

Abbreviations
- ABCB11
adenosine triphosphate–binding cassette family B member 11
- ATP8B1
adenosine triphosphatase phospholipid transporting 8B1
- BRIC/PFIC
benign recurrent and progressive forms of familial intrahepatic cholestasis
- BSEP
bile salt export pump
- CLD
cholestatic liver disease
- GGT
gamma‐glutamyltransferase
- MRP2
multidrug resistance–associated protein 2
- MVID
microvillus inclusion disease
- MYO5B
myosin 5B
- PTC
premature termination codon
- TPN
total parenteral nutrition
Myosin 5B–Associated Cholestatic Liver Disease
Cholestatic liver disease (CLD) is characterized by an increase in the serum concentrations of compounds that are normally excreted with bile, such as bile acids and bilirubin.( 1 ) CLD clinically manifest with cholestasis, jaundice, and pruritis. Diagnosis involves evaluation of the patient’s clinical manifestations, exclusion of common causes of childhood cholestasis, and analyses of blood biochemistry and liver histology.( 1 ) One blood marker that aids in the differential diagnosis of liver diseases is gamma‐glutamyltransferase (GGT), a liver enzyme which is typically elevated in serum upon liver damage. The best known CLDs characterized by low/normal GGT are the benign recurrent and progressive forms of familial intrahepatic cholestasis (BRIC/PFIC), which are caused by mutations in the adenosine triphosphatase phospholipid transporting 8B1 (ATP8B1) gene encoding the ATP8B1 protein (BRIC/PFIC1) or in the adenosine triphosphate–binding cassette family B member 11 (ABCB11) gene encoding the canalicular bile salt export pump (BSEP) (BRIC/PFIC2).( 2 , 3 ) In hepatocytes, ATP8B1 and BSEP are localized to the apical bile canalicular domain, where they contribute to biliary secretion. When mutated as in PFIC1/2 patients, the respective proteins are less expressed or mislocalized and/or display impaired activity, leading to defective biliary secretion and, consequently, cholestasis.( 2 , 3 )
Since 2017 four independent reports have identified in total 30 different myosin 5B (MYO5B) mutations in 22 patients with non–microvillus inclusion disease (MVID) who were diagnosed with intermittent, recurrent, or progressive cholestasis presenting with jaundice, pruritus, hepatomegaly, and low/normal serum levels of GGT (Fig. 1; Supporting Table S1). These patients tested negative for mutations in other PFIC‐associated genes.( 4 , 5 , 6 , 7 ) MYO5B mutations may account for approximately 20% of pediatric patients with idiopathic low‐GGT intrahepatic cholestasis.( 8 ) Based on microscopic analyses of liver biopsies, a reduced expression or mislocalization of BSEP was reported( 4 , 5 ) but not in all patients.( 7 )
Fig. 1.
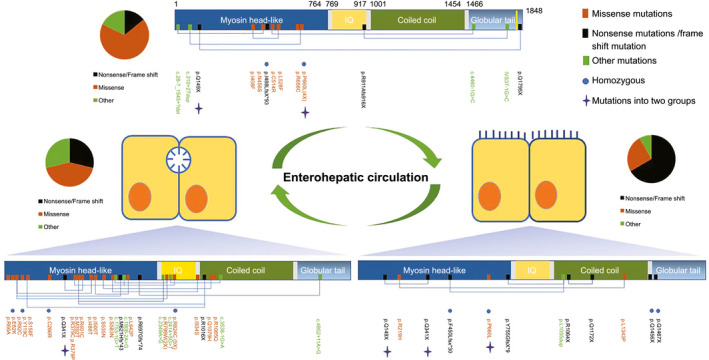
Cartoon showing MYO5B mutations associated with isolated CLD, isolated MVID, or MVID + CLD.
MYO5B mutations were earlier reported to cause MVID.( 9 ) MVID is a severe congenital enteropathy, characterized by severe intractable secretory diarrhea from the moment of birth and the inability to absorb any nutrients.( 10 ) Diagnosis involves evaluation of the patient’s clinical manifestations, exclusion of common causes of diarrhea, and inspection of small intestinal biopsies. MVID small intestinal tissue reveals (near‐) total villus atrophy and, at the cellular level, microvillus atrophy and mislocalization of many apical brush border proteins including the diagnostic brush border protein cluster of differentiation 10.( 10 , 11 ) The presence of microvillus inclusion bodies in the enterocytes by electron microscopy confirms the diagnosis. Tissue and cellular defects are seen throughout the intestine( 12 ) and have been causally related to the loss of function of the encoded myosin Vb protein. For their survival, patients with MVID require lifelong total parenteral nutrition (TPN) and, in some cases, intestinal transplantation.( 13 , 14 )
A significant number of patients with MVID present CLD with low/normal serum GGT levels, manifesting as jaundice (associated with elevated levels of conjugated serum bilirubin), pruritus, and hepatomegaly.( 13 , 15 ) The prevalence of CLD in patients with MVID has been estimated at ~30% in a local cohort of 28 patients.( 15 ) We have extracted data from all published MVID case reports until 2020 (reporting on 133 patients with MVID) (Supporting Table S1) and calculated the prevalence of MVID‐associated CLD to be 37%. When we restricted the analyses to only patients with MVID and reported MYO5B mutations, the prevalence was 54%. It is possible that this is an underestimation as patients with MVID who were still alive at the moment of publication of their case report could have developed CLD later on.
Microscopic analyses of liver biopsies of patients with MVID and MYO5B mutations showed a reduced expression and/or mislocalization of BSEP, which is considered a major factor in the severity of cholestasis.( 16 ) Further, a reduced expression and/or mislocalization of the bilirubin and bile acid transporter multidrug resistance–associated protein 2 (MRP2) was shown in some,( 17 ) but not all,( 14 ) patients with MVID. Mutations in the MRP2‐coding adenosine triphosphate–binding cassette family C member 2 gene cause Dubin‐Johnson syndrome associated with elevated conjugated bilirubin levels in serum.( 18 , 19 ) The expression or canalicular localization of ATP8B1 in liver biopsies of patients with MVID has not been studied. The mislocalization of MRP2, which is not typically observed in patients with ABCB11 mutations, indicates that MYO5B mutations can affect multiple canalicular transporters in parallel.
Because MYO5B mutations disrupt the intracellular trafficking of brush border proteins in the intestine,( 20 , 21 ) it has been assumed that the same MYO5B mutations similarly disrupt the trafficking of the canalicular bile acid–transporting and bilirubin‐transporting proteins in the liver.( 9 ) While this seems plausible and is supported by cell culture studies,( 22 ) not all data are in support of this. For example, in other patients with MVID and MYO5B mutations and presenting with CLD no mislocalization of these canalicular transporters was reported, and CLD later resolved.( 23 ) In another patient without MVID diagnosed with CLD and MYO5B mutations no mislocalization of BSEP was observed.( 7 ) Furthermore, 5 patients with MVID showed reduction of cholestasis and pruritus when soybean oil–based lipids in the TPN were replaced by fish oil–based lipids, indicating TPN as a contributing factor in cholestasis and pruritus in at least these patients with MVID.( 8 , 24 , 25 ) The prevalence of TPN‐associated CLD in pediatric cohorts was estimated to be between 20% (< 1 month of TPN) and 70% (> 2 months of TPN).( 26 , 27 ) Given their lifelong TPN dependency,( 6 ) a prevalence of CLD of 37%‐54% in patients with MVID seems rather low. Clearly, with these prevalence numbers at hand, it is difficult to conclude whether CLD in TPN‐dependent patients with MVID is caused by the TPN or the patient’s MYO5B mutations. Nonetheless, it appears that pathogenic MYO5B mutations may not always affect bile canalicular protein localization and/or cause CLD.
Correlations Between MYO5B Genotype and CLD
Similar to the 37%‐54% of patients diagnosed with MVID presenting symptoms of CLD, 21% of patients diagnosed with low/normal‐GGT CLD were reported to present loose stools or diarrheal episodes.( 4 , 5 , 7 ) Overlap of symptoms thus occurs, but there are more MVID patients who present cholestasis than there are patients with CLD who present diarrhea. An outstanding question is why MYO5B mutations cause either CLD or MVID with or without symptoms of MVID or CLD, respectively. Possibly, the type of MYO5B mutation may play a role. While the many private MYO5B mutations running in the affected families complicate a detailed explanation, some generalizations can be made. For example, it was suggested that in patients with isolated CLD MYO5B mutations may not result in sufficient loss of myosin Vb protein function to cause intestinal failure. This was supported by in silico analyses based on the myosin Vb homology protein structure. These revealed that the compound heterozygous patients with isolated CLD often carried in one allele a mutation that corresponded to more peripheral residues of the motor domain that were predicted to be less damaging for the protein’s motor function.( 28 ) Further, several MYO5B mutations associated with isolated CLD give rise to amino acid substitutions that normally occur in other species, suggesting that these do not significantly affect the protein function. Thus, CLD in patients without MVID appears to be a manifestation of relatively mild MYO5B mutations.( 4 , 28 ) Why relatively mild MYO5B mutations that cause a liver defect would not cause intestinal defects is not known. Possibly, the higher expression level of MYO5B in the intestine (https://www.gtexportal.org/home/gene/MYO5B) may compensate for a lower per‐protein activity.
An additional correlation between MYO5B genotype and isolated CLD has been observed. That is, isolated CLD did not involve any patient with biallelic nonsense or frameshift mutations in MYO5B that induce a premature termination codon (PTC) and affect the C‐terminal rab11a‐binding domain.( 4 , 28 ) A molecular mechanism to explain this genotype–phenotype correlation in CLD was recently proposed.( 29 ) Thus, the expression of an MVID‐associated and CLD‐associated MYO5B missense mutation (c.1979C > T), but not nonsense mutations, in a MYO5B‐depleted human hepatoma cell line caused the mislocalization of MRP2 and other bile canalicular transporter proteins. Depletion of MYO5B in these cells or in the mouse liver as such did not result in the mislocalization of these canalicular transporters. The discordant effect of missense versus PTC‐inducing MYO5B mutations suggested that the mutant myosin Vb protein exerted a gain‐of‐toxic function. It was shown that this gain‐of‐toxic function required the carboxyl terminus of the mutant myosin Vb protein. Protein‐truncating mutations cause loss of the myosin Vb protein or synthesis of a protein that lacks this carboxyl terminus, which explains, at least from the perspective of the studied canalicular transporters, why isolated CLD does not appear to correlate well with PTC‐inducing MYO5B mutations.( 29 )
The relationship between PTC‐inducing MYO5B mutations and the occurrence of CLD in patients with MVID has not been studied. Therefore, we examined the published MVID case reports (Supporting Table S1) and the international MVID registry (www.mvid‐central.org) and extracted information about the mutation and presence of CLD. Details regarding the mutations and the presence or absence of CLD were reported for 22 patients with MVID and biallelic MYO5B mutations (Table 1). One of the 11 patients (18%) with at least one PTC‐inducing mutation and 9 of the 11 patients (82%) without a PTC mutation developed CLD. There was a significant relationship between the two variables (chi squared [1, n = 22] = 8.9, P < 0.01). There was also a significant relationship between the two variables when omitting multiple patients with the same mutation (chi square [1, n = 17] = 4.5, P < 0.05). While the inclusion of more patients will tell whether this relationship holds, the current numbers suggest that patients with PTC‐inducing MYO5B mutations have a reduced risk of developing CLD. Conceivably, with MYO5B expression abolished on one allele, the c.3163‐3165dup (p.Leu1055dup) mutation (a common single‐nucleotide polymorphism [rs200219597; gnomad.broadinstitute.org]) in the MYO5B gene on the other allele (Table 1) may not provide a strong enough effect to cause CLD.
Table 1.
Individual Nontransplanted Patients With MVID and Reported MYO5B Variations and Presence or Absence of CLD
| Variations of the Patient | With CLD | |
|---|---|---|
| Patients with MVID and at least one PTC‐inducing MYO5B mutation | c.5139‐1G > C(het)/c.2731delC (p.Arg911Alafs*6, het) † | No |
| c.445C > T (p.Gln149Ter, het)/c.5383C > T (p.Arg1795Ter, het) † | Yes | |
| c.3163_3165dup (p.Leu1055dup, het) ‡ /c.2259_2262dup (p.Tyr755Glyfs*9, het) † | No | |
| c.3163_3165dup (p.Leu1055dup, het) ‡ /c.2259_2262dup (p.Tyr755Glyfs*9, het) † | No | |
| c.4399C > T (p.Gln1467Ter, hom*) | No | |
| c.1347delC (p.Phe450Leufs*30, het)2/c.3163_3165dup (p.Leu1055dup, het) ‡ | No | |
| c.1347delC (p.Phe450Leufs*30, het)2/c.3163_3165dup (p.Leu1055dup, het) ‡ | No | |
| c.445C > T (p.Gln149Ter, het) † /c.1021C > T (p.Gln341Ter, het) † | No | |
| c.1390C > T (p.Arg1064Ter, het) † /c.3514C > T (p.Gln1172Ter, het) † | No | |
| c.4366C > T (p.Gln1456Ter, hom) † | No | |
| c.4399C > T (p.Gln1467Ter, hom) | No | |
| Patients with MVID and no PTC‐inducing MYO5B mutations | c.1222A > T (p.Ile408Phe, het)/c.1582C > T (p.Leu528Phe, het) | Yes |
| c.1222A > T (p.Ile408Phe, het)/c.1582C > T (p.Leu528Phe, het) | Yes | |
| c.310 + 2dupT, het/c.1966C > T (p.Arg656Cys,het) | Yes | |
| c.1540T > C (p.Cys514Arg, het)/c.4460‐1G > C, het | Yes | |
| c.28‐?_1545+?del, het/c.1367A > G (p.Asn456Ser, het) | Yes | |
| c.1979C > T (p.Pro660Leu) | Yes | |
| c.1979C > T (p.Pro660Leu) | Yes | |
| c.1979C > T (p.Pro660Leu) | Yes | |
| c.1979C > T (p.Pro660Leu) | Yes | |
| c.656G > T (p.Arg219His, het)/c.4028T > C (p.Leu1343Pro, het) | No | |
| c.1979C > T (p.Pro660Leu, hom) | No |
This mutation is expected to result in the expression of a truncated protein that retained its rab8‐binding and rab11a‐binding sites.
This variation is a common single‐nucleotide polymorphism (rs200219597).
Abbreviations: het, heterozygous mutation; hom, homozygous mutation.
MYO5B Mutations Can Protect Against CLD in Patients With MVID
Intriguingly, 3 out of a group of 8 patients with MVID and MYO5B mutations who underwent intestinal transplantation presented with posttransplantation onset of CLD, and others showed exacerbation of preexisting CLD.( 13 , 15 ) Also, in our medical center, a patient with MVID developed cholestasis and pruritus after receiving an intestinal transplant. His CLD resolved following rejection and subsequent removal of the graft. In addition, a patient with late‐onset MVID presented with CLD after birth, which resolved at the moment when his enteral symptoms appeared. CLD did not return following a combined liver and intestinal transplantation. These observations led to the suggestion that the onset or severity of CLD may be inversely related to intestinal function.( 15 , 28 )
Because MYO5B mutations cause total malabsorption due to villus and brush border atrophy and the mislocalization of many brush border proteins,( 10 , 11 ) a reduced bile acid absorption capacity in the ileum (through the solute carrier family 10 member 2–encoded apical sodium‐bile acid transporter [ASBT])( 30 , 31 ) can be expected. The consequently expected reduction of recirculating bile acids may (partially) diminish the clinical CLD symptoms. Indeed, the interruption of the bile acid circulation between liver and intestine, through either intestinal graft removal, ileal exclusion, nasobiliary drainage, or partial external bile diversion, led to a (partial) remission of CLD in patients with MVID,( 15 ) which is in line with the effectivity of these treatments in other patients with PFIC.( 32 ) Thus, MYO5B mutations that reduce the bile acid absorption capacity of the intestine may provide some protection against CLD.( 15 ) Future studies are needed to address the bile acid pool and bile flow dynamics in the various patients with MYO5B mutations.
Conclusions and Future Perspectives
MYO5B is a new kid on the block in CLD. Different mutations in MYO5B display seemingly contradictory effects on the clinical presentation of CLD. It appears that variations in the clinical presentation of MYO5B‐related low‐GGT CLD can be attributed to the coincident expression but unequal effects of MYO5B mutations in enterocytes versus hepatocytes (Fig. 1). Thus, (a combination of) MYO5B mutations with a predicted mild effect on myosin Vb protein function can cause canalicular transport defects in hepatocytes but not (serious) problems in enterocytes, resulting in a BRIC/PFIC‐like CLD. MYO5B missense mutations that have a more profound impact on myosin Vb protein function can cause similar (or stronger) canalicular transport defects in hepatocytes. However, these additionally cause absorptive defects in the intestine and, in case of an expected mislocalization of ASBT, likely reduce ileal bile acid recirculation and thereby diminish the clinical presentation of CLD. PTC‐inducing mutations cause absorptive defects in the intestine but, as such, no apparent canalicular defects.
Of interest, the amount of bile acid that is absorbed in the intestine likely affects the bile acid synthesis and pool size through farnesoid X receptor–fibroblast growth factor 19 signaling.( 33 ) Future studies should address the size of the bile acid pool and bile flow dynamics in patients with MYO5B mutations.
For most patients with MVID, it remains uncertain whether CLD results from TPN and/or their MYO5B mutations. Current prevalence data on CLD in MVID (this study) versus that of non‐MVID pediatric TPN cohorts suggest that MYO5B mutations in general do not predispose to TPN‐associated CLD. Changing to a TPN formula with fish oil–based lipids may provide insight into the contribution of the TPN to MVID‐associated CLD. Knowledge as to whether given MYO5B mutations disrupt canalicular transport in hepatocytes will aid family counseling and therapeutic decision‐making. Currently, a combined bowel–liver transplantation is considered for children with MVID to reduce the risk of posttransplantation exacerbation or onset of CLD.( 13 , 15 ) However, if not necessary, the scarce donor liver may be used for another patient in need. Immunohistochemical analyses of the localization of bile canalicular transporters in patients’ liver biopsies may prove informative. Patient stem cell–derived hepatocytes( 34 ) and liver–gut on‐a‐chip models( 35 ) may provide powerful preclinical tools to assess the impact of the patient’s mutation(s) on bile canalicular transport. As a cautionary note, however, it should be emphasized that gene mutations are not likely to be the sole determinant of CLD as siblings with the same MYO5B mutations showed different disease courses.( 4 )
In contrast to familial CLDs that are caused by predominantly liver‐specific genes, i.e., ABCB11 (https://www.gtexportal.org/home/gene/ABCB11) and ABCB4 (https://www.gtexportal.org/home/gene/ABCB4),(32 ) MYO5B‐associated CLD should be viewed as a disease of the enterohepatic circulation, rather than of solely the liver. We expect that this may also apply to other familial CLDs caused by genes that are expressed in both liver and intestine (e.g., ATP8B1 or nuclear receptor subfamily 1 group H member 4( 32 )).
Clearly, a given (set of) MYO5B mutation(s) unequally affects two organs which cooperatively control bile flow complicates the disease presentation. Therefore, to understand the development and natural course of the disease as well as postintervention outcomes, a personalized patient/mutation‐specific multiorgan system approach will be required.
Search Strategy and Selection Criteria
References for this review were identified through searches of PubMed with the search terms “MYO5B,” “cholestasis,” “microvillus inclusion disease,” and “liver” from the earliest records until May 2020. The final reference list was generated on the basis of originality and relevance to the broad scope of this review.
Author Contributions
All authors contributed to the content and writing of this article.
Supporting information
Supplementary Material
Acknowledgment
We thank Li Wang for assistance with tables and gene mutation nomenclature.
Potential conflict of interest: Nothing to report.
References
- 1. Vleggaar FP, Van Ooteghem NA, Van Buuren HR, Van Berge Henegouwen HR. Cholestatic liver diseases: slow progress in understanding and treating slowly progressive disorders. Scand J Gastroenterol Suppl 2000:86‐92. [PubMed] [Google Scholar]
- 2. Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, et al. A gene encoding a liver‐specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet 1998;20:233‐238. [DOI] [PubMed] [Google Scholar]
- 3. Bull LN, Thompson RJ. Progressive familial intrahepatic cholestasis. Clin Liver Dis 2018;22:657‐669. [DOI] [PubMed] [Google Scholar]
- 4. Qiu Y‐L, Gong J‐Y, Feng J‐Y, Wang R‐X, Han J, Liu T, et al. Defects in myosin VB are associated with a spectrum of previously undiagnosed low γ‐glutamyltransferase cholestasis. Hepatology 2017;65:1655‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gonzales E, Taylor SA, Davit‐Spraul A, Thébaut A, Thomassin N, Guettier C, et al. MYO5B mutations cause cholestasis with normal serum gamma‐glutamyl transferase activity in children without microvillous inclusion disease. Hepatology 2017;65:164‐173. [DOI] [PubMed] [Google Scholar]
- 6. Sangkhathat S, Laochareonsuk W, Maneechay W, Kayasut K, Chiengkriwate P. Variants associated with infantile cholestatic syndromes detected in extrahepatic biliary atresia by whole exome studies: a 20‐case series from Thailand. J Pediatr Genet 2018;7:67‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cockar I, Foskett P, Strautnieks S, Clinch Y, Fustok J, Rahman O, et al. Mutations in myosin 5B (MYO5B) in children with early onset cholestasis. J Pediatr Gastroenterol Nutr 2020. 10.1097/MPG.0000000000002740. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 8. Fuchs J, Fallon EM, Gura KM, Puder M. Use of an omega‐3 fatty acid‐based emulsion in the treatment of parenteral nutrition‐induced cholestasis in patients with microvillous inclusion disease. J Pediatr Surg 2011;46:2376‐2382. [DOI] [PubMed] [Google Scholar]
- 9. Müller T, Hess MW, Schiefermeier N, Pfaller K, Ebner HL, Heinz‐Erian P, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet 2008;40:1163‐1165. [DOI] [PubMed] [Google Scholar]
- 10. Davidson GP, Cutz E, Hamilton JR, Gall DG. Familial enteropathy: a syndrome of protracted diarrhea from birth, failure to thrive, and hypoplastic villus atrophy. Gastroenterology 1978;75:783‐790. [PubMed] [Google Scholar]
- 11. Cutz E, Rhoads JM, Drumm B, Sherman PM, Durie PR, Forstner GG. Microvillus inclusion disease: an inherited defect of brush‐border assembly and differentiation. N Engl J Med 1989;320:646‐651. [DOI] [PubMed] [Google Scholar]
- 12. Golachowska MR, van Dael CML, Keuning H, Karrenbeld A, Hoekstra D, Gijsbers CFM, et al. MYO5B mutations in patients with microvillus inclusion disease presenting with transient renal Fanconi syndrome. J Pediatr Gastroenterol Nutr 2012;54:491‐498. [DOI] [PubMed] [Google Scholar]
- 13. Halac U, Lacaille F, Joly F, Hugot J‐P, Talbotec C, Colomb V, et al. Microvillous inclusion disease: how to improve the prognosis of a severe congenital enterocyte disorder. J Pediatr Gastroenterol Nutr 2011;52:460‐465. [DOI] [PubMed] [Google Scholar]
- 14. Ruemmele FM, Jan D, Lacaille F, Cézard J‐P, Canioni D, Phillips AD, et al. New perspectives for children with microvillous inclusion disease: early small bowel transplantation. Transplantation 2004;77:1024‐1028. [DOI] [PubMed] [Google Scholar]
- 15. Girard M, Lacaille F, Verkarre V, Mategot R, Feldmann G, Grodet A, et al. MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatology 2014;60:301‐310. [DOI] [PubMed] [Google Scholar]
- 16. Lam P, Pearson CL, Soroka CJ, Xu S, Mennone A, Boyer JL. Levels of plasma membrane expression in progressive and benign mutations of the bile salt export pump (Bsep/Abcb11) correlate with severity of cholestatic diseases. Am J Physiol Cell Physiol 2007;293:C1709‐C1716. [DOI] [PubMed] [Google Scholar]
- 17. Schlegel C, Weis VG, Knowles BC, Lapierre LA, Martin MG, Dickman P, et al. Apical membrane alterations in non‐intestinal organs in microvillus inclusion disease. Dig Dis Sci 2018;63:356‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wada M, Toh S, Taniguchi K, Nakamura T, Uchiumi T, Kohno K, et al. Mutations in the canilicular multispecific organic anion transporter (cMOAT) gene, a novel ABC transporter, in patients with hyperbilirubinemia II/Dubin‐Johnson syndrome. Hum Mol Genet 1998;7:203‐207. [DOI] [PubMed] [Google Scholar]
- 19. Keppler D. The roles of MRP2, MRP3, OATP1B1, and OATP1B3 in conjugated hyperbilirubinemia. Drug Metab Dispos 2014;42:561‐565. [DOI] [PubMed] [Google Scholar]
- 20. Knowles BC, Roland JT, Krishnan M, Tyska MJ, Lapierre LA, Dickman PS, et al. Myosin Vb uncoupling from RAB8A and RAB11A elicits microvillus inclusion disease. J Clin Invest 2014;124:2947‐2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roland JT, Bryant DM, Datta A, Itzen A, Mostov KE, Goldenring JR. Rab GTPase–Myo5B complexes control membrane recycling and epithelial polarization. Proc Natl Acad Sci USA 2011;108:2789‐2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wakabayashi Y, Dutt P, Lippincott‐Schwartz J, Arias IM. Rab11a and myosin Vb are required for bile canalicular formation in WIF‐B9 cells. Proc Natl Acad Sci USA 2005;102:15087‐15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fernández Caamaño B, Quiles Blanco MJ, Fernández Tomé L, Burgos Lizáldez E, Sarría Osés J, Molina Arias M, et al. Intestinal failure and transplantation in microvillous inclusion disease. [in Spanish] An Pediatr (Barc) 2015;83:160‐165. [DOI] [PubMed] [Google Scholar]
- 24. Anez‐Bustillos L, Dao DT, Potemkin AK, Perez‐Atayde AR, Raphael BP, Carey AN, et al. An intravenous fish oil‐based lipid emulsion successfully treats intractable pruritus and cholestasis in a patient with microvillous inclusion disease. Hepatology 2019;69:1353‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Siahanidou T, Koutsounaki E, Skiathitou A‐V, Stefanaki K, Marinos E, Panajiotou I, et al. Extraintestinal manifestations in an infant with microvillus inclusion disease: complications or features of the disease? Eur J Pediatr 2013;172:1271‐1275. [DOI] [PubMed] [Google Scholar]
- 26. Wales PW, de Silva N, Kim JH, Lecce L, Sandhu A, Moore AM. Neonatal short bowel syndrome: a cohort study. J Pediatr Surg 2005;40:755‐762. [DOI] [PubMed] [Google Scholar]
- 27. Lauriti G, Zani A, Aufieri R, Cananzi M, Chiesa PL, Eaton S, et al. Incidence, prevention, and treatment of parenteral nutrition‐associated cholestasis and intestinal failure‐associated liver disease in infants and children: a systematic review. J Parenter Enteral Nutr 2014;38:70‐85. [DOI] [PubMed] [Google Scholar]
- 28. Dhekne HS, Pylypenko O, Overeem AW, Zibouche M, Ferreira RJ, van der Velde KJ, et al. MYO5B, STX3, and STXBP2 mutations reveal a common disease mechanism that unifies a subset of congenital diarrheal disorders: a mutation update. Hum Mutat 2018;39:333‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Overeem AW, Li Q, Qiu Y‐L, Cartón‐García F, Leng C, Klappe K, et al. A molecular mechanism underlying genotype‐specific intrahepatic cholestasis resulting from MYO5B mutations. Hepatology 2019. doi:10.1002/hep.31002. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jansen PLM. New therapies target the toxic consequences of cholestatic liver disease. Expert Rev Gastroenterol Hepatol 2018;12:277‐285. [DOI] [PubMed] [Google Scholar]
- 31. Xiao L, Pan G. An important intestinal transporter that regulates the enterohepatic circulation of bile acids and cholesterol homeostasis: the apical sodium‐dependent bile acid transporter (SLC10A2/ASBT). Clin Res Hepatol Gastroenterol 2017;41:509‐515. [DOI] [PubMed] [Google Scholar]
- 32. Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol 2012;36(Suppl. 1):S26‐S35. [DOI] [PubMed] [Google Scholar]
- 33. Kliewer SA, Mangelsdorf DJ. Bile acids as hormones: the FXR‐FGF15/19 pathway. Dig Dis 2015;33:327‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Overeem AW, Klappe K, Parisi S, Klöters‐Planchy P, Mataković L, du Teil EM, et al. Pluripotent stem cell‐derived bile canaliculi‐forming hepatocytes to study genetic liver diseases involving hepatocyte polarity. J. Hepatol 2019;71:344‐356. [DOI] [PubMed] [Google Scholar]
- 35. Boeri L, Izzo L, Sardelli L, Tunesi M, Albani D, Giordano C. Advanced organ‐on‐a‐chip devices to investigate liver multi‐organ communication: focus on gut, microbiota and brain. Bioengineering (Basel) 2019;6:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material