solomon S. A., Caras-Quintero D., Ingleson M. J., Chem. Commun. 2011, 47, 12459–12461; [DOI] [PubMed] [Google Scholar]
- 10c. Solomon S. A., Del Grosso A., Clark E. R., Bagutski V., McDouall J. J. W., Ingleson M. J., Organometallics 2012, 31, 1908–1916; [Google Scholar]
- 10d. Yin Q., Klare H. F. T., Oestreich M., Angew. Chem. Int. Ed. 2017, 56, 3712–3717; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3766–3771; [Google Scholar]
- 10e. Tanaka S., Saito Y., Yamamoto T., Hattori T., Org. Lett. 2018, 20, 1828–1831; [DOI] [PubMed] [Google Scholar]
- 10f. Iqbal S. A., Cid J., Procter R. J., Uzelac M., Yuan K., Ingleson M. J., Angew. Chem. Int. Ed. 2019, 58, 15381–15385; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 15525–15529; [Google Scholar]
- 10g. Lv J., Chen X., Xue X.-S., Zhao B., Liang Y., Wang M., Jin L., Yuan Y., Han Y., Zhao Y., Lu Y., Zhao J., Sun W.-Y., Houk K. N., Shi Z., Nature 2019, 575, 336–340. [DOI] [PubMed] [Google Scholar]
- 11. Wang D.-Y., Minami H., Wang C., Uchiyama M., Chemistry Letters 2015, 44, 1380–1382. [Google Scholar]
- 12.
- 12a. Del Grosso A., Pritchard R. G., Muryn C. A., Ingleson M. J., Organometallics 2010, 29, 241–249; [Google Scholar]
- 12b. Del Grosso A., Singleton P. J., Muryn C. A., Ingleson M. J., Angew. Chem. Int. Ed. 2011, 50, 2102–2106; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 2150–2154. [Google Scholar]
- 13. Iashin V., Chernichenko K., Pápai I., Repo T., Angew. Chem. Int. Ed. 2016, 55, 14146–14150; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14352–14356. [Google Scholar]
- 14.DFT calculations were carried out using the ωB97X-D functional with the 6–311G(d,p) basis set as implemented in Gaussian 16. The electronic energies were refined by single-point energy calculations using the 6–311++G(3df,3pd) basis set. The solvent effects were estimated via the SMD continuum model using benzene as the solvent. The reported energies refer to solvent-phase Gibbs free energies (T=333 K, c=1 mol L−1). For further details, see Supporting Information.
- 15. 1 a-BF3 intermediate was also proposed in BF3⋅OEt2 catalyzed borylation of indoles to the 2nd position. For details, see: Zhong Q., Qin S., Yin Y., Hu J., Zhang H., Angew. Chem. Int. Ed. 2018, 57, 14891–14895; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15107–15111. [Google Scholar]
- 16.The FLP-type C−H activation pathway discussed in our paper can be regarded as a concerted SEAr mechanism. For related contributions, see:
- 16a.ref. [11];
- 16b. Yang S., Bour C., Gandon V., ACS Catal. 2020, 10, 3027–3033. [Google Scholar]
- 17. Hartman J. S., Schrobilgen G. J., Stilbs P., Can. J. Chem. 1976, 54, 1121–1129. [Google Scholar]
- 18.Interestingly, stabilization of borocations by resonance in nitrogen and sulfur containing ligands has been shown with benzothiazoles. For details, see: Crossley D. L., Cid J., Curless L. D., Turner M. L., Ingleson M. J., Organometallics 2015, 34, 5767–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.V. Iashin, K. Chernichenko, T. Repo, unpublished results.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary