

SUMMARY
Acer truncatum (purpleblow maple) is a woody tree species that produces seeds with high levels of valuable fatty acids (especially nervonic acid). However, the lack of a complete genome sequence has limited both basic and applied research on A. truncatum. We describe a high‐quality draft genome assembly comprising 633.28 Mb (contig N50 = 773.17 kb; scaffold N50 = 46.36 Mb) with at least 28 438 predicted genes. The genome underwent an ancient triplication, similar to the core eudicots, but there have been no recent whole‐genome duplication events. Acer yangbiense and A. truncatum are estimated to have diverged about 9.4 million years ago. A combined genomic, transcriptomic, metabonomic, and cell ultrastructural analysis provided new insights into the biosynthesis of very long‐chain monounsaturated fatty acids. In addition, three KCS genes were found that may contribute to regulating nervonic acid biosynthesis. The KCS paralogous gene family expanded to 28 members, with 10 genes clustered together and distributed in the 0.27‐Mb region of pseudochromosome 4. Our chromosome‐scale genomic characterization may facilitate the discovery of agronomically important genes and stimulate functional genetic research on A. truncatum. Furthermore, the data presented also offer important foundations from which to study the molecular mechanisms influencing the production of nervonic acids.
Keywords: Acer truncatum, nervonic acid, de novo assembly, very long‐chain monounsaturated fatty acid, KCS
Significance Statement
Here, we provide the first report on the Acer truncatum genome. Our work provides extensive genetic resources necessary for very long‐chain monounsaturated fatty acid biosynthesis.
INTRODUCTION
Purpleblow maple (Acer truncatum Bunge, 2n = 2x = 26) is a diploid monoecious tree species of the family Aceraceae. It is a versatile oil‐producing woody tree that is a native species widely distributed in northern China, Korea, and Japan, but it has also been identified in Europe and North America (More et al., 2003; Guo et al., 2014). In China, this tree species is referred to as ‘yuan bao feng’ because of its gold ingot‐shaped fruits (Figure 1). It has historically been used for landscaping. Potential further uses of the various tree parts have been revealed through years of research. For example, A. truncatum leaves are used to produce health‐promoting tea and folk medicines for treating cerebrovascular diseases and angina pectoris (Ma et al., 2005) because of their substantial abundance in tannins, flavonoids, and chlorogenic acid (Lingguang et al., 2017). Moreover, A. truncatum seed oil is extracted by pressing the kernels, and is composed mainly of triacylglycerols with approximately 90% unsaturated fatty acids (including oleic acid: 25.8%, linoleic acid: 37.3%, and nervonic acid [NA]: 5.5%) (Wang et al., 2006; Liu et al., 2013).
Figure 1.

Photographs of A. truncatum. (a) Mature tree. (b) Flowers. (c) Immature fruits. (d) Mature fruits. (e) Seeds.
NA (24:1Δ15, cis‐15‐tetracosenoic acid, n‐9) is a very long‐chain monounsaturated fatty acid (VLCFA) that was first discovered in the brain of sharks. It has been detected mainly in the brain and nervous system tissues, where it is considered an important component that contributes to myelinated nerve fiber biosynthesis (Poulos, 1995; Merrill et al., 1997). In humans, abnormal NA levels can lead to several neurological disorders or mental illnesses, including schizophrenia, psychosis, and attention deficit disorder (Chen et al., 2004; Pamplona et al., 2005; Tanaka et al., 2007; Amminger et al., 2012). NA‐containing oils have been important agents for preventing and treating neurological disorders and diseases. The beneficial attributes of NA include its ability to inhibit the reverse transcriptase activity of human immunodeficiency virus type‐1 (Kasai et al., 2002). Thus, NA oils are important bioactive lipid supplements for promoting human health. Although the availability of NA sources is limited, the seeds of several plant species reportedly contain relatively large amounts of NA within storage lipids (e.g., Cardamine graeca, Malania oleifera, A. truncatum, Lunaria annua, Borago officinalis, and Tropaeolum speciosum) (Bettger et al., 2001; Wang and Wang, 2005; Guo et al., 2009; Taylor et al., 2009; Yang et al., 2018). Additional natural plant resources rich in NAs are needed for large‐scale NA production to satisfy the high market demand, but these plant species may be of limited utility because of issues related to their NA contents and growth adaptability. A. truncatum might be a viable NA resource for commercial production, with features such as rapid growth, wide geographic distribution, and high adaptability. In 2011, A. truncatum seed oils were certified as a new food resource by the Ministry of Health of the People’s Republic of China, with potentially important implications for the fields of food and medicine.
NA is synthesized in the endoplasmic reticulum by a process involving the following four key enzymes: 3‐ketoacyl CoA synthetase (KCS), 3‐ketoacyl CoA reductase (KCR), 3‐hydroxyacyl CoA dehydratase (HCD), and trans‐2,3‐enoyl‐CoA reductase (ECR) (Fehling and Mukherjee, 1991; Samuels et al., 2008; Wang et al., 2018). KCS is a key rate‐limiting enzyme that determines the tissue specificity and the substrate for fatty acid elongation (Mietkiewska et al., 2007; James et al., 1995; Mietkiewska et al., 2007). Previous studies revealed that NA levels were raised in the seed oil of KCS‐expressing transgenic plants and NA was found among the fatty acids of KCS‐expressing transgenic yeast (Taylor et al., 2009; Dongxin et al., 2015).
The genus Acer L. comprises more than 200 species that grow in China, including many with considerable medicinal, ornamental, and economic value (Xu, 1998). To date, there is a lack of a fully sequenced genomes among the species in this genus. In the present study, we completed a de novo sequence and assembly of the genome of A. truncatum. An analysis of this genome revealed features such as high heterozygosity and highly similar repeats. Moreover, examination of the transposable elements (TEs) of A. truncatum suggested a TE burst occurred 1–2 million years ago (Mya), which is relatively recent in evolution, while A. truncatum diverged from Acer yangbiense about 9.4 Mya. Comprehensive comparisons between the A. truncatum and A. yangbiense genomes were also conducted, including the identification of centromeric regions and expanded and contracted gene families. Moreover, comparative transcriptomic and metabonomic analyses were completed to obtain new insights into the characterization of the late embryogenesis abundant (LEA) gene family and NA biosynthesis. The genome sequence represents a valuable resource for genetic studies and for accelerating the breeding of new lines with increased production of bioactive compounds, particularly NA.
RESULTS
Genome sequencing, assembly, and annotation
The A. truncatum genome was sequenced using a combination of PacBio single‐molecule long reads and Illumina short reads. We improved the assembly by adding 10× Genomics linked reads. On the basis of a K‐mer analysis, the A. truncatum genome size was estimated to be 653.44 Mb, with a heterozygosity of 1.12% (Table S1 and Figure S1). High‐quality consensus sequences were assembled from PacBio long reads (115.85 × coverage) (Table S2) with the Falcon program (Chin et al., 2016), after which errors in the assembly were corrected with Illumina short reads (180.48 × coverage) and the Pilon program (Walker et al., 2014). We generated a genome assembly comprising 628.84 Mb with a contig N50 of ~773.17 kb. The 10 × Genomics data (124.79 × coverage) were used to scaffold the genome with the FragScaff program (Mostovoy et al., 2016). The chromosome‐scale scaffolds were finally assembled based on Hi‐C data (72.21 Gb; Figure S2). A total of 13 long super‐scaffolds (hereafter denoted as pseudochromosomes) were generated, representing 99.44% (Table S3) of the final genome assembly with a total genome size of 633.28 Mb and a scaffold N50 of 46.36 Mb (Figure 2 and Table S4). Regarding the constructed pseudochromosomes, the Hi‐C interaction matrices displayed a distinct diagonal pattern for the intrachromosomal interactions (Figure S2), indicating that most of the contigs were accurately oriented on the pseudochromosomes. The mapping rate reached 97.24% when we aligned the Illumina reads to the genome assembly (Table S5). In addition, the completeness of the A. truncatum genome assembly was evaluated with the Core Eukaryotic Genes Mapping Approach (CEGMA) and the Benchmarking Universal Single‐Copy Orthologs (BUSCO) assessment. Accordingly, 239 of 248 core eukaryotic genes (96.37%) and 1342 complete gene models among 1440 conserved genes (93.2%) were identified based on the CEGMA and BUSCO analyses, respectively (Tables S6 and S7). In addition, 100 924 (95.16%) of unigenes (>500 bp) assembled by Trinity mapped to our genome assembly (Table S8) and the long terminal repeat (LTR) Assembly Index (LAI) score was 16.4 (Ou et al., 2018). These results for the evaluation of genome quality verified that our genome assembly was accurate and complete at the chromosome scale.
Figure 2.
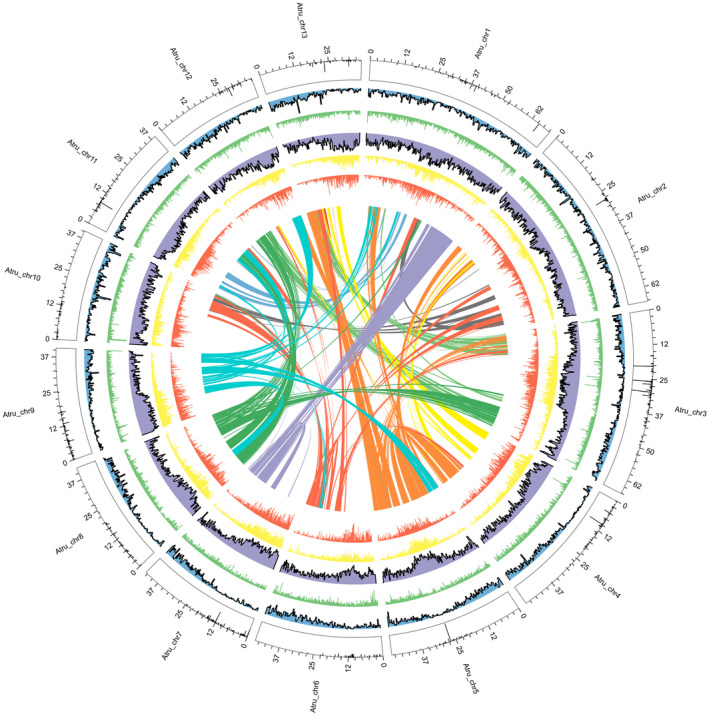
Circos plot of the genomic features of Acer truncatum. Concentric circles, from innermost to outermost, show: intra‐genome collinear blocks connected by curved lines, LTR‐Gypsy density (red), LTR‐Copia density (yellow), transposon element density (purple), tandem repeats density (green), and gene density (blue). The outermost circle displays the narrow peaks which represent centromeric regions. All distributions are drawn in a window size of 300 kb, chromosomes_units = 500 000.
We applied a combination of de novo, homology, and transcript‐based approaches to predict the genes in the A. truncatum genome. Approximately 48 Gb RNA sequencing data were generated from five libraries representing major tissue types (root, stem, young leaf, flower bud, and mature seed). In all, 28 438 genes were predicted, with average coding sequence and intron lengths of 1111.6 and 636 bp, respectively. Of these genes, 28 290 genes (99.48%) were anchored to 13 pseudochromosomes (Table S4). Moreover, 28 023 (98.5%) were similar to known genes and were functionally annotated in at least one of the following databases: NR, Swiss‐Prot, KEGG, and InterPro (Table S9). Of the predicted gene models, 21 579 genes (75.87%) contained Pfam domains and 25 432 genes (89.43%) were assigned to Gene ontology (GO) categories. Additionally, non‐coding RNA sequences were predicted in the A. truncatum genome, including 1345 miRNAs, 744 tRNAs, 368 rRNAs, and 868 snRNAs, with an average length of 132.48, 75.11, 169.31, and 113 bp, respectively (Table S10).
Repetitive content and structure of centromeres
Using a combination of approaches, the repetitive elements were determined to represent approximately 391.05 Mb (61.75%) of the assembled A. truncatum genome (Tables 1 and S11). Most of these repetitive elements were TEs (approximately 379.62 Mb; 59.95%; Table 1). Similar to other plants, LTRs were the predominant retrotransposon in A. truncatum (approximately 287.49 Mb; 45.40%; Table S11) and most of the LTRs were Copia and Gypsy. The retroelements with the highest copy numbers were the Tork subfamily members (2200) of the Ty1/Copia family, followed by the Tat subfamily members (1789) of the Ty3/Gypsy family. Similar to A. truncatum, Tork (668) also represented the highest copy numbers in the Ty1/Copia subfamilies of Camellia sinensis. However, in the A. yangbiense and M. oleifera genomes, Sire has the highest copy numbers, with 6624 and 8373 copies, respectively. Athila (751), Tat (1831), and Del (10 005) were the Ty3/Gypsy subfamilies with the highest copy numbers in C. sinensis, A. yangbiense, and M. oleifera, respectively (Table S12). An examination of the recent TE activities among A. truncatum, A. yangbiense, C. sinensis, and M. oleifera revealed a recent burst in A. truncatum and A. yangbiense around 1–2 Mya, whereas a substantial abundance of TEs was inserted into the M. oleifera genome about 4–5 Mya (Figure S3). These results provide important information regarding the genome evolution of Sapindales species.
Table 1.
Global statistics of Acer truncatum genome assembly and annotation
| Number | Size | |
|---|---|---|
| Assembly | ||
| Estimated genome size | 653.44 Mb | |
| Scaffolds | 34 | 633.28Mb |
| N50 of scaffolds | 6 | 46.36 Mb |
| Longest scaffolds | 70.31 Mb | |
| Contigs | 1453 | 628.84 Mb |
| N50 of contigs | 239 | 773.17 kb |
| Longest contigs | 4.57 Mb | |
| Pseudochromosomes | 13 | 629.76 Mb |
| Annotation | ||
| Repetitive sequences | 61.75% | 391.05Mb |
| Transposable element | 59.95% | 379.62 Mb |
| Protein‐coding genes | 28 438 | |
| Mean gene length | 3457.58 bp | |
| Mean coding sequence length | 1111.28 bp | |
| Mean intron length | 636.49 bp | |
| Mean exon length | 237.15 bp | |
| Non‐coding RNAs | 3325 | 394, 462 bp |
We also identified centromeric regions in A. truncatum and A. yangbiense by searching highly clustered tandem repeats (VanBuren et al., 2015). Most chromosomes of A. truncatum (except Chr10) and A. yangbiense (except Chr5) had narrow peaks (Figures 2 and S9), representing clustered satellites with a base monomer length of 161 bp (GC content: 53%) and 160 bp (GC content: 41.25%), respectively (Table S13). These clustered satellites were located in Gypsy‐ and Copia‐rich regions, also indicating they are putative centromeric regions of A. truncatum and A. yangbiense chromosomes. The putative centromeric regions in the A. truncatum chromosomes verified the completeness of our genome assembly.
Comparative genomic analysis
To investigate the relationship between gene families and specific A. truncatum traits, we clustered the orthologs of A. truncatum and 14 other sequenced plant species, yielding 18 616 gene groups, including 109 single‐copy gene families (Figure S4). We analyzed the phylogenetic relationships among A. truncatum and 14 other plant species with 109 single‐copy orthologs. The results revealed that A. truncatum clustered with A. yangbiense and both species were located close to C. sinensis, which was the expected result, as the three species belong to the order Sapindales (Xu et al., 2013). Moreover, an examination of divergence times indicated that A. truncatum diverged from A. yangbiense and C. sinensis about 9.4 and 68.6 Mya, respectively, after the common ancestor of both Sapindales species diverged (approximately 92.8 Mya) (Figure S5). Meanwhile, to determine the phylogenetic position of A. truncatum in Aceraceae, 17 complete chloroplast genome sequences of Aceraceae species were obtained (Tables S14) and used to construct phylogenetic trees (Figures S6 and S7). It showed that A. truncatum is closely related to Acer catalpifolium and Acer miaotaiense.
A further comparison of A. truncatum, C. sinensis, Gossypium raimondii, Arabidopsis thaliana, and A. yangbiense revealed 10 033 gene clusters shared by the five malvid species. In addition, we found 504 gene families that were unique to the A. truncatum genome when compared with the other four genomes (Figure 3(b)). An investigation of gene family evolution indicated that 262 gene families expanded in A. truncatum, whereas 513 gene families contracted (Figure 3(c), Tables S15–S18). The number of expanded gene families (587) was higher than that of contracted gene families (225) in A. yangbiense (Tables S19 and S20). The expanded gene families of two genomes were both mapped to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. In the A. truncatum genome, we observed significant enrichment (P < 0.05) in environment adaptation, flavonoid biosynthesis, phenylpropanoid biosynthesis, fatty acid elongation, and linoleic acid and nitrogen metabolism (Tables S18). Compared with the A. truncatum genome, the A. yangbiense genome is significantly enriched (P < 0.05) in environment adaptation, cell cycle, cyanoamino acid metabolism, phenylpropanoid biosynthesis, and pentose and glucuronate interconversions (Tables S21). These functions might be related to plant defense responses and the production of secondary metabolites.
Figure 3.
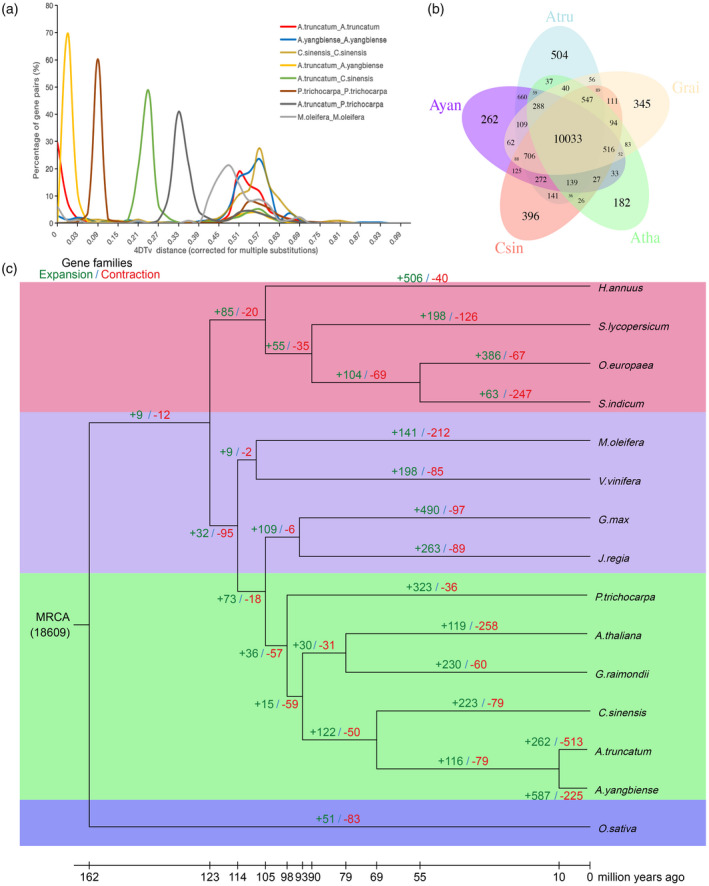
Genome evolution and comparative analysis of the Acer truncatum genome. (a) WGD analysis of A. truncatum, where the x‐axis indicates 4DTv distribution of transversion substitutions at fourfold degenerate sites. (b) Venn diagram of A. truncatum (Atru), C. sinensis (Csin), G. raimondii (Grai), A. thaliana (Atha), and A. yangbiense (Ayan). Each number in the diagram is the number of gene families within a group. (c) Gene family expansion and contraction among 15 plants. The green and red values above each branch represent the expanded and contracted gene families after the diversification from the most common ancestor, respectively.
On the basis of the accumulated rate at fourfold synonymous third‐codon transversion (4DTv) of the duplicate gene pairs, the peak values in A. truncatum, A. yangbiense, and C. sinensis were 0.51, 0.57, and 0.57, respectively (Figure 3(a)). These similar peaks indicated that they underwent the same whole‐genome duplication (WGD), the γ event. Moreover, we identified 159 syntenic blocks comprising 3891 collinear genes (13.75% of 28 290 genes) in A. truncatum. A total of 396 syntenic blocks were detected between A. truncatum and C. sinensis with 1:1 syntenic patterns (cscore >0.7), and a total of 171 syntenic blocks were detected between A. truncatum and A. yangbiense (Figures S8–S10). The results also showed that A. truncatum lacks a recent independent WGD in its evolutionary history, similar to A. yangbiense (Yang et al., 2019) and C. sinensis (Xu et al., 2013); only an ancient γ event occurred in A. truncatum.
Analysis of LEA protein family in A. truncatum
Abiotic stresses, including drought, extreme temperatures, and salinity, restrict plant growth and development. A. truncatum is drought‐tolerant and can survive in severely barren environments. In our study, 433 genes and 132 transcription factors (TFs) related to drought resistance were identified in A. truncatum and a comparative analysis with five other plant species with annotated genomes was also completed (A. thaliana, C. sinensis, M. oleifera, A. yangbiense, and Solanum lycopersicum) (Tables S22 and S23). The results suggested that a set of genes related to drought resistance had expanded, including members of the MSR, AQP1, LEA, LEW, TIP, and SUS gene families (Table S22). These expanded gene families might contribute to enhanced resistance in A. truncatum. The LEAs have received increasing attentions in recent years and have been shown to play important roles in the responses against various stresses, including drought and salinity (Hincha and Thalhammer, 2012; Gao and Lan, 2016; Magwanga et al., 2018).
We analyzed the characteristics of the LEA gene family in A. truncatum. The results showed that LEAs expanded to 82 genes in A. truncatum, which is more than the number of LEAs detected in the A. thaliana (50), C. sinensis (72), M. oleifera (55), A. yangbiense (68), and S. lycopersicum (74) genomes (Table S22). From the results of a phylogenetic analysis, the A. truncatum LEA genes were further classified into the following seven groups: LEA1 (4), LEA2 (43), LEA3 (6), LEA4 (8), LEA5 (3), SMP (7), and dehydrin (11) (Figure S11). The LEA genes are distributed across every pseudochromosome except 11. The majority of genes are located on pseudochromosomes 1, 3, and 5 (Figure S12). In previous studies, LEA proteins were found to be expressed in seedlings, stems, roots and other organs throughout all developmental stages (Shao et al., 2005; Du et al., 2013; Pedrosa et al., 2015). As shown in Figure S13, the expression of LEAs in A. truncatum was very diverse in five organs. Most of the LEA2 group members were highly expressed in the root or stem. However, the SMP and dehydrin members were most highly expressed in the seed. Ten LEA genes from different groups were selected to determine their expression pattern by real‐time PCR (RT‐PCR) analysis. The results were mostly consistent with the RNA‐seq data (Figures S13–S15). Moreover, the expression levels of most genes gradually increased in the later stages of seed development (Figure S14). Our results will pave the way for future functional analyses to unravel the role of LEA genes in drought resistance in A. truncatum.
Analysis of fatty acid biosynthesis in A. truncatum
Fatty acid biosynthesis, which is one of the major steps involved in the production of complex oils, is completed via the activities of fatty acid synthases, elongases, desaturases, and carboxylases. In our study, the combined results of genomic, transcriptomic, cell ultrastructural, and gas chromatography–mass spectrometry analyses provided some new insights regarding this biosynthesis pathway in A. truncatum. The fatty acid contents and cell ultrastructures were analyzed during six seed development stages (i.e., 70, 85, 100, 115, 145, and 180 days after flowering [DAF]). Fatty acids were undetectable at 70 DAF, but the oleic acid (18C:1) and linoleic acid (18C:2) contents increased rapidly from 85 DAF (0.46% and 0.63%, respectively) to 115 DAF (20.93% and 30.21%, respectively) (Figure 4(b)). In contrast, the other fatty acids, including erucic acid (22C:1) and NA (24C:1), apparently accumulated after 85 DAF. NA was undetectable at 85 DAF but increased to 1.96% at 100 DAF (Figure 4(b)). Lipids that accumulate in seeds are generally stored in oil bodies (Gu et al., 2017), The lipid bodies of the cotyledons during various seed development stages were examined using the cell ultrastructural analysis. The results confirmed that the key period for oil production was between 85 DAF (no oil bodies) and 100 DAF (oil bodies detected) (Figure 4(a)). These observations are consistent with the results of our NA content analysis. Moreover, the lipid bodies gradually increased in size and filled the endosperm cells over the subsequent 80 days (Figure 4(a)).
Figure 4.
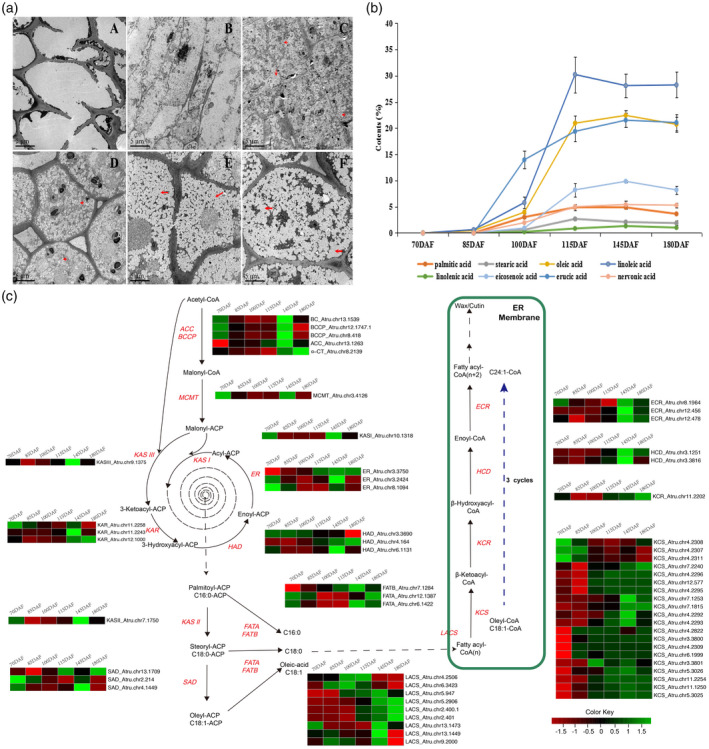
Detailed biosynthetic pathways of VLCFA in A. truncatum. (a) Oil bodies in seed cells, as observed by electron microscopy, at (A) 70, (B) 85, (C) 100, (D) 115, (E) 145, and (F) 180 DAF. (b) The extraction and determination of fatty acid contents in A. truncatum seeds after flowering. The x‐axis indicates the time after flowering (in days) and the y‐axis indicates the oil content (in %). (c) Different columns of each heat map indicate different numbers of days of seed development (70–180 DAF). Gene expression levels (log10(RPKM + 1)) in five tissues are represented by color gradation. Gene expression with RPKM ≤ 1 was set to 0 after log10 transformation. Genes with more than one homologous gene are represented by equally colored horizontal stripes and are termed from top to bottom. ACC: acetyl‐CoA carboxylase; BC: biotin carboxylase; BCCP: biotin carboxyl carrier protein; ER: enoyl‐ACP reductase; HAD: hydroxyacyl‐ACP dehydrase; KAR: ketoacyl‐ACP reductase; KASI, II, and III: ketoacyl‐ACP synthase I, II, and III; MCMT: malonyl‐CoA:acyl carrier protein (ACP) malonyltransferase; α‐CT: α‐carboxyltransferase; SAD: stearoyl‐ACP desaturase; FATA (B): fatty acyl thioesterase A (B); LACS: long‐chain acyl‐CoA synthetase; KCS: ketoacyl‐CoA synthase; KCR: ketoacyl‐CoA reductase; HCD: hydroxyacyl‐CoA dehydrase; ECR: enoyl‐CoA reductase.
To elucidate the molecular mechanisms underlying VLCFA production, especially NA biosynthesis, in A. truncatum, we identified 68 genes that are important for the VLCFA biosynthesis pathway (Table S24). There were some differences in the number of duplications of genes encoding enzymes involved in the fatty acid biosynthesis pathway (Figure 4(c)). A few enzyme‐encoding gene families related to lipid metabolism (ECR, ER, HAD, KAR, SAD, LACS, and KCS) underwent more than three duplications (Figure 4(c) and Table S20). NAs (C24:1) can be synthesized from oleyl‐CoA (C18:1‐CoA) by the four enzyme‐catalyzed reactions of the elongation cycle on the endoplasmic reticulum via three additions of C2 moieties (Yang et al., 2018). In the A. truncatum genome, 34 genes were predicted to affect the four reactions of the elongation cycle, including three genes encoding ECR, two encoding HCD, one encoding KCR, and 28 encoding KCS (Figure 4(c) and Table S24). Among these enzymes, KCS is considered to be the rate‐limiting enzyme during fatty acid elongation because it determines the substrate and tissue specificities. Previous studies confirmed that the KCS gene is important for NA biosynthesis (Taylor et al., 2009; Dongxin et al., 2015). In the current study, we determined that the A. truncatum KCS gene family expanded to 28 genes. A phylogenetic analysis of A. truncatum, A. yangbiense, A. thaliana, and M. oleifera showed that their KCS gene families consist of 28, 22, 21, and 19 members, respectively. The results indicated that 10 of the A. truncatum KCS genes and 6 A. yangbiense KCS genes were classified in the same group, and closely related to the three clustered genes (KCS_Maole_016461.T1, KCS_Maole_016463.T1, KCS_Maole_016467.T1). Xu et al. (2019) identified three genes that were duplicated and predicted to be important in regulating the VLCFA biosynthesis pathway in M. oleifera. Those genes were also closely related to three A. thaliana KCS genes (KCS11, KCS2, and KCS20) (Figures 5(a) and S16). Interestingly, the 10 A. truncatum KCS genes underwent sequential tandem duplications and were clustered in the 0.27‐Mb region of pseudochromosome 4 (Figure 5(b)). In our study, the expression levels of most of the KCS genes were high during the early seed development stages and then gradually decreased. However, the expression patterns of Chr4.2308.KCS, Chr4.2307.KCS, and Chr4.2311.KCS differed from those of the other genes analyzed (Figure S17), with expression level trends that were generally consistent with the accumulation of NA (Figure 4(c)). Interestingly, they are all clustered in a small branch (Figure 5(a)). To confirm the expression level trends at six seed development stages, Chr4.2308.KCS, Chr4.2307.KCS, and Chr4.2311.KCS, including two randomly selected KCS genes (Chr4.2309.KCS and Chr4.2822.KCS) in the clustered group, were used to confirm the expression patterns by RT‐PCR analysis. We found that the expression levels of Chr4.2308.KCS, Chr4.2307.KCS, and Chr4.2311.KCS were all much higher than those of Chr4.2309.KCS and Chr4.2822.KCS, and the expression of the latter two did not changed more at different seed development stages (Figure S18).
Figure 5.
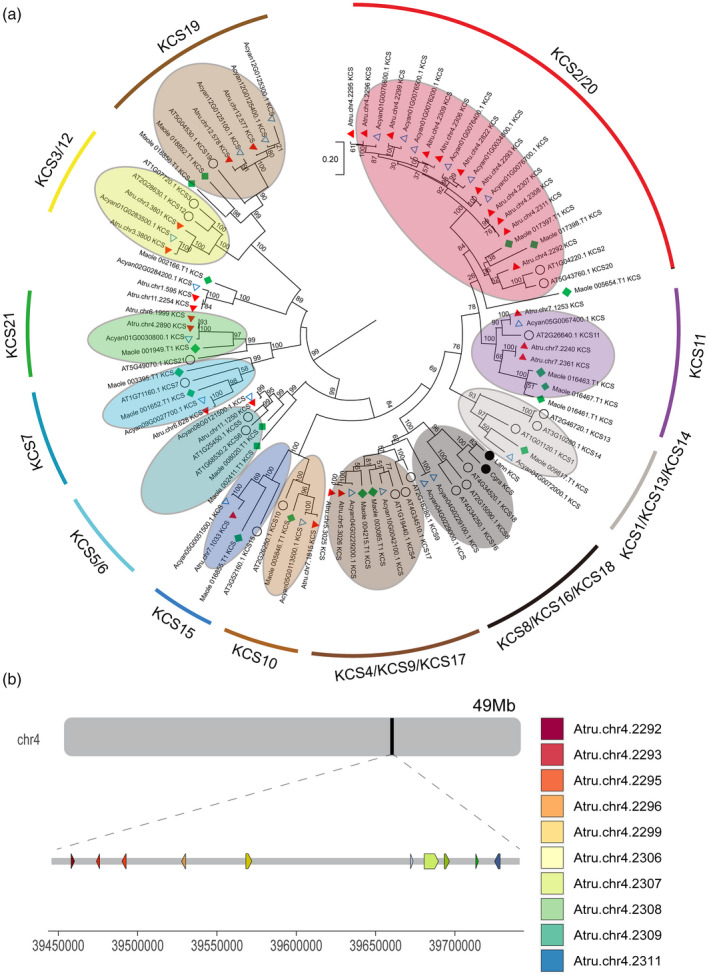
The phylogenetic tree and distribution of KCS genes based on protein sequences of Acer truncatum. The phylogenetic tree and distribution of KCS genes. (a) The maximum likelihood tree of KCS genes. A. truncatum (red triangle), A. yangbiense (write triangle), M. oleifera (blue square), A. thaliana (write circle), C. graeca (Cgra), and L. annua (Lann) genes are shown in the tree with corresponding gene ID nomenclature. (b) the distribution of 10 KCS genes on chromosome 4 of A. truncatum. Each arrow represents a KCS gene.
The proportion of NA in seed oil increased from 0% at 85 DAF to 1.96% at 100 DAF (Figure 4(c)), suggesting that this period marks the critical time for NA biosynthesis. We therefore compared these two time points, and found that the expression levels of Chr4.2307.KCS, Chr4.2308.KCS, and Chr4.2311.KCS were significantly upregulated (4.74‐, 4.25‐, and 3.28‐fold, respectively; P < 0.01) between 85 and 100 DAF (Table S25). These results were consistent with the RT‐PCR analysis at six seed development stages. The expression levels of Chr4.2307.KCS, Chr4.2308.KCS, ad Chr4.2311.KCS were indeed significantly upregulated from 85 DAF to 100 DAF (Figure S18). Furthermore, they were more highly expressed in the mature seeds than in the other plant tissues examined (root, stem, leaf, and flower) (Figure S19). These results suggested that these three genes are critically important in lipid metabolism and that they might be mainly responsible for regulating NA biosynthesis in A. truncatum.
A weighted gene co‐expression network analysis (WGCNA) was used to search for candidate genes associated with NA biosynthesis regulators. A total of 23 773 genes that are expressed among lines during seed development were chosen as probes for the WGCNA. The genes in one module (black module) were positively related to NA biosynthesis (P < 0.09) (Figure S20). The well‐known MYB and bZIP TFs were found to be involved in regulating NA biosynthesis (Table S26). Previous studies showed that MYB and bZIP TFs participate in regulating the synthesis of fatty acids (Wang et al., 2007; Yamamoto et al., 2009), and in particular, MYB had been found to play important roles in VLCFA biosynthesis (Raffaele et al., 2008).
DISCUSSION
Aceraceae species are important ornamental foliage plants (Yang and Liu, 1998) that are distributed worldwide. High‐quality genome sequencing represents a key research option for studying specialized metabolites and the evolution of their biosynthesis (Afendi et al., 2012). A. truncatum is a versatile woody oil tree crop that produces NA. However, to date, the complete genome sequence of A. truncatum has not been reported. A sequence assembly and high‐quality sequencing based on PacBio RS II sequencing data combined with 10 × Genomics data represents a viable alternative to conventional genome sequencing. In this study, the A. truncatum genome was assembled into 13 pseudochromosomes based on Hi‐C technology, ultimately resulting in a high‐quality reference genome sequence of 633.28 Mb. The integrated strategy described herein was highly effective for the assembly of the complex A. truncatum genome.
The evolutionary history of the A. truncatum genome
The characterization and annotation of the A. truncatum genome and a comparison with other plant genomes generated new information relevant for future investigations of the evolutionary history of A. truncatum. We updated the evolution of A. truncatum in Aceraceae species. In previous studies, Chen et al. (2019) and Ma et al. (2019) proved that A. truncatum and A. miaotaiense are closely related. In our study, we found that A. truncatum was not only closely related to A. miaotaiense, but also to A. catalpifolium, based on phylogeny of the chloroplast genomes. Up to now, the A. yangbiense genome was the only reported genome among Aceraceae species, so we compared the A. truncatum and A. yangbiense genomes. The phylogenetic analysis revealed they diverged about 9.4 Mya. WGDs are important for plant genome evolution (Panchy et al., 2016). We also determined that an ancient duplication occurred in A. truncatum, similar to most plant species (Jiao et al., 2011). There were no recent WGDs, with the exception of the γ event shared by all core eudicots (Jiao et al., 2011; Mcgrath et al., 2014), which is consistent with the results of the earlier evolution of Sapindales species, such as A. yangbiense (Yang et al., 2019), C. sinensis (Xu et al., 2013) and Dimocarpus longan (Lin et al., 2017).
The A. truncatum genome contains a large number of repeat sequences
Repeat sequences are major components of eukaryotic genomes, and their activation might have caused the duplication and insertion events, leading to an increase in genome size (Levin and Moran, 2011; Kajitani et al., 2014). A previous study reported that C. sinensis (sweet orange) has a relatively compact genome among Sapindales species, in which the TEs account for 20.5% (Xu et al., 2013). We found that the number of TEs in A. truncatum is about threefold higher than in C. sinensis. Similar to the A. truncatum genome, the repeat sequences of most reported genomes in Sapindales species accounted for relatively high proportions, for example, 68.0% in A. yangbiense (Yang et al., 2019), 52.87% in D. longan (Lin et al., 2017), and 56.39 in Xanthoceras sorbifolium (Liang et al., 2019). Most plant genomes appear to contain abundant LTR retrotransposons. Similar to other genomes, LTRs were also dominant in the A. truncatum genome, especially Copia and Gypsy. These results will lay important foundations for studying the genome evolution of Acer species.
The A. truncatum genome benefits NA research
Previous research reported that NA can be helpful in the treatment of brain diseases (Pamplona et al., 2005; Tanaka et al., 2007; Amminger et al., 2012). The NA biosynthesis pathway has been studied and some key components and enzyme‐catalyzed steps have been characterized (Taylor et al., 2009; Guo et al., 2009). Nevertheless, the mechanism underlying NA biosynthesis needs to be more thoroughly elucidated. In previous studies, L. annua and C. graeca KCS genes were cloned and heterologously expressed, resulting in an increase in the NA content (Guo et al., 2009; Taylor et al., 2009). However, the NA content was not significantly different between plants heterologously expressing KCS alone, KCS with KCR, KCS with HCD, KCS with ECR, or KCS with KCR and HCD (Dongxin et al., 2015), suggesting that KCS alone is crucial for NA biosynthesis. However, in the present study we detected no genes that were similar to the abovementioned cloned and heterologously expressed KCS genes (Guo, et al., 2009; Taylor et al., 2009).
Our further genomic analysis of paralogous KCS genes revealed that these genes are usually located within syntenic genomic blocks, which are the result of WGDs or segmental chromosomal duplications (Guo et al., 2016). A survey of tandem‐arrayed KCS genes was performed; the results showed that 10 of 28 KCS genes exhibited tandem arrays in A. truncatum. Interestingly, 10 and 6 KCS genes in A. yangbiense were clustered into a big branch. They are closely related to three A. thaliana KCS genes, namely, KCS11, KCS2, and KCS20, the latter two associated with cuticular wax biosynthesis (Saet‐Buyl et al., 2010). Although the KCS paralogous gene family expanded to 28 members, the evolution of the KCS gene family is essentially a history of gene duplications. The variations of different members resulted in the generation of product specificity and the emergence of completely new functions involved in other physiological processes (Vavouri et al., 2008; Guo et al., 2016). In our study, we found that only three of them (Chr4.2307.KCS, Chr4.2308.KCS, and Chr4.2311.KCS) were significantly upregulated at the critical time periods (from nothing to something) for NA biosynthesis. Moreover, their extremely high expression in seeds also indicated that these three KCS genes play an important role in seed development or fatty acid synthesis. Although family members share significant protein similarity, each may have partially overlapping or even distinct biological functions (Danilevskaya et al., 2007). The expansion and retention of these genes might facilitate natural selection of different catalytic functions and end products. Therefore, we speculate that the key genes of A. truncatum and M. oleifera play important roles in NA biosynthesis, which differ from those of L. annua and C. graeca.
A comparative transcriptome analysis, a WGCNA, and an examination of NA accumulation in developing seeds were combined to clarify which KCS genes contribute to NA biosynthesis. Finally, three KCS genes and related TFs were identified that might contribute to the regulation of NA biosynthesis. However, future transgenic studies will be needed to confirm the roles of the KCS genes in NA biosynthesis in A. truncatum, to elucidate whether a major single gene or the cooperation of three KCS genes is centrally important.
In summary, our study provides critical information regarding Aceraceae genomes, which has been limited to date. The elucidation of the A. truncatum genome sequence described herein and the sequence details deposited in the NCBI database will benefit future research on A. truncatum flowering, seed production, and resistance to biotic and abiotic stresses. Therefore, our findings can be expected to contribute to the further characterization of A. truncatum as an economically important tree species that produces NA in its seed oil.
EXPERIMENTAL PROCEDURES
Plant materials
The diploid monoecious A. truncatum, originally collected in Nanjing, Jiangsu Province, was preserved in the Jiangsu Province Aceraceae Germplasm Repository (Lishui, Nanjing). The materials used for genome sequencing and assembly were healthy and young leaves.
DNA extraction and sequencing
Genomic DNA was extracted from leaves of A. truncatum using the DNAsecure Plant Kit (TIANGEN, Biotech Co., Ltd., Beijing, China). Sequencing libraries with an insert size of 350 bp were constructed using a library construction kit (Illumina, CA, USA). These libraries were then sequenced using an Illumina HiSeq X10 platform. Raw reads were filtered according to sequencing quality, the presence of adapter contamination, and duplication. Only high‐quality reads were used for genome assembly. For a PacBio 20‐kb insert size library, at least 10 μg of sheared DNA was required. SMRTbell template preparation involved DNA concentration, damage repair, end repair, ligation of hairpin adapters, and template purification. Finally, we carried out 20‐kb single‐molecule real‐time DNA sequencing by PacBio and sequenced the DNA library on the Pacbio Sequel platform. DNA sample preparation, indexing, and barcoding were done using the GemCode Instrument from 10x Genomics. About 1 ng input DNA was used for the GEM reaction procedure during PCR, and 16‐bp barcodes were introduced into droplets. Then, the droplets were fractured following the purification of the intermediate DNA library. 10x Genomics libraries were finally sequenced on the Illumina Hiseq X10.
For the Hi‐C library, chromatin was fixed in place with formaldehyde in the nucleus. Fixed chromatin was digested, 5′ overhangs were filled in with biotinylated nucleotides, and free blunt ends were ligated. After ligation, cross‐links were reversed and the DNA was purified from protein. Purified DNA was treated to remove biotin that was not internal to the ligated fragments. The DNA was then sheared to an average fragment size of ~350 bp, and sequencing libraries were generated using NEBNext Ultra enzymes and Illumina‐compatible adapters. Biotin‐containing fragments were isolated using streptavidin beads before PCR enrichment of each library. The libraries were sequenced on an Illumina Novaseq platform.
Genome assembly
The long subreads generated by the PacBio platform were assembled by the following procedures: The initial assembly was first generated by Falcon (Chin et al., 2016) using pre‐assemble reads after error correction (up to 99.999%). After the initial assembly, FALCON‐Unzip (Chin et al., 2016) was used to produce primary contigs (p‐contigs), which were polished using Quiver (https://github.com/PacificBiosciences/GenomicConsensus). Lastly, Pilon (Walker et al., 2014) was used to perform error correction of p‐contigs with the short paired‐end reads and high‐quality consensus sequences were obtained. Finally, scaffolding was performed by FragScaff (Mostovoy et al., 2016) with the barcoded sequencing reads generated by 10x Genomics.
To obtain chromosome‐level assembly, the Hi‐C clean data were aligned to the preceding assembly using BWA software (Li and Durbin, 2009). Only the read pairs with both reads aligned to contigs were considered for scaffolding. For each read pair, its physical coverage was defined as the total bases spanned by the sequence of reads and the gap between the two reads when mapped to contigs. Per‐base physical coverage for each base in the contig was defined as the number of read pairs’ physical coverage. Mis‐assembly could be detected by the sudden drop in per‐base physical coverage in a contig. According to physical coverage of the alignment result, the mis‐assemblies would be sheared to correct the mis‐assemble errors by SALSA (Jay et al., 2017). According to the linkage information and restriction enzyme site, the string graph formulation was used to construct the scaffold graph with LACHESIS (Burton et al., 2013).
To assess the accuracy and completeness of the assemblies, the Illumina clean reads were mapped to the corresponding assembly using BWA (Li and Durbin, 2009). CEGMA (http://korflab.ucdavis.edu/dataseda/cegma/) (Parra et al., 2007) and BUSCO (http://busco.ezlab.org/) (Simao et al., 2015) were also used to assess the completeness of our assemblies. The assembly quality of gene‐coding regions was evaluated by aligning the transcriptome sequenced from whole‐plant RNAs using BLAT (Kent, 2002). In addition, the LAI score (Ou et al., 2018), a standard for evaluating the assembly of repeat sequences, was used to evaluate the assembly continuity.
Repetitive elements annotation
A combined strategy based on sequence homology and de novo prediction was employed to identify the repetitive elements of our assemblies. Tandem repeats were also identified by the software Tandem Repeats Finder (http://tandem.bu.edu/trf/trf.html). For the de novo search, we searched repeat elements by RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html), RepeatScout (Price et al., 2005), Piler (Edgar and Myers, 2005), and LTR‐Finder (Xu and Wang, 2007), with default parameters. The non‐redundancy repeat library was finally put into Repeatmasker (http://www.repeatmasker.org) to predict repeat elements. For the homology‐based prediction, RepeatProteinMask (http://www.repeatmasker.org) was used to identify repeats compared with the known consensus sequences of the Repbase library (Jurka et al., 2005).
To identify complete LTR‐retrotransposons, LTRfinder (Xu and Wang, 2007) (parameters: ‐l 1000 ‐L 20 000 ‐d 100 ‐D 5000 ‐M 0.3) and LTRharvest (Ellinghaus, et al., 2008) (parameters: ‐v ‐mintsd 4 ‐maxtsd 6) were used to identify the candidate LTR‐retrotransposons. Then LTRdigest (Steinbiss et al., 2009) was used to identify candidate retrotransposons by searching for known protein domains (parameters: ‐trnas ‐hmms). Elements containing GAG domains, protease domains, reverse transcriptase domains, and integrase domains were considered as intact. The predicted LTRs were extracted and aligned with muscle (Edgar, 2004), and the distance K was calculated with the Jukes–Cantor model by an in‐house Perl script and the insert time of each LTR‐retrotransposon was calculated by the following formula: T = K/(2 × r), where r refers to a general substitution rate of 1.3 × 10−8 per site per year.
Centromere region identification
The centromeric regions of A. truncatum were identified using an approach described by VanBuren et al. (2015). Tandem Repeat Finder (Benson, 1999) was used to identify all tandem repeats. The base centromere repeats were identified by trf_cluster (downloaded from http://korflab.ucdavis.edu/datasets/Centromere_data/). To determine the location of centromeres, the base centromere repeat was aligned to the genome using BLASTN (E‐value < 1E−5, overlap > 90%). The above approaches were applied to detect the centromere regions of A. yangbiense.
Gene prediction and annotation
To predict genes in the A. truncatum genome, we performed a combination of de novo, homology, and transcript‐based approaches. For the homology‐based prediction, the protein sequences of A. thaliana (TAIR10), C. sinensis (Xu et al., 2013), Glycine max (V1.0), G. raimondii (GCF_000327365), Populus trichocarpa (JGI2.0), S. lycopersicum (SL2.50), and Oryza sativa (IRGSP‐1.0) were downloaded from the NCBI, Ensemble, and JGI databases, and aligned to the A. truncatum genome by TBLASTN with E‐value < 1E−5. GeneWise (version 2.4.1) (Birney et al., 2004) was used to annotate genes with the alignments. The clean RNA‐seq data (root, stem, leaf, flower, and mature seed) were aligned to the genome by Tophat (version 2.0.13) (Trapnell et al., 2009) and assembled into gene models by Cufflinks (version 2.1.1) (http://cole‐trapnell‐lab.github.io/cufflinks/releases/v2.1.1/). Besides, we applied Trinity (version 2.0) (Grabherr et al., 2011) to assemble the RNA‐seq data, and then pasa software (Haas et al., 2003) was used to improve the gene structures. For the de novo prediction, we annotated genes by augustus (version 2.5.5) (Stanke and Morgenstern, 2005), GlimmHMM (version 3.0.1) (Majoros et al., 2004), SNAP (Korf, 2004), GeneScan (version 1.0) (Aggarwal and Ramaswamy, 2002), and GeneID (v1.4) (Parra, et al., 2000). The gene models retrieved from all methods were integrated by EvidenceModeler (Haas et al., 2008). The software pasa (Haas et al., 2003) was then used to obtain the information of genes’ untranslated regions and alternative splicing variations. We applied InterProScan (Zdobnov and Apweiler, 2001) to determine the domains and motifs of the gene set. GO IDs were derived from the InterPro entry. Gene functions were obtained according to the best match of the search against the NCBI NR, Swiss‐Prot, and KEGG databases, using BLASTP (E‐value < 1E−5).
Genome comparison and evolution
We performed a comparative genome analysis to identify gene family clusters among A. truncatum, A. yangbiense (Yang et al., 2019), O. sativa (IRGSP‐1.0), A. thaliana (TAIR10), C. sinensis (Xu et al., 2013), G. raimondii (GCF_000327365), Juglans regia (GCF_001411555.1_wgs.5d), Glycine max (v1.0), Vitis vinifera (IGGP_12x), Olea europae (GCF_002742605.1_O_europaea_v1), Sesamum indicum (GCF_000512975.1_S_indicum_v1.0), Helianthus annuus (HanXRQr1.0), P. trichocarpa (JGI2.0), S. lycopersicum (SL2.50), and M. Oleifera (Xu et al., 2019). The longest protein sequences (longer than 50 amino acids) of all species were searched against each other using BLASTP (E‐value < 1E−7) (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and clustered using the OrthoFinder algorithm (inflation parameter: 1.5) (version 2.2.6) (Emms and Kelly, 2019). A dataset of single‐copy orthologous genes (only one gene copy per species in the cluster) was then used for muscle (Edgar, 2004) alignment, and the phylogenetic tree was constructed using RAxML (Stamatakis, 2014). Mcmctree in PAML packages (Yang, 1997) was performed to estimate the divergence time, with four corrected divergence time points from the TimeTree website (http://www.timetree.org/): Monocots versus Dicots (173–148 Mya), A. thaliana versus P. trichocarpa (109–97 Mya), A. thaliana versus V. vinifera (115–105 Mya), and H. annuus L. versus S. lycopersicum (107–93 Mya). café (De et al., 2006) was used to analyze the expansion/contraction of gene families. In addition, the chloroplast sequences of 17 Aceraceae species and D. longan were downloaded from the NCBI database and clustered using OrthoFinder (inflation parameter: 1.5) (version 2.2.6) (Emms and Kelly, 2019). A dataset of single‐copy orthologous genes (only one gene copy per species in the cluster) was then used for muscle (Edgar, 2004) alignment, and the phylogenetic tree was constructed using RAxML (Stamatakis, 2014).
The whole‐genome duplication and synteny analysis
Syntenic blocks in the A. truncatum genome were detected using MCScanX (http://chibba.pgml.uga.edu/mcscan2) (Wang et al., 2012), after an all‐to‐all BLASTP (https://blast.ncbi.nlm.nih.gov/Blast.cgi) search for the best hit. To estimate WGD events, we calculated 4DTv values of paralogous genes of A. truncatum using in‐house Perl scripts. 4DTv was the return value of 4DT (third codon transversions within these fourfold degenerate sites) divided by 4D (the fourfold degenerate sites) and corrected by the HKY substitution model. The syntenic blocks within A. yangbiense, C. sinensis, P. trichocarpa, and M. oleifera were identified and 4DTv values were calculated by the same method. The syntenic regions between A. truncatum, A. yangbiense, P. trichocarpa, and C. sinensis were also searched by MCScanX (Wang et al., 2012) and the 4DTv value of orthologous genes was calculated.
Lipid analysis and oil body obesrvation
The 50 kernels from different developmental stages (70, 85, 100, 115, 145, and 180 DAF) were dried at 65°C for 72 h. The seed oil was extracted with petroleum ether (30–60°C) using a Soxhlet apparatus and the distillation temperature was kept at 65°C for 24 h. Then lipid was separated using a rotary vacuum evaporator at 65°C. To make fatty acid methyl esters (FAMEs), about 0.2 ml oil was dissolved in 2 ml of benzene/petroleumether (1:1, v/v), which was then mixed with 2 ml of KOH‐CH3OH (0.4 m) as described by Gao (2006). The oil was extracted, and the esterification reactions were directly performed in a rotary vacuum evaporator. The product was diluted to 10 ml with deionized water, and the upper phase was used to analyze fatty acid composition by gas chromatography‐mass spectrometry (GC/MS) using a TRACE DSQ GC/MS (Thermo Fisher Scientific Inc., Waltham, MA, USA). The procedures were performed as described by Zhang et al. (2018). The standards (Seebio Biotech Co., Ltd, Shanghai, China) were used to make standard curves and perform sample analysis. The amounts of fatty acids were calculated by comparing the peak areas of FAMEs with the standard (Kim et al., 2015). Transmission electron microscopy samples were cut from the middle section of each embryo (JEM‐1400; JEOL, Tokyo, Japan) and prepared following the procedure described by Chen et al. (2012). Brief, the samples were immediately fixed with 4% (v/v) glutaraldehyde in sodium phosphate buffer (pH 7.2), post‐fixed in osmium tetroxide, dehydrated, and embedded in Spurr’s resin. The sections were observed at 60 kV with an electron microscope (JEM‐1200; JEOL).
Transcriptome library preparation and gene expression analysis
We collected A. truncatum roots, stems, leaves, flowers, and developing seeds (70, 85, 100, 115, 145 and 180 DAF) from three plants (with three biological repeats each). Total RNA was extracted using TRIzol® Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions, and the RNA‐seq libraries were constructed using the NEBNext Ultra Directional RNA Library Prep Kit (NEB, USA). The RNA‐seq libraries were then sequenced on an Illumina Novaseq platform and 150‐bp paired‐end reads were generated. After the quality control (reads that containing adapters, reads containing poly‐N, and low‐quality reads were removed) by an in‐house Perl script, the clean reads were mapped to the genome using HISTA2 (version 2.0.4) (Kim et al., 2015). The Reads Per Kilobase per Million mapped reads (RPKM) values were used to analyze the expression level of each gene for further calculation of the differentially expressed genes using DESeq2 (https://bioconductor.org/packages/release/bioc/html/DESeq2.html).
The validation of differentially expressed genes was performed by quantitative RT‐PCR (qRT‐PCR). Total RNA was extracted as described above, and cDNA was reverse transcribed from 1 μg total RNA using M‐MLV reverse transcriptase (Promega, Madison, WI). The cDNA templates were diluted 20‐fold before use. qRT‐PCR was performed with in a Step One Plus Real‐Time PCR System (Applied Biosystems) using SYBR Premix ExTaqTM (TaKaRa) according to the manufacturer’s protocol. The specific primers were designed with Primer5 (Tables S27 and S28). Actin was selected as the internal reference gene (Wang et al., 2018). The expression of each gene was analyzed in three biological replicates and three technical repetitions. Corresponding gene expression levels were analyzed with the 2−∆∆Ct method.
The identification and phylogenetic analyses of NA biosynthesis genes and LEA genes in A. truncatum
We used different methods to identify NA biosynthesis genes and LEA genes. To identify homologous genes in NA biosynthesis pathways, the sequences of proteins from the A. thaliana fatty acid biosynthesis pathway were downloaded (http://aralip.plantbiology.msu.edu/) and used for BLASTP (https://blast.ncbi.nlm.nih.gov/Blast.cgi) queries. The genes were selected if the following requirements were met: (i) filtering for hits with E‐value ≤ 1E−5, alignment identity ≥ 50%, and alignment coverage ≥ 50% of the query genes; (ii) having the same Pfam domains as the query genes. These genes were also searched in A. yangbiense, C. sinensis, and M. oleifera by the same method. The Hidden Markov Model profiles of the LEA protein domains PF03760 (LEA1), PF03168 (LEA2), PF03242 (LEA3), PF02987 (LEA4), PF00477 (LEA5), PF00257 (DEHYDRIN), and PF04927 (SMP) were used to query the protein dataset of A. truncatum by HMMER (http://hmmer.janelia.org/) with E‐value < 0.01. Multiple sequence alignment of the amino acid sequences of NA biosynthesis genes and LEA genes were performed respectively using the default parameters of ClustalW, and then phylogenetic analyses were conducted by RAxML (Stamatakis, 2014).
WGCNA (Weighted Gene Co‐Expression) network analysis and the identification of transcription factors
The datasets of the six seed development stages were averaged and filtered to remove non‐expressed genes (RPKM < 1). The co‐expression network was generated using the RPKM values of the filtered genes by the WGCNA package in R (Langfelder and Horvath, 2008; Team, 2016). To relate the physiology measurements with the network, the module eigengenes were correlated with the fatty acid contents data. TTFs within modules were identified and classified into different families using iTAK software (Zheng et al., 2016).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
QYM, QZL, XZ, and SXJ designed the project and the strategy; SXL, JW, LZ, KYY, and SXL contributed to plant sample collection, DNA/RNA preparation, library construction, and sequencing; QYM, TLS, QZL, JFM, and XX worked on genome assembly and annotation and comparative analyses; QYM, TLS, SXL, JW, YNW, and LZ performed transcriptome and genetic analyses and identified candidate genes; QYM, TLS, TMY, QZL, XZ, and SXJ wrote and revised the manuscript.
Supporting information
Figure S1. The K‐mer analysis of A. truncatum. The frequency of 17‐mers is exhibited by 17‐bp sequences within filtered reads of the 350‐bp inset size library. As the curve shows, the peak depth is 76, and the genome size was estimated as 662.08 Mb (653.44 Mb after revision).
Figure S2. The Hi‐C chromatin interaction map of 13 pseudomolecules in A. truncatum.
Figure S3. The insert time of LTR retrotransposons in A. truncatum, A. yangbiense, C. sinensis, and M. oleifera. The red, green, and blue regions represent the LTR insertion time (million years ago) of Atru: A. truncatum, Ayan: A. yangbiense; Csin: C. sinensis, and Mole: M. oleifera.
Figure S4. Gene numbers in each group defined by OrthoMCL.
Figure S5. The phylogeny and divergence time of A. truncatum and 14 other plants.
Figure S6. The phylogeny based on coding sequences of single‐copy orthologous genes shared among 17 chloroplast genomes in Aceraceae species. Dimocarpus longan served as the outgroup.
Figure S7. The phylogeny based on protein sequences of single‐copy orthologous genes shared among 17 chloroplast genomes in Aceraceae species. Dimocarpus longan served as the outgroup.
Figure S8. Synteny comparison of A. truncatum and C. sinensis. The outermost circle (blue) displays ideograms of the pseudochromosomes of the genomes. Atru: A. truncatum, Csin: C. sinensis. The second circle displays gene density. The innermost circle displays homologies between A. truncatum and C. sinensis. All distributions are drawn in a window size of 300 kb, chromosomes_units = 500 000.
Figure S9. Synteny comparison of A. truncatum and A. yangbiense. The outermost circle (blue) displays ideograms of the pseudochromosomes of the genomes. Atru: A. truncatum, Ayan: A. yangbiense. The second circle displays gene density. The innermost circle displays homologies between A. truncatum and A. yangbiense. All distributions are drawn in a window size of 300 kb, chromosomes_units = 500 000.
Figure S10. Syntenic pattern of A. truncatum with A. yangbiense and C. sinensis. The x‐axis indicates that each gene of one genome has no orthologous region, one orthologous region, two orthologous regions, three orthologous regions, or four orthologous regions with another genome. The y‐axis indicates the percentage of the gene blocks in genomes.
Figure S11. The phylogeny analysis of LEA genes in A. truncatum.
Figure S12. The distribution of LEA genes in A. truncatum.
Figure S13. Heat map of gene expression profiles of LEA genes. (a) Differentially expressed LEA genes during seed development. (b) Differentially expressed LEA genes in five tissues.
Figure S14. Quantitative PCR of LEA genes in different tissues. Error bars indicate standard errors of the mean from three technical replicates.
Figure S15. Quantitative PCR of LEA genes in six seed development stages. Error bars indicate standard errors of the mean from three technical replicates.
Figure S16. The phylogenetic tree and distribution of KCS genes based on coding sequences of A. truncatum. (a) The maximum likelihood tree of KCS genes from A. truncatum (red triangle), A. yangbiense (write triangle), M. oleifera (blue square), and A. thaliana (write circle).
Figure S17. The expression profiles of KCS genes in different tissues of A. truncatum. Z‐scores were calculated after log10 transformation of FPKM values plus 1.
Figure S18. Quantitative PCR of KCS genes in six seed development stages. Error bars indicate standard errors of the mean from three technical replicates.
Figure S19. Quantitative PCR of KCS genes in different tissues. Error bars indicate standard errors of the mean from three technical replicates.
Figure S20. WGCNA of fatty acid contents with RNA‐seq data of seed development stages in A. truncatum. The numbers within the heat map represent correlations (red, positively correlated; blue, negatively correlated) and P‐values (in parentheses) for the module–trait associations.
Table S1. K‐mer statistics of A. truncatum.
Table S2. Sequencing data of A. truncatum.
Table S3. Summary of A. truncatum pseudomolecules.
Table S4. The Hi‐C assembly results of A. truncatum.
Table S5. Coverage statistics of the A. truncatum genome.
Table S6. CEGMA evaluation results.
Table S7. BUSCO notation assessment of A. truncatum genome and annotation.
Table S8. Assessing the gene region assembly by mapping the RNA assembly to the A. truncatum genome.
Table S9. Statistical results of gene functional annotations.
Table S10. Statistical results of non‐coding RNAs.
Table S11. Classification of repetitive elements in the A. truncatum genome.
Table S12. The LTR subfamilies in A. truncatum and other published genomes.
Table S13. The seed sequences of centromeric regions of A. truncatum and A. yangbiense chromosomes.
Table S14. Details of the chloroplast genome sequences used for the phylogenetic analysis.
Table S15. GO enrichment results for expanded genes in A. truncatum.
Table S16. GO enrichment results for contracted genes in A. truncatum.
Table S17. GO enrichment results for the genes in A. truncatum‐specific gene families.
Table S18. KEGG enrichment results for expanded genes in A. truncatum.
Table S19. GO enrichment results for expanded genes in A. yangbiense.
Table S20. GO enrichment results for contracted genes in A. yangbiense.
Table S21. KEGG enrichment results for expanded genes in A. yangbiense.
Table S22. A summary of drought‐tolerant genes identified in A. truncatum and other published genomes.
Table S23. A summary of drought tolerance‐related transcription factors identified in A. truncatum and other published genomes.
Table S24. The copy numbers of VLCFA biosynthesis‐related genes in 12 plants.
Table S25. Expression levels of VLCFA biosynthesis‐related genes that are significantly upregulated between 85 DAF and 100 DAF and are involved in fatty acid elongation.
Table S26. The transcription factors (TFs)/transcription regulatory factors (TRs) in the black module of WGCNA analysis.
Table S27. List of KCS gene‐specific primers (5’–3’) used for RT‐PCR.
Table S28. List of LEA gene‐specific primers (5’–3’) used for RT‐PCR.
ACKNOWLEDGMENTS
This work was funded by the Natural Science Foundation of China (31700628), the Natural Science Foundation of Jiangsu Province (BK20170602), the Independent Innovation Fund Project of Agricultural Science and Technology in Jiangsu Province (CX(17)1004), and the Technology Innovation and Extension Project of Forestry Science in Jiangsu Province (LYKJ[2018]14).
Contributor Information
Xing Zhao, Email: zhaoxing@novogene.com.
Qianzhong Li, Email: qianzhongli@jaas.ac.cn.
Data Availability Statement
The A. truncatum genome has been deposited under BioProject accession number PRJNA557096 and BioSample accession number SAMN12389479. PacBio and Illumina reads, resequencing sequences reads and Hi‐C data have been submitted in the Sequence Read Archive (SRA) under study accession number SUB6287730. The final assembled version(s) must be available in https://doi.org/10.6084/m9.figshare.12986237.v2.
References
- Aggarwal, G. and Ramaswamy, R. (2002) Ab initio gene identification: prokaryote genome annotation with GeneScan and GLIMMER. J. Biosci. 27, 7–14. [DOI] [PubMed] [Google Scholar]
- Afendi, F.M. , Okada, T. , Yamazaki, M. et al (2012) KNApSAcK family databases: integrated metabolite‐plant species databases for multifaceted plant research. Plant Cell Physiol. 53, 10.1093/pcp/pcr165. [DOI] [PubMed] [Google Scholar]
- Amminger, G.P. , Schäfer, M.R. , Klier, C.M. , Slavik, J.M. , Holzer, I. , Holub, M. , Goldstone, S. , Whitford, T.J. , McGorry, P.D. and Berk, M. (2012) Decreased nervonic acid levels in erythrocyte membranes predict psychosis in help‐seeking ultra‐high‐risk individuals. Mol. Psychiatr. 17, 1150–1167. [DOI] [PubMed] [Google Scholar]
- Benson, G. (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettger, W.J. , McCorquodale, M.L. and Blackadar, C.B. (2001) The effect of a Tropaeolum speciosum oil supplement on the nervonic acid content of sphingomyelin in rat tissues. J. Nutr. Biochem. 12, 492–496. [DOI] [PubMed] [Google Scholar]
- Birney, E. , Clamp, M. and Durbin, R. (2004) GeneWise and genomewise. Genome Res. 14, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton, J.N. , Adey, A. , Patwardhan, R.P. , Qiu, R. , Kitaman, J.O. and Shendure, J. (2013) Chromosome‐scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J.R. , Hsu, S.F. , Hsu, C.D. , Hwang, L.H. and Yang, S.C. (2004) Dietary patterns and blood fatty acid composition in children with attention‐deficit hyperactivity disorder in Taiwan. J. Nutr. Biochem. 15, 467–472. [DOI] [PubMed] [Google Scholar]
- Chen, S. , Liu, B. , Zhang, S. and Huang, J. (2019) Complete chloroplast genome of Acer truncatum bunge (Aceraceae). Mitochondrial DNA Part B, 4, 607–608. [Google Scholar]
- Chen, M. , Wang, Z. , Zhu, Y. et al (2012) The effect of TRANSPARENT TESTAR2 on seed fatty acid biosynthesis and tolerance to environmental stresses during young seedling establishment in Arabidopsis. Plant Physiol. 160, 1023–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, C. , Peluso, P. , Sedlazeck, F.J. , Nattestad, M. , Concepcion, G.T. , Clum, A. , Dunn, C. , O’Malley, R. , Figueroa‐Balderas, R. and Morales‐Cruz, A. (2016) Phased diploid genome assembly with single‐molecule real‐time sequencing. Nat. Methods, 13, 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilevskaya, O.N. , Meng, X. , Hou, Z. , Danilevskaya, O.N. , Meng, X. , Hou, Z. , Ananiev, E.V. and Simmons, C.R. (2007) A genomic and expression compendium of the expanded PEBP gene family from maize. Plant Physiol. 146(1), 250–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongxin, H. , Yuanyuan, Z. , Chunyu, Z. , Cahoon, E.B. , Yongming, Z. and Thierry, C. (2015) Combinatorial effects of fatty acid elongase enzymes on nervonic acid production in Camelina sativa . PLoS One, 10, e0131755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, D. , Zhang, Q. , Cheng, T. , Pan, H. , Yang, W. and Sun, L. (2013) Genome‐wide identification and analysis of late embryogenesis abundant (LEA) genes in Prunus mume . Mol. Biol. Rep. 40(2), 1937–1946. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. and Myers, E.W. (2005) PILER: identification and classification of genomic repeats. Bioinformatics, 21, i152–i158. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms, D. and Kelly, S. (2019) OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinghaus, D. , Kurtz, S. and Willhoeft, U. (2008) LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehling, E. and Mukherjee, K.D. (1991) Acyl‐CoA elongase from a higher plant (Lunaria annua): metabolic intermediates of very‐long‐chain acyl‐CoA products and substrate specificity. Biochim. Biophys. Acta (BBA)‐Lipids Lipid Metab. 1082, 239–246. [DOI] [PubMed] [Google Scholar]
- Gao, J. (2006) Experimental guidance for plant physiology. Beijing, Chinese: China Higher Education Press. [Google Scholar]
- Gao, J. and Lan, T. (2016) Functional characterization of the late embryogenesis abundant (LEA) protein gene family from Pinus tabuliformis (Pinaceae) in Escherichia coli . Sci. Rep. 6, 19467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, J.W. , Chao, H.B. , Wang, H. et al (2017) Identification of the relationship between oil body morphology and oil content by microstructure comparison combining with QTL analysis in Brassica napus . Front. Plant Sci. 7, 10.3389/fpls.2016.01989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y. , Mietkiewska, E. , Francis, T. , Katavic, V. , Brost, J.M. , Giblin, M. , Barton, D.L. and Taylor, D.C. (2009) Increase in nervonic acid content in transformed yeast and transgenic plants by introduction of a Lunaria annua L. 3‐ketoacyl‐ CoA synthase (KCS) gene. Plant Mol. Biol. 69, 565–575. [DOI] [PubMed] [Google Scholar]
- Guo, X. , Wang, R. , Chang, R. , Liang, X. , Wang, C. , Luo, Y. , Yuan, Y. and Guo, W. (2014) Effects of nitrogen addition on growth and photosynthetic characteristics of Acer truncatum seedlings. Dendrobiology, 72, 151–161. [Google Scholar]
- Guo, H.S. , Zhang, Y.M. , Sun, X.Q. , Li, M.M. , Hang, Y.Y. and Xue, J.Y. (2016) Evolution of the kcs gene family in plants: the history of gene duplication, sub/neofunctionalization and redundancy. Mol. Genet. Genom. 291, 739–752. [DOI] [PubMed] [Google Scholar]
- Grabherr, M.G. , Haas, B.J. , Yassour, M. et al (2011) Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J. , Salzberg, S.L. , Zhu, W. , Pertea, M. , Allen, J.E. , Orvis, J. , White, O. , Buell, C.R. and Wortman, J.R. (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J. , Delcher, A.L. , Mount, S.M. et al (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hincha, D.K. and Thalhammer, A. (2012) LEA proteins: IDPs with versatile functions in cellular dehydration tolerance. Biochem. Soc. Trans. 40, 1000–1003. [DOI] [PubMed] [Google Scholar]
- James, D.W. , Lim, E. , Keller, J. , Plooy, I. , Ralston, E. and Dooner, H.K. (1995) Directed tagging of the Arabidopsis FATTY ACID ELONGATION1 (FAE1) gene with the maize transposon activator . Plant Cell, 7, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay, G. , Pop, M. , Koren, S. , Bickhart, D. and Chin, C.‐S. (2017) Scaffolding of long read assemblies using long range contact information. BMC Genom. 18, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, Y. , Wickett, N.J. , Ayyampalayam, S. , Chanderbali, A.S. , Landherr, L. , Ralph, R.P.E. , Tomsho, L.P. and Hu, Y. (2011) Ancestral polyploidy in seed plants and angiosperms. Nature, 473, 97–100. [DOI] [PubMed] [Google Scholar]
- Jiao, Y. (2011) Phylogenomic analysis of ancient genome duplications in the history of plants. Dissertations & Theses ‐ Gradworks. [Google Scholar]
- Jurka, J. , Kapitonov, V.V. , Pavlicek, A. , Klonowski, P. , Kohany, O. and Walichiewicz, J. (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. [DOI] [PubMed] [Google Scholar]
- Kasai, N. , Mizushina, Y. , Sugawara, F. and Sakaguchi, K. (2002) Three‐dimensional structural model analysis of the binding site of an inhibitor, nervonic acid, of both DNA polymerase? and HIV‐1 reverse transcriptase. J. Biochem. 132, 819–828. [DOI] [PubMed] [Google Scholar]
- Kajitani, R. , Toshimoto, K. , Noguchi, H. , Toyoda, A. , Ogura, Y. , Okuno, M. , Yabana, M. , Harada, M. , Nagayasu, E. and Maruyama, H. (2014) Efficient de novo assembly of highly heterozygous genomesfrom whole‐genome shotgun short reads. Genome Res. 24, 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J. (2002) BLAT–the BLAST‐like alignment tool. Genome Res. 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. and Salzberg, S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nat. Methods, 12(4), 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinform. 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder, P. and Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saet‐Buyl, L. , Su‐Jin, J. , Young‐Sam, G.o. , Jeong‐Kook, K. , Hong‐Joo, C. , Park Ohkmae, K. and Mi, C.S. (2010) Two Arabidopsis 3‐ketoacyl CoA synthase genes, KCS20 and KCS2/DAISY, are functionally redundant in cuticular wax and root suberin biosynthesis, but differentially controlled by osmotic stress. Plant J. 60, 462–475. [DOI] [PubMed] [Google Scholar]
- Levin, H.L. and Moran, J.V. (2011) Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 12, 615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, Q. , Li, H.Y. , Sk, L.i. et al (2019) The genome assembly and annotation of yellowhorn (Xanthoceras sorbifolium Bunge). GigaScience, 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X.Y. , Fu, H. and Chen, Y.H. (2013) Study on the physico‐chemical properties and fatty acid composition of Acer Truncatum Buge oil. China Oils Fats, 28, 66–67. [Google Scholar]
- Lin, Y.L. , Min, J.M. , Lai, R.L. et al (2017) Genome‐wide sequencing of longan (dimocarpus longan lour.) provides insights into molecular basis of its polyphenol‐rich characteristics. GigaScience, 6, 10.1093/gigascience/gix023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingguang, Y. , Peipei, Y. , Hang, F. , Qiang, X. , Ke, L. , Xiang, L. , Liwei, S. and Yujun, L. (2017) Response surface methodology optimization of ultrasonic‐assisted extraction of Acer Truncatum leaves for maximal phenolic yield and antioxidant activity. Molecules, 22, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, X.F. , Tian, W.X. , Wu, L.H. , Cao, X.L. and Ito, Y. (2005) Isolation of quercetin‐3‐O‐L‐rhamnoside from Acer truncatum Bunge by high‐speed counter‐current chromatography. J. Chromatogr. A, 1070, 211–214. [DOI] [PubMed] [Google Scholar]
- Ma, Q.Y. , Wang, Y.N. , Zhu, L. , Li, B.C.W. , Li, S. , Li, S. , Wen, J. , Yan, K.Y. and Li, Q.Z. (2019) Characterization of the complete chloroplast genome of Acer truncatum bunge (sapindales: aceraceae): a new woody oil tree species producing nervonic acid. Biomed. Res. Int. 7417239, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magwanga, R.O. , Lu, P. , Kirungu, J.N. et al (2018) Characterization of the late embryogenesis abundant (LEA) proteins family and their role in drought stress tolerance in upland cotton. BMC Genet. 19, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros, W.H. , Pertea, M. and Salzberg, S.L. (2004) TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene‐finders. Bioinformatics, 20, 2878–2879. [DOI] [PubMed] [Google Scholar]
- Mcgrath, C.L. , Gout, J.F. , Johri, P. , Doak, T.G. and Lynch, M. (2014) Differential retention and divergent resolution of duplicate genes following whole‐genome duplication. Genome Res. 24, 1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill, A.H. , Schmelz, E.‐M. , Wang, E. , Dillehay, D.L. , Rice, L.G. , Meridith, F. and Riley, R.T. (1997) Importance of sphingolipids and inhibitors of sphingolipid metabolism as components of animal diets. J. Nutr. 127, 830S–833S. [DOI] [PubMed] [Google Scholar]
- Mietkiewska, E. , Brost, J.M. , Giblin, E.M. , Barton, D.L. and Taylor, D.C. (2007a) Cloning and functional characterization of the fatty acid elongase 1 (FAE1) gene from high erucic Crambe abyssinica cv. Prophet. Plant Biotechnol. J. 5, 636–645. [DOI] [PubMed] [Google Scholar]
- Mietkiewska, E. , Brost, J.M. , Giblin, E.M. , Barton, D.L. and Taylor, D.C. (2007b) A Teesdalia nudicaulis FAE1 complements the fae1 mutation in transgenic Arabidopsis thaliana plants and shows a preference for elongating oleic acid to eicosenoic acid. Plant Sci. 173, 198–205. [Google Scholar]
- More, D. , White, J. , More, D. and White, J. (2003) Cassell's trees of Britain and northern Europe. Cassells Trees of Britain & Northern Europe.
- Mostovoy, Y. , Levy‐Sakin, M. , Lam, J. , Lam, E.T. , Hastie, A.R. , Marks, P. , Lee, J. , Chu, C. , Lin, C. and Džakula, Ž. (2016) A hybrid approach for de novo human genome sequence assembly and phasing. Nat. Methods, 13, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, S. , Chen, J. and Jiang, N. (2018) Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46(21), e126‐e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamplona, R. , Dalfó, E. , Ayala, V. , Bellmunt, M.J. , Prat, J. , Ferrer, I. and Portero‐Otín, M. (2005) Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation: effects of Alzheimer disease and identification of lipoxidation targets. J. Biol. Chem. 280, 21522–21530. [DOI] [PubMed] [Google Scholar]
- Panchy, N. , Lehti‐Shiu, M.D. and Shiu, S.‐H. (2016) Evolution of gene duplication in plants. Plant Physiol. 171, 2294–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra, G. , Bradnam, K. and Korf, I. (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Parra, G. , Blanco, E. and Guigo, R. (2000) GeneID in Drosophila. Genome Res. 10, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrosa, A.M. , Martins, C.D.P.S. , Gonçalves, L.P. and Costa, M.G.C. (2015) Late embryogenesis abundant (LEA) constitutes a large and diverse family of proteins involved in development and abiotic stress responses in sweet orange (citrus sinensis L. osb.). PLoS One, 10(12), e0145785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos, A. (1995) Very long chain fatty acids in higher animals‐a review. Lipids, 30, 1–14. [DOI] [PubMed] [Google Scholar]
- Price, A.L. , Jones, N.C. and Pevzner, P.A. (2005) De novo identification of repeat families in large genomes. Bioinformatics, 21, i351–i358. [DOI] [PubMed] [Google Scholar]
- Ma, Q. , Li, S. , Ma, X. , Jing Wen, L.u. , Zhu, L.T. , Li, R. , Guo, W. and Li, Q. (2018) Estimation of genome size of two Acer Linn. by flow cytometry. J. Nanjing Fores. Univ. (Natural Sciences Edition), 42, 201–205. [Google Scholar]
- Raffaele, S. , Vailleau, F. , Léger, A. , Miersch, O. , Huard, C. , Blée, E. , Mongrand, S. , Domergue, F. and Roby, D. (2008) A MYB transcription factor regulates very long‐chain fatty acid biosynthesis for activation of the hypersensitive cell death response in Arabidopsis. Plant Cell, 20, 752–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels, L. , Kunst, L. and Jetter, R. (2008) Sealing plant surfaces: cuticular wax formation by epidermal cells. Annu. Rev. Plant Biol. 59, 683–707. [DOI] [PubMed] [Google Scholar]
- Shao, H.B. , Liang, Z.S. and Shao, M.A. (2005) LEA proteins in higher plants: structure, function, gene expression and regulation. Colloids Surf. B, 45, 131–135. [DOI] [PubMed] [Google Scholar]
- Simao, F.A. , Waterhouse, R.M. , Ioannidis, P. et al (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. and Morgenstern, B. (2005) AUGUSTUS: a web server for gene prediction in eukaryotes that allows user‐defined constraints. Nucleic Acids Res. 33, W466–W467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbiss, S. , Willhoeft, U. , Gremme, G. and Kurtz, S. (2009) Fine‐grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 37, 7002–7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, D.C. , Francis, T. , Guo, Y. , Brost, J.M. , Katavic, V. , Mietkiewska, E. , Michael, G.E. , Lozinsky, S. and Hoffman, T. (2009) Molecular cloning and characterization of a KCS gene from Cardamine graeca and its heterologous expression in Bracssica oilseeds to engineer high nervonic acid oils for potential medical and industrial use. Plant Biotechnol. J. 7, 925–938. [DOI] [PubMed] [Google Scholar]
- Tanaka, K. , Shimizu, T. , Ohtsuka, Y. , Yamashiro, Y. and Oshida, K. (2007) Early dietary treatments with Lorenzo’s oil and docosahexaenoic acid for neurological development in a case with Zellweger syndrome. Brain Dev. 29, 586–589. [DOI] [PubMed] [Google Scholar]
- Team RC (2016) R: a language and environment for statistical computing. http://www.R‐project.org/.
- Trapnell, C. , Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanburen, R. , Bryant, D. , Edger, P.P. et al (2015) Single‐molecule sequencing of the desiccation‐tolerant grass oropetium thomaeum . Nature, 527, 508–511. [DOI] [PubMed] [Google Scholar]
- Vavouri, T. , Semple, J.I. and Lehner, B. (2008) Widespread conservation of genetic redundancy during a billion years of eukaryotic evolution. Trends Genet. 24, 485–488. [DOI] [PubMed] [Google Scholar]
- Walker, B.J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. , Cuomo, C.A. , Zeng, Q. , Wortman, J. and Young, S.K. (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One, 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X.Y. , Fan, J.S. , Wang, S.Y. and Sun, R.C. (2006) A new resource of nervonic acid from purpleblow maple (Acer truncatum) seed oil. Forest Products J. 56, 147–150. [Google Scholar]
- Wang, X.Y. and Wang, S.Q. (2005) A new resource of nervonic acid: purpleblow maple oil. China Oils Fats, 9, 021. [Google Scholar]
- Wang, R. , Liu, P. , Fan, J. and Li, L. (2018) Comparative transcriptome analysis two genotypes of Acer truncatum Bunge seeds reveals candidate genes that influences seed VLCFAs accumulation. Sci. Rep. 8, 15504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , DeBarry, J.D. et al (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, T.Z. (1998) The systematic evolution and distribution of the genus Acer. Acta Botan. Yunnanica. 20(4), 383–393. [Google Scholar]
- Xu, C.Q. , Liu, H. , Zhou, S.S. et al (2019) Genome sequence of Malania oleifera, a tree with great value for nervonic acid production. GigaScience 8, giy164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Q. , Chen, L.L. , Ruan, X. et al (2013) The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 45, 59–66. [DOI] [PubMed] [Google Scholar]
- Xu, Z. and Wang, H. (2007) LTR_FINDER: an efficient tool for the prediction of full‐length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, A. , Kagaya, Y. , Toyoshima, R. , Kagaya, M. , Takeda, S. and Hattori, T. (2009) Arabidopsis NF‐YB subunits LEC1 and LEC1‐LIKE activate transcription by interacting with seed‐specific ABRE‐binding factors. Plant J. 58, 843–856. [DOI] [PubMed] [Google Scholar]
- Yang, C. and Liu, X. (1998) The Resources of maple in China and its ornamental utilization. J. Southwest Agr. Univ. 20(1), 68–71. [Google Scholar]
- Yang, J. , Wariss, H.M. , Tao, L.D. et al (2019) De novo genome assembly of the endangered Acer yangbiense, a plant species with extremely small populations endemic to Yunnan Province, China. GigaScience, 8, giz085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, T.Q. , Yu, Q. , Xu, W. , Li, D.‐Z. , Chen, F. and Liu, A.Z. (2018) Transcriptome analysis reveals crucial genes involved in the biosynthesis of nervonic acid in woody Malania oleifera oilseeds. BMC Plant Biol. 18, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Bioinformatics, 13, 555–556. [DOI] [PubMed] [Google Scholar]
- Zdobnov, E.M. and Apweiler, R. (2001) InterProScan–an integration platform for the signature‐recognition methods in InterPro. Bioinformatics, 17, 847–848. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , Luo, Y. , Wang, X. and Yu, F. (2018) Fruit spray of 24‐epibrassinolide and fruit shade alter pericarp photosynthesis activity and seed lipid accumulation in Styrax tonkinensis. J. Plant Growth Regul. 37(4), 1066–1084. [Google Scholar]
- Zheng, Y. , Jiao, C. , Sun, H. et al (2016) iTAK: a program for genome‐wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Molocelar Plant, 9, 1667–1670. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The K‐mer analysis of A. truncatum. The frequency of 17‐mers is exhibited by 17‐bp sequences within filtered reads of the 350‐bp inset size library. As the curve shows, the peak depth is 76, and the genome size was estimated as 662.08 Mb (653.44 Mb after revision).
Figure S2. The Hi‐C chromatin interaction map of 13 pseudomolecules in A. truncatum.
Figure S3. The insert time of LTR retrotransposons in A. truncatum, A. yangbiense, C. sinensis, and M. oleifera. The red, green, and blue regions represent the LTR insertion time (million years ago) of Atru: A. truncatum, Ayan: A. yangbiense; Csin: C. sinensis, and Mole: M. oleifera.
Figure S4. Gene numbers in each group defined by OrthoMCL.
Figure S5. The phylogeny and divergence time of A. truncatum and 14 other plants.
Figure S6. The phylogeny based on coding sequences of single‐copy orthologous genes shared among 17 chloroplast genomes in Aceraceae species. Dimocarpus longan served as the outgroup.
Figure S7. The phylogeny based on protein sequences of single‐copy orthologous genes shared among 17 chloroplast genomes in Aceraceae species. Dimocarpus longan served as the outgroup.
Figure S8. Synteny comparison of A. truncatum and C. sinensis. The outermost circle (blue) displays ideograms of the pseudochromosomes of the genomes. Atru: A. truncatum, Csin: C. sinensis. The second circle displays gene density. The innermost circle displays homologies between A. truncatum and C. sinensis. All distributions are drawn in a window size of 300 kb, chromosomes_units = 500 000.
Figure S9. Synteny comparison of A. truncatum and A. yangbiense. The outermost circle (blue) displays ideograms of the pseudochromosomes of the genomes. Atru: A. truncatum, Ayan: A. yangbiense. The second circle displays gene density. The innermost circle displays homologies between A. truncatum and A. yangbiense. All distributions are drawn in a window size of 300 kb, chromosomes_units = 500 000.
Figure S10. Syntenic pattern of A. truncatum with A. yangbiense and C. sinensis. The x‐axis indicates that each gene of one genome has no orthologous region, one orthologous region, two orthologous regions, three orthologous regions, or four orthologous regions with another genome. The y‐axis indicates the percentage of the gene blocks in genomes.
Figure S11. The phylogeny analysis of LEA genes in A. truncatum.
Figure S12. The distribution of LEA genes in A. truncatum.
Figure S13. Heat map of gene expression profiles of LEA genes. (a) Differentially expressed LEA genes during seed development. (b) Differentially expressed LEA genes in five tissues.
Figure S14. Quantitative PCR of LEA genes in different tissues. Error bars indicate standard errors of the mean from three technical replicates.
Figure S15. Quantitative PCR of LEA genes in six seed development stages. Error bars indicate standard errors of the mean from three technical replicates.
Figure S16. The phylogenetic tree and distribution of KCS genes based on coding sequences of A. truncatum. (a) The maximum likelihood tree of KCS genes from A. truncatum (red triangle), A. yangbiense (write triangle), M. oleifera (blue square), and A. thaliana (write circle).
Figure S17. The expression profiles of KCS genes in different tissues of A. truncatum. Z‐scores were calculated after log10 transformation of FPKM values plus 1.
Figure S18. Quantitative PCR of KCS genes in six seed development stages. Error bars indicate standard errors of the mean from three technical replicates.
Figure S19. Quantitative PCR of KCS genes in different tissues. Error bars indicate standard errors of the mean from three technical replicates.
Figure S20. WGCNA of fatty acid contents with RNA‐seq data of seed development stages in A. truncatum. The numbers within the heat map represent correlations (red, positively correlated; blue, negatively correlated) and P‐values (in parentheses) for the module–trait associations.
Table S1. K‐mer statistics of A. truncatum.
Table S2. Sequencing data of A. truncatum.
Table S3. Summary of A. truncatum pseudomolecules.
Table S4. The Hi‐C assembly results of A. truncatum.
Table S5. Coverage statistics of the A. truncatum genome.
Table S6. CEGMA evaluation results.
Table S7. BUSCO notation assessment of A. truncatum genome and annotation.
Table S8. Assessing the gene region assembly by mapping the RNA assembly to the A. truncatum genome.
Table S9. Statistical results of gene functional annotations.
Table S10. Statistical results of non‐coding RNAs.
Table S11. Classification of repetitive elements in the A. truncatum genome.
Table S12. The LTR subfamilies in A. truncatum and other published genomes.
Table S13. The seed sequences of centromeric regions of A. truncatum and A. yangbiense chromosomes.
Table S14. Details of the chloroplast genome sequences used for the phylogenetic analysis.
Table S15. GO enrichment results for expanded genes in A. truncatum.
Table S16. GO enrichment results for contracted genes in A. truncatum.
Table S17. GO enrichment results for the genes in A. truncatum‐specific gene families.
Table S18. KEGG enrichment results for expanded genes in A. truncatum.
Table S19. GO enrichment results for expanded genes in A. yangbiense.
Table S20. GO enrichment results for contracted genes in A. yangbiense.
Table S21. KEGG enrichment results for expanded genes in A. yangbiense.
Table S22. A summary of drought‐tolerant genes identified in A. truncatum and other published genomes.
Table S23. A summary of drought tolerance‐related transcription factors identified in A. truncatum and other published genomes.
Table S24. The copy numbers of VLCFA biosynthesis‐related genes in 12 plants.
Table S25. Expression levels of VLCFA biosynthesis‐related genes that are significantly upregulated between 85 DAF and 100 DAF and are involved in fatty acid elongation.
Table S26. The transcription factors (TFs)/transcription regulatory factors (TRs) in the black module of WGCNA analysis.
Table S27. List of KCS gene‐specific primers (5’–3’) used for RT‐PCR.
Table S28. List of LEA gene‐specific primers (5’–3’) used for RT‐PCR.
Data Availability Statement
The A. truncatum genome has been deposited under BioProject accession number PRJNA557096 and BioSample accession number SAMN12389479. PacBio and Illumina reads, resequencing sequences reads and Hi‐C data have been submitted in the Sequence Read Archive (SRA) under study accession number SUB6287730. The final assembled version(s) must be available in https://doi.org/10.6084/m9.figshare.12986237.v2.