

Abstract
Introduction
Local DNA hypermethylation is a potential source of cancer biomarkers. While the evaluation of single gene methylation has limited value, their selected panel may provide better information.
Aims
This study aimed to analyze the promoter methylation level in a 7‐gene panel in brain tumors and verifies the usefulness of methylation‐sensitive high‐resolution melting (MS‐HRM) for this purpose.
Methods
Forty‐six glioma samples and one non‐neoplastic brain sample were analyzed by MS‐HRM in terms of SFRP1, SFRP2, RUNX3, CBLN4, INA, MGMT, and RASSF1A promoter methylation. The results were correlated with patients’ clinicopathological features.
Results
DNA methylation level of all analyzed genes was significantly higher in brain tumor samples as compared to non‐neoplastic brain and commercial, unmethylated DNA control. RASSF1A was the most frequently methylated gene, with statistically significant differences depending on the tumor WHO grade. Higher MGMT methylation levels were observed in females, whereas the levels of SFRP1 and INA promoter methylation significantly increased with patients’ age. A positive correlation of promoter methylation levels was observed between pairs of genes, for example, CBLN4 and INA or MGMT and RASSF1A.
Conclusions
Our 7‐gene panel of promoter methylation can be helpful in brain tumor diagnosis or characterization, and MS‐HRM is a suitable method for its analysis.
Keywords: biomarker, DNA methylation, gliomas, MS‐HRM
Promoter methylation of a 7‐gene panel can be helpful in brain tumor diagnosis or characterization, and MS‐HRM is a suitable method for its analysis.
![]()
1. INTRODUCTION
Aberrant DNA methylation is one of the hallmarks of cancer. Hypermethylation of tumor suppressor gene promoter sequences, as well as global DNA hypomethylation, characterizes all tumors, including CNS cancers. Distinctive epigenetic changes alter gene expression, contributing to disease pathogenesis, but can also be helpful in diagnosis and prediction of prognosis or treatment response. The updated 2016 World Health Organization (WHO) classification of CNS tumors indicated that beyond clinical and symptom‐based examinations, also molecular characterization analysis should be applied. 1 It did not include epigenetic tumor characteristics yet, but recent data presented by Capper et al show that classification of CNS tumors based on DNA methylation profiling results in the revision of the initial histopathological diagnosis in 12% of cases (of 2,801 reference samples analyzed). 2 Moreover, DNA methylation analysis can also serve as a prognostic indicator. In this context, G‐CIMP (Glioma CpG island methylator phenotype)–positive phenotype correlates with the presence of IDH mutation and is associated with a good prognosis in GBM (glioblastoma multiforme). 3 Eventually, methylation biomarkers can also predict the response to specific therapy and guide patients’ treatment. Multiple large‐scale clinical studies proved that GBM patients with O6‐methylguanine‐DNA methyltransferase (MGMT) promoter hypermethylation benefit from alkylating agent therapy, especially temozolomide (TMZ). 4 , 5 , 6 , 7 Thus, MGMT is now the most frequently used methylation‐associated biomarker in neuro‐oncology. However, to date, there is no consensus on the optimal method for the detection of MGMT as well as other DNA methylation biomarkers in the clinical setting. 8
There is a variety of analytical methods for DNA methylation analysis, allowing both whole‐genome methylation profiling and locus‐specific DNA methylation assays. The first approach mentioned is often used in the discovery phase allowing the identification of differentially methylated regions and their relevance to the disease. On the other hand, the second approach—locus‐specific DNA methylation analysis, is an optimal technique in the clinics, since most of the established biomarkers rely on DNA methylation differences in the limited number of CpG dinucleotides. 9 Overall, the optimal DNA methylation detection method should be sensitive, specific, quick, cost‐effective, and suitable for screening of large sets of clinical samples. 10 Methylation‐sensitive high‐resolution melting (MS‐HRM) has all of these features. 11 , 12 , 13
In this method, establishing the DNA methylation level of a particular sequence is based on the differences in the melting profiles of its PCR amplicon. Bisulfite treatment, preceding PCR amplification, creates the difference in the nucleotide sequence corresponding to the presence or absence of methyl groups. In this regard, unmethylated cytosine is oxidatively deaminated to uracyl (read as thymine during PCR), whereas methylated cytosine remains cytosine. Cytosine‐guanine pair melts in higher temperatures, as compared to the adenine‐thymine pair, resulting in markedly different melting profiles. Due to its simplicity and high reproducibility, MS‐HRM is now gaining importance both in screening and in determining new molecular biomarkers. 13 , 14
The DNA methylation studies in CNS neoplasms usually concentrate on single genes 15 or evaluate methylomes or methylation profiles using microarrays. 16 The first approach carries a disadvantage, as cancer is not a single gene disease. 17 The second methodology provides an enormous amount of data that create a complex picture of a disease, but fail to serve as a simple test. 2 , 18 , 19 , 20 The golden mean would be defining a set of genes crucial for diagnostic and therapeutic purposes. Thus, the aim of this study was to explore the biomarker potential of the promoter methylation level of a 7‐gene panel that we propose for CNS cancer detection and characterization. In this regard, the obtained data were correlated with patients’ clinicopathological features such as gender, age, histological tumor type, and WHO grade, as well as overall survival time (OS). The idea behind the gene set was to represent the crucial processes disturbed in gliomagenesis, that is, signal transduction (Wnt/β‐catenin pathway) or neuronal development and organization. In the panel, we included SFRP1, SFRP2, RUNX3, CBLN4, INA, MGMT, and RASSF1A. The choice of genes was determined based on the established or potential role of promoter methylation of these genes in glioma detection and patients' management. SFRP1, SFRP2, and RUNX3 are negative regulators of the Wnt/β‐catenin pathway. Secreted frizzled‐related protein (SFRP) genes, SFRP1 and SFRP2, belong to the secreted glycoprotein family. Epigenetic silencing of SFRP1 may cause dysregulation of cell proliferation, migration, and invasion, which leads to cancer cell formation, disease progression, poor prognosis, and treatment resistance. 21 SFRP2 is regarded as the most potent antagonist of Wnt signaling. However, SFRP2 can also exert an angiogenic effect in renal and lung cancer. 22 Runt‐related transcription factor functions as a tumor suppressor, and the gene (RUNX3) is frequently deleted or transcriptionally silenced in cancer. 23 , 24 Its inactivation is frequently caused by promoter methylation. 21 , 25 , 26 Previously, we found that RUNX3 methylation correlates with WHO tumor grade and glioma patients’ age. 27 , 28 Moreover, SFRP1 methylation predicts shorter survival of patients with gliomas. 26 INA (internexin neuronal intermediate filament protein α) is a major component of the intermediate filament network in the cytoplasm of small interneurons and cerebellar granule cells and plays a role in neuronal development. 29 It is implicated to contribute to neurodegenerative disorders. 29 INA is overexpressed mostly in oligodendroglial phenotype gliomas and is related to 1p/19q codeletion with >70% specificity. Therefore, it is a favorable prognostic marker. 30 Recently, a set of CpG loci differentially hypermethylated in GBM short‐term survivors (overall survival < 1 year) vs. long‐term survivors (overall survival > 3 years) was identified. 31 According to this report, methylation of INA was one of the top hypermethylated loci and indicated short‐term survival. Another differentially hypermethylated gene was CBLN4 (cerebellin 4 precursor), a trans‐synaptic cell adhesion molecule, which is important for the synaptic organization of specific subsets of neurons 32 and it was not previously linked to brain tumors or other cancers. 6 MGMT is already established as a predictive factor in patients with GBM treated with temozolomide (TMZ). 4 , 33 It encodes O6‐methylguanine‐DNA methyltransferase, which acts by removing alkyl adducts from the O6 position of guanine at DNA level, thus antagonizing the cytotoxic effects of alkylating agents, including TMZ, gold standard anti‐GBM therapeutic. 7 Recently, also the prognostic value of that marker was established, making it a great descriptor of a link between disease character and therapy response. 16 Loss or altered expression of RASSF1A, Ras association domain family 1 isoform A encoding gene, has been associated with the pathogenesis of a variety of cancers. 34 In our previous study, hypermethylation of RASSF1A analyzed in circulating tumor‐derived DNA differentiated primary from metastatic brain cancers. 35 The summary of the proposed panel characteristics is presented in Table S1.
2. METHODS
2.1. Sample characteristics
This study was performed on brain glioma samples obtained from 46 patients during surgery at the Department of Neurosurgery and Neurotraumatology, Poznan University of Medical Sciences, and on one non‐neoplastic brain tissue. Patients’ age ranged from 16 to 83 years, whereas the largest subgroup consisted of patients within the age range of 51‐60 years (18 subjects). The median age of patients at the time of tumor diagnosis was 50.8 ± 15.5 years. In the group, there were 27 (58.7%) males and 19 (41.3%) females. The most abundant histological subgroups were as follows: anaplastic astrocytomas (13 patients—28.3%, 2 of them with recurrent tumors), and glioblastomas (19 patients—41.3%, 7 of them with recurrent tumors). Single cases represented other histological types. Twenty patients were described as WHO grade IV, 19—grade III, 5—grade II, and 2—grade I. The histological types and grades (from I—highly differentiated, least malignant to IV—low differentiated, most malignant), age, and gender are shown in Table 1.
TABLE 1.
The list of patients with brain gliomas evaluated in the present study with their basic characteristics (histopathological diagnosis, WHO grade, gender, age, and overall survival time), as well as promoter DNA methylation of analyzed genes (SFRP1, SFRP2, RUNX3, CBLN4, INA, MGMT, RASSF1A)
| Indication | |||||||
|---|---|---|---|---|---|---|---|
| Methylation level [%] | 0 | >0, ≤5 | >5, ≤10 | >10, ≤25 | >25, ≤50 | >50, ≤75 | >75, ≤100 |
| No | Histopathological classification | WHO grade | Gender | Age [years] | Overall survival [months] | SFRP1 | SFRP2 | RUNX3 | CBLN4 | INA | MGMT | RASSF1A |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GBM | IV | M | 60 | 8 | >10, ≤25% | >0, ≤5% | >0, ≤5% | >0, ≤5% | >0, ≤5% | 0% | >10, ≤25% |
| 2 | GBM | IV | M | 58 | 17 | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| 3 | GBM | IV | M | 58 | 10 | >0, ≤5% | >0, ≤5% | 0% | 0% | 0% | 0% | 0% |
| 4 | GBM | IV | M | 58 | 9 | >10, ≤25% | >0, ≤5% | >0, ≤5% | >0, ≤5% | 0% | 0% | >10, ≤25% |
| 5 | GBM | IV | M | 56 | nk | >10, ≤25% | 0% | 0% | >0, ≤5% | >5, ≤10% | 0% | >10, ≤25% |
| 6 | GBM | IV | M | 54 | 16 | >25, ≤50% | >0, ≤5% | 0% | >0, ≤5% | 0% | 0% | 0% |
| 7 | GBM | IV | M | 47 | 10 | 0% | >0, ≤5% | >0, ≤5% | 0% | 0% | 0% | >0, ≤5% |
| 8 | GBM | IV | M | 45 | 21 | 0% | nk | 0% | 0% | 0% | 0% | 0% |
| 9 | GBM | IV | M | 41 | nk | >25, ≤50% | 0% | 0% | >0, ≤5% | 0% | 0% | 0% |
| 10 | GBM | IV | F | 70 | 3 | >25, ≤50% | 0% | 0% | 0% | 0% | >25, ≤50% | 0% |
| 11 | GBM | IV | F | 65 | nk | >10, ≤25% | 0% | 0% | 0% | >0, ≤5% | 0% | >25, ≤50% |
| 12 | GBM | IV | F | 60 | 36 | >25, ≤50% | 0% | 0% | >10, ≤25% | >0, ≤5% | >10, ≤25% | >25, ≤50% |
| 13 | GBM | IV | F | 52 | nk | >0, ≤5% | >0, ≤5% | 0% | 0% | 0% | >10, ≤25% | 0% |
| 14 | GBM recidivans | IV | M | 31 | 8 | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| 15 | GBM recidivans | IV | M | 31 | 8 | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| 16 | GBM recidivans | IV | F | 64 | nk | >10, ≤25% | 0% | >10, ≤25% | 0% | 0% | 0% | 0% |
| 17 | GBM recidivans | IV | F | 60 | nk | 0% | 0% | 0% | 0% | 0% | >0, ≤5% | >0, ≤5% |
| 18 | GBM recidivans | IV | F | 59 | 4 | 0% | >0, ≤5% | 0% | 0% | >0, ≤5% | 0% | 0% |
| 19 | GBM recidivans | IV | F | 50 | nk | >10, ≤25% | 0% | 0% | 0% | 0% | >5, ≤10% | 0% |
| 20 | Gliosarcoma | IV | F | 75 | 10 | >10, ≤25% | >5, ≤10% | >25, ≤50% | >0, ≤5% | >0, ≤5% | >10, ≤25% | >0, ≤5% |
| 21 | Anaplastic astrocytoma | III | M | 83 | nk | >25, ≤50% | >0, ≤5% | 0% | >0, ≤5% | >0, ≤5% | >10, ≤25% | >10, ≤25% |
| 22 | Anaplastic astrocytoma | III | M | 60 | 10 | 0% | >0, ≤5% | 0% | 0% | >0, ≤5% | >25, ≤50% | >10, ≤25% |
| 23 | Anaplastic astrocytoma | III | M | 59 | nk | >10, ≤25% | >0, ≤5% | >5, ≤10% | >0, ≤5% | >0, ≤5% | 0% | 0% |
| 24 | Anaplastic astrocytoma | III | M | 58 | 11 | 0% | 0% | >5, ≤10% | 0% | 0% | 0% | >0, ≤5% |
| 25 | Anaplastic astrocytoma | III | M | 45 | nk | >5, ≤10% | >0, ≤5% | >5, ≤10% | >0, ≤5% | >0, ≤5% | 0% | >10, ≤25% |
| 26 | Anaplastic astrocytoma | III | M | 29 | 18 | >25, ≤50% | >0, ≤5% | >0, ≤5% | >0, ≤5% | 0% | 0% | >5, ≤10% |
| 27 | Anaplastic astrocytoma | III | F | 73 | nk | >25, ≤50% | >5, ≤10% | >10, ≤25% | >0, ≤5% | >10, ≤25% | >50, ≤75% | >0, ≤5% |
| 28 | Anaplastic astrocytoma | III | F | 66 | nk | >10, ≤25% | >0, ≤5% | >5, ≤10% | 0% | >0, ≤5% | 0% | 0% |
| 29 | Anaplastic astrocytoma | III | F | 48 | nk | 0% | >0, ≤5% | >0, ≤5% | >0, ≤5% | >0, ≤5% | 0% | 0% |
| 30 | Anaplastic astrocytoma | III | F | 48 | nk | 0% | 0% | 0% | >0, ≤5% | >0, ≤5% | >0, ≤5% | 0% |
| 31 | Anaplastic astrocytoma | III | F | 41 | 42 | 0% | 0% | 0% | 0% | 0% | >10, ≤25% | >50, ≤75% |
| 32 | Anaplastic astrocytoma recidivans | III | M | 53 | nk | 0% | >0, ≤5% | 0% | 0% | 0% | 0% | >25, ≤50% |
| 33 | Anaplastic astrocytoma recidivans | III | F | 47 | 8.5 | >25, ≤50% | 0% | 0% | >0, ≤5% | 0% | >10, ≤25% | >50, ≤75% |
| 34 | Astrocytoma gemistocyticum in astrocytoma angioplasticum vertens | III | F | 28 | nk | 0% | 0% | 0% | 0% | 0% | >10, ≤25% | >25, ≤50% |
| 35 | Glioma anaplasticum NOS | III | M | 58 | 23 | 0% | 0% | 0% | 0% | 0% | >10, ≤25% | >25, ≤50% |
| 36 | Glioma anaplasticum recidivans | III | M | 59 | 3 | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| 37 | Oligoastrocytoma anaplasticum | III | M | 29 | 23 | >25, ≤50% | 0% | 0% | 0% | 0% | >10, ≤25% | >50, ≤75% |
| 38 | Oligoastrocytoma anaplasticum | III | F | 77 | 3.5 | >25, ≤50% | 0% | 0% | >0, ≤5% | >0, ≤5% | 0% | >25, ≤50% |
| 39 | Clear cell ependymoma | III | F | 19 | 3 | 0% | >0, ≤5% | 0% | 0% | 0% | 0% | >25, ≤50% |
| 40 | Astrocytoma fibrillare partim protoplasmaticum | II | M | 51 | 12.5 | >10, ≤25% | >0, ≤5% | 0% | >0, ≤5% | 0% | >10, ≤25% | >50, ≤75% |
| 41 | Astrocytoma fibrillare partim gemistocyticum | II | M | 37 | nk | >10, ≤25% | 0% | 0% | 0% | 0% | >0, ≤5% | >50, ≤75% |
| 42 | Astrocytoma fibrillare | II | F | 38 | 18 | >10, ≤25% | 0% | >0, ≤5% | nk | nk | >0, ≤5% | >10, ≤25% |
| 43 | Oligodendroglioma isomorphum | II | M | 59 | 6 | >25, ≤50% | >0, ≤5% | >5, ≤10% | 0% | 0% | >10, ≤25% | >50, ≤75% |
| 44 | Ganglioglioma | II | M | 37 | nk | >25, ≤50% | nk | >0, ≤5% | 0% | 0% | >10, ≤25% | >25, ≤50% |
| 45 | Subependymal giant cell astrocytoma | I | M | 23 | 24 | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| 46 | Ganglioglioma | I | M | 16 | nk | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
The percentage of methylation is expressed with different color intensity (lowest DNA methylation level—brightest color, highest—the darkest one). Indication "nk", not known—indicates samples for which the results were not obtained or were ambiguous.
Tumor samples were collected between January 2010 and September 2013, and they were stored at −80°C. The samples were evaluated at the Laboratory of Neuropathology and grouped according to the histological type and the 2007 WHO classification criteria. Overall survival (OS) time was known for more than half of the patients. All the patients gave informed consent for the analyses to be undertaken, and the study protocol was approved by the Clinical Research Ethics Committee (number 505/12).
2.2. DNA isolation and bisulfite conversion
GeneMATRIX Tissue DNA Purification Kit (EurX, Gdańsk, Poland) was used for DNA isolation. DNA concentration and purity were verified using NanoDrop spectrophotometer. Bisulfite conversion of 500 ng of genomic DNA was performed using EZ DNA Methylation Kit (Zymo Research, USA), following the manufacturer's protocol. The elution volume after bisulfite conversion was 10 µL, and 1 µL of each converted DNA was taken for the subsequent MS‐HRM analysis.
2.3. Primer sequences and design
Data on primers for SFRP1, SFRP2, RUNX3, MGMT, and RASSF1A were taken from the literature, 36 , 37 , 38 , 39 , 40 whereas those for INA and CBLN4 were designed using Methyl Primer Express v 1.0 software (Applied Biosystems) (Figure S1). The synthesis of primers took place at the Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw, Poland. Their sequences, together with the annealing temperature, number of CpGs evaluated, and amplicon size, are presented in Table [Link], [Link]. Primer design was performed according to the guidelines of Wojdacz et al 39 and recommendations provided by Applied Biosystems. 41 Optimal annealing temperatures (60‐65℃), and optimal amplicon length (100‐200 bp), as well as the recommended number of potential methylation sites in the amplicons (3‐22), were considered during primer design. 42 , 43 , 44 Each primer of the two primer pairs also contained 1 CpG site, to increase the binding to methylated DNA and to overcome the PCR bias.
2.4. Standards
CpG Methylated HeLa Genomic DNA (New England Biolabs, USA) and CpGenome Universal Unmethylated DNA Set (Merck, Germany) of equal concentration were mixed in different ratios (0%, 5%, 10%, 25%, 50%, 75%, 100% methylated DNA) to mimic DNA samples with defined levels of DNA methylation. These standards were used for the evaluation of the sensitivity of the assay and the semi‐quantitative estimation of gene promoter methylation in the clinical samples. The assays were optimized in terms of primer annealing temperature to obtain the best possible resolution and the highest sensitivity.
Moreover, the whole‐genome amplified DNA from pooled peripheral blood lymphocytes was prepared with GenomePlex® Whole Genome Amplification Kit (Merck, Germany), according to manufacturer's protocol, and was used for each run.
2.5. MS‐HRM analysis
MS‐HRM analysis was performed using Light Cycler® 96 (Roche Diagnostics GmbH, Germany). Bisulfite‐converted DNA was amplified using 5x Hot FIREPol EvaGreen HRM Mix (Solis BioDyne Co., Estonia). Reactions were carried out in a total volume of 20 µL containing 5× HOT FIREPol EvaGreen HRM Mix, 10 pmol/µL of each primer, depending on the gene assayed and 1µl of the template. Samples were run in duplicate in each experiment to verify the reproducibility, and each experiment was repeated twice. The protocol involved 15 minutes of preincubation at 95℃ and 40 cycles of three‐step amplification (15 seconds/95℃, 20 seconds/Ta, 20 seconds/72℃), and obtained amplicons were melted in a temperature gradient to max 95℃ (each Ta is presented in Table [Link], [Link]).
The obtained melting curves were normalized automatically by the calculation of the "line of the best fit" in between two normalization regions before and after the significant fluorescence decrease. The methylation level of each sample was assessed by comparison of the PCR product normalized melting curve/peak with the normalized melting curves/peaks of the controls. Data interpretation was performed according to the guidelines of Smith et al. 42
2.6. Pyrosequencing
Pyrosequencing of MGMT gene was performed on Pyrosequencer PSQ™ 96 ID system, and the data were analyzed on PyroQ CpG™ software 1.0.9 (Biotage, Uppsala, Sweden), as described previously [28]. With the use of PyroMark™ MGMT Kit (Biotage, Uppsala, Sweden), we analyzed five CpG sites in exon 1 (positions 17 to 39, Ensemble ID: OTTHUMT00000051009) of the MGMT promoter. The temperature profile was the following: 95°C for 15 minutes; 45 cycles of 94°C for 30 seconds, 53°C for 30 seconds, and 72°C for 30 seconds; followed by 72°C for 10 minutes. The amplicon length was 104 bp. A cytosine not followed by a guanine, and which is therefore not methylated, served as an internal control for the completion of bisulfite conversion reaction. The mean methylation across all CpG sites analyzed was calculated for each sample and used for comparison with MS‐HRM.
2.7. Statistical analysis
All variables were measured on an ordinal scale, so they did not require any test to assess normality distribution. Nonparametric tests, Mann‐Whitney U test, Kruskal‐Wallis, or Wilcoxon tests were used to analyze data, and the results with P‐value < 0.05 were considered significant. The descriptive statistics were performed, and methylation levels of selected genes were examined using STATISTICA 13.1 software, depending on patients’ gender, age, survival time, histological type, and the WHO grade of the tumor. Genes were also analyzed in pairs, for the estimation of the interrelation between their methylation profiles. DNA methylation in cancerous and non‐cancerous samples was additionally determined based on GraphPad Instant 3 and PQStat v 1.6.8 statistical tools. It has to be mentioned that WHO grade I tumors were excluded from the statistical analysis due to the low number of samples (2/46). For comparison between MS‐HRM and pyrosequencing results, Cohen's kappa coefficient was applied to measure the ordinal association between measured quantities, when data were dichotomized. Moreover, Kendall's coefficient of concordance was used when the results of both methods were categorized as percent ranges.
3. RESULTS
3.1. MS‐HRM analysis of a gene panel of promoter DNA methylation
The normalized melting curves and normalized melting peaks of the analyzed genes obtained from standards and representative samples are presented in Figure S2 (SFRP1), Figure S3 (SFRP2), Figure S4 (RUNX3), Figure S5 (CBLN4), Figure S6 (INA), Figure S7 (MGMT), and Figure S8 (RASSF1A). The methylation level of each sample was assessed by comparison of the PCR product and standards melting profiles with a known ratio of methylated and unmethylated templates. The melting profiles were displayed as both, normalized melting curves and normalized melting peaks, to ease the distinction of each sample melting profile characteristics.
Data were analyzed using the "Tm calling" module of Light Cycler 96 SW 1.1 Software. The algorithm used in data calculation provided default settings for the temperature ranges specifying the normalization areas. Premelting signals were uniformly set to a relative value of 100%, while postmelting signals were set to a relative value of 0%.
The results of the MS‐HRM analysis of all analyzed genes (SFRP1, SFRP2, RUNX3, CBLN4, INA, MGMT, and RASSF1A) showed that their promoter sequence methylation level is significantly higher (P < 0.0001) in DNA samples from brain tumor patients than in the non‐neoplastic brain sample (Figure 1). The highest possible level of gene promoter methylation (>75, ≤100%) was not reached in any sample or gene, while the methylation level of >50, ≤75% was observed for RASSF1A (6/46 samples; 13.04%). The level of >25, ≤50% was achieved frequently for SFRP1 (12/46 samples; 26.09%) and RASSF1A (8/46 samples; 17.39%), followed by MGMT (2/46 samples; 4.35%) (Table 1). RUNX3 was unmethylated in the majority of cases (31/46; 67.39%), but the methylation level of >25, ≤50% was reached in 1 sample, and the level of >10, ≤ 25% was observed in 4 samples, whereas 5 samples were scored as >5, ≤ 10% methylation. More than half of the samples were unmethylated (0% methylation) in regard to SFRP2 (24/44; 54.54%), while 2 samples reached the level >5, ≤ 10% methylation and 18 samples scored >0, <5% methylation. Both CBLN4 and INA were mostly unmethylated; only single samples reached the level of >10, ≤ 25% methylation.
FIGURE 1.
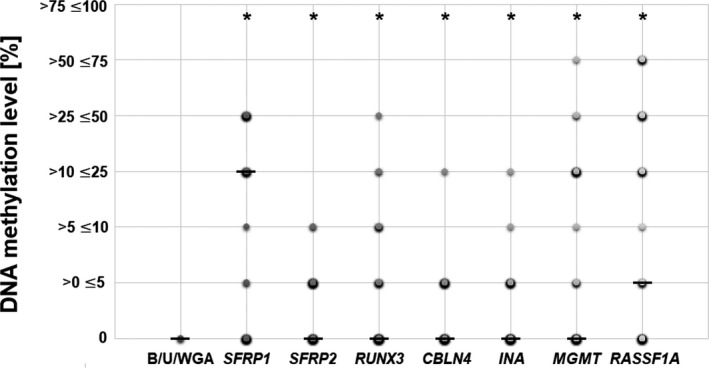
DNA methylation level [%] of the analyzed 7‐gene panel in relation to non‐neoplastic, control brain sample (indicated with "B"), unmethylated DNA control (indicated with "U"), and whole‐genome amplified DNA from pooled peripheral blood lymphocytes (indicated with "WGA"). The size and color intensity of each dot represents the number of samples with the particular methylation range. Median is indicated with a horizontal line "–" whereas * represents statistically significant difference (P < 0.05) in regard to the results obtained for sample "B," "U," and "WGA."
RASSF1A had significantly higher methylation level in lower‐grade tumors (Figure 2). A similar relation was observed for SFRP1, MGMT, and RUNX3, but these results did not reach the statistical significance. The distribution of the obtained methylation levels of each gene regarding patients’ gender is presented in Figure 3. The statistical significance (P < 0.05) was found between males and females in the case of MGMT. The positive correlation of promoter DNA methylation and patients’ age was found in SFRP1 and INA (Figure 4). The analysis of the interrelation between MGMT,SFRP2, and RUNX3 methylation levels and patients' age also revealed a trend toward higher methylation levels in older patients, however, without statistical significance (Figure S9). Finally, the overall survival time of patients ranged from 3 to 42 months. The average survival time was 13.5 months, and the median was estimated as 10 months. No correlation was noted between gene promoter methylation level and patients’ survival time (Table S3).
FIGURE 2.
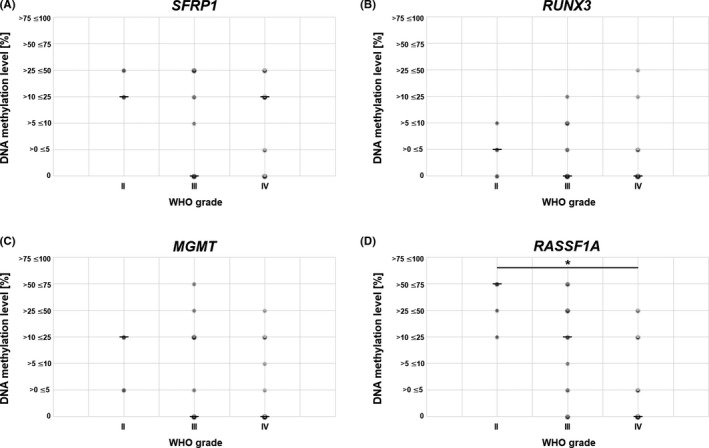
DNA methylation level [%] of SFRP1, RUNX3, MGMT, and RASF1A in relation to tumor WHO grade. The size and color intensity of each dot indicates the number of samples representing the particular methylation range. Median is indicated with a horizontal line "–". RASSF1A promoter methylation level is significantly higher in lower‐grade gliomas (P < 0.05)
FIGURE 3.
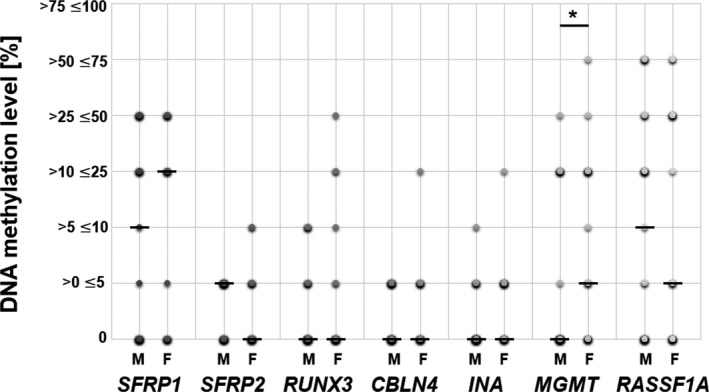
DNA methylation level [%] of the analyzed 7‐gene panel in relation to patients’ gender. M indicates male, F—female. The size and color intensity of each dot indicates the number of samples representing the particular methylation range. Median is indicated with a horizontal line "–". MGMT promoter methylation level is significantly higher in female patients, as compared to the value in males (P < 0.05)
FIGURE 4.
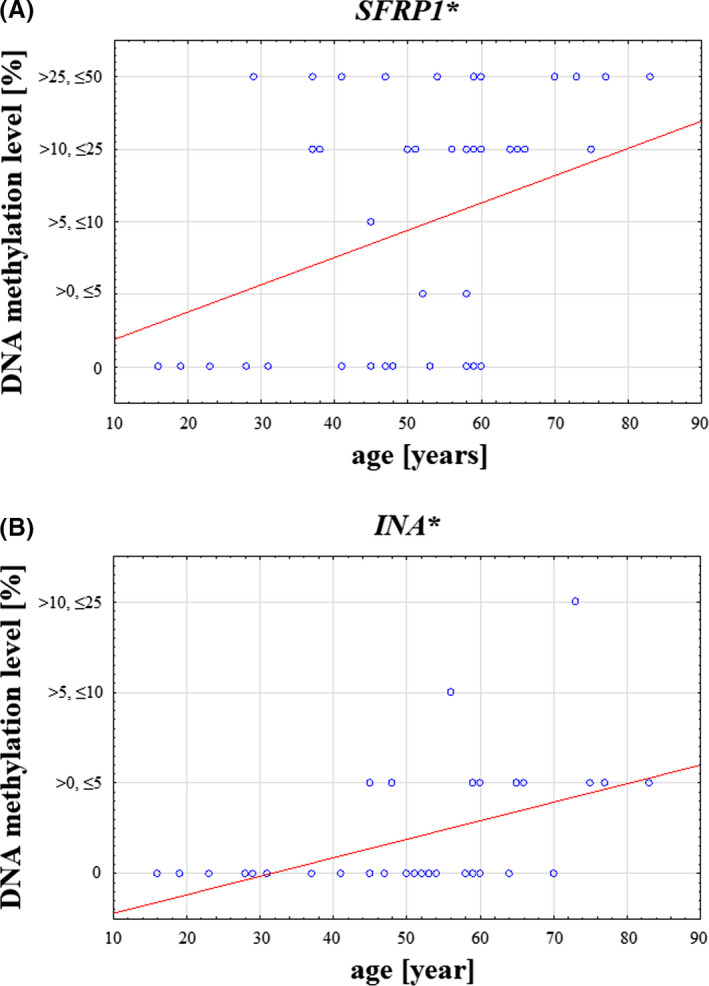
DNA methylation level [%] of SFRP1 and INA in relation to patients’ age [years]. DNA methylation levels of both genes are significantly higher in older patients (P < 0.05)
3.2. Interrelations between promoter methylation levels of genes in the analyzed panel
The strong interrelationship between promoter methylation levels was detected between pairs of genes: SFRP1 and CBLN4 (P = 0.00031), MGMT and RASSF1A (P = 0.002337), SFRP1 and MGMT (P = 0.035), SFRP2 and RUNX3 (P = 0.000773), SFRP2 and CBLN4 (P = 0.049108), SFRP2 and INA (P = 0.02783), and CBLN4 and INA (P = 0.000137) (Figure 5). For a particular patient, an increased DNA methylation level in one of the paired genes was associated with higher methylation in the other gene promoter, and a lack of methylation in one gene correlated with a lack of methylation in another. This finding proves our concept of choosing the panel of genes which represent different aspects of CNS carcinogenesis, but show relations to each other.
FIGURE 5.
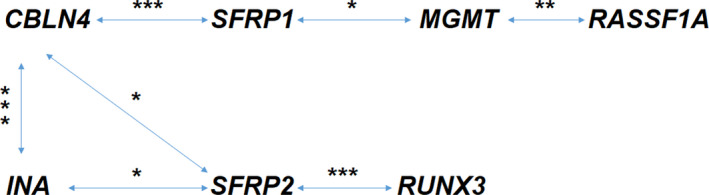
The interrelation between the methylation levels of genes analyzed in pairs. * indicates statistical significance at P < 0.05, **P < 0.005, and ***P < 0.0005
3.3. Comparison of MS‐HRM and pyrosequencing of MGMT gene
In order to validate the results of MS‐HRM, pyrosequencing of MGMT promoter region was applied to all 46 samples analyzed. Ten representative pyrograms and corresponding MS‐HRM normalized melting curves are presented in Figure S10. Both methods were highly reproducible, and when the results of both methods were dichotomized (>10% regarded as methylated, ≤ 10% regarded as unmethylated), they were fully concordant (Cohen's kappa coefficient κ = 1, P < 0.000001). There were 15/46 (32.61%) methylated samples and 31/46 (67.39%) unmethylated samples. When pyrosequencing data (mean methylation percentage of five CpG dinucleotides) were grouped into percentage ranges, following MS‐HRM categorization, Kendall's coefficient of concordance was still showing good correlation between the ranges (Kendall's W = 0.92, P = 0.00051).
4. CONCLUSIONS
New epigenetic biomarkers associated with CNS tumors are constantly being sought and tested. Their identification would enable more rational selections of strategies to cure patients and prevent disease relapse. However, it is already known that probably there cannot be a single gene test for glioma detection, as well as for predictive and prognostic purposes, because of the multifactorial process of carcinogenesis. 17 Epigenetics is another area for exploring the disease background and biomarkers, as well as the search for potential therapeutic targets. 45
Additionally, the debate over the best analytical method for methylation markers detection is still ongoing.
We proposed a 7 gene panel for brain glioma characterization. It consists of genes involved in carcinogenesis in general and CNS pathology in particular, as well as having a proven impact on therapeutic response. We confirmed a significantly higher DNA methylation level for each of the 7 analyzed genes in glioma patients compared with the non‐neoplastic brain and unmethylated control. Similar results were obtained by Shinawi et al 31 and Lai et al, 46 who differentiated DNA methylation levels in CNS cancer patients and healthy, cancer‐free individuals. Thus, this panel of genes could be used for the differentiation of the CNS tumors from the tissue without signs of tumor growth. Moreover, we observed a significant positive correlation between promoter methylation levels in gene pairs: SFRP1/MGMT, SFRP2/RUNX3, SFRP2/CBLN4, SFRP2/INA, CBLN4/INA, and MGMT/RASSF1A. Increased DNA methylation level in one gene was accompanied by a higher methylation level in another and vice versa. Such relationships are valuable markers since they increase screening cost‐effectiveness by narrowing the molecular diagnostic panel of genes.
In this study, the highest median of promoter methylation (>25 ≤50%) was obtained for RASSF1A analysis. Interestingly, we observed a general decrease in promoter DNA methylation of RASSF1A with increasing glioma malignancy (this effect was also observed to a lesser extent for SFRP1, MGMT, and RUNX3). This result is concordant with previous findings on total DNA demethylation with increasing glioma grade, 47 being the effect of oxidative DNA damage. 48 In that context, the interesting aspect is a stable level of CBLN4, INA, and SFRP2 promoter methylation, which can make them promising biomarkers of neuro‐oncogenesis in general.
Reports published to date provide moderate evidence of the correlation between DNA methylation level and the CNS cancer histological type. Gao et al 49 examined 28 samples of glial tumors. They found that RASSF1A hypermethylation is associated with a loss of expression, but it is not statistically significant in regard to histological type and malignancy of CNS tumors. In turn, Muñoz et al 50 observed the relationship between RASSF1A promoter methylation and a secondary glioma phenotype. In our previous study, we showed that RASSF1A hypermethylation differentiated primary from metastatic brain cancers. 35 Thus, this gene should further be tested and validated in regard to CNS tumors.
Among many assays for DNA methylation analysis, MS‐HRM is one of the most frequently recommended methods. 13 , 14 It has already been proved useful in the assessment of cancer biomarkers in bladder, colorectal, and breast cancer patients. 11 Our study is the first report presenting the application of MS‐HRM for CNS tumor analysis. One of the possible disadvantages of MS‐HRM is that the reaction is semi‐quantitative since the results of the analysis are presented as a methylation range. Nevertheless, such data presentation is commonly regarded as sufficient for the sample evaluation in the clinics. 51 Many advantages of the method, including its sensitivity as well as specificity, cost‐effectiveness, and no sophisticated equipment needed, make this method suitable for the routine clinical application. The fact that MS‐HRM analysis requires no manual post‐PCR processing and is performed in a closed‐tube system, with minimal risk of contamination, is equally important.
In order to validate MS‐HRM results, we performed additional pyrosequencing analysis of the most important biomarker from our panel, namely MGMT. For the purpose of this comparison, the pyrosequencing data were categorized into methylation ranges, same as in MS‐HRM method. The obtained data showed high agreement among methylation ranges, even though both methods do not analyze exactly the same CpG dinucleotides (Figure S10). However, when pyrosequencing and MS‐HRM results were dichotomized into methylated and unmethylated groups (taking 10% methylation as a cutoff value) the results were fully concordant. Similar results were reported by several groups. 52 , 53 , 54
In this study, we also established and optimized protocols for MS‐HRM analysis of CBLN4 and INA genes, which were, according to our best knowledge, not reported so far. The reaction was optimized in terms of, for example, primer sequences and primer annealing temperature, so that the fluorescent melting profiles from subsequent methylated DNA standards exhibit significant differences. The sensitivity of both assays was high, allowing the detection of as low as 5% methylation. Indeed, in our tested group, the fluorescent curves of almost all of the samples were found between 0% and 5% methylated standards. INA encodes a protein that is a member of the intermediate filament family and is involved in the morphogenesis of neurons. It is a novel candidate for a CNS biomarker, indicating GBM with a worse prognosis. 31 In turn, CBLN4 encodes a cerebellin 4 precursor, involved in the regulation of neurexin signaling during synapse development. 55 Their clinically relevant level of promoter methylation and potential role as CNS cancer biomarkers should be determined in further studies.
In the current study, not only INA and CBLN4 but also other genes from the chosen panel did not show any association between DNA methylation level and patients’ OS. Thus, their prognostic potential is minimal. However, in our previous study, SFRP1 methylation was associated with shorter OS. 26 Also, MGMT methylation status represents one of the most relevant prognostic factors in GBMs. 4 , 33 Therefore, the results of our study should be verified on larger patients' cohorts.
CNS tumors have two morbidity peaks. The first one occurs in childhood, and the second one is between 55 and 65 years of age. 56 In this study, patients’ age ranged between 16 and 83 years of age, and higher methylation levels of two genes, namely SFRP1 and INA, were observed in older patients, which confirmed our previous observation, 26 indicating a more frequent occurrence of SFRP1 26 and RUNX3 27 , 28 promoter methylation in older CNS tumor patients. In turn, lack of association between age and SFRP1 methylation level was proved by Chang 57 and Kafka, 58 while no correlation for MGMT in the analysis of 58 anaplastic astrocytomas was reported by Bell et al. 59
The patient's sex is another recognized prognostic factor for brain malignancies. Epidemiological data show that glial tumors are slightly more common (about 1.2×) in men than women. 56 In our study, RASSF1A was the most frequently methylated gene in both female and male patients. This observation was also confirmed by Muñoz et al 50 and in our previous studies. 27 , 28 Moreover, the results of our current study, showing higher MGMT promoter methylation in female as compared to male patients, are in line with Franceschi et al 60 results. Moreover, in this study, also the patients' sex and methylated MGMT occurrence were significantly related to patients' survival time. CNS cancer female patients with methylated DNA lived significantly longer as compared to men with the methylated MGMT, showing the importance of sex as a prognostic factor. 60 Tian et al 61 also confirmed the prognostic significance of sex in women with GBM. Therefore, the role of female hormones in glial tumor pathogenesis should be further clarified.
The development of diagnostic methods and discovering new biomarkers gain importance in neuro‐oncology. 62 , 63 It is necessary to introduce epigenetic biomarker panels for CNS tumors, enabling earlier cancer detection or better therapy monitoring. Our study verifies MS‐HRM usefulness for the assessment of DNA methylation in CNS tumors. We conclude that our 7‐gene panel of promoter methylation can be helpful in brain tumor diagnosis or characterization, and MS‐HRM can play a crucial role in the development of cost‐efficient personalized patient care.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Fig S10
Table S1
Table S2
Table S3
ACKNOWLEDGMENT
This research was funded by Poznan University of Medical Sciences. We thank