

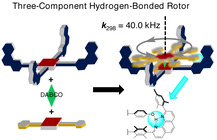
Abstract
The dynamics of hydrogen bonding do not only play an important role in many biochemical processes but also in Nature's multicomponent machines. Here, a three‐component nanorotor is presented where both the self‐assembly and rotational dynamics are guided by hydrogen bonding. In the rate‐limiting step of the rotational exchange, two phenolic O‐H–N,N(phenanthroline) hydrogen bonds are cleaved, a process that was followed by variable‐temperature 1H NMR spectroscopy. Activation data (ΔG ≠ 298=46.7 kJ mol−1 at 298 K, ΔH ≠=55.3 kJ mol−1, and ΔS ≠=28.8 J mol−1 K−1) were determined, furnishing a rotational exchange frequency of k 298=40.0 kHz. Fully reversible disassembly/assembly of the nanorotor was achieved by addition of 5.0 equivalents of trifluoroacetic acid (TFA)/1,8‐diazabicyclo[5.4.0]undec‐7‐ene (DBU) over three cycles.
Keywords: hydrogen bonding, multicomponent assembly, rotational dynamics, self-assembly, self-sorting
In a spin: Two three‐component nanorotors are presented where both the reversible formation and rotational dynamics decisively depend on the proper geometric fit of intra‐supramolecular hydrogen bonding. In the rate‐limiting step of the rotational exchange, two phenolic O‐H–N,N(phenanthroline) hydrogen bonds were cleaved, a process that was investigated by variable‐temperature (VT) 1H NMR spectroscopy.
Both the construction and dynamics of molecular machines may be based on hydrogen bonding, an exciting field of supramolecular chemistry strongly influenced by the works of Leigh and Paolucci. [1] Hydrogen bonding has demonstrated its utility in the shuttling of rotaxanes[ 1 , 2 , 3 ] and its acceleration, [4] the lubrication of rotaxane shuttling,[ 5 , 6 ] as a template in the synthesis of interlocked molecules, [7] in host–guest interactions,[ 11 , 12 , 13 ] in the modulation of rotational speed, [3] and in other molecular machines.[ 6 , 14 ]
Over the years, comprehending the role of hydrogen bonding for regulating motion in molecular rotors has received ample interest.[ 2 , 15 , 16 , 17 ] Recently, speed control in a molecular rotor was achieved by using hydrogen‐bonded guests. [18] In most of the cases, intramolecular hydrogen bonding slows down the rate of rotation; [16] however, addition of a guest may also disrupt intramolecular hydrogen bonds and enhance the rotational speed, thus acting as a molecular grease.[ 19 , 20 ] Notably, all of these examples represent covalent or mechanically interlocked systems. [18]
In contrast, most of the biological rotors, such as ATP synthase[ 21 , 22 ] and bacterial flagella, [23] are multicomponent self‐assembled machines held together by weak noncovalent interactions, often to some extent by hydrogen bonding. It would be thus highly desirable to combine, for the first time, hydrogen bonding and metal–ligand binding in the preparation of multicomponent dynamic devices. Here, we present the three‐component nanorotors [1⋅3⋅DABCO] and [2⋅4⋅DABCO] (DABCO=1,4‐diazabicyclo[2.2.2]octane) where two aspects, self‐assembly and rotational dynamics, are critically hinged on the same hydrogen bond (Figure 1). This approach requires identification of the ideal balance between binding strength and dynamics. [24] Hydrogen bonding in the rotor system suggested further benefits of this approach, that is, to reversibly test disassembly and reassembly over several cycles by acid/base changes for probing the robustness of the chosen self‐sorting strategy. [25]
Figure 1.
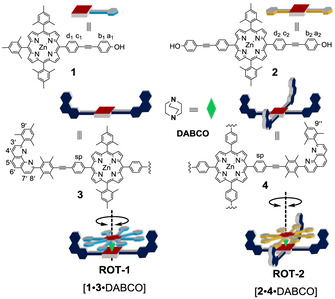
Cartoon representation of rotor components and rotors.
The key challenge in constructing multicomponent devices is to connect dynamic binding motifs that are fully orthogonal to each other.[ 26 , 27 , 28 , 29 ] In our previous work, four‐component nanorotors were made from zinc porphyrin (ZnPor) appended phenanthrolines as stator, ZnPor‐appended pyridines as rotators, copper(I) ion to link pyridine and phenanthrolines (phen), and DABCO (1,4‐diazabicyclo[2.2.2]octane) as the axle. Two metal–ligand binding motifs, axial NDABCO→ZnPor [30] and Npy→[Cu(phenAr2)]+ coordination (pyridine→copper phenanthroline=HETPYP motif), [31] were chosen to force the stator and rotator into a heteromeric bis(zinc porphyrin) sandwich rotor structure. However, phenanthrolines do not only bind to metal ions, [32] they are also suitable as hydrogen‐bond acceptors. We thus asked ourselves whether hydrogen bonding between a phenolic OH and the shielded phenanthroline would be strong enough and orthogonal to the NDABCO→ZnPor [33] interaction.
We first began our study by examining hydrogen bonding in model complexes of phenanthroline 5 and phenol 6 (Figure 2). After mixing 5 and 6 in CDCl3, the hydrogen‐bonded complex C1 was identified by 1H NMR spectroscopy. In the 1H NMR spectrum (Supporting Information, Figure S19), phenanthroline 5 surprisingly remained unaffected upon formation of C1, whereas compound 6 exhibited some diagnostic chemical shift changes, for example, the signal of hydrogen a‐H moved from 6.72 to 6.61 ppm and that of b‐H shifted from 7.34 to 7.22 ppm. These shift changes in C1 were a consequence of compound 6 experiencing strong shielding effects from the mesityl groups of phenanthroline 5. Moreover, the phenolic proton displayed a diagnostic downfield shift from 4.75 to 5.30 ppm because of hydrogen bond formation [34] to the phenanthroline's nitrogen atoms. The binding constant, determined by 1H NMR spectroscopy, turned out to be log K 5⋅6=2.50±0.02 (Supporting Information, Figure S31).
Figure 2.
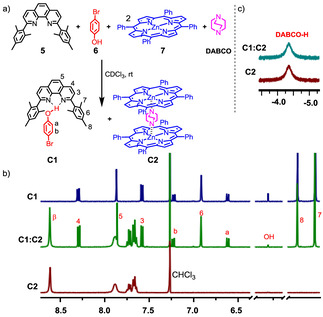
a) Testing orthogonality of hydrogen‐bonding and NDABCO→ZnPor interaction. b) Partial 1H NMR (CDCl3, 400 MHz, 298 K) and comparison of signals from C1 (1.83 mm) and C2 (1.83 mm). c) The unaltered positions of the signals of DABCO, C1, and C2 in the mixture corroborate the orthogonality of C1 and C2 (1.83 mm).
In the next step, we checked the interference‐free formation of hydrogen bonding in C1 and the axial NDABCO→ZnPor coordination in C2 by mixing 5, 6, 7, and DABCO in CDCl3. Rewardingly, the 1H NMR signals of the mixture indicated clean orthogonal formation of C1 and C2 (Figure 2).
After proving the orthogonality between hydrogen bonding and NDABCO→ZnPor, we implemented the concept to furnish a sandwich‐type of multicomponent nanorotor where hydrogen bonding guides the rotor formation as well as rotational dynamics. We have chosen ligands 3 and 4 as potential stators, [35] whereas the phenols 1 and 2 were selected as rotators. Rotator 1 was synthesized by Sonogashira coupling of the corresponding zinc(II) (4‐iodophenyl)trimesitylporphyrin and 4‐ethynylphenol by using Pd(PPh3)4 as the catalyst (Supporting Information, Scheme S1).
At first, rotator 1 and stator 3 (1:1) were mixed to check whether intermolecular hydrogen bonding between stator 3 and rotator 1 was sufficient to provide a stable assembly. The 1H NMR analysis suggested that the desired assembly did not form quantitatively (Supporting Information, Figure S13 b). However, when 1.0 equivalent of DABCO was added to the above mixture, ROT‐1=[1⋅3⋅DABCO] self‐assembled quantitatively (Figure 3 a, Supporting Information Figure S13a). The phenol ring hydrogens a1‐H and b1‐H displayed drastic upfield shifts upon formation of ROT‐1 owing to the shielding effects exerted by the aryl groups of the phenanthroline stations (Supporting Information, Figures S11 and S13 a) suggesting close proximity and thus hydrogen bonding between the phenol and phenanthroline units. The 1H NMR spectra showed a broad singlet at −4.08 ppm indicative of sandwich complex formation (Figure 3 c). The broadness of the signal may be due to fast tumbling of DABCO [36] inside the sandwich complex ROT‐1 on the NMR timescale. For a hetero‐sandwich assembly though, one would expect the DABCO hydrogens to appear as two sets of signals. Indeed, at lower temperature, the 1H NMR signal of DABCO at −4.08 ppm split into two signals in agreement with a hetero‐sandwich structure (Figure 3 c). This finding clearly proves the existence of the hetero‐assembly because otherwise multiple signals for DABCO‐H (mixture of homo and hetero assemblies) would appear at low temperature, as demonstrated in the formation of multiple assemblies without hydrogen bonding (Supporting Information, Figure S28).
Figure 3.
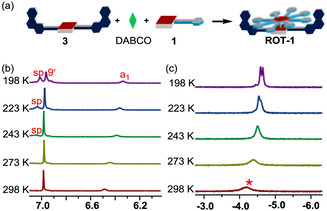
a) Cartoon representation of preparation of ROT‐1=[1⋅3⋅DABCO]. b) Partial 1H NMR spectrum of ROT‐1 (CD2Cl2, 600 MHz; 1.83 mm). c) Peak pattern of DABCO‐H in ROT‐1 (1.83 mm). Upon lowering the temperature, it first sharpened and then split into two multiplets (1:1), confirming the formation of the hetero‐sandwich.
1H‐DOSY corroborated that ROT‐1 had formed as a single species in solution (Supporting Information, Figure S24). Additionally, ROT‐1 was characterized unambiguously by 1H‐1H COSY (Supporting Information, Figure S12) and elemental analysis. Importantly, the fact that at 25 °C all phenanthroline protons appeared as a single set of signals in ROT‐1 indicating that rotator 1 rapidly oscillated between both degenerate phenanthroline stations of stator 3 with DABCO acting as an axle. Even when the temperature was lowered to −75 °C, none of the phenanthroline station signals showed splitting or coalescence (Supporting Information, Figure S26) indicating that the rotation still occurred at high speed. Accordingly, the corresponding activation barrier for rotation should be quite low. As the only effect seen in the VT‐1H NMR, proton signal a1‐H showed upfield shifts upon lowering the temperature owing to the increasing π–π stacking from both aryl groups connected to the phenanthroline station of stator 3 (Supporting Information, Figure S26).
As rotational motion in ROT‐1 requires only cleavage of one hydrogen bond, we considered increasing the activation barrier by doubling the intramolecular hydrogen‐bonding interactions. Hereunto, the 5,15‐disubstituted rotator 2 with two phenolic groups at the terminals was synthesized (Supporting Information, Scheme S1) and mixed with stator 4. The 1H NMR results suggested that the desired complex did not form quantitatively (Supporting Information, Figure S18 b). Hence, addition of DABCO became mandatory. In the presence of 1.0 equivalent of DABCO, the hetero‐sandwich complex ROT‐2=[2⋅4⋅DABCO] was furnished quantitatively as supported by two sets of DABCO signals. Furthermore, the diagnostic upfield chemical shift of the ortho‐phenol proton a2‐H from 6.89 to 6.41 ppm also indicated hydrogen bond formation (Figure 4 b and Supporting Information, Figure S18 a). Analogous to ROT‐1, the single 1H NMR signal set for all phenanthroline stations suggested rapid exchange of the phenol terminals between all phenanthroline stations in ROT‐2. VT‐1H NMR was performed from +25 °C to 0 °C showing the splitting of proton signal 9′′‐H into two singlets (1:1 ratio) with coalescence broadening at 15 °C (Figure 4 d). At 0 °C [37] the singlet separated into two sets (1:1) at 6.98 and 6.99 ppm, which were assigned to phenanthroline protons 9c′′‐H (for the hydrogen‐bonded phenanthroline station) and 9u′′‐H (for the free phenanthroline station), respectively (Figure 4 d). Simulation of the VT‐1H NMR spectra by winDNMR provided the exchange frequency k 298=40.0 kHz at 298 K with the corresponding free activation data (ΔG ≠ 298=46.7 kJ mol−1, ΔH ≠=55.3 kJ mol−1, ΔS ≠=28.8 J mol−1 K−1, see the Supporting Information Figure S30). As the rate‐determining step for rotation involves breaking of the hydrogen bond (Supporting Information, page S33), the positive value for ΔS ≠ was expected. Considering the kinetic data for ROT‐2, rotor ROT‐1 should have a barrier ΔG ≠ 198 of approximately 0.5×49.6 kJ mol−1 explaining why we could not freeze its motion even at 198 K as the rotational exchange would still occur at 1.2×106 Hz.
Figure 4.
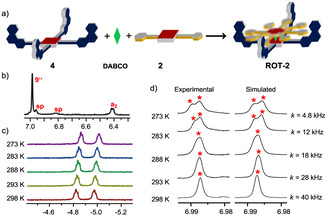
a) Formation of ROT‐2=[2⋅4⋅DABCO] by mixing all three components (cartoon representation). b) Partial 1H NMR spectrum of ROT‐2 (600 MHz, CD2Cl2, 298 K; 1.83 mm), showing the position of the proton signal a2‐H, confirming HETPYP complex formation. c) Partial VT‐1H NMR (600 MHz, CD2Cl2) of ROT‐2. Characteristic signals for DABCO‐H in this region indicate that the assembly is intact throughout the temperature range. d) Partial VT‐1H NMR spectrum (600 MHz, CD2Cl2) of ROT‐2 (1.83 mm), showing the splitting of proton 9′′‐H (marked with red asterisk). The solubility was insufficient to further lower the temperature.
DFT (B3LYP/6‐31G) calculations suggested that both nitrogen atoms of the phenanthroline did not participate equally in the hydrogen bonding (Figure 5). In fact, in both rotors, the nitrogen next to the mesityl group (N1) is the spatially better located hydrogen‐bond acceptor as suggested from bond length values: for ROT‐1, N1–H‐O=1.78 Å and N2–H‐O=2.41 Å; for ROT‐2, N1–H‐O=1.79 Å, N2–H‐O=2.40 Å, N3–H‐O=1.78 Å, N4–H‐O=2.40 Å (Supporting Information, chapter 8). Therefore, one would expect to see a temperature‐dependent wing‐like flapping motion of the OH between both phenanthroline nitrogen atoms in the rotors. Indeed, in the VT‐1H NMR analysis, the phenanthroline protons 4,7‐H in ROT‐1 and ROT‐2, which serve to monitor changes at the nitrogen atoms, separated upon lowering the temperature (Supporting Information, Figure S29) owing to the loss of local symmetry.
Figure 5.
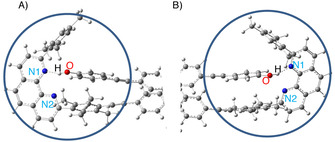
DFT (B3LYP/6‐31G) optimized structures of: a) ROT‐1 and b) ROT‐2 (only partial view) showing hydrogen bonding of the phenolic OH to nitrogen N1 of the phenanthroline. For N1–H‐O and N2–H‐O distances, see text.
Finally, to check the disassembly/re‐assembly of the hydrogen‐bonded nanorotor in the presence of acid and base, we titrated nanorotor ROT‐2 with trifluoroacetic acid (TFA) and 1,8‐diazabicyclo[5.4.0]undec‐7‐ene (DBU). When TFA was added (Supporting Information, Figure S23), the rotor assembly remained intact up to 2.0 equivalents of TFA. Upon addition of more than 2.0 equivalents of TFA, DABCO was protonated and hence the hetero‐sandwich increasingly disassembled. Eventually, addition of 5.0 equivalents of TFA furnished the fully disassembled state (DS). Conversely, addition of DBU to ROT‐2 deprotonated the hydroxyl group of the rotator rupturing the hydrogen bonding responsible for ROT‐2 formation and led to disassembly of the rotor as indicated by 1H NMR evidence (Supporting Information, Figure S23).
We derive from the results of the titration that the first two equivalents of acid protonate free phenanthroline sites and that only further addition interferes with the sandwich complexation by protonating DABCO. Finally, 5.0 equivalents of TFA fully disassembled the rotor, whereas addition of 5.0 equivalents of DBU to that solution reassembled the nanorotor.
Eventually, the reversible disassembly/assembly of the rotor over three cycles was achieved by addition of TFA/DBU as monitored by 1H NMR, UV/Vis, and fluorescence spectroscopy (see Figure 6; Supporting Information, Figures S32 and S33). In the UV/Vis channel, the porphyrin Q‐bands of ROT‐2 at λ max=563 and 605 nm shifted to λ max=553 and 598 nm upon addition of 5.0 equivalents of TFA, which indicated the loss of axial coordination (NDABCO→ZnPor) and disassembly (Figure 6 c, d). Addition of 5.0 equivalents of DBU removed the proton from [DABCO⋅H]+ thus regenerating ROT‐2 as observed by the Q‐band absorptions shifting back to λ max=563 and 605 nm (Supporting Information, Figure S32a). Analogously, the process was followed by emission spectroscopy. ROT‐2 showed luminescence (Supporting Information, Figure S33a) at λ=624 and 662 nm (λ exc=400 nm). Upon addition of 5.0 equivalents of TFA, rotor ROT‐2 disassembled as indicated by the emission at λ=604 and 647 nm. The emission reverted to its original position upon addition of 5.0 equivalents of DBU. Two full switching cycles were monitored following the luminescence changes.
Figure 6.
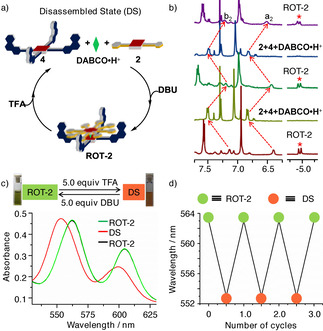
a) Cartoon representation of toggling between ROT‐2 and the disassembled state (DS). b) Partial 1H NMR spectrum showing the switching by sequential addition of 5.0 equivalents of TFA and DBU; red asterisk shows the disappearance and reappearance of DABCO‐H. c) Change in the UV/Vis spectra (6.5×10−5 m). d) Change of wavelength of the maxima in the UV/Vis spectra (6.5×10−5 m) for three switching cycles.
In conclusion, we have fabricated two three‐component nanorotors where both the formation and rotational dynamics decisively depend on the proper geometric fit of intra‐supramolecular hydrogen bonding. This approach calls for a fine balance between the thermodynamics and dynamics of the hydrogen bond. The rotational speed in ROT‐2, requiring cleavage of two hydrogen bonds, was experimentally determined as k 298=40.0 kHz. Finally, the reversible disassembly/assembly of the nanorotor was accomplished by addition/removal of 5.0 equivalents of acid/base over three cycles, demonstrating a high robustness for the self‐sorting strategy towards multicomponent devices. The present results thus open the door for designing assembly/disassembly processes of multicomponent hydrogen‐bonded rotors by using acids as chemical fuel. [38]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful to the DFG (SCHM 647/20‐2) and the University of Siegen for financial support. We thank Dr. T. Paululat (Siegen) for recording the VT‐1H NMR spectra. Open access funding enabled and organized by Projekt DEAL.
P. K. Biswas, A. Goswami, S. Saha, M. Schmittel, Chem. Eur. J. 2020, 26, 14095.
Dedicated to Professor Harald Günther on the occasion of his 85th birthday
References
- 1. Altieri A., Gatti F. G., Kay E. R., Leigh D. A., Martel D., Paolucci F., Slawin A. M. Z., Wong J. K. Y., J. Am. Chem. Soc. 2003, 125, 8644–8654. [DOI] [PubMed] [Google Scholar]
- 2. Kay E. R., Leigh D. A., Top. Curr. Chem. 2005, 262, 133–177. [Google Scholar]
- 3. Zhang Q.-C., Wu F.-T., Hao H.-M., Xu H., Zhao H.-X., Long L.-S., Huang R.-B., Zheng L.-S., Angew. Chem. Int. Ed. 2013, 52, 12602–12605; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12834–12837. [Google Scholar]
- 4. Kumpulainen T., Panman M. R., Bakker B. H., Hilbers M., Woutersen S., Brouwer A. M., J. Am. Chem. Soc. 2019, 141, 19118–19129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fu H., Shao X., Chipot C., Cai W., Chem. Sci. 2017, 8, 5087–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Panman M. R., Bakker B. H., den Uyl D., Kay E. R., Leigh D. A., Buma W. J., Brouwer A. M., Geenevasen J. A. J., Woutersen S., Nat. Chem. 2013, 5, 929–934. [DOI] [PubMed] [Google Scholar]
- 7. Schalley C. A., Weilandt T., Brüggemann J., Vögtle F., Top. Curr. Chem. 2004, 248, 141–200. [Google Scholar]
- 8. Ahmed R., Altieri A., D'Souza D. M., Leigh D. A., Mullen K. M., Papmeyer M., Slawin A. M. Z., Wong J. K., Woollins J. D., J. Am. Chem. Soc. 2011, 133, 12304–12310. [DOI] [PubMed] [Google Scholar]
- 9. Evans N. H., Akien G. R., Supramol. Chem. 2018, 30, 758–764. [Google Scholar]
- 10. Evans N. H., Eur. J. Org. Chem. 2019, 3320–3343. [Google Scholar]
- 11. Cooke G., Rotello V. M., Chem. Soc. Rev. 2002, 31, 275–286. [DOI] [PubMed] [Google Scholar]
- 12. Beatty A. M., Coord. Chem. Rev. 2003, 246, 131–143. [Google Scholar]
- 13. Naranjo T., Cerron F., Nieto-Ortega B., Latorre A., Somoza A., Ibarra B., Perez E. M., Chem. Sci. 2017, 8, 6037–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zigon N., Hosseini M. W., Chem. Commun. 2015, 51, 12486–12489. [DOI] [PubMed] [Google Scholar]
- 15. Jiang X., Duan H.-B., Jellen M. J., Chen Y., Chung T. S., Liang Y., Garcia-Garibay M. A., J. Am. Chem. Soc. 2019, 141, 16802–16809. [DOI] [PubMed] [Google Scholar]
- 16. Dial B. E., Pellechia P. J., Smith M. D., Shimizu K. D., J. Am. Chem. Soc. 2012, 134, 3675–3678. [DOI] [PubMed] [Google Scholar]
- 17. Alfonso I., Burguete M. I., Luis S. V., J. Org. Chem. 2006, 71, 2242–2250. [DOI] [PubMed] [Google Scholar]
- 18. Rushton G. T., Vik E. C., Burns W. G., Rasberry R. D., Shimizu K. D., Chem. Commun. 2017, 53, 12469–12472. [DOI] [PubMed] [Google Scholar]
- 19. Wu Y., Wang G., Li Q., Xiang J., Jiang H., Wang Y., Nat. Commun. 2018, 9, 1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dial B. E., Rasberry R. D., Bullock B. N., Smith M. D., Pellechia P. J., Profeta S., Shimizu K. D., Org. Lett. 2011, 13, 244–247. [DOI] [PubMed] [Google Scholar]
- 21. Yoshida M., Muneyuki E., Hisabori T., Nat. Rev. Mol. Cell Biol. 2001, 2, 669–677. [DOI] [PubMed] [Google Scholar]
- 22. Schliwa M., Woehlke G., Nature 2003, 422, 759–765. [DOI] [PubMed] [Google Scholar]
- 23. Dreyfus R., Baudry J., Roper M. L., Fermigier M., Stone H. A., Bibette J., Nature 2005, 437, 862–865. [DOI] [PubMed] [Google Scholar]
- 24. Nikitin K., O'Gara R., Chem. Eur. J. 2019, 25, 4551–4589. [DOI] [PubMed] [Google Scholar]
- 25. Schmittel M., Chem. Commun. 2015, 51, 14956–14968. [DOI] [PubMed] [Google Scholar]
- 26. Kishore R. S. K., Paululat T., Schmittel M., Chem. Eur. J. 2006, 12, 8136–8149. [DOI] [PubMed] [Google Scholar]
- 27. Saha M. L., De S., Pramanik S., Schmittel M., Chem. Soc. Rev. 2013, 42, 6860–6909. [DOI] [PubMed] [Google Scholar]
- 28. De S., Pramanik S., Schmittel M., Angew. Chem. Int. Ed. 2014, 53, 14255–14259; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14480–14484. [DOI] [PubMed] [Google Scholar]
- 29. Shi Q., Zhou X., Yuan W., Su X., Neniškis A., Wei X., Taujenis L., Snarskis G., Ward J. S., Rissanen K., de Mendoza J., Orentas E., J. Am. Chem. Soc. 2020, 142, 3658–3670. [DOI] [PubMed] [Google Scholar]
- 30. Samanta S. K., Bats J. W., Schmittel M., Chem. Commun. 2014, 50, 2364–2366. [DOI] [PubMed] [Google Scholar]
- 31. Schmittel M., He B., Fan J., Bats J. W., Engeser M., Schlosser M., Deiseroth H. J., Inorg. Chem. 2009, 48, 8192–8200. [DOI] [PubMed] [Google Scholar]
- 32. Schmittel M., Ammon H., Kalsani V., Wiegrefe A., Michel C., Chem. Commun. 2002, 97, 2566–2567. [Google Scholar]
- 33. Liu S., Kondratuk D. V., Rousseaux S. A. L., Gil-Ramirez G., O'Sullivan M. C., Cremers J., Claridge T. D. W., Anderson H. L., Angew. Chem. Int. Ed. 2015, 54, 5355–5359; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5445–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baures P. W., Rush J. R., Wiznycia A. V., Desper J., Helfrich B. A., Beatty A. M., Cryst. Growth Des. 2002, 2, 653–664. [Google Scholar]
- 35. Samanta S. K., Schmittel M., J. Am. Chem. Soc. 2013, 135, 18794–18797. [DOI] [PubMed] [Google Scholar]
- 36. Samanta S. K., Samanta D., Bats J. W., Schmittel M., J. Org. Chem. 2011, 76, 7466–7473. [DOI] [PubMed] [Google Scholar]
- 37.Lowering the temperature below 0 °C led to precipitation.
- 38. Biagini C., Capocasa G., Cataldi V., Del Giudice D., Mandolini L., Di Stefano S., Chem. Eur. J. 2019, 25, 15205–15211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary