

Abstract
Quantum chemical studies using density functional theory and ab initio methods have been carried out for the molecules L‐C3‐L with L=PPh3 (1), NHCMe (2, NHC=N‐heterocyclic carbene), and cAACMe (3, cAAC=cyclic (alkyl)(amino) carbene). The calculations predict that 1 and 2 have equilibrium geometries where the ligands are bonded with rather acute bonding angles at the linear C3 moiety. The phosphine adduct 1 has a synclinal (gauche) conformation whereas 2 exhibits a trans conformation of the ligands. In contrast, the compound 3 possesses a nearly linear arrangement of the carbene ligands at the C3 fragment. The bond dissociation energies of the ligands have the order 1<2<3. The bonding analysis using charge and energy decomposition methods suggests that 3 is best described as a cumulene with electron‐sharing double bonds between neutral fragments (cAACMe)2 and C3 in the respective electronic quintet state yielding (cAACMe)=C3=(cAACMe). In contrast, 1 and 2 possess electron‐sharing and dative bonds between positively charged ligands [(PPh3)2]+ or [(NHCMe)2]+ and negatively charged [C3]− fragments in the respective doublet state.
Keywords: coordination chemistry, dative bonding, electron-sharing bonding, ligand stabilization, physical chemistry
C3 stabilization: Quantum chemical studies using density functional theory and ab initio methods have been carried out for the molecules L‐C3‐L with L=PPh3 (1), NHCMe (2, NHC=N‐heterocyclic carbene), and cAACMe (3, cAAC= cyclic (alkyl)(amino) carbene). The calculations predict that 1 and 2 have equilibrium geometries where the ligands are bonded with rather acute bonding angles at the linear C3 moiety.

Introduction
The stabilization of transient species, small clusters, and bare atoms through coordination with Lewis bases has become a highly prolific discipline in modern chemistry. [1] Stable singlet carbenes like cyclic (alkyl)(amino) carbene (cAAC) [2] and N‐heterocyclic carbene (NHC) [3] have turned out to be excellent ligands for this purpose [4] besides the well‐known phosphine groups. Focusing on the carbon atoms as the species to be stabilized, the first divalent carbon(0) complex, carbodiphosphorane C(PPh3)2 was synthesized by Ramirez et al. [5] in 1961, followed by a proper structural characterization through X‐ray analysis in 1978 by Hardy et al. [6] Later, theoretical studies on the bonding situation revealed that this complex can be best described in terms of donor–acceptor interactions Ph3P→C(0)←PPh3 between the phosphine ligands and a bare carbon atom in the excited 1D state. [7] This finding led to the theoretical prediction of stable carbodicarbene C(NHC)2 in 2007, [8] a benzoannulated derivative C(NHCBz)2 that was shortly afterwards synthesized by the groups of Bertrand and Fürstner. [9] Then, gradually the bonding concept of carbones [10] L→C(0)←L was extended for other donor ligands L. [11] The notion of dative bonding in carbones was not undisputed, [12] but it has been generally accepted now as a valid description of main‐group compounds. [13] It should be noted that the use of arrows for dative bonds was originally introduced by Sidgwick in the 1920s [14] and that the suggestion of divalent carbon(0) compounds L→C←L was suggested already by Varshavskii in 1980. [15]
The bonding analysis also showed that molecules like (R2N)2C=C=C(NR2)2 and (cAAC)=C=(cAAC) can be better represented with classical electron‐sharing double bonds rather than dative bonds.[ 11d , 16 ] It should be noted that the donor–acceptor interactions comprise σ donation L→C(0)←L as well as π back‐donation L←C(0)→L, where the extent of back‐donation depends on the availability of low‐lying π acceptor orbitals of the ligand L. For example, a strong π acceptor such as CO induces strong π back‐donation, which means that the dative bonds in the bent equilibrium structure of carbon suboxide C3O2 [17] should be described with the formula OC⇄C(0)⇄CO.[ 12b , 13b ]
Regarding its next larger carbon homolog, Bestmann and co‐workers prepared in 1989 a push–pull molecule, R3P‐C2‐BR3, with a dicarbon core. [18] In retrospect, it is interesting that the authors suggested in that paper that carbodiphosphorane C(PPh3)2 can be formally regarded as a “complex” consisting of two donor molecules and an electron‐rich, excited carbon atom citing earlier work with similar suggestions, [19] which were then thought to be a purely hypothetical formulation. The authors postulated that carbon chains with an even number n of carbon atoms would be stabilized by a push–pull mechanism D→Cn→A with a donor ligand D and acceptor ligand A at either end, whereas carbon chains with an odd number of carbon atoms are stabilized by two donor ligands D→Cn+1←D. The hypothesis was later confirmed by the synthesis of the C3 complex Ph3P‐C3‐PPh3 and the C4 adduct Ph2(Me)P‐C4‐B(CH2Ph)3. [20] The latter species could be structurally characterized by X‐ray analysis whereas the former C3 complex could only be identified by NMR and IR spectroscopy, which were interpreted in terms of a trans‐bent geometry.
The possibility of stabilizing C2 by two donor NHC ligands was theoretically explored in 2012 by Dutton and Wilson. [21] They also considered the PMe3 ligand in their study and reported that, although in (NHC)2C2 the central C4 unit is almost linear, in (PMe3)2C2, P‐C‐C‐P has a trans‐bent geometry. More recently, successful attempts to synthesize cAAC‐stabilized C2 as neutral species (cAAC)C2 were reported independently by the Roesky and Bertrand groups. [22] A similar effort to synthesize NHCBz‐stabilized C2, by the deprotonation of a doubly protonated precursor, [(NHCBz)2C2H2]2+, failed presumably because of the small HOMO–LUMO energy gap of (NHCBz)2C2. [23] Similar to its calculated NHC analog, the C4 core in (cAAC)C2 only slightly deviates from linearity. Although a comparative bonding analysis for donor–acceptor versus electron‐sharing is still missing, the linear C4 core moiety suggests an electron‐sharing C4 unit with electron‐sharing double bonds in (NHC)=C=C=(NHC) and (cAAC)=C=C=(cAAC) rather than dative bonds (NHC)→C2←(NHC) and (cAAC)→C2←(cAAC). Later, Wu et al. [24] isolated the 4‐pyridylidene‐supported C2 species, which exhibits a more significant deviation from linearity by 24.2° than that observed in NHC or cAAC analogs.
The stabilization of longer carbon chains Cn where n>2 in compounds R‐Cn‐R′ with various groups R has been the topic of numerous studies. The synthetic efforts to isolate polyynes that are stabilized by metal or non‐metal end groups R has been summarized in a seminal review by Szafert and Gladys in 2003. [25] Research about longer cumulenes has been reviewed more recently by Tykwinski and co‐workers. [26] Very little is known about ligand‐stabilized Cn species with donor ligands beyond the C2 unit. As mentioned above, the complex Ph3P‐C3‐PPh3 has been synthesized in the equilibrium geometry and the bonding situation is not known so far. [20] Free C3 is known as an interstellar species having 1Σg + ground state, [27] which was spectroscopically studied in the gas phase. [28]
In the present study, we carried out a density functional theory (DFT) study on (L)2C3 complexes where L=PPh3, NHCMe, and cAACMe. The bonding situation is analyzed through natural bond orbital (NBO) and charge and energy decomposition analyses (EDA‐NOCV). The present results suggest that the phosphine complex Ph3P‐C3‐PPh3 unexpectedly has a synclinal (gauche) conformation and not a trans conformation as originally assumed. [20] The calculations also suggest that the title molecules L‐C3‐L with L=NHCMe and cAACMe are also viable to be realized experimentally at ambient conditions. The complex (NHCMe)2C3 has a trans‐bent equilibrium geometry whereas the adduct (cAACMe)2C3 possesses a nearly linear arrangement of the central C4 core. The bonding situation of the latter complex is distinctively different from the other two adducts.
Computational Details
The geometry optimization followed by the vibrational frequencies calculations of (L)2C3 molecules where L=NHCMe, cAACMe, and PPh3 in their respective singlet and triplet electronic states were performed at the BP86‐D3(BJ)/def2‐TZVPP level. [29] All the molecules are minima on the potential energy surfaces as ensured by the absence of any imaginary frequencies. For some molecules, additional calculations were carried out with the M06‐2X functional developed by Truhlar and co‐workers. [30] These calculations were carried out with the Gaussian 16 program package. [31] The NBO analysis [32] was performed to evaluate the partial charge, Wiberg bond indices (WBI), and natural bond orbitals by using the NBO 6.0 program. [33] Unless otherwise noted, we discuss geometries and energies at the BP86‐D3(BJ)/def2‐TZVPP level. Some ab initio calculations were carried out with the DLPNO‐CCSD(T) approach [34] in conjunction with the cc‐pVTZ basis set [35] by using the program package ORCA. [36]
The bonding situations were analyzed by means of an energy decomposition analysis (EDA) [37] together with the natural orbitals for chemical valence (NOCV) [38] method by using the ADF 2018.105 program package. [39] The EDA‐NOCV calculations were carried out at the BP86‐D3(BJ)/TZ2P level [40] by using the BP86‐D3(BJ)/def2‐TZVPP optimized geometries. In this analysis, the intrinsic interaction energy (ΔΕ int) between two fragments can be divided into four energy components as follows [Eq. 1]:
| (1) |
Although the electrostatic ΔE elstat term represents the quasi‐classical electrostatic interaction between the unperturbed charge distributions of the prepared fragments, the Pauli repulsion ΔE Pauli corresponds to the energy change associated with the transformation from the superposition of the unperturbed electron densities of the isolated fragments to the wavefunction, which properly obeys the Pauli principle through explicit antisymmetrization and renormalization of the production wavefunction. As we included D3(BJ), it provides us with the dispersion interaction energy between two interacting fragments ΔE disp. The orbital term ΔE orb originates from the mixing of orbitals, charge transfer, and polarization between the isolated fragments, which can be further decomposed into contributions from each irreducible representation of the point group of the interacting system as follows [Eq. 2]:
| (2) |
The combination of the EDA with NOCV enables the partition of the total orbital interactions into pairwise contributions of the orbital interactions, which is vital to get a complete picture of the bonding. The charge deformation Δρk(r), resulting from the mixing of the orbital pairs ψ k(r) and ψ −k(r) of the interacting fragments presents the amount and the shape of the charge flow owing to the orbital interactions [Eq. 3], and the associated energy term ΔE orb provides the size of stabilizing orbital energy originating from such interactions [Eq. 4].
| (3) |
| (4) |
More details about the EDA‐NOCV method and its application are given in recent review articles. [41]
Results and Discussion
Figure 1 shows the optimized geometries of the three (L)2C3 molecules with L=PPh3 (1), NHCMe (2), cAACMe (3), and the three free ligands L and the C3 moiety in the electronic singlet ground states. The three compounds show distinct differences in their equilibrium geometries. The molecules 1 and 2 have rather acute bonding angles L‐C1‐C2 of 124° and 139° whereas 3 exhibits a nearly linear arrangement with a bending angle L‐C1‐C2 of 171°. Another difference between the carbene complexes concerns the planes of the ligands in 2 and 3. The two NHCMe ligands in 2 are in the same plane as C3, whereas the two cAACMe ligands in 3 are perpendicular to each other, as can be seen from the torsional angle, t(N′‐C′‐C′′‐N′′) of 178.7° in the first molecule and 99.9° in the second complex.
Figure 1.

Optimized geometries of (L)2C3 (L=PPh3, NHCMe, and cAACMe) and fragments at the BP86‐D3(BJ)/def2‐TZVPP level. Bond lengths are given in Å, angles in degrees. Calculated bond dissociation energies D e and free energies ΔG 298 for the reactions (L)2C3→2 L+C3.
The phosphine ligands L around the central C3 component in the energetically lowest lying form of 1 unexpectedly have a synclinal (gauche) conformation and not a trans conformation as originally suggested. [20] The calculations at the BP86‐D3(BJ)/def2‐TZVPP level predict that the trans conformation 1 a is 8.9 kcal mol−1 higher in energy than the gauche form 1. The optimization of both isomers without the dispersion term at the BP86/def2‐TZVPP level suggests that 1 is only 0.4 kcal mol−1 lower in energy than 1 a, which suggests that dispersion interactions significantly contribute to the stability of 1. We further optimized both conformations by using the meta‐hybrid functional M06‐2X using a def2‐TZVPP basis set with and without explicit consideration of dispersion interactions using the D3 term of Grimme. [42] The calculations at the M06‐2X‐D3/def2‐TZVPP level also predict that 1 is 7.0 kcal mol−1 lower in energy than 1 a whereas M06‐2X/def2‐TZVPP favors 1 over 1 a by 6.0 kcal mol−1. According to the DFT calculations, (PPh3)2C3 (1) has a gauche equilibrium geometry whereas (NHCMe)2C3 (2) has a trans‐bent conformation as the energetically lowest‐lying form. The same energy ordering is predicted by ab initio calculations at the DLPNO‐CCSD(T)/cc‐pVTZ level using the BP86‐D3(BJ)/def2‐TZVPP optimized geometries, which give the gauche isomer 1 as being 7.7 kcal mol−1 lower in energy than the trans isomer 1 a. For all three molecules, the corresponding triplet state is considerably higher in energy by 30.8–41.7 kcal mol−1 than the singlet state (see Figure S1 in the Supporting Information)
The experimental work on (PPh3)2C3 (1) reported the 31P NMR spectrum of the complex, which has a signal at δ=−10.4 ppm. [20] We calculated the 31P NMR chemical shift values of 1 and 1 a by using the GIAO approach at the BP86/def2‐TZVPP level. The calculated chemical shifts for the 31P NMR spectrum are δ=+11.3 and +17.5 ppm for the non‐degenerate P atoms of 1 and δ=−5.6 ppm for the degenerate P of 1 a. The latter value is in much better agreement with the experimental result, which was recorded in C6D6. It is possible that in solution 1 a prevails over 1 or that a fast equilibrium between the gauche conformations leads to a 31P NMR spectrum that mimics a trans form. The calculation of 1 and 1 a with inclusion of the solvent effect of C6D6 by using the CPCM [43] approach at the BP86‐D3(BJ)/def2‐TZVPP level even showed a slightly higher preference of 1 over 1 a by 9.2 kcal mol−1.
The comparison of the equilibrium geometries of 1–3 with the free ligands shows that the C−C bonds of the central C3 moiety in 1 (1.299/1.305 Å) and 2 (1.299 Å) have essentially the same length as in free C3 (1.301 Å) whereas 3 possesses somewhat shorter bonds (1.282 Å). The geometries suggest that the l−C3 bonds in 1 and 2 may have a different character than in 3. This is supported by the calculated bond strength of the three different ligands to the C3 fragment, which is also given in Figure 1. The cAACMe ligands in 3 are much more strongly bonded (D e=263.9 kcal mol−1; ΔG 298=233.5 kcal mol−1) than the NHCMe ligands in 2 (D e=166.9 kcal mol−1; ΔG 298=137.6 kcal mol−1) and the PPh3 ligands in 1 (D e=126.5 kcal mol−1; ΔG 298=92.7 kcal mol−1). The calculated bond dissociation energies suggest that 2 and particularly 3 are thermodynamically more stable than 1.
We analyzed the electronic structure and bonding situation of the three molecules with charge and energy decomposition methods. Table 1 shows the NBO results of the adducts. The NBO method finds a single P−C1 σ‐bond between the phosphine ligands and C3 in 1 whereas double bonds CL=C1 are calculated for the ligands in 2 and 3. The P−C1 σ‐bond in 1 is polarized toward the carbon end whereas the CL=C1 σ‐bonds in 2 and 3 are slightly polarized toward the ligand atom. The CL=C1 π‐bonds in 2 and 3 exhibit a small polarization in the opposite direction towards C3. The charge distribution suggests that the central carbon atom C2 of the central C3 moiety is essentially neutral whereas the terminal carbon atoms carry negative charges, which indicates the total charge donation L→C3←L in the order 1>2>3.
Table 1.
NBO results of the compounds (L)2C3 at the BP86/def2‐TZVPP level of theory. Occupation number ON, polarization, and hybridization of the L−C1 bonds and partial charges q.
|
Complex |
Bond |
ON |
l−C1 polarization |
q |
|||
|---|---|---|---|---|---|---|---|
|
|
|
|
and hybridization [%] |
C1 |
C2 |
C3 |
|
|
1 |
P−C1 σ |
1.96 |
P: 40.6 s(30.0), p(69.4) |
C1: 59.4 s(49.0), p(51.0) |
−0.80 |
0.09 |
−0.79 |
|
2 |
CL−C1 σ |
1.97 |
CL: 52.6 s(42.1), p(57.9) |
C1: 47.4 s(47.6), p(52.4) |
−0.42 |
0.05 |
−0.42 |
|
CL−C1 π |
1.65 |
CL: 46.7 s(0.1), p(99.9) |
C1: 53.3 s(0.1), p(99.9) |
||||
|
3 |
CL−C1 σ |
1.97 |
CL: 51.2 s(38.6), p(61.4) |
C1: 48.8 s(50.1), p(49.9) |
−0.19 |
−0.01 |
−0.19 |
|
CL−C1 π |
1.80 |
CL: 46.1 s(0.1), p(99.9) |
C1: 53.9 s(0.1), p(99.9) |
||||
Detailed insight into the nature of the ligand–C3 interactions is provided by the EDA‐NOCV approach. The versatility of the method makes it possible to distinguish (a) between the total interactions of the isolated fragments in their original electronic states, charges, and equilibrium geometries, and (b) between the interactions of the fragments in the electronic states, charges, and geometries that are present in the finally formed molecule. For example, the electronic structure of diatomic LiF can be analyzed either by using the neutral atoms Li and F as interacting fragments in the EDA‐NOCV calculations, taking into account the charge migration Li→F during bond formation. Alternatively, one can use the finally formed ions Li+ and F− as fragments, which are better suited to calculate the interactions in the eventually formed bond. Those fragments that give the smallest change in the orbital interactions ΔE orb are best suited to describe the final bond. This approach has proven to be very useful in cases where the electronic state or charge of the fragments is not directly apparent. [44]
In the present case of the molecules l‐C3‐L, we focus on the best description of the l−C bonds in 1–3, which can be sketched with dative or electron‐sharing bonds between neutral or charged fragments. We considered four different pairs of interacting species L2 q and C3 q in different electronic states, which may be considered as chemically reasonable species, which lead to the molecules l‐C3‐L. These are (a) the neutral fragments L2 and C3 in the electronic singlet (S) state forming dative bonds; (b) the neutral fragments L2 and C3 in the electronic quintet (Q) state forming electron‐sharing double bonds; (c) the singly charged fragments L2 + and C3 − in the electronic doublet (D) state forming electron‐sharing and dative bonds; (d) the doubly charged fragments L2 2+ and C3 2− in the electronic triplet (T) state forming electron‐sharing and dative bonds. Table 2 gives the numerical results of the EDA‐NOCV calculations of 1–3 using the four fragments pairs (a)–(d) as interacting species.
Table 2.
EDA‐NOCV results of (L)2C3 molecules using four different sets of fragments with different charges and electronic states (S=singlet, D=doublet, T=triplet, Q=quintet) and associated bond types at the BP86‐D3(BJ)/TZ2P level. Energies are in kcal mol−1. The most favorable fragmentation scheme and bond type is given by the smallest ΔE orb value written in bold.
|
Molecule |
Bond type[a] |
Fragments |
ΔE int |
ΔE Pauli |
ΔE elstat |
ΔE disp |
ΔE orb |
|---|---|---|---|---|---|---|---|
|
(PPh3)2C3 |
D |
(PPh3)2(S)+C3 (S) |
−488.5 |
674.6 |
−428.1 |
−16.3 |
−718.6 |
|
E |
(PPh3)2(Q)+C3 (Q) |
−461.1 |
630.4 |
−371.4 |
−16.3 |
−703.7 |
|
|
D+E |
[(PPh3)2]+(D)+[C3]− (D) |
−493.8 |
766.5 |
−565.5 |
−16.3 |
−678.5 |
|
|
D+E |
[(PPh3)2]2+(T)+[C3]2− (T) |
−824.4 |
967.8 |
−918.8 |
−16.3 |
−857.4 |
|
|
(NHCMe)2C3 |
D |
(NHCMe)2(S)+C3 (S) |
−541.0 |
634.7 |
−466.2 |
−9.4 |
−700.1 |
|
E |
(NHCMe)2(Q)+C3 (Q) |
−544.8 |
506.4 |
−373.8 |
−9.4 |
−667.9 |
|
|
D+E |
[(NHCMe)2]+(D)+[C3]− (D) |
−551.9 |
621.5 |
−517.2 |
−9.4 |
−646.7 |
|
|
D+E |
[(NHCMe)2]2+(T)+[C3]2− (T) |
−875.9 |
659.2 |
−798.2 |
−9.4 |
−727.5 |
|
|
(cAACMe)2C3 |
D |
(cAACMe)2(S)+C3 (S) |
−594.2 |
419.5 |
−359.6 |
−10.6 |
−643.5 |
|
E |
(cAACMe)2(Q)+C3 (Q) |
−407.7 |
519.1 |
−348.5 |
−10.6 |
−567.6 |
|
|
D+E |
[(cAACMe)2]+(D)+[C3]− (D) |
−586.8 |
555.8 |
−445.4 |
−10.6 |
−686.5 |
|
|
D+E |
[(cAACMe)2]2+(T)+[C3]2− (T) |
−930.1 |
873.0 |
−819.7 |
−10.6 |
−972.7 |
[a] D=Dative bond; E=electron‐sharing bond.
The results in Table 2 suggest that the best description of the l‐C3‐L bonds for L=PPh3 (1) and L=NHCMe (2) comes from the interactions of the singly charged fragments L2 + and C3 − in the electronic doublet (D) state forming electron‐sharing and dative bonds. In contrast, the smallest ΔE orb value and thus the best description of the l‐C3‐L bonds for L=cAACMe (3) is provided by the neutral fragments L2 and C3 in the electronic quintet (Q) state forming electron‐sharing double bonds. This agrees with the NBO charges in Table 1, which suggest that the partial charges of the interacting fragments in 1 and 2 are clearly higher than in 3. We further analyzed the orbital interactions ΔE orb of 1–3 by using the most favorable fragments and studied the main pairwise orbital contributions ΔE orb(n). The numerical results are given in Table 3.
Table 3.
The EDA‐NOCV results at the BP86‐D3(BJ)/TZ2P level of L2C3 molecules 1–3 using (L2)+ and (C3)− in the electronic doublet (D) states as interacting fragments for 1 and 2 and neutral L2 and C3 in the quintet (Q) states as interacting fragments for 3. Energies are in kcal mol−1.
|
Energy |
Interaction[c] |
[(PPh3)2]+(D) +[C3]−(D) |
[(NHCMe)2]+ (D) +[C3]−(D) |
(cAACMe)2(Q) +C3 (Q) |
|---|---|---|---|---|
|
ΔE int |
|
−493.8 |
−551.9 |
−407.7 |
|
ΔE Pauli |
|
766.5 |
621.5 |
519.1 |
|
ΔE disp [a] |
|
−16.3 (1.2 %) |
−9.4 (0.8 %) |
−10.6 (1.1 %) |
|
ΔE elstat [a] |
|
−565.5 (44.8 %) |
−517.2 (44.0 %) |
−348.5 (37.6 %) |
|
ΔE orb [a] |
|
−678.5 (53.8 %) |
−646.7 (55.1 %) |
−567.6 (61.2 %) |
|
ΔE orb(1) [b] |
l‐C3‐L σ e− sharing (+,+) |
|
|
−187.6 (33.1 %) |
|
|
L→C3←L σ donation (+,+) |
−286.3 (42.1 %) |
−235.1 (36.3 %) |
– |
|
ΔE orb(2) [b] |
l‐C3‐L σ e− sharing (+,−) |
−231.3 (34.1 %) |
−208.7 (32.2 %) |
−180.3 (31.8 %) |
|
|
L→C3←L σ donation (+,−) |
|
|
– |
|
ΔE orb(3) [b] |
l‐C3‐L π/σ e− sharing |
|
|
−77.7 (13.7 %) |
|
|
L←C3→L σ back donation |
−51.8 (7.6 %) |
−88.3 (13.6 %) |
– |
|
ΔE orb(4) [b] |
l‐C3‐L π e− sharing |
|
|
−76.7 (13.5 %) |
|
|
L←C3→L π back‐donation |
−40.4 (5.9 %) |
−52.9 (8.1 %) |
– |
|
ΔE orb(5) [b] |
L←C3→L π back‐donation |
−25.0 (3.6 %) |
−18.4 (2.8 %) |
– |
|
ΔE orb(rest) [b] |
|
−43.6 (6.4 %) |
−43.3 (6.6 %) |
−45.3 (8.0 %) |
[a] The values in the parentheses show the contribution to the total attractive interaction ΔE elstat+ΔE orb+ΔE disp. [b] The values in parentheses show the contribution to the total orbital interaction ΔE orb.
There are five relevant orbital interactions ΔE orb(1)–ΔE orb(5) for molecules 1 and 2 and four terms ΔE orb(1)–ΔE orb(4) for complex 3. The nature of the orbital terms and the involved fragments orbitals can be identified with the help of the associate deformation densities Δρn and connected MOs that are shown in Figure 2, Figure 3, and Figure 4. The strongest orbital interaction ΔE orb(1) in 1 and 2 is due to the σ donation of the HOMO of the ligands L2 + into the LUMO of C3 −. It is remarkable and counter‐intuitive that the cation serves as donor and the anion as acceptor. However, this must be seen as only one part of the total orbital interactions. The slightly weaker orbital term ΔE orb(2) in 1 and 2 comes from the electron‐sharing interactions of the unpaired electrons in the SOMO of the fragments. The remaining three orbital terms ΔE orb(3)–ΔE orb(5) in the two molecules are due the π back‐donation of the degenerate HOMO and the HOMO−1 of C3 − to vacant orbitals of L2 +. [45] The strength of the π back‐donation is much less than that of the σ donation ΔE orb(1) but the amount of charge migration, which is given by the eigenvalues υn, shows that π back‐donation has a similar magnitude as the σ donation, which is consistent with the NBO charges (Table 1). The amount of charge migration given by υn does not correlate with the associated stabilization energy ΔE orb(n).
Figure 2.

The shape of the deformation densities Δρ (1)–(5) that correspond to ΔE orb(1)–(5), and the associated MOs of (PPh3)2C3 and the fragments orbitals of [(PPh3)2]+ and [C3]− in the doublet state at the BP86‐D3(BJ)/TZ2P level. Isosurface values are 0.003 au. The eigenvalues |νn| give the size of the charge migration in e. The direction of the charge flow of the deformation densities is red→blue.
Figure 3.

The shape of the deformation densities Δρ (1)–(5) that correspond to ΔE orb(1)–(5), and the associated MOs of (NHCMe)2C3 and the fragments orbitals of [(NHCMe)2]+ and [C3]− in the doublet state at the BP86‐D3(BJ)/TZ2P level. Isosurface values are 0.003 au. The eigenvalues |νn| give the size of the charge migration in e. The direction of the charge flow of the deformation densities is red→blue.
Figure 4.
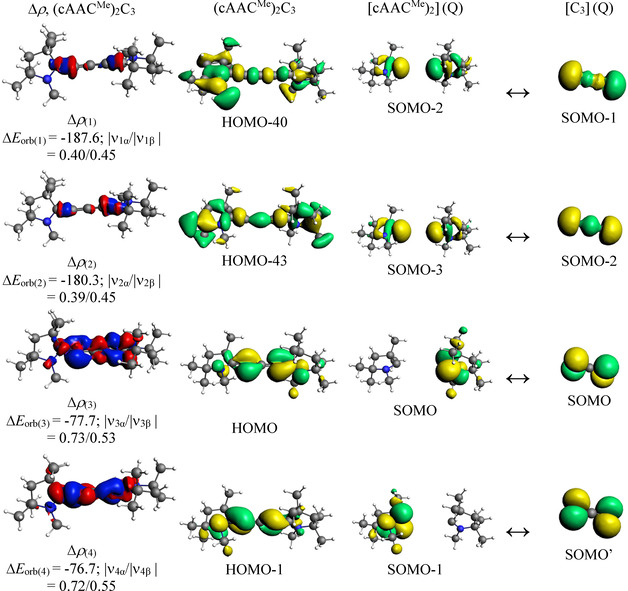
The shape of the deformation densities Δρ (1)–(5) that correspond to ΔE orb(1)–(5), and the associated MOs of (cAACMe)2C3 and the fragments orbitals of (cAACMe) and C3 in the quintet state at the BP86‐D3(BJ)/TZ2P level. Isosurface values are 0.003 au. The eigenvalues |νn| give the size of the charge migration in e. The direction of the charge flow of the deformation densities is red→blue.
The four significant orbital terms ΔE orb(1)–ΔE orb(4) between the neutral fragments in the quintet states in complex 3 are easily recognized as the strong in‐phase (+,+) and out‐of‐phase (+,−) electron‐sharing interactions ΔE orb(1) and ΔE orb(2) and the weaker but still rather strong orthogonal π interactions [45] ΔE orb(3) and ΔE orb(4), which are slightly different, because the molecular planes of the cAACMe ligands are not perfectly orthogonal to each other.
Summary and Conclusion
The present study may be summarized as follows. Quantum chemical calculations suggest that the molecules l‐C3‐L with L=PPh3 (1) and NHCMe (2) have equilibrium geometries where the ligands are bonded with rather acute bonding angles at the linear C3 moiety. The phosphine adduct 1 has a synclinal (gauche) conformation whereas 2 exhibits a trans conformation of the ligands. In contrast, the complex with L=cAACMe (3) possesses a nearly linear arrangement of the carbene ligands at the C3 fragment. The bond dissociation energies of the ligands have the order 1<2<3. The bonding analysis using charge and energy decomposition methods suggests that 3 is best described as a cumulene with electron‐sharing double bonds between neutral fragments (cAACMe)2 and C3 in the respective electronic quintet state yielding (cAACMe)=C3=(cAACMe). In contrast, 1 and 2 possess electron‐sharing and dative bonds between positively charged ligands [(PPh3)2]+ or [(NHCMe)2]+ and negatively charged [C3]− fragments in the respective doublet state.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
G.F. and S.P. thank the Deutsche Forschungsgemeinschaft and Nanjing Tech University for financial support. K.C.M. thanks DST and IIT‐Madras and G.N.V.T.S.M. thanks CSIR for a fellowship. Open access funding enabled and organized by Projekt DEAL.
S. M. N. V. T. Gorantla, S. Pan, K. C. Mondal, G. Frenking, Chem. Eur. J. 2020, 26, 14211.
NHC=N‐heterocyclic carbene, cAAC=cyclic (alkyl)(amino) carbene.
Contributor Information
Prof. Dr. Kartik Chandra Mondal, Email: csdkartik@iitm.ac.in.
Prof. Dr. Gernot Frenking, Email: frenking@chemie.uni-marburg.de.
References
- 1.
- 1a. Frenking G., Hermann M., Andrada D. M., Holzmann N., Chem. Soc. Rev. 2015, 44, 1129; [DOI] [PubMed] [Google Scholar]
- 1b. Xiong Y., Yao S., Inoue S., Epping J. D., Driess M., Angew. Chem. Int. Ed. 2013, 52, 7147; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7287; [Google Scholar]
- 1c. Frenking G., Tonner R., Klein S., Takagi N., Shimizu T., Krapp A., Pandey K. K., Parameswaran P., Chem. Soc. Rev. 2014, 43, 5106; [DOI] [PubMed] [Google Scholar]
- 1d. Niepötter B., Herbst-Irmer R., Kratzert D., Samuel P. P., Mondal K. C., Roesky H. W., Jerabek P., Frenking G., Stalke D., Angew. Chem. Int. Ed. 2014, 53, 2766; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2806; [Google Scholar]
- 1e. Xiong Y., Yao S., Tan G., Inoue S., Driess M., J. Am. Chem. Soc. 2013, 135, 5004; [DOI] [PubMed] [Google Scholar]
- 1f. Braunschweig H., Dewhurst R. D., Hammond K., Mies J., Radacki K., Vargas A., Science 2012, 336, 1420; [DOI] [PubMed] [Google Scholar]
- 1g. Wang Y., Xie Y., Wei P., King R. B., H. F. Schaefer III , Schleyer P. von R., Robinson G. H., Science 2008, 321, 1069; [DOI] [PubMed] [Google Scholar]
- 1h. Sidiropoulos A., Jones C., Stasch A., Klein S., Frenking G., Angew. Chem. Int. Ed. 2009, 48, 9701; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9881; [Google Scholar]
- 1i. Jones C., Sidiropoulos A., Holzmann N., Frenking G., Stasch A., Chem. Commun. 2012, 48, 9955; [DOI] [PubMed] [Google Scholar]
- 1j. Appel R., Schöllhorn R., Angew. Chem. Int. Ed. Engl. 1964, 3, 805; [Google Scholar]; Angew. Chem. 1964, 76, 991; [Google Scholar]
- 1k. Wang Y., Xie Y., Wei P., King R. B., H. F. Schaefer III , Schleyer P. von R., Robinson G. H., J. Am. Chem. Soc. 2008, 130, 14970; [DOI] [PubMed] [Google Scholar]
- 1l. Abraham M. Y., Wang Y., Xie Y., Wei P., H. F. Schaefer III , Schleyer P. von R., Robinson G. H., Chem. Eur. J. 2010, 16, 432; [DOI] [PubMed] [Google Scholar]
- 1m. Kinjo R., Donnadieu B., Bertrand G., Angew. Chem. Int. Ed. 2010, 49, 5930; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6066; [Google Scholar]
- 1n. Jana A., Huch V., Rzepa H. S., Scheschkewitz D., Angew. Chem. Int. Ed. 2015, 54, 289; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 291. [Google Scholar]
- 2.
- 2a. Bourissou D., Guerret O., Gabbai F. P., Bertrand G., Chem. Rev. 2000, 100, 39; [DOI] [PubMed] [Google Scholar]
- 2b. Lavallo V., Canac Y., Prasang C., Donnadieu B., Bertrand G., Angew. Chem. Int. Ed. 2005, 44, 5705; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5851; [Google Scholar]
- 2c. Soleilhavoup M., Bertrand G., Acc. Chem. Res. 2015, 48, 256. [DOI] [PubMed] [Google Scholar]
- 3. A. J. Arduengo III , Harlow R. L., Kline M., J. Am. Chem. Soc. 1991, 113, 2801. [Google Scholar]
- 4.
- 4a. Mondal K. C., Roesky H. W., Schwarzer M. C., Frenking G., Niepåtter B., Wolf H., Herbst-Irmer R., Stalke D., Angew. Chem. Int. Ed. 2013, 52, 2963; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3036; [Google Scholar]
- 4b. Li Y., Mondal K. C., Roesky H. W., Zhu H., Stollberg P., Herbst-Irmer R., Stalke D., Andrada D. M., J. Am. Chem. Soc. 2013, 135, 12422; [DOI] [PubMed] [Google Scholar]
- 4c. Chu T., Belding L., van der Est A., Dudding T., Korobkov I., Nikonov G. I., Angew. Chem. Int. Ed. 2014, 53, 2711; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2749; [Google Scholar]
- 4d. Mondal K. C., Samuel P. P., Roesky H. W., Aysin R. R., Leites L. A., Neudeck S., Luebben J., Dittrich B., Holzmann N., Hermann M., Frenking G., J. Am. Chem. Soc. 2014, 136, 8919; [DOI] [PubMed] [Google Scholar]
- 4e. Holzmann N., Dange D., Jones C., Frenking G., Angew. Chem. Int. Ed. 2013, 52, 3004; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3078; [Google Scholar]
- 4f. Holzmann N., Stasch A., Jones C., Frenking G., Chem. Eur. J. 2011, 17, 13517; [DOI] [PubMed] [Google Scholar]
- 4g. Scheschkewitz D., Angew. Chem. Int. Ed. 2005, 44, 2954; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3014; [Google Scholar]
- 4h. Scheschkewitz D., Chem. Eur. J. 2009, 15, 2476; [DOI] [PubMed] [Google Scholar]
- 4i. Bejan I., Scheschkewitz D., Angew. Chem. Int. Ed. 2007, 46, 5783; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5885; [Google Scholar]
- 4j. Abersfelder K., White A. J. P., Rzepa H. S., Scheschkewitz D., Science 2010, 327, 564; [DOI] [PubMed] [Google Scholar]
- 4k. Abersfelder K., White A. J. P., Berger R. J. F., Rzepa H. S., Scheschkewitz D., Angew. Chem. Int. Ed. 2011, 50, 7936; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8082; [Google Scholar]
- 4l. Mondal K. C., Roy S., Dittrich B., Andrada D. M., Frenking G., Roesky H. W., Angew. Chem. Int. Ed. 2016, 55, 3158; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3210; [Google Scholar]
- 4m. Pan S., Saha R., Osorio E., Chattaraj P. K., Frenking G., Merino G., Chem. Eur. J. 2017, 23, 7463; [DOI] [PubMed] [Google Scholar]
- 4n. Saha R., Pan S., Chattaraj P. K., ACS Omega 2018, 3, 13720; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4o. Ghara M., Pan S., Chattaraj P. K., J. Phys. Chem. A 2019, 123, 10764; [DOI] [PubMed] [Google Scholar]
- 4p. Arrowsmith M., Braunschweig H., Celik M., Celik M. A., Dellermann T., Dewhurst R. D., Ewing W. C., Hammond K., Kramer T., Krummenacher I., Mies J., Radacki K., Schuster J. K., Nat. Chem. 2016, 8, 890; [DOI] [PubMed] [Google Scholar]
- 4q. Wang G., Walley J. E., Dickie D. A., Pan S., Frenking G., R. J. Gilliard, Jr. , J. Am. Chem. Soc. 2020, 142, 4560. [DOI] [PubMed] [Google Scholar]
- 5. Ramirez F., Desai N. B., Hansen B., McKelvie N., J. Am. Chem. Soc. 1961, 83, 3539. [Google Scholar]
- 6. Hardy G. E., Zink J. I., Kaska W. C., Baldwin J. C., J. Am. Chem. Soc. 1978, 100, 8001. [Google Scholar]
- 7. Tonner R., Öxler F., Neumüller B., Petz W., Frenking G., Angew. Chem. Int. Ed. 2006, 45, 8038; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 8206. [Google Scholar]
- 8. Tonner R., Frenking G., Angew. Chem. Int. Ed. 2007, 46, 8695; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 8850. [Google Scholar]
- 9.
