

Abstract
Intermolecular hydroaminoalkylation reactions of propadiene with selected secondary amines take place in the presence of a 2,6‐bis(phenylamino)pyridinato titanium catalyst. The corresponding products, synthetically useful allylamines, are formed in convincing yields and with high selectivities. In addition, propadiene easily inserts into the titanium‐carbon bond of a titanaaziridine.
Keywords: allenes, amines, C−H activation, hydroaminoalkylation, titanium
Propadiene undergoes regioselective hydroaminoalkylation reactions with N‐aryl‐ and N‐alkyl‐substituted benzylamines in the presence of an aminopyridinato titanium catalyst. The corresponding products, synthetically useful allylamines, are formed in very good yields (up to 97 %) and with regioselectivities as high as 99:1.

Nitrogen‐containing compounds like secondary amines are on the forefront of pharmaceutical and agrochemical industries. [1] The most established and widely used method to synthesize amines is based on a two‐step strategy, namely hydroformylation followed by reductive amination, starting from alkenes. [2] Apart from the disadvantage that two transformations are necessary to isolate the target amines, the hydroformylation step is commonly catalyzed by late and increasingly rare and expensive transition metals like rhodium. [2] In recent years, the intermolecular hydroaminoalkylation of alkenes [3] enabled by transition metals of groups 4 [4] and 5 [4] has developed enormously into an efficient one‐step alternative with theoretically 100 % atom economy.[ 5 , 6 ] This reaction yields functionalized amines by a C−C bond forming process in the α‐position to an amine nitrogen atom, employing alkenes and secondary amines as starting materials. In this context, the titanium catalyzed variant is particularly noteworthy, given that titanium is the second most abundant transition metal in the Earth's crust. [7] Recent advances report both the successful use of industrially important small amines (e.g. dimethylamine) [8] and ethylene [9] in this reaction and the performance of this reaction under milder conditions and in significantly shorter reaction times. [10] Another highly desirable continuation in the field of hydroaminoalkylation reactions is the extension of the unsaturated substrate scope to compounds beyond alkenes to gain access to novel functionalized amine scaffolds. Herein, we report the intermolecular hydroaminoalkylation of propadiene, the simplest representative of the allene substance class, with secondary amines to yield corresponding allylamines in high chemoselectivities and yields. These allylamines themselves are very important building blocks for the synthesis of manifold natural products or pharmaceuticals, and the terminal double bond offers a variety of possible subsequent transformations. [11] In this context, it should be noted that a few examples of Rh/Ir‐catalyzed hydroaminoalkylation reactions of substituted allenes with a tertiary 1,2,3,4‐tetrahydroisoquinoline were recently reported by the Breit group. [12]
Previous work has demonstrated that the titanium‐catalyzed intermolecular hydroaminoalkylation of propadiene with N‐methylaniline (Scheme 1) did not lead to the formation of the envisaged homocoupling products, for example, II [branched (b)] or III [linear (l)]. Instead, the aminocyclopentane derivative I was formed from N‐methylaniline and two equivalents of propadiene in low yields. [13] The formation of I is presumably based on the generation of an allylic cation intermediate from the initially formed allylamine product (II, R1=Ph, R2=H) caused by the presence of the Lewis acidic titanium catalyst. [13]
Scheme 1.

Previous results on intermolecular hydroaminoalkylation reactions of propadiene with N‐methylaniline, [13] and targeted products II and III (this work).
As a starting point, the reaction between N‐methylaniline (1 a) and propadiene (2, synthesized according to a literature procedure[ 13 , 14 ] and used as a solution in toluene, c=0.4 molL−1) was chosen as the benchmark reaction and a catalyst screening was performed (Scheme 2, reaction conditions: 140 °C, 30 min, 10 mol % catalyst, scale: 0.1 mmol, control by GC analysis). [15] Due to propadiene's boiling point of −34 °C, the reactions were performed in sealed ampoules (V=5 mL). Unfortunately, the screening revealed that most of the investigated catalyst systems did not convert the starting materials into any products. The only active catalyst systems were found to be the formamidinato titanium systems V [4g] and VI. [10] However, in these cases, only the already reported formation of the aminocyclopentane derivative I could be verified. In this context, it should also be mentioned that the use of fewer equivalents of propadiene (2) did not inhibit the formation of I. A detailed summary of this initial catalyst screening can be found in the Supporting Information (Table S1).
Scheme 2.

Investigated intermolecular hydroaminoalkylation reaction between N‐methylaniline (1 a) and propadiene (2, top) and part of the investigated catalyst systems IV‐VI (bottom).
Since all investigated catalyst systems either did not catalyze the formation of one of the desired simple hydroaminoalkylation products (3 b or 3 l) or initiated unwanted subsequent reactions, the hypothesis was put forward as to whether the use of sterically more demanding secondary amine substrates could suppress the formation of by‐products. In this case, the interaction between Lewis‐acidic titanium species and the nitrogen center of the initially formed hydroaminoalkylation products should be suppressed and as a consequence, formation of allylic cations which initiate the unwanted side reactions should be avoided. For that purpose, N‐benzylaniline (1 b) was tested in reactions with propadiene (2) under the same reaction conditions and with the same scope of catalysts (Table S2). [15] Intriguingly, the use of the aminopyridinato based catalyst IVa, [4e] which was inactive in the corresponding reaction with N‐methylaniline (1 a), showed significant conversion of N‐benzylaniline (1 b) and formation of the desired coupling products 4 b and 4 l in a ratio of 93:7 (GC analysis, Figures S64 and S65) in favor of the branched product (Scheme 3). Noteworthy, in the case of sterically more demanding catalyst IVb, [4f] there is no conversion of the starting material 1 b (Figure S66). On the other hand, usage of the formamidinato based catalysts V and VI resulted in the conversion of 1 b but also in the formation of undesired by‐products (Figures S67 and S68), so that catalyst system IVa proved to be best for this transformation.
Scheme 3.

Intermolecular hydroaminoalkylation of propadiene (2) with N‐benzylaniline (1 b) catalyzed by IVa.
Subsequent optimization of the reaction conditions (Table S3) showed that the best results are obtained in sealed ampoules (V=5 mL) with 10 mol % catalyst loading (IVa), 140 °C, 4 h, and using 1.2 equivalents of propadiene (2, Figure S69). [15] Under these optimized reaction conditions, a mixture of the allylamines 4 b and 4 l could be isolated in very good yield of 92 % and with a selectivity of 4 b:4 l=93:7. [16] Because all screening and optimization experiments were carried out on a 0.1 mmol scale, an upscaled experiment (1 mmol) was conducted to isolate larger amounts of the products for characterization purposes. This experiment verified that both the branched product 4 b and the linear product 4 l are formed under the reaction conditions (Figures S3–S5) and that after column chromatography, the pure major product 4 b could be isolated in 87 % yield (Figure S70). In this case, prior to chromatography, the selectivity in favor of the branched isomer 4 b was determined by GC analysis to be 92:8 (Scheme 4).
Scheme 4.

Ti‐catalyzed synthesis of allylamines from propadiene (2) and N‐benzyl anilines. Reaction conditions: amine (1.0 mmol), propadiene (2, c=0.4 mol L−1 in toluene, 3.0 mL, 1.20 mmol), IVa (0.10 mmol, 10 mol %), 140 °C, 4 h, sealed ampoule. [a] The isolated product contained trace amounts of the linear isomer.
To improve reproducibility, we also investigated the sensitivity of the synthesis of N‐(2‐methyl‐1‐phenylallyl)aniline (4 b) to operational variation in reaction conditions (Figure 1). [17] The reaction turned out to be highly sensitive towards oxygen and moisture while light intensity, and scale have no influence on the reaction. The temperature has only a slight influence on the yield of the reaction.
Figure 1.
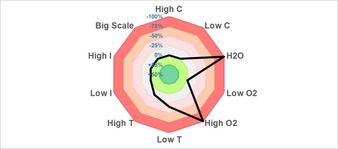
Sensitivity assessment of the synthesis of N‐(2‐methyl‐1‐phenylallyl)aniline (4 b, c=concentration, T=temperature, I=light intensity).
Subsequently, the scope of N‐benzyl anilines was investigated and the results are summarized in Scheme 4. During initial experiments with substrates that contain para‐ and meta‐substituents on the N‐phenyl ring, it was found that chloro (13 b), methyl (5 b, 6 b) and methoxy substitution (9 b, 10 b) is well tolerated. Notably, ortho‐substituted substrates did not show any reactivity which can be explained by steric hindrance. [4e] In addition, other pharmacologically promising substituents such as trifluoromethyl (12 b) or trifluoromethoxy groups (11 b) are also very well tolerated. The reaction was also successful in the presence of bromo substituents (7 b, 8 b) which offer various possibilities for further functionalization (e.g., Pd‐catalyzed cross coupling). [4n] Furthermore, we functionalized the N‐benzyl substituent. One limitation here is that again, a reaction with ortho‐substituted substrates is not possible while meta‐ and para‐substituents are well tolerated and in all corresponding cases, the products (14 b–21 b) were obtained in good to excellent yields. Notably, various pharmacologically interesting substructures such as trifluoromethyl (18 b) or fluoro (21 b) substituents could also be employed without any erosion in yield. In all cases, the hydroaminoalkylation reaction took place with very good to excellent regioselectivity in favour of the branched product.
With a reliable set of conditions for the hydroaminoalkylation of propadiene (2) with N‐benzyl anilines in hand, we wondered whether our protocol could also work with other amine substrates (Scheme 5). For that reason, we initially explored three benzylamines with sterically differently demanding N‐alkyl substituents which all reacted smoothly to give the desired methyl‐ (22 b), ethyl‐ (23 b) and isopropyl‐substituted (24 b) allylamine products in good yields (≥67 %) and with very good regioselectivity (≥94:6). In this context, it should be mentioned that these products could only be purified by distillation. We then turned our attention towards N‐alkyl anilines and found that in these cases, the hydroaminoalkylation reactions are significantly slower than with benzylamines. After the typical reaction time of 4 h, GC analysis revealed that the conversion of N‐isobutylaniline, N‐propylaniline, and N‐ethylaniline was unsatisfactory (Figures S71–S73) and even after 16 h (Figures S74–S76), only slightly better results were obtained. As a consequence, the isopropyl‐substituted product (27 b) could not be isolated in pure form while the methyl‐ (25 b) and the ethyl‐substituted product (26 b) were only be obtained in poor yields of 18 % and 23 %, respectively. Interestingly, 1,2,3,4‐tetrahydroquinoline was found to give a mixture of the expected branched product 28 b and the linear product 28 l in a surprisingly low ratio of only 59:41. In this case, the combined yield was 21 %. Additional experiments performed with dialkylamines like N‐methylcyclohexylamine or N‐ethylcyclohexylamine were not successful due to poor conversion of the starting materials as well as the formation of a number of side products.
Scheme 5.

Ti‐catalyzed hydroaminoalkylation reactions of propadiene (2) with N‐alkyl benzylamines or N‐alkyl anilines. Reaction conditions: amine (1.0 mmol), propadiene (2, c=0.4 mol L−1 in toluene, 3.0 mL, 1.20 mmol), IVa (0.10 mmol, 10 mol %), 140 °C, 4 h or 16 h, sealed ampoule. [a] 4 h. [b] 16 h.
From a mechanistical point of view, the migratory insertion of a C−C multiple bond of an unsaturated substrate into the Ti−C bond of a titanaaziridine (formed by N−H and C−H activation of the secondary amine) is generally accepted to be the C−C bond forming step of the hydroaminoalkylation reaction. To get insights into the possibility of a migratory insertion reaction of propadiene (2) into the Ti−C bond of a titanaaziridine, the reaction of the model titanaaziridine 29 with 2 was performed (Scheme 6), which smoothly gave access to the expected titanapyrrolidines 30 and 31 in 73 % combined yield. Titanaaziridines of type 29 are known to lack any catalytic activity in hydroaminoalkylation reactions and are therefore perfectly suited for the synthesis and characterization of stable five‐membered intermediates of the expected catalytic cycle.[ 9 , 18 ] Due to similar solubility properties of 30 and 31 in polar and non‐polar solvents (e.g., n‐hexane, tetrahydrofuran, benzene), 30 and 31 could not be separated. However, results obtained from multinuclear and 2D NMR spectroscopy unambiguously allowed for the assignment of the connectivity (Figures S60–S63). The NMR‐studies also revealed that the titanapyrrolidines 30 and 31 are surprisingly formed in a ratio of 10:1 in favor of 30 which corresponds to a linear hydroaminoalkylation product. This finding is in sharp contrast to the preferred formation of the branched products in the catalytic hydroaminoalkylation reactions described above. Unfortunately, at the moment, we do not have a plausible explanation for the different behavior of propadiene (2) in the catalytic and stoichiometric experiments. However, the reaction in Scheme 6 was performed at room temperature, while the catalytic process occurs at 140 °C and as a consequence, it is possible that this is a kinetic versus thermodynamic product issue. Furthermore, titanaaziridine 29 is not an active hydroaminoalkylation catalyst and the selectivity preferences for the insertion reaction may differ from catalyst IVa. In this context, it should also be noted that the insertion of 1‐hexene into the Ti−C bond of an analogous titanaaziridine has already been reported to give a titanapyrrolidine at room temperature that corresponds to a branched hydroaminoalkylation product. [18] Obviously, the reactivity of allenes towards titanaaziridines differs dramatically from the reactivity of alkenes. While on the one hand, this finding can serve as an explanation for the fact that titanium‐catalyzed hydroaminoalkylation reactions of allenes are extremely rare in the literature, [13] on the other hand, it becomes clear that a lot of additional, future studies with various allenes are required to understand the differences between allenes and alkenes in hydroaminoalkylation chemistry. Noteworthy, attempts to release the hydroaminoalkylation products from the mixture of the titanapyrrolidines 30 and 31 by simple hydrolysis (wet CH2Cl2, rt, 1 h) or heating failed. This finding is again in sharp contrast to a recent study with a titanapyrrolidine obtained from 29 and ethylene from which the hydroaminoalkylation product could easily be released in the presence of wet CH2Cl2. [9]
Scheme 6.

Migratory insertion of propadiene (2) into the Ti−C bond of titanaaziridine 29 giving the corresponding titanapyrrolidines 30 and 31.
In summary, successful intermolecular hydroaminoalkylation reactions of propadiene with secondary amines giving access to allylamines could be achieved for the first time. While best results were obtained with various N‐aryl‐ and N‐alkyl‐substituted benzylamines using the aminopyridinato titanium catalyst IVa, dialkylamines, N‐alkyl anilines and especially N‐methylaniline turned out to be poor substrates. Additional studies with substituted allenes which also undergo successful intermolecular hydroaminoalkylation reactions are presently underway in our laboratories. These results will be published in due course.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Research Training Group “Chemical Bond Activation” (GRK 2226) funded by the Deutsche Forschungsgemeinschaft and the Heinz Neumüller Stiftung for financial support of this project. We also thank Jessica Reimer for experimental assistance and Kirstin Glaser, Karin Grittner, and Frank Fleischer for supplying the ampoules. Open access funding enabled and organized by Projekt DEAL.
T. Kaper, M. Fischer, M. Warsitz, R. Zimmering, R. Beckhaus, S. Doye, Chem. Eur. J. 2020, 26, 14300.
Contributor Information
Prof. Dr. Ruediger Beckhaus, Email: ruediger.beckhaus@uni-oldenburg.de.
Prof. Dr. Sven Doye, Email: doye@uni-oldenburg.de.
References
- 1. Salvatore R. N., Yoon C. H., Jung K. W., Tetrahedron 2001, 57, 7785–7811. [Google Scholar]
- 2.For selected examples, see:
- 2a. Franke R., Selent D., Börner A., Chem. Rev. 2012, 112, 5675–5732; [DOI] [PubMed] [Google Scholar]
- 2b. Clarker M. L., Hydroformylation, Vol. 2, Wiley-VCH, Weinheim, 2016. [Google Scholar]
- 3.For reviews on hydroaminoalkylation reactions of alkenes, see:
- 3a. Chong E., Garcia P., Schafer L. L., Synthesis 2014, 46, 2884–2896; [Google Scholar]
- 3b. Dong Z., Ren Z., Thompson S. J., Xu Y., Dong G., Chem. Rev. 2017, 117, 9333–9403; [DOI] [PubMed] [Google Scholar]
- 3c. Edwards P. M., Schafer L. L., Chem. Commun. 2018, 54, 12543–12560. [DOI] [PubMed] [Google Scholar]
- 4.For selected examples of group 4 and 5 catalysts, see:
- 4a. Herzon S. B., Hartwig J. F., J. Am. Chem. Soc. 2007, 129, 6690–6691; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Kubiak R., Prochnow I., Doye S., Angew. Chem. Int. Ed. 2009, 48, 1153–1156; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1173–1176; [Google Scholar]
- 4c. Prochnow I., Zark P., Müller T., Doye S., Angew. Chem. Int. Ed. 2011, 50, 6401–6405; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6525–6529; [Google Scholar]
- 4d. Dörfler J., Doye S., Angew. Chem. Int. Ed. 2013, 52, 1806–1809; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1851–1854; [Google Scholar]
- 4e. Dörfler J., Preuß T., Schischko A., Schmidtmann M., Doye S., Angew. Chem. Int. Ed. 2014, 53, 7918–7922; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8052–8056; [Google Scholar]
- 4f. Lühning L. H., Brahms C., Nimoth J. P., Schmidtmann M., Doye S., Z. Anorg. Allg. Chem. 2015, 641, 2071–2082; [Google Scholar]
- 4g. Dörfler J., Preuß T., Brahms C., Scheuer D., Doye S., Dalton Trans. 2015, 44, 12149–12168; [DOI] [PubMed] [Google Scholar]
- 4h. Hamzaoui B., Pelletier J. D. A., El Eter M., Chen Y., Abou-Hamad E., Basset J. M., Adv. Synth. Catal. 2015, 357, 3148–3154; [Google Scholar]
- 4i. Braun C., Bräse S., Schafer L. L., Eur. J. Org. Chem. 2017, 1760–1764; [Google Scholar]
- 4j. DiPucchio R. C., Roşca S.-C., Schafer L. L., Angew. Chem. Int. Ed. 2018, 57, 3469–3472; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3527–3530; [Google Scholar]
- 4k. DiPucchio R. C., Roşca S.-C., Athavan G., Schafer L. L., ChemCatChem 2019, 11, 3871–3876; [Google Scholar]
- 4l. Koperniku A., Foth P. J., Sammis G. M., Schafer L. L., J. Am. Chem. Soc. 2019, 141, 18944–18948; [DOI] [PubMed] [Google Scholar]
- 4m. Perry M. R., Gilmour D. J., Schafer L. L., Dalton Trans. 2019, 48, 9782–9790; [DOI] [PubMed] [Google Scholar]
- 4n. Kaper T., Doye S., Tetrahedron 2019, 75, 4343–4350; [Google Scholar]
- 4o. Zi G., Zhang F., Song H., Chem. Commun. 2010, 46, 6296–6298; [DOI] [PubMed] [Google Scholar]
- 4p. Reznichenko A. L., Emge T. J., Audörsch S., Klauber E. G., Hultzsch K. C., Schmidt B., Organometallics 2011, 30, 921–924; [Google Scholar]
- 4q. Reznichenko A. L., Hultzsch K. C., J. Am. Chem. Soc. 2012, 134, 3300–3311. [DOI] [PubMed] [Google Scholar]
- 5.For selected examples of late transition metal catalysts, see:
- 5a. Schmitt D. C., Lee J., Dechert-Schmitt A.-M. R., Yamaguchi E., Krische M. J., Chem. Commun. 2013, 49, 6096–6098; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Lahm G., Opatz T., Org. Lett. 2014, 16, 4201–4203; [DOI] [PubMed] [Google Scholar]
- 5c. Chen T.-Y., Tsutsumi R., Montgomery T. P., Volchkov I., Krische M. J., J. Am. Chem. Soc. 2015, 137, 1798–1801; [DOI] [PubMed] [Google Scholar]
- 5d. Thullen S. M., Rovis T., J. Am. Chem. Soc. 2017, 139, 15504–15508. [DOI] [PubMed] [Google Scholar]
- 6.For selected examples of scandium-catalyzed alkene hydroaminoalkylation with tertiary amines, see:
- 6a. Nako A. E., Oyamada J., Nishiura M., Hou Z., Chem. Sci. 2016, 7, 6429–6434; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Liu F., Luo G., Hou Z., Luo Y., Organometallics 2017, 36, 1557–1565; [Google Scholar]
- 6c. Gao H., Su J., Xu P., Xu X., Org. Chem. Front. 2018, 5, 59–63. [Google Scholar]
- 7.
- 7a. Wedepohl K. H., Geochim. Cosmochim. Acta 1995, 59, 1217–1232; [Google Scholar]
- 7b. Hunt A. J., Farmer T. F., Clark J. H. in Element Recovery and Sustainability (Ed.: Hunt A. J.), Royal Society of Chemistry, Cambridge, 2013, pp. 1–28; [Google Scholar]
- 7c. Hunt A. J., Farmer T. J. in Sustainable Catalysis: With Nonendangered Metals, Part 1, (Ed.: North M.), Royal Society of Chemistry, Cambridge, 2015, pp. 1–14. [Google Scholar]
- 8. Bielefeld J., Doye S., Angew. Chem. Int. Ed. 2017, 56, 15155–15158; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15352–15355. [Google Scholar]
- 9. Rosien M., Töben I., Schmidtmann M., Beckhaus R., Doye S., Chem. Eur. J. 2020, 26, 2138–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bielefeld J., Doye S., Angew. Chem. Int. Ed. 2020, 59, 6138–6143; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 6194–6199. [Google Scholar]
- 11.For selected recent examples of allylamines as starting materials, see:
- 11a. Yu H., Zhang G., Huang H., Angew. Chem. Int. Ed. 2015, 54, 10912–10916; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11062–11066; [Google Scholar]
- 11b. Wu Z., Laffoon S. D., Nguyen T. T., McAlpin J. D., Hull K. L., Angew. Chem. Int. Ed. 2017, 56, 1371–1375; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1391–1395; [Google Scholar]
- 11c. Vanable E. P., Kennemur J. L., Joyce L. A., Ruck R. T., Schultz D. M., Hull K. L., J. Am. Chem. Soc. 2019, 141, 739–742; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Fan C., Lv X.-Y., Xiao L.-J., Xie J.-H., Zhou Q.-L., J. Am. Chem. Soc. 2019, 141, 2889–2893. [DOI] [PubMed] [Google Scholar]
- 12. Zheng J., Breit B., Angew. Chem. Int. Ed. 2019, 58, 3392–3397; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3430–3435. [Google Scholar]
- 13. Bielefeld J., Mannhaupt S., Schmidtmann M., Doye S., Synlett 2019, 30, 967–971. [Google Scholar]
- 14. Cabezas J. A., Poveda R. R., Brenes J. A., Synthesis 2018, 50, 3307–3321. [Google Scholar]
- 15.For details, see the Supporting Information.
- 16.A control experiment performed under identical conditions in a Schlenk tube (V=80 mL) equipped with a Teflon stopcock did not show any conversion of the starting materials.
- 17. Pitzer L., Schäfers F., Glorius F., Angew. Chem. Int. Ed. 2019, 58, 8572–8576; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8660–8664. [Google Scholar]
- 18. Manßen M., Lauterbach N., Dörfler J., Schmidtmann M., Saak W., Doye S., Beckhaus R., Angew. Chem. Int. Ed. 2015, 54, 4383–4387; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4458–4462. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary