

Summary
Deviation from Mendelian inheritance expectations (transmission ratio distortion, TRD) has been observed in several species, including the mouse and humans. In this study, TRD was characterized in the turkey genome using both allelic (specific‐ and unspecific‐parent TRD) and genotypic (additive‐ and dominance‐TRD) parameterizations within a Bayesian framework. In this study, we evaluated TRD for 23 243 genotyped Turkeys across 56 393 autosomal SNPs. The analyses included 500 sires, 2013 dams and 11 047 offspring (trios). Three different haplotype sliding windows of 4, 10 and 20 SNPs were used across the autosomal chromosomes. Based on the genotypic parameterizations, 14 haplotypes showed additive and dominance TRD effects highlighting regions with a recessive TRD pattern. In contrast, the allelic model uncovered 12 haplotype alleles with the allelic TRD pattern which showed an underrepresentation of heterozygous offspring in addition to the absence of homozygous animals. For regions with the allelic pattern, only one particular region showed a parent‐specific TRD where the penetrance was high via the dam, but low via the sire. The gene set analysis uncovered several gene ontology functional terms, Reactome pathways and several Medical Subject Headings that showed significant enrichment of genes associated with TRD. Many of these gene ontology functional terms (e.g. mitotic spindle assembly checkpoint, DRM complex and Aneuploidy), Reactome pathways (e.g. Mismatch repair) and Medical Subject Headings (e.g. Adenosine monophosphate) are known to be related to fertility, embryo development and lethality. The results of this study revealed potential novel candidate lethal haplotypes, functional terms and pathways that may enhance breeding programs in Turkeys through reducing mortality and improving reproduction rate.
Keywords: fertility, functional analysis, gene set enrichment, lethal haplotypes, transmission ratio distortion
Introduction
Owing to its considerable economic impact, reproduction has drawn the attention of turkey breeders and producers (Saif & Nestor 2002; Huff et al. 2005; Emamgholi Begli et al. 2019). Lethal alleles may cause mortality before, during or after the embryonic stage, and hence reduce reproductive performance. By their nature, livestock breeding programs tend to increase inbreeding levels among individuals, and consequently the probability of mating parents carrying lethal alleles may increase (Granleese et al. 2015). Turkeys are not an exception, thus identifying genetic regions that influence reproductive efficiency and mortality is relevant and may enhance breeding programs in this species.
Many autosomal recessive lethal loci have been distinguished in livestock species such as cattle (e.g. Dong et al. 2019; Guarini et al. 2019) and the correct mate allocation is expected to reduce the economic losses (Cole et al. 2016). Several methods, such as screening for the absence of homozygous haplotypes (e.g. VanRaden et al. 2011; Hoff et al. 2017; Jenko et al. 2018) and transmission ratio distortion (TRD; Casellas et al. 2012, 2014), can be used to discover genomic regions with potentially lethal alleles. TRD is a process whereby the transmission of alleles from heterozygous parents to offspring deviates from Mendelian ratios, regardless of the cause (Crow 1999; Pardo‐Manuel De Villena et al. 2000; Huang et al. 2013). Thus, TRD reveals locus‐specific signals that provide insight into genetics and evolutionary processes of individual fitness variation, population divergence and speciation (Fishman & McIntosh 2019).
The availability of genomic markers has facilitated the task of investigating lethal alleles. The decline in reproductive performance and ability of parents to contribute equally to next generations may alter the expected Mendelian inheritance patterns, resulting in an observable TRD. For instance, it is possible to trace back the inheritance of each allele as well as the combination of two alleles inherited from parents to offspring using genotype trios with high accuracy. Several Bayesian models have been developed to detect and analyze all types of TRD based on allelic and genotypic parameterizations (Casellas et al. 2012, 2014). These two parametrizations have been already evaluated in a previous study (Casellas et al. In press) reporting the relevance of implementing and comparing the different parametrizations to capture all types of TRD. The objective of this study was to assess TRD based on allelic and genotypic parameterizations and perform a functional gene set enrichment analysis to uncover biological pathways associated with TRD in a purebred line of turkeys.
Materials and methods
Data
In this study, we used a turkey population with 23 243 (6867 males and 16 376 females) genotyped animals for 61 705 SNPs. The animals were hatched between late 2010 and early 2018, and genomic and pedigree information was provided by Hybrid Turkeys, Kitchener, Canada. The whole dataset combines 11 047 parent‐offspring genotyped trios, including 500 sires and 2013 dams. All birds were genotyped with the same SNP genotyping platform array (65 000 SNP; Illumina, Inc.) and mapped to the Turkey 5.0 Meleagris gallopavo assembly (Dalloul et al. 2010). Quality control analyses were performed and resulted in the removal of non‐autosomal SNP markers and those with a call rate below 90%. Whereas all birds had a call rate higher than 90% and passed the quality control criteria, the number of SNPs retained for analysis was 56 393 out of the 61 705 markers. beagle 5.0 (Browning et al. 2018) was used to phase genotypes and impute the missing genotypes.
Statistical analyses
To trace the haplotype allele inheritance from parents to offspring in this turkey population, two parametrizations were considered in this study.
Allelic parametrization
Following Casellas et al. (2014, 2017), the probability of allele transmission (p) from heterozygous parents to offspring can be parameterized, including TRD effects on allelic basis, as:
where A is the particular haplotype allele j being analysed, B represents the remaining haplotype alleles and αj is the overall TRD for the allele j. To capture parent‐specific TRD origin, a parent‐specific model was also implemented on the basis of allelic parametrization, but including two different parameters:
where s and d represent sire and dam respectively, and α s j and α d j are sire‐ and dam‐specific TRD for allele j. For all TRD parameters, flat priors were assumed within a parametric space ranging from −0.5 to 0.5. Under a Bayesian implementation, the conditional posterior probabilities of the TRD parameters are defined as:
for overall and parent‐specific TRD respectively, where y is a column vector of genotypes of the offspring generation. The likelihood of data consists of a straightforward multiplication of the corresponding probabilities for each offspring (i.e. ), where n is the total number of offspring and p off and yi are the probability and the genotype of the ith offspring respectively. The software trdscan version 1.0 (Id‐Lahoucine et al. 2019) uses a multinomial process, hence the likelihood of the data becomes:
In the above, ni is the sum of n AA, i, n AB, i and n BB, i offspring genotypes, p off, i is the probability of an offspring genotype from the ith mating and n AA, i, n AB, i and n BB, i are the number of AA, AB and BB offspring genotypes from the specific ith mating respectively. For the parent‐specific TRD model, five kinds of matings were differentiated in the multinomial expression (i.e. ). For the sampling process, uniform proposal distributions (flat priors) were used for both α g and δ g within a deepened parametric space ranging from −1 to 1 (Casellas et al. 2012).
Genotypic parameterization
This parameterization captures the interaction between alleles of offspring genotypes. Additive (α g) and dominance (δ g) or over‐dominance, both positive or negative, TRD parameters are considered regardless of the origin of the allele. As described by Casellas et al. (2012, In press), the probability of observing offspring (p off) from heterozygous‐by‐heterozygous mating can be estimated as follows:
where α g j and δ g j are additive and dominance TRD parameters for the specific allele j respectively.
Under a Bayesian implementation, the conditional posterior probabilities of the TRD parameters are defined as:
where y is a column vector of genotypes of the offspring generation. For the sampling process, uniform proposal distributions (flat priors) were used for both α g and δ g within an extended parametric space ranging from −1 to 1 (Casellas et al. 2012). Thus, as the parametric space for α g is initially [−1, 1], the parametric space for δ g is restricted to [−1, |α g|]. Moreover, the parametric space of α g itself is restricted to [−1 + δ g to 1 − δ g] if δ g > 0 and this guarantees that the sum of offspring’s genotypes probabilities for specific mating is equal to 1.
Transmission ratio distortion was evaluated using three sliding windows: 4, 10, and 20 SNP. The analyses were carried out using trdscan version 1.0 software (Id‐Lahoucine et al. 2019) based on a MCMC and the Metropolis–Hastings’ algorithm (Hastings 1970). A single MCMC of 100 000 iterations was run for each analysis with the first 10 000 iterations being discarded as burn‐in. Bayes factor (BF; Kass & Raftery 1995), which is a ratio of probabilities between full and null TRD models, was used to determine significant TRD (BF ≥ 100; decisive evidence according to Jeffreys’ (1984) scale). To obtain a reasonable statistical power and to minimize false TRD as a result of genotyping errors, only haplotype alleles with a minimum number of 50 informative offspring (i.e. from heterozygous parents) and five heterozygous sires and/or dams were analyzed.
The identified TRD regions were then filtered to minimize genotypic errors and to eliminate regions with random TRD. First, an approximate empirical null distribution of TRD (Id‐Lahoucine et al. 2019) at less than 0.001% margin error was used to remove TRD generated by chance. Also, a minimal number of informative parents (≥5 heterozygous sires and/or heterozygous dams) were considered to minimize possible false TRD from genotyping errors. Similarly, regions with few heterozygous parents fully explaining the observed TRD in the corresponding region were discarded as potential genotyping errors. Moreover, for the allelic parametrization, an arbitrary minimum magnitude of TRD at least 0.20 and the number of underrepresented offspring at least 1000 were considered to identify haplotype alleles with strong allelic TRD patterns. The number of underrepresented offspring is the total number of offspring expected, but not observed for a particular allele, which is also approximately equal to the number of informative offspring multiplied by twice the magnitude of the TRD. These thresholds ensured the identification of target haplotypes with a moderate‐to‐high level of TRD (i.e. moderate‐to‐high penetrance) as well as a reasonable number of offspring that are expected, but not observed (i.e. a minimum frequency for the allele in the population). For the genotypic parametrization, an arbitrary minimum number of 10 non‐observed homozygous offspring from heterozygous‐by‐heterozygous matings with additive TRD effect less than −0.50 and dominance TRD effect greater than 0.10 was considered to determine haplotype alleles with a substantial recessive TRD pattern. This threshold was considered to maintain the most important regions. It should also be emphasized that a well‐known lethal haplotype in cattle industry (Holstein haplotype 3) was initially identified by VanRaden et al. (2011) with only seven non‐observed homozygous offspring from heterozygous sires and heterozygous maternal grandsires matings. In addition, as different sliding windows were used, only the haplotype alleles with the largest BF within a region (with many physically linked haplotypes) were selected as the best candidates to explain the observed TRD in the region and potentially harbor the causal mutations. Thus, it is important to mention that the patterns of TRD observed in short windows are displayed in larger haplotypes including the same allele and also on other physically linked haplotypes, supporting the relevance of TRD in the corresponding particular locus.
Functional and gene set enrichment
Assignment of lethal haplotypes to genes
Genes associated with complex traits are expected to represent only a small fraction of the genetic variation and, hence, some genetic variants with small effects and disease risks may not ever be detected (Peng et al. 2010; Abdalla et al. 2016). To further investigate the potential lethal haplotypes identified with TRD, the coordinates of these haplotype regions were used to mine for annotated genes using the Turkey 5.0 (release 102) assembly (Dalloul et al. 2010). It has been reported that strong LD may extend up to 10–30 kb in chickens (Rao et al. 2008; Megens et al. 2009; Qanbari et al. 2010). Thus, haplotypes were assigned to genes if they were located within the genomic sequence of an annotated gene or within 15 kb of the 5′ or 3′ ends of the first and last exons respectively. The 15 kb distance was used to capture proximal regulatory regions and other functional sites that may lie outside (e.g. promoter regions) but close to each gene. If a haplotype was found to be located within or close to more than one gene, all of these genes were included in the subsequent analyses.
Assignment of genes to functional categories
Gene Ontology (GO; Ashburner et al. 2000), Pathway Knowledgebase (Reactome; Fabregat et al. 2018) and Medical Subject Headings (MeSH; Coletti & Bleich 2001; Nelson et al. 2004) databases were used to define functional sets of genes. Biological descriptors, known as GO terms, fall into three categories: biological process, molecular functions and cellular components. Reactome, on the other hand, provides several biochemical networks including metabolic and regulatory pathways. Finally, MeSH is a collection of descriptors or headings representing key topics discussed in the papers indexed in the MEDLINE database. Whereas MeSH terms are classified into 19 categories, in this study, we were interested in only four: anatomy, disease, phenomena and processes, and lastly chemicals and drugs.
Pathway‐based association analysis
The Fisher’s exact test was used to declare the association of a given GO term, Reactome pathway and MeSH heading with TRD. This test was performed to search for an overrepresentation of significant genes in a given functional category among all genes. The P‐value of observing g significant genes in the term was calculated as follows:
where N is the total number of genes analyzed in the study, S is the total number of genes that were deemed significantly associated with TRD and k is the total number of genes in the functional category in the database under consideration.
Owing to a lack of biological information related to turkeys, both the turkey 5.0 assembly (Dalloul et al. 2010) and the GRCg6a chicken assembly (International Chicken Genome Sequencing Consortium 2004) were used to perform the pathway analysis. It is important to emphasize that all potential genes defined in this study are conserved across many organisms, including humans and chickens. The GO and Reactome enrichment analyses were carried out using GO (Ashburner et al. 2000) and panther (Mi et al. 2016) respectively, whereas the MeSH enrichment analysis was performed using the meshr package (Tsuyuzaki et al. 2015) available in the r environment (R Core Team 2019).
Gene network
The overlaps among significant genes associated with GO terms and Reactome pathways and their functional networks were also examined. We retrieved close neighbor genes and then generated an aggregate interaction network based on physical protein interaction and co‐expression of those genes using genemania software (Warde‐Farley et al. 2010).
Results and discussion
Prevalence of TRD across the turkey genome
The prevalence of TRD was widely distributed across the turkey genome as shown in Fig. 1. The initial numbers of haplotype alleles detected with decisive significant evidence (BF ≥ 100) of TRD, according to Jeffreys’ scale (Jeffreys 1984), were 48 951, 52 896 and 54 287 for 4, 10 and 20 SNP haplotype windows respectively. Despite this high number of regions, some of them had a large BF (10100) suggesting virtually 0 probability of the null TRD model. In addition, most of the detected TRD regions had low frequencies (i.e. rare variants); however, these haplotype alleles were supported by the large dataset. It is important to mention that it has been suggested that rare variants are more likely to be functional than common variants (Gorlov et al. 2008; Karaca et al. 2015), emphasizing the importance of TRD regions despite their low frequencies. It is noteworthy to mention that the majority of the regions were detected with more than one of the models applied, but with different fits and statistical significance. Thus, after the characterization of the TRD across the whole genome with the filtration criteria provided in the ‘Materials and methods’ section, the list of the most relevant haplotypes is provided in Tables 1 and 2 for regions with allelic and recessive TRD patterns respectively. These haplotypes indicate candidate regions potentially carrying deleterious alleles or genes affecting reproduction. However, these TRD findings were obtained under the assumption of no selection of offspring within a family to be genotyped. The violation of this must be taken into consideration by further investigating the source of the observed TRD signals. This is because the pre‐selection of offspring to be genotyped within families (Id‐Lahoucine & Casellas 2017) could be a source of bias on TRD analyses as discussed by Id‐Lahoucine et al. (2019). It is well known that selection of data has been a concern and a limitation for many types of analyses. In particular, for TRD analyses, major genes that present crucial and large impact in reproductive performance are targeted. The genetic selection performed in turkey populations is based on selection indexes targeting a multiple‐trait breeding objective (mainly highly polygenic production traits). Thus, the chance to observe TRD signals with absence of homozygous offspring as a result of selection is less likely. Here, we are using the TRD method as an alternative strategy to first scan possible relevant regions harboring lethal alleles that will require further research to exclude both genotyping errors and biases from pre‐selection in the offspring generation.
Figure 1.
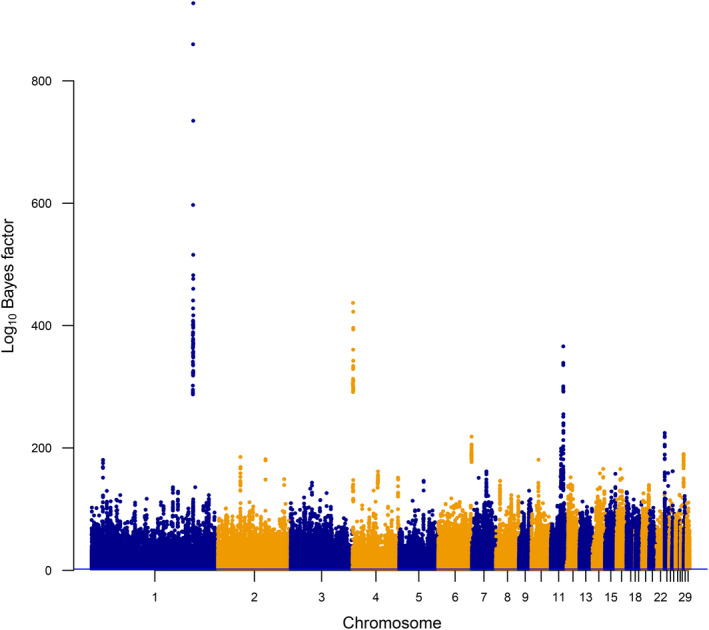
Bayes factor for haplotypes with transmission ratio distortion across the turkey genome. Significant haplotype alleles were determined based on log10 Bayes factor ≥2 according to Jeffreys’ scale (Jeffreys 1984)
Table 1.
Potential candidate lethal or semi‐lethal alleles identified with allelic transmission ratio distortion patterns by the allelic model
| Chromosome | Region (kbp) | SNP 1 | Hetero sires 2 | Hetero dams 3 | Frequency (%) | AB 4 × AA | AB × BB | AB × AB | TRD effects 6 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AA 5 | AB | AB | BB | AA | AB | BB | α | α s | α d | ||||||
| 1 | 173 127–173 367 | 10 | 17 | 360 | 4.54 | 0 | 0 | 641 | 1688 | 1 | 5 | 15 | −0.23 | −0.33 | −0.23 |
| 2 | 71 543–71 622 | 4 | 21 | 305 | 2.93 | 0 | 0 | 397 | 1664 | 0 | 7 | 2 | −0.31 | −0.21 | −0.32 |
| 2 | 99 815–99 884 | 4 | 67 | 384 | 4.44 | 0 | 0 | 654 | 1915 | 0 | 27 | 39 | −0.25 | −0.25 | −0.25 |
| 5 | 36 746–36 854 | 4 | 15 | 262 | 2.77 | 0 | 0 | 403 | 1505 | 0 | 2 | 3 | −0.29 | −0.31 | −0.29 |
| 6 | 50 141–50 184 | 4 | 22 | 182 | 1.37 | 0 | 0 | 146 | 1242 | 0 | 1 | 11 | −0.40 | −0.44 | −0.39 |
| 7 | 10 128–10 171 | 4 | 61 | 277 | 2.66 | 0 | 0 | 342 | 1344 | 0 | 14 | 42 | −0.31 | −0.26 | −0.32 |
| 10 | 10 424–10 465 | 4 | 15 | 235 | 2.34 | 0 | 0 | 327 | 1338 | 0 | 5 | 6 | −0.31 | −0.38 | −0.30 |
| 11 | 15 529–15 551 | 4 | 25 | 410 | 4.41 | 0 | 0 | 639 | 2157 | 0 | 10 | 22 | −0.28 | −0.16 | −0.29 |
| 14 | 16 726–16 743 | 4 | 32 | 287 | 2.96 | 0 | 0 | 383 | 1557 | 0 | 9 | 9 | −0.31 | −0.31 | −0.31 |
| 16 | 10 429–10 450 | 4 | 95 | 457 | 8.21 | 4 | 15 | 1136 | 2025 | 34 | 189 | 145 | −0.15 | −0.03 | −0.22 |
| 23 | 69–81 | 4 | 27 | 92 | 1.31 | 0 | 0 | 189 | 1271 | 0 | 8 | 30 | −0.38 | −0.42 | −0.30 |
| 28 | 4507–4549 | 10 | 29 | 264 | 2.8 | 0 | 0 | 372 | 1597 | 0 | 8 | 21 | −0.32 | −0.27 | −0.32 |
Number of SNP in a haplotype window.
Number of heterozygous sires.
Number of heterozygous dams.
Genotypes of parents.
Genotypes of offspring.
α, α s and α d are overall, sire and dam allelic transmission ratio distortion respectively.
Table 2.
Potential candidate lethal alleles identified with recessive transmission ratio distortion patterns by the genotypic model
| Chr 1 | Region (Kbp) | SNP 2 | Hetero sires 3 | Hetero dams 4 | Frequency (%) | AB 5 × AA | AB × BB | AB × AB | TRD effects 7 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AA 6 | AB | AB | BB | AA | AB | BB | α g | δ g | ||||||
| 1 | 27 680–27 924 | 10 | 24 | 101 | 2.51 | 0 | 0 | 450 | 447 | 0 | 17 | 10 | −0.58 | 0.29 |
| 1 | 29 253–29 484 | 10 | 30 | 108 | 4.33 | 0 | 0 | 305 | 265 | 0 | 20 | 11 | −0.54 | 0.35 |
| 2 | 40 083–40 199 | 4 | 13 | 86 | 1.83 | 0 | 0 | 336 | 481 | 0 | 23 | 15 | −0.77 | 0.14 |
| 3 | 29 955–30 188 | 10 | 5 | 39 | 0.67 | 0 | 0 | 106 | 142 | 0 | 15 | 10 | −0.71 | 0.16 |
| 4 | 60 400–60 941 | 20 | 9 | 64 | 1.93 | 0 | 0 | 252 | 321 | 0 | 32 | 16 | −0.72 | 0.21 |
| 4 | 65 823–65 908 | 4 | 10 | 48 | 0.91 | 0 | 0 | 153 | 232 | 0 | 20 | 11 | −0.77 | 0.11 |
| 5 | 3900–3975 | 4 | 50 | 170 | 4.92 | 0 | 0 | 950 | 967 | 0 | 26 | 17 | −0.62 | 0.30 |
| 6 | 8973–9097 | 10 | 19 | 81 | 2.48 | 0 | 0 | 431 | 458 | 0 | 21 | 11 | −0.63 | 0.27 |
| 11 | 23 084–23 137 | 4 | 31 | 107 | 3.04 | 0 | 0 | 573 | 673 | 0 | 22 | 12 | −0.67 | 0.23 |
| 12 | 15 689–15 829 | 20 | 9 | 49 | 1.84 | 0 | 0 | 285 | 304 | 0 | 15 | 10 | −0.61 | 0.26 |
| 19 | 4054–4196 | 20 | 43 | 156 | 4.39 | 0 | 0 | 763 | 756 | 0 | 62 | 18 | −0.63 | 0.33 |
| 21 | 9664–9680 | 4 | 38 | 129 | 3.03 | 0 | 0 | 525 | 573 | 0 | 52 | 22 | −0.67 | 0.29 |
| 24 | 672–742 | 10 | 13 | 54 | 1.86 | 0 | 0 | 323 | 384 | 0 | 17 | 10 | −0.66 | 0.22 |
| 28 | 1640–1794 | 20 | 20 | 107 | 2.96 | 0 | 0 | 484 | 476 | 0 | 15 | 10 | −0.57 | 0.29 |
Chromosome.
Number of SNP on a haplotype window.
Number of heterozygous sires.
Number of heterozygous dams.
Genotypes of parents.
Genotypes of offspring.
α g and δ g are additive and dominance transmission ratio distortion respectively.
Haplotypes with allelic TRD pattern using the allelic parametrization model
Across the turkey genome, 12 potentially lethal haplotype candidates were detected with allelic TRD, as shown in Table 1. The number of informative offspring detected in those 12 potentially lethal haplotype regions ranged between 1400 and 3548, whereas the number of underrepresented offspring reached 1564. Most of the haplotypes showed relatively small differences between male‐ and female‐specific TRD, supporting their unspecific‐parent TRD pattern. In contrast, one haplotype allele located on chromosome 16 showed parent‐specific TRD, where it was high for the dam (−0.22) and low via the sire (−0.03). Although this region had some offspring (4 and 15) from homozygous‐by‐heterozygous (AB × AA) matings (i.e. the homozygous parent carries two copies of the lethal allele), the numbers of offspring were extremely small compared with the expectations. Similarly, the matings of heterozygous‐by‐heterozygous (AB × AB) in this region produced some (34) homozygous (AA) individuals. However, this number is still substantially lower than expected, indicating lower viability. This could be due to the variation in specific TRD between males and females. As the probability of transmitting this lethal allele from heterozygous sires is close to the Mendelian expectation (0.5–0.03 = 0.47), the probability of observing live homozygous parents/offspring increases (4 instead of 0). Matings with either parent carrying the lethal allele in the homozygous state for the remaining 11 regions were not observed, supporting the lethal effect of this haplotype allele. On the other hand, it must be emphasized that these regions were also detected by the genotypic model. Nevertheless, the allelic model had better goodness‐of‐fit than the genotypic model in terms of deviance information criterion units and accurate TRD estimates with short credible intervals.
Haplotypes with recessive TRD pattern using the genotypic parametrization model
Based on the genotypic parametrization model, 14 potentially lethal haplotypes were identified with additive‐ and dominance‐TRD resulting in lethal homozygous offspring from heterozygous‐by‐heterozygous matings as shown in Table 2. The additive TRD component of these regions ranged from −0.54 to −0.94, and interestingly, the negative effects of these haplotypes are counteracted by the dominance TRD effects, which ranged between 0.11 and 0.35. The interaction of additive and dominance TRD effects provides an equal chance for heterozygous (carriers) offspring to survive as non‐carrier birds. From the heterozygous‐by‐heterozygous (AB × AB) matings, no homozygous (AA) individuals were observed, indicating the lethality of the haplotypes in the homozygous state. For these regions, the numbers of carrier parents were between five and 50 for sires and between 39 and 170 for dams. The number of informative offspring ranged from 273 to 1960, whereas the number of expected homozygotes that were not observed ranged from 10 to 22. For a fully recessive TRD pattern, it is expected that there will be similar numbers of heterozygous (AB) and homozygous (BB) offspring from heterozygous‐by‐homozygous matings (AB × BB). Nevertheless, slightly more heterozygous offspring (AB) in heterozygous‐by‐homozygous matings were observed, which could be partly explained as a result of a random TRD (i.e. TRD generated by chance; Id‐Lahoucine et al. 2019).
It is worth noting that most of these regions were only detected with the genotypic model whereas no statistical evidence was found using the allelic model. This is due to their different parameterizations, where the genotypic model includes the interaction between alleles, allowing detection of recessive TRD patterns. Specifically, the negative effect of the additive TRD component can be contrasted with the positive effect of the dominance TRD component in heterozygous offspring, allowing for capture of recessive TRD patterns. In contrast, the fact that the allelic parametrization is based on targeting the transmission of an allele from a specific parent to offspring, which is separate from possible interaction in offspring generation, prevents its ability to capture recessive TRD patterns.
Functional analysis and gene set enrichment
Sixty‐seven biological process, 17 cellular components, 35 molecular function GO terms, in addition to 19 Reactome pathways and nine MeSH terms showed a significant overrepresentation (P‐value < 0.05) of genes associated with TRD in turkeys. All of these significant terms and pathways are listed in Tables S1–S5. It is important to mention that these terms, pathways and genes should be further investigated and validated to avoid false positives. Four biological process GO terms, in a close GO hierarchy relationship and related to mitosis, showed a significant overrepresentation (P‐value < 0.05) of genes statistically associated with TRD (Table 3). Mitotic spindle assembly checkpoint (GO:0007094) is a mitotic spindle checkpoint (GO:0071174) and a negative regulation of mitotic metaphase/anaphase transition (GO:0045841), which is, in turn, a negative regulation of mitotic sister chromatid separation (GO:2000816). Mitosis, which is associated with TRD, is a specialized division of chromosomes that occur during the formation of reproductive cells (Nicklas 1971). A similar significant (P‐value < 0.05) cellular component GO term, Mitotic spindle pole (GO:0097431), was also associated with TRD (Table 4).
Table 3.
Biological process function terms significantly overrepresented with genes statistically associated with transmission ratio distortion
| GO ID | Term (GO hierarchy level) | Number of genes in the GO term | Number of significant genes | P‐value 1 |
|---|---|---|---|---|
| 0051315 | Attachment of mitotic spindle microtubules to kinetochore (10) | 10 | 1 | 0.023 |
| 0033567 | DNA replication, Okazaki fragment processing (11) | 3 | 1 | 0.008 |
| 1902969 | Mitotic DNA replication (11) | 8 | 1 | 0.019 |
| 0007094 | Mitotic spindle assembly checkpoint (14) | 18 | 1 | 0.039 |
| 0071174 | Mitotic spindle checkpoint (9) | 18 | 1 | 0.039 |
| 2000697 | Negative regulation of epithelial cell differentiation involved in kidney development (12) | 2 | 1 | 0.006 |
| 2000094 | Negative regulation of mesonephric nephron tubule epithelial cell differentiation (15) | 1 | 1 | 0.004 |
| 0061218 | Negative regulation of mesonephros development (11) | 4 | 1 | 0.001 |
| 0045841 | Negative regulation of mitotic metaphase/anaphase transition (12) | 20 | 1 | 0.044 |
| 2000816 | Negative regulation of mitotic sister chromatid separation (11) | 21 | 1 | 0.046 |
| 0072183 | Negative regulation of nephron tubule epithelial cell differentiation (14) | 1 | 1 | 0.004 |
| 1903461 | Okazaki fragment processing involved in mitotic DNA replication (12) | 1 | 1 | 0.004 |
| 2000093 | Regulation of mesonephric nephron tubule epithelial cell differentiation (12) | 1 | 1 | 0.004 |
Significance declared at P < 0.05.
Table 4.
Cellular component function terms significantly overrepresented in genes statistically associated with transmission ratio distortion
| GO ID | Term (GO hierarchy level) | Number of genes in the GO term | Number of significant genes | P‐value 1 |
|---|---|---|---|---|
| 0044444 | Cytoplasmic part (7) | 5503 | 18 | 0.038 |
| 0070176 | DRM complex (15) | 1 | 1 | 0.004 |
| 0034709 | Methylosome (8) | 6 | 1 | 0.014 |
| 0097431 | Mitotic spindle pole (12) | 18 | 1 | 0.039 |
| 0032021 | NELF complex (14) | 2 | 1 | 0.006 |
| 0090568 | Nuclear transcriptional repressor complex (13) | 19 | 2 | 0.000 |
| 0090571 | RNA polymerase II transcription repressor complex (14) | 4 | 2 | 0.000 |
Significance declared at P < 0.05.
Three GO terms detected in the analysis classified into the biological process domain showed significant association with TRD (Table 3). In addition to their similar functions in mitosis, these three terms are close in the GO hierarchy. Okazaki fragment processing involved in mitotic DNA replication (GO:1903461) is a DNA replication, Okazaki fragment processing (GO:0033567) and part of mitotic DNA replication (GO:1902969). In cell biology, Okazaki fragments (Sakabe & Okazaki 1966) comprise the processes involved in any mitotic cell cycle DNA replication, a necessary step in the cell cycle.
The Negative regulation of mesonephric nephron tubule epithelial cell differentiation (GO:2000094) term was detected as significantly enriched (P‐value < 0.01) with genes associated with TRD and related to cell differentiation. This GO term is close in the GO hierarchy and in function to four other GO terms, which all can be linked to TRD: GO:2000093, GO:0061218, GO:0072183 and GO:2000697. Cellular differentiation is the process in which a simple cell changes from one cell type to a more specialized type, which may occur in numerous steps (Jones & Taylor 1980; Slack 2007). In a study on the development of the kidney in mice, McCright et al (2001) reported that animals died perinatally owing to the lack of normal capillary tufts, which is a result of defects in kidney development.
The terms NELF complex (GO:0032021) and two similar GO terms, methylosome (GO:0034709) and cytoplasmic part (GO:0044444), are classified under the cellular component domain and show a significant overrepresentation of genes statistically associated with TRD (Table 4). The NELF complex is a key regulatory step of the transcription cycle (Tamborrini & Piatti 2019). The GO term DRM complex (GO:0070176), located in the cellular component category, is an RNA polymerase II transcription repressor complex (GO:0090571), which is, in turn, a nuclear transcriptional repressor complex (GO:0090568). Interestingly, a connection between RNA polymerase II and TRD has been previously reported (Paterson et al. 2009). Moreover, Harrison et al. (2006) reported that DRM complex is a transcriptional repressor complex and involved in cell fate specification.
Three GO terms, which are close in the GO hierarchy (Table 5), were classified into the molecular function category and showed a significant overrepresentation of genes statistically associated with TRD (Table 4). Glycine:sodium symporter activity (GO:0015375) is a glycine transmembrane transporter activity (GO:0015187), which is in turn a neutral amino acid transmembrane transporter activity (GO:0015175). Genetic factors that stop coding glycine cause late‐stage embryo lethality and hence TRD (Seidel et al. 2011). Protein binding, bridging involved in substrate recognition for ubiquitination (GO:1990756) is a protein binding, bridging (GO:0030674). The molecular function of binding and sperm motility was reported by Bauer et al. (2007). As the authors mentioned, the binding partner for Ropporin and Ras proteins to the outer surface of the dense fiber proteins plays a crucial function in sperm motility.
Table 5.
Molecular function terms significantly overrepresented in genes statistically associated with transmission ratio distortion
| GO ID | Term (GO hierarchy level) | Number of genes in the GO term | Number of significant genes | P‐value 1 |
|---|---|---|---|---|
| 0005488 | Binding (2) | 9725 | 28 | 0.017 |
| 0015187 | Glycine transmembrane transporter activity (13) | 5 | 1 | 0.012 |
| 0015375 | Glycine: sodium symporter activity (16) | 1 | 1 | 0.004 |
| 0060090 | Molecular adaptor activity (3) | 124 | 2 | 0.028 |
| 0015175 | Neutral amino acid transmembrane transporter activity (12) | 23 | 1 | 0.048 |
| 0030674 | Protein binding, bridging (4) | 112 | 2 | 0.023 |
| 1990756 | Protein binding, bridging involved in substrate recognition for Ubiquitination (11) | 3 | 1 | 0.008 |
Significance declared at P < 0.05.
Two Reactome pathways were detected as significantly (P‐value < 0.01) enriched with genes related to TRD (Table 6). Mismatch repair (MMR)directed by MSH2:MSH3 (MutSbeta; R‐GGA‐5358606) and Mismatch Repair (R‐GGA‐5358508) are similar in their activities to the GO term Mitotic spindle assembly checkpoint (GO:0007094) and represent the GO biological process mismatch repair (GO:0006298). Analogous to the GO terms related to mitosis and cell cycle, these two pathways are associated with DNA mismatch repair. It has been demonstrated that their activity increases and reaches the highest levels during the S phase of the cell cycle (Edelbrock et al. 2009). Mismatch Repair corrects single base mismatches and small insertion and deletion loops of unpaired bases. Mismatch repair directed by MSH2:MSH3 (MutSbeta) binds unpaired loops of two or more nucleotides (Palombo et al. 1996).
Table 6.
Reactome terms significantly overrepresented in genes statistically associated with transmission ratio distortion.
| Reactome ID | Reactome term name | Number of genes in the pathway | Number of significant genes | P‐value 1 |
|---|---|---|---|---|
| R‐GGA‐5358508 | Mismatch repair | 11 | 1 | 0.025 |
| R‐GGA‐5358606 | Mismatch repair (MMR) directed by MSH2:MSH3 (MutSbeta) | 3 | 1 | 0.008 |
Significance declared at P < 0.05.
A MeSH term, Adenosine Monophosphate (D000249), classified into the chemicals and drugs category, showed a significant overrepresentation of genes statistically associated with TRD (Table 7). Interestingly, various reproductive functions, such as those requiring hormone synthesis and maintenance of fluid composition, are modulated by adenosine (Zhou et al. 2006; Aliagas et al. 2010). Another significant (P‐value < 0.05) MeSH term, Anaplasia (D000708), is classified into the disease domain (Table 7). Anaplasia is related to neoplastic cells and refers to the loss of mature or specialized features of differentiated neoplastic cells (Rodriguez et al. 2010; Pujadas et al. 2019). Previous studies have shown that genetic variation among inbred mice as a result of continuous uploading and removal of rare variation (new mutations) may generate TRD (Casellas & Medrano 2008; Niu & Liang 2009).
Table 7.
Disease and chemicals and drugs MeSH terms significantly overrepresented in genes statistically associated with transmission ratio distortion
| Category | MeSH term ID | MeSH term name | P‐value 1 |
|---|---|---|---|
| Chemicals and drugs | D000249 | Adenosine monophosphate | 0.012 |
| Disease | D000708 | Anaplasia | 0.048 |
Significance declared at P < 0.05.
Gene network
The Venn diagram (Fig. 2) depicted the intersections between the significant genes associated with the terms of three GO domains (Tables 3, 4, 5) and the Reactome pathways (Table 6). Notably, the gene MAD1L1 is significant across the three GO domains and two of these GO domains (biological process and molecular function) overlap with the Reactome pathways in the gene LIG1. The aggregate interaction networks for MAD1L1 and LIG1 genes, based on physical protein interaction and co‐expression, are shown in Figs 3 and 4 respectively. The functional networks of gene MAD1L1 with 19 other genes indicate that this group of genes is involved in several activates related to mitosis such as regulation of mitosis and negative regulation of mitosis cell cycle phase transition. The gene APITD1 was not linked to MAD1L1 via physical protein interaction or co‐expression networks, but through the pathways network (this connection is not shown in Fig. 3). Similarly, the functional network for gene LIG1 revealed 19 genes associated with it through physical protein interaction and co‐expression networks (Fig. 4) and one gene (i.e. UBE2R2) through prediction. The functions for these genes include DNA replication and cell cycle DNA replication, which are among the most important cell activaties concerning reproduction and cell deviation.
Figure 2.
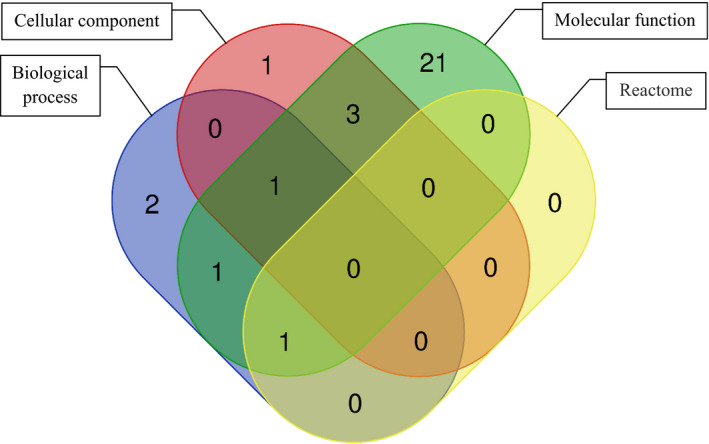
Venn diagram showing overlaps between significant genes associated with the terms of three GO domains (presented in Tables 3, 4, 5) and the Reactome pathways (presented in Table 6)
Figure 3.
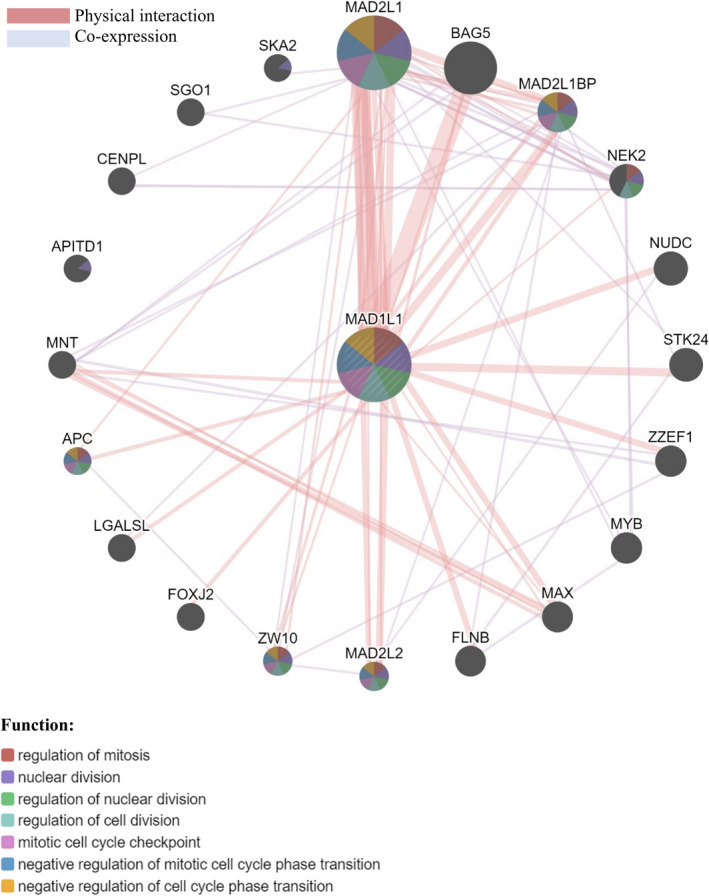
Network integration of gene MAD1L1 based on physical protein interaction and co‐expression. The gene APITD1 is associated with MAD1L1 through pathways (links are not shown)
Figure 4.
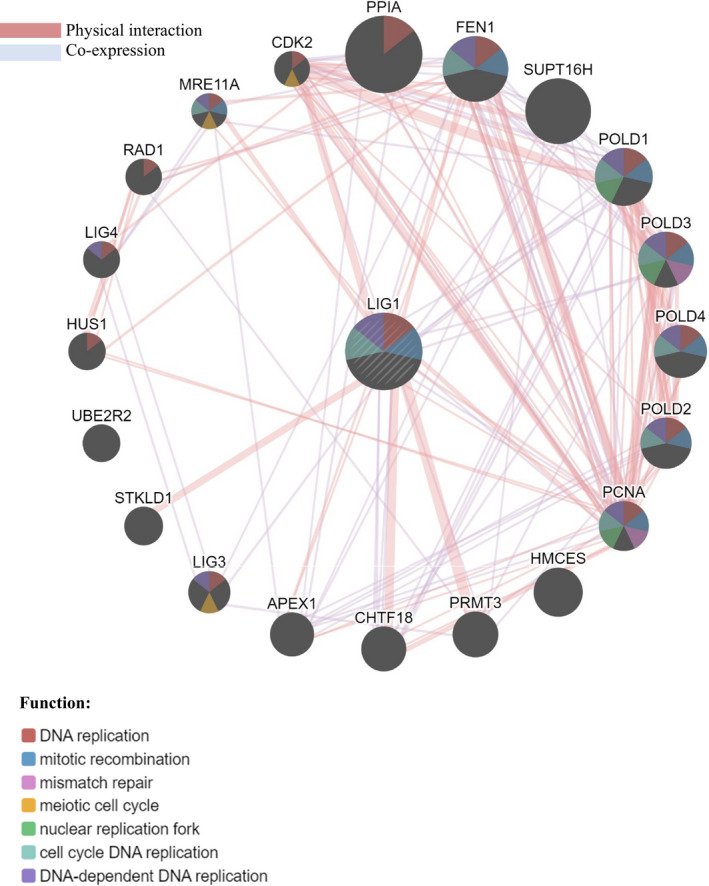
Network integration of gene LIG1 based on physical protein interaction and co‐expression. The gene UBE2R2 is associated with LIG1 through prediction (links are not shown)
Conclusions
In this study, we applied allelic and genotypic parameterizations of TRD to detect potential lethal haplotypes in turkeys. The two methods revealed relevant regions across the turkey genome with either a classical recessive inheritance pattern (i.e. lethal only in the homozygous state) or allelic patterns (i.e. reduced viability of the carrier offspring). In addition to 19 Reactome pathways and nine MeSH terms, 67 biological process, 17 cellular components and 35 molecular function GO terms showed a significant (P‐value < 0.05) overrepresentation of genes statistically associated with TRD. Functional networks among several significant genes also showed links to mitosis and cell replication. These highlighted pathways and gene ontologies, along with the overall findings of this study, will contribute in developing novel turkey breeding and management strategies, as well as to specific mating programs for turkeys.
Conflict of interests
All authors declare no conflict of interest.
Funding
This study was conducted as part of the project entitled ‘Application of genomic selection in turkeys for health, welfare, efficiency and production traits’ funded by the Government of Canada through the Genome Canada Genomic Application Partnership Program and administered by Ontario Genomics (recipients: C.F. Baes (Academic), B.J. Wood (Industry)). This study was also financially supported by Hybrid Turkeys (Kitchener, Canada). The authors declare that this study received funding from Hybrid Turkeys. The funder was involved in providing the datasets used in this study.
Supporting information
Table S1 Biological process terms significantly overrepresented with genes statistically associated with TRD
Table S2 Cellular component function terms significantly overrepresented with genes statistically associated with TRD
Table S3 Molecular function terms significantly overrepresented with genes statistically associated with TRD
Table S4 Reactome pathways significantly overrepresented with genes statistically associated with TRD
Table S5 MeSH terms significantly overrepresented with genes statistically associated with TRD
Acknowledgements
The authors extend their gratitude to Jeff Mohr and personnel of the Hybrid Turkeys pedigree farm (Kitchener, Canada) for collecting and providing data used in this study.
Data availability statement
Data that support the findings of this study are available from Hybrid Turkeys upon reasonable request and were used under license for the current study, and thus are not publicly available.
References
- Abdalla E.A., Peñagaricano F., Byrem T.M., Weigel K.A. & Rosa G.J.M. (2016) Genome‐wide association mapping and pathway analysis of leukosis incidence in a US Holstein cattle population. Animal Genetics 47, 395–407. [DOI] [PubMed] [Google Scholar]
- Aliagas E., Torrejón‐Escribano B., Lavoie E.G., De Aranda I.G., Sévigny J., Solsona C. & Martín‐Satué M. (2010) Changes in expression and activity levels of ecto‐5′‐nucleotidase/CD73 along the mouse female estrous cycle. Acta Physiologica 199, 191–7. [DOI] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A. et al (2000) Gene ontology: tool for the unification of biology. Nature Genetics 25, 25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer H., Véron N., Willert J. & Herrmann B.G. (2007) The t‐complex‐encoded guanine nucleotide exchange factor Fgd2 reveals that two opposing signaling pathways promote transmission ratio distortion in the mouse. Genes & Development 21, 143–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning B.L., Zhou Y. & Browning S.R. (2018) A one‐penny imputed genome from next‐generation reference panels. American Journal of Human Genetics 103, 338–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas J., Cañas‐Álvarez J.J., González‐Rodríguez A., Puig‐Oliveras A., Fina M., Piedrafita J., Molina A., Díaz C., Baró J.A. & Varona L. (2017) Bayesian analysis of parent‐specific transmission ratio distortion in seven Spanish beef cattle breeds. Animal Genetics 48, 93–6. [DOI] [PubMed] [Google Scholar]
- Casellas J., Gularte R.J., Farber C.R., Varona L., Mehrabian M., Schadt E.E., Lusis A.J., Attie A.D., Yandell B.S. & Medrano J.F. (2012) Genome scans for transmission ratio distortion regions in mice. Genetics 191, 247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas J., Id‐Lahoucine S., & Cánovas A. (In press). Discriminating between allele‐ and genotype‐specific transmission ratio distortion. Animal Genetics. 10.1111/age.13007 [DOI] [PubMed] [Google Scholar]
- Casellas J., Manunza A., Mercader A., Quintanilla R. & Amills M. (2014) A flexible Bayesian model for testing for transmission ratio distortion. Genetics 198, 1357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas J. & Medrano J.F. (2008) Within‐generation mutation variance for litter size in inbred mice. Genetics 179, 2147–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole J.B., Null D.J. & VanRaden P.M. (2016) Phenotypic and genetic effects of recessive haplotypes on yield, longevity, and fertility. Journal of Dairy Science 99, 7274–88. [DOI] [PubMed] [Google Scholar]
- Coletti M.H. & Bleich H.L. (2001) Medical subject headings used to search the biomedical literature. Journal of the American Medical Informatics Association 8, 317–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow J.F. (1999) Unmasking a cheating gene. Science 283, 1651–2. [DOI] [PubMed] [Google Scholar]
- Dalloul R.A., Long J.A., Zimin A.V. et al (2010) Multi‐platform next‐generation sequencing of the domestic Turkey (Meleagris gallopavo): genome assembly and analysis. PLoS Biology 8, e1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C., Yuanjun H.U., Yurui W.U. & Xiaoying L.I. (2019) Risk factors of death in newborns with congenital diaphragmatic hernia. Journal of Zhejiang University (Medical Science) 48, 83–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelbrock M.A., Kaliyaperumal S. & Williams K.J. (2009) DNA mismatch repair efficiency and fidelity are elevated during DNA synthesis in human cells. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 662, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emamgholi Begli H., Wood B.J., Abdalla E.A., Balzani A., Willems O., Schenkel F., Harlander‐Matauschek A. & Baes C.F. (2019) Genetic parameters for clutch and broodiness traits in turkeys (Meleagris gallopavo) and their relationship with body weight and egg production. Poultry Science 98, 6263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A., Jupe S., Matthews L. et al (2018) The reactome pathway knowledgebase. Nucleic Acids Research 46, D649–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman L. & McIntosh M. (2019) Standard deviations: the biological bases of transmission ratio distortion. Annual Review of Genetics 53, 347–72. [DOI] [PubMed] [Google Scholar]
- Gorlov I.P., Gorlova O.Y., Sunyaev S.R., Spitz M.R. & Amos C.I. (2008) Shifting paradigm of association studies: value of rare single‐nucleotide polymorphisms. American Journal of Human Genetics 82, 100–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granleese T., Clark S.A., Swan A.A. & van der Werf J.H.J. (2015) Increased genetic gains in sheep, beef and dairy breeding programs from using female reproductive technologies combined with optimal contribution selection and genomic breeding values. Genetics Selection Evolution 47, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarini A.R., Sargolzaei M., Brito L.F., Kroezen V., Lourenco D.A.L., Baes C.F., Miglior F., Cole J.B. & Schenkel F.S. (2019) Estimating the effect of the deleterious recessive haplotypes AH1 and AH2 on reproduction performance of Ayrshire cattle. Journal of Dairy Science 102, 5315–22. [DOI] [PubMed] [Google Scholar]
- Harrison M.M., Ceol C.J., Lu X. & Horvitz H.R. (2006) Some C. elegans class B synthetic multivulva proteins encode a conserved LIN‐35 Rb‐containing complex distinct from a NuRD‐like complex. Proceedings of the National Academy of Sciences of the United States of America, 103, 16782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings W.K. (1970) Monte Carlo sampling methods using Markov chains and their applications. Biometrika 57, 97–109. [Google Scholar]
- Hoff J.L., Decker J.E., Schnabel R.D. & Taylor J.F. (2017) Candidate lethal haplotypes and causal mutations in Angus cattle. BMC Genomics 18, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.O., Labbe A. & Infante‐Rivard C. (2013) Transmission ratio distortion: review of concept and implications for genetic association studies. Human Genetics 132, 245–63. [DOI] [PubMed] [Google Scholar]
- Huff G.R., Huff W.E., Balog J.M., Rath N.C., Anthony N.B. & Nestor K.E. (2005) Stress response differences and disease susceptibility reflected by heterophil to lymphocyte ratio in turkeys selected for increased body weight. Poultry Science 84, 709–17. [DOI] [PubMed] [Google Scholar]
- Id‐Lahoucine S., Cánovas A., Jaton C., Miglior F., Fonseca P., Miller S., Sargolzaei M., Schenkel F., Medrano F. & Casellas J. (2019) Implementation of Bayesian methods to identify SNP and haplotype regions with transmission ratio distortion across the whole genome: TRDscan v. 1.0. Journal of Dairy Science 102, 3175–88. [DOI] [PubMed] [Google Scholar]
- Id‐Lahoucine S. & Casellas J. (2017) Impact of incomplete pedigree data and independent culling level pre‐selection on the genetic evaluation of livestock: a simulation study on lamb growth. Livestock Science 198, 76–81. [Google Scholar]
- International Chicken Genome Sequencing Consortium (2004) Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716. [DOI] [PubMed] [Google Scholar]
- Jeffreys H. (1984) Theory of Probability. Oxford, UK: Clarendon Press. [Google Scholar]
- Jenko J., McClure M.C., Matthews D., McClure J., Johnsson M., Gorjanc G. & Hickey J.M. (2018) Analysis of a large data set reveals haplotypes carrying putatively recessive lethal alleles with pleiotropic effects on economically important traits in beef cattle. Genetics Selection Evolution, 51, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.A. & Taylor S.M. (1980) Cellular differentiation, cytidine analogs and DNA methylation. Cell 20, 85–93. [DOI] [PubMed] [Google Scholar]
- Karaca E., Harel T., Pehlivan D., Jhangiani S.N., Gambin T., Akdemir Z.C., Gonzaga‐Jauregui C., Erdin S., Bayram Y. & Campbell I.M. (2015) Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron 88, 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass R. & Raftery A. (1995) Bayes factors. Journal of the American Statistical Association 90, 773–95. [Google Scholar]
- McCright B., Gao X., Shen L., Lozier J., Lan Y., Maguire M., Herzlinger D., Weinmaster G., Jiang R., & Gridley T. (2001). Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development, 128(4), 491‐502. [DOI] [PubMed] [Google Scholar]
- Megens H.‐J., Crooijmans R.P.M.A., Bastiaansen J.W.M., Kerstens H.H.D., Coster A., Jalving R., Vereijken A., Silva P., Muir W.M. & Cheng H.H. (2009) Comparison of linkage disequilibrium and haplotype diversity on macro‐and microchromosomes in chicken. BMC Genetics 10, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H., Poudel S., Muruganujan A., Casagrande J.T. & Thomas P.D. (2016) PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Research 44(D1), D336–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson S.J., Schopen M., Savage A.G., Schulman J.‐L. & Arluk N. (2004) The MeSH translation maintenance system: structure, interface design, and implementation. Studies in Health Technology and Informatics 107(Pt 1), 67–9. [PubMed] [Google Scholar]
- Nicklas R.B. (1971) Mitosis In: Advances in Cell Biology (Ed. by Prescott D. M.), pp. 225–97. Boston, MA: Springer US. [DOI] [PubMed] [Google Scholar]
- Niu Y. & Liang S. (2009) Genetic differentiation within the inbred C57BL/6J mouse strain. Journal of Zoology 278, 42–7. [Google Scholar]
- Palombo F., Iaccarino I., Nakajima E., Ikejima M., Shimada T. & Jiricny J. (1996) hMutSbeta, a heterodimer of hMSH2 and hMSH3, binds to insertion/deletion loops in DNA. Current Biology 6, 1181–4. [DOI] [PubMed] [Google Scholar]
- Pardo‐Manuel De Villena F., De La Casa‐Esperó E., Briscoe T.L., Sapienza C. (2000) A genetic test to determine the origin of maternal transmission ratio distortion: meiotic drive at the mouse Om locus. Genetics, 154, 333–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson A.D., Waggott D., Schillert A., Infante‐Rivard C., Bull S.B., Yoo Y.J. & Pinnaduwage D. (2009) Transmission‐ratio distortion in the Framingham Heart Study. BMC Proceedings 3(S7), S51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G., Luo L., Siu H. et al (2010) Gene and pathway‐based second‐wave analysis of genome‐wide association studies. European Journal of Human Genetics 18, 111–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujadas E., Chen L. & Rodriguez F.J. (2019) Pathologic and molecular aspects of anaplasia in circumscribed gliomas and glioneuronal tumors. Brain Tumor Pathology 36, 40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qanbari S., Hansen M., Weigend S., Preisinger R. & Simianer H. (2010) Linkage disequilibrium reveals different demographic history in egg laying chickens. BMC Genetics 11, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y.S., Liang Y., Na Xia M., Shen X., Du Jun Y., Glong Luo C., Hua Nie Q., Zeng H. & Quan Zhang X. (2008) Extent of linkage disequilibrium in wild and domestic chicken populations. Hereditas 145, 251–7. [DOI] [PubMed] [Google Scholar]
- R Core Team (2019) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. [Google Scholar]
- Rodriguez F.J., Scheithauer B.W., Burger P.C., Jenkins S. & Giannini C. (2010) Anaplasia in pilocytic astrocytoma predicts aggressive behavior. The American Journal of Surgical Pathology 34, 147–60. [DOI] [PubMed] [Google Scholar]
- Saif Y.M. & Nestor K.E. (2002) Increased mortality in turkeys selected for increased body weight following vaccination with a live Newcastle disease virus vaccine. Avian Diseases 46, 505–8. [DOI] [PubMed] [Google Scholar]
- Sakabe K. & Okazaki R. (1966) A unique property of the replicating region of chromosomal DNA. Biochimica et Biophysica Acta 129, 651–4. [DOI] [PubMed] [Google Scholar]
- Seidel H.S., Ailion M., Li J., van Oudenaarden A., Rockman M.V. & Kruglyak L. (2011) A novel sperm‐delivered toxin causes late‐stage embryo lethality and transmission ratio distortion in C. elegans . PLoS Biology 9, e1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack J.M.W. (2007) Metaplasia and transdifferentiation: from pure biology to the clinic. Nature Reviews Molecular Cell Biology 8, 369–78. [DOI] [PubMed] [Google Scholar]
- Tamborrini D. & Piatti S. (2019) Septin clearance from the division site triggers cytokinesis in budding yeast. Microbial Cell 6, 296–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuyuzaki K., Morota G., Ishii M., Nakazato T., Miyazaki S. & Nikaido I. (2015) MeSH ORA framework: R/Bioconductor packages to support MeSH over‐representation analysis. BMC Bioinformatics 16, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRaden P.M., Olson K.M., Null D.J. & Hutchison J.L. (2011) Harmful recessive effects on fertility detected by absence of homozygous haplotypes. Journal of Dairy Science 94, 6153–61. [DOI] [PubMed] [Google Scholar]
- Warde‐Farley D., Donaldson S.L., Comes O. et al (2010) The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Research 38(Suppl. 2), W214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.‐J., Li J., Zou J.‐C., Liang F.‐S., Ye C.‐J., Jin D.‐M., Weng M.‐L. & Wang B. (2006) Cloning and characterization of a second form of the rice adenine phosphoribosyl transferase gene (OsAPT2) and its association with TGMS. Plant Molecular Biology 60, 365–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Biological process terms significantly overrepresented with genes statistically associated with TRD
Table S2 Cellular component function terms significantly overrepresented with genes statistically associated with TRD
Table S3 Molecular function terms significantly overrepresented with genes statistically associated with TRD
Table S4 Reactome pathways significantly overrepresented with genes statistically associated with TRD
Table S5 MeSH terms significantly overrepresented with genes statistically associated with TRD
Data Availability Statement
Data that support the findings of this study are available from Hybrid Turkeys upon reasonable request and were used under license for the current study, and thus are not publicly available.