- 9a. Dyker C. A., Lavallo V., Donnadieu B., Bertrand G., Angew. Chem. Int. Ed. 2008, 47, 3206; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3250; [Google Scholar]
- 9b. Fürstner A., Alcarazo M., Goddard R., Lehmann C. W., Angew. Chem. Int. Ed. 2008, 47, 3210; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3254. [Google Scholar]
- 10. Frenking G., Tonner R., Pure Appl. Chem. 2009, 81, 597. [Google Scholar]
- 11.
- 11a. Tonner R., Frenking G., Chem. Eur. J. 2008, 14, 326; [Google Scholar]
- 11b. Tonner R., Frenking G., Chem. Eur. J. 2008, 14, 3273; [DOI] [PubMed] [Google Scholar]
- 11c. Tonner R., Heydenrych G., Frenking G., ChemPhysChem 2008, 9, 1474; [DOI] [PubMed] [Google Scholar]
- 11d. Klein S., Tonner R., Frenking G., Chem. Eur. J. 2010, 16, 10160; [DOI] [PubMed] [Google Scholar]
- 11e. Esterhuysen C., Frenking G., Chem. Eur. J. 2011, 17, 9944; [DOI] [PubMed] [Google Scholar]
- 11f. Klein S., Frenking G., Angew. Chem. Int. Ed. 2010, 49, 7106; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7260; [Google Scholar]
- 11g. Esterhuysen C., Frenking G., Dalton Trans. 2013, 42, 13349; [DOI] [PubMed] [Google Scholar]
- 11h. Kaufhold O., Hahn F. E., Angew. Chem. Int. Ed. 2008, 47, 4057; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4122; [Google Scholar]
- 11i. Phukan A. K., Guha A. K., Dalton Trans. 2012, 41, 8973. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Himmel D., Krossing I., Schnepf A., Angew. Chem. Int. Ed. 2014, 53, 370; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 378; [Google Scholar]
- 12b. Frenking G., Angew. Chem. Int. Ed. 2014, 53, 6040; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6152; [Google Scholar]
- 12c. Himmel D., Krossing I., Schnepf A., Angew. Chem. Int. Ed. 2014, 53, 6047; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6159. [Google Scholar]
- 13.
- 13a. Frenking G., Hermann M., Andrada D. M., Holzmann N., Chem. Soc. Rev. 2016, 45, 1129; [DOI] [PubMed] [Google Scholar]
- 13b. Zhao L., Hermann M., Holzmann N., Frenking G., Coord. Chem. Rev. 2017, 344, 163. [Google Scholar]
- 14. Sidgwick N. V., The Electronic Theory of Valency, Clarendon, Oxford, 1927. [Google Scholar]
- 15. Varshavskii Yu. S., Russ. J. Gen. Chem. 1980, 50, 406; [Google Scholar]; Varshavskii Yu. S., Zhurnal Obshchei Khimii 1980, 50, 514. [Google Scholar]
- 16. Andrada D. M., Holzmann N., Frenking G., Can. J. Chem. 2016, 94, 1006. [Google Scholar]
- 17. Jensen P., Johns J. W. C., J. Mol. Spectrosc. 1986, 118, 248. [Google Scholar]
- 18. Bestmann H. J., Behl H., Bremer M., Angew. Chem. Int. Ed. Engl. 1989, 28, 1219; [Google Scholar]; Angew. Chem. 1989, 101, 1303. [Google Scholar]
- 19.
- 19a. Kaska W. C., Mitchell D. K., Reichelderfer R. F., J. Organomet. Chem. 1973, 47, 391; [Google Scholar]
- 19b. Schmidbaur H., Angew. Chem. Int. Ed. Engl. 1983, 22, 907; [Google Scholar]; Angew. Chem. 1983, 95, 980. [Google Scholar]
- 20. Bestmann H. J., Hadawi D., Behl H., Bremer M., Humpel F., Angew. Chem. Int. Ed. Engl. 1993, 32, 1205; [Google Scholar]; Angew. Chem. 1993, 105, 1198. [Google Scholar]
- 21. Dutton J. L., Wilson D. J. D., Angew. Chem. Int. Ed. 2012, 51, 1477; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1506. [Google Scholar]
- 22.
- 22a. Li Y., Mondal K. C., Samuel P. P., Zhu H., Orben C. M., Panneerselvam S., Dittrich B., Schwederski B., Kaim W., Mondal T., Koley D., Roesky H. W., Angew. Chem. Int. Ed. 2014, 53, 4168; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4252; [Google Scholar]
- 22b. Jin L., Melaimi M., Liu L., Bertrand G., Org. Chem. Front. 2014, 1, 351. [Google Scholar]
- 23. Georgiou D. C., Stringer B. D., Hogan C. F., Barnard P. J., Wilson D. J. D., Holzmann N., Frenking G., Dutton J. L., Chem. Eur. J. 2015, 21, 3377. [DOI] [PubMed] [Google Scholar]
- 24. Wu D., Li Y., Ganguly R., Kinjo R., Chem. Commun. 2014, 50, 12378. [DOI] [PubMed] [Google Scholar]
- 25. Szafert S., Gladysz J. A., Chem. Rev. 2003, 103, 4175. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Januszewski J. A., Tykwinski R. R., Chem. Soc. Rev. 2014, 43, 3184; [DOI] [PubMed] [Google Scholar]
- 26b. Casari C. S., Tommasini M., Tykwinski R. R., Milani A., Nanoscale 2016, 8, 4414; [DOI] [PubMed] [Google Scholar]
- 26c. Wendinger D., Tykwinski R. R., Acc. Chem. Res. 2017, 50, 1468. [DOI] [PubMed] [Google Scholar]
- 27. Nicolas C., Shu J., Peterka D. S., Hochlaf M., Poisson L., Leone S. R., Ahmed M., J. Am. Chem. Soc. 2006, 128, 220. [DOI] [PubMed] [Google Scholar]
- 28. Hinkle K. W., Keady J. J., Bernath P. F., Science 1988, 241, 1319. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Becke A. D., Phys. Rev. A 1988, 38, 3098; [DOI] [PubMed] [Google Scholar]
- 29b. Perdew J. P., Phys. Rev. B 1986, 33, 8822; [DOI] [PubMed] [Google Scholar]
- 29c. Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem. 2011, 32, 1456; [DOI] [PubMed] [Google Scholar]
- 29d. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104; [DOI] [PubMed] [Google Scholar]
- 29e. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297; [DOI] [PubMed] [Google Scholar]
- 29f. Weigend F., Phys. Chem. Chem. Phys. 2006, 8, 1057. [DOI] [PubMed] [Google Scholar]
- 30. Zhao Y., Truhlar D. G., Theor. Chem. Acc. 2008, 120, 215. [Google Scholar]
- 31.Gaussian 16, Revision A.03, M. J. Frisch et al., Gaussian Inc., Wallingford, CT, 2016.
- 32.
- 32a. Weinhold F., Landis C., Valency and Bonding, A Natural Bond Orbital Donor–Acceptor Perspective, Cambridge University Press, Cambridge, 2005; [Google Scholar]
- 32b. Landis C. R., Weinhold F., The NBO View of Chemical Bonding, in The Chemical Bond: Fundamental Aspects of Chemical Bonding (Eds.: Frenking G., Shaik S.), Wiley-VCH, Weinheim, 2014, pp. 91–120. [Google Scholar]
- 33. Glendening E. D., Landis C. R., Weinhold F., J. Comput. Chem. 2013, 34, 1429. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Riplinger C., Neese F., J. Chem. Phys. 2013, 138, 034106; [DOI] [PubMed] [Google Scholar]
- 34b. Riplinger C., Sandhoefer B., Hansen A., Neese F., J. Chem. Phys. 2013, 139, 134101. [DOI] [PubMed] [Google Scholar]
- 35. T. H. Dunning, Jr. , J. Chem. Phys. 1989, 90, 1007. [Google Scholar]
- 36. Neese F., WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar]
- 37. Ziegler T., Rauk A., Theor. Chim. Acta 1977, 46, 1. [Google Scholar]
- 38.
- 38a. Mitoraj M., Michalak A., Organometallics 2007, 26, 6576; [Google Scholar]
- 38b. Mitoraj M., Michalak A., J. Mol. Model. 2008, 14, 681. [DOI] [PubMed] [Google Scholar]
- 39.
- 39a.ADF2017, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands http://www.scm.com;
- 39b. te Velde G., Bickelhaupt F. M., Baerends E. J., Guerra C. F., van Gisbergen S. J. A., Snijders J. G., Ziegler T., J. Comput. Chem. 2001, 22, 931. [Google Scholar]
- 40.
- 40a. van Lenthe E., Baerends E. J., J. Comput. Chem. 2003, 24, 1142; [DOI] [PubMed] [Google Scholar]
- 40b. van Lenthe E., Baerends E. J., Snijders J. G., J. Chem. Phys. 1993, 99, 4597; [Google Scholar]
- 40c. van Lenthe E., Baerends E. J., Snijders J. G., J. Chem. Phys. 1994, 101, 9783. [Google Scholar]
- 41.
- 41a. Frenking G., Bickelhaupt F. M., The Chemical Bond 1: Fundamental Aspects of Chemical Bonding, chapter: The EDA Perspective of Chemical Bonding, 121, Wiley-VCH, Weinheim, 2014; [Google Scholar]
- 41b. Zhao L. M., von Hopffgarten M., Andrada D. M., Frenking G., WIREs Comput. Mol. Sci. 2018, 8, 1345; [Google Scholar]
- 41c. Zhao L., Hermann M., Schwarz W. H. E., Frenking G., Nat. Rev. Chem. 2019, 3, 48; [Google Scholar]
- 41d. Zhao L., Pan S., Holzmann N., Schwerdtfeger P., Frenking G., Chem. Rev. 2019, 119, 8781. [DOI] [PubMed] [Google Scholar]
- 42.The M06-2X functional takes into account short-range dispersion interactions but not long-range dispersion, which is corrected by the specific D3 term of Grimme in ref. [29d].
- 43.
- 43a. Klamt A., Schüürmann G., J. Chem. Soc. Perkin Trans. 1993, 2, 799; [Google Scholar]
- 43b. Andzelm J., Kölmel C., Klamt A., J. Chem. Phys. 1995, 103, 9312; [Google Scholar]
- 43c. Barone V., Cossi M., J. Phys. Chem. A 1998, 102, 1995; [Google Scholar]
- 43d. Cossi M., Rega N., Scalmani G., Barone V., J. Comput. Chem. 2003, 24, 669. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. Zhang Q., Li W.-L., Xu C., Chen M., Zhou M., Li J., Andrada D. M., Frenking G., Angew. Chem. Int. Ed. 2015, 54, 11078; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11230; [Google Scholar]
- 44b. Andrada D. M., Frenking G., Angew. Chem. Int. Ed. 2015, 54, 12319; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12494; [Google Scholar]
- 44c. Mohapatra C., Kundu S., Paesch A. N., Herbst-Irmer R., Stalke D., Andrada D. M., Frenking G., Roesky H. W., J. Am. Chem. Soc. 2016, 138, 10429; [DOI] [PubMed] [Google Scholar]
- 44d. Scharf L. T., Andrada M., Frenking G., Gessner V. H., Chem. Eur. J. 2017, 23, 4422; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44e. Hermann M., Frenking G., Chem. Eur. J. 2017, 23, 3347; [DOI] [PubMed] [Google Scholar]
- 44f. Georgiou D. C., Zhao L., Wilson D. J. D., Frenking G., Dutton J. L., Chem. Eur. J. 2017, 23, 2926; [DOI] [PubMed] [Google Scholar]
- 44g. Wu Z., Xu J., Sokolenko L., Yagupolskii Y. L., Feng R., Liu Q., Lu Y., Zhao L., Fernández I., Frenking G., Trabelsi T., Francisco J. S., Zeng X., Chem. Eur. J. 2017, 23, 16566; [DOI] [PubMed] [Google Scholar]
- 44h. Petz W., Andrada D. M., Hermann M., Frenking G., Neumüller B., Z. Anorg. Allg. Chem. 2017, 643, 1096; [Google Scholar]
- 44i. Andrada D. M., Casalz-Sainz J. L., Pendás A. M., Frenking G., Chem. Eur. J. 2018, 24, 9083; [DOI] [PubMed] [Google Scholar]
- 44j. Saha R., Pan S., Merino G., Chattaraj P. K., Angew. Chem. Int. Ed. 2019, 58, 8372; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17848; [Google Scholar]
- 44k. Wang Q., Pan S., Wu Y., Deng G., Wang G., Zhao L., Zhou M., Frenking G., Angew. Chem. Int. Ed. 2019, 58, 17365; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17526; [Google Scholar]
- 44l. Yang T., Andrada D. M., Frenking G., Mol. Phys. 2019, 117, 1306. [Google Scholar]
- 45.The symmetry assignments σ and π refer to the orbitals of the C3 fragment.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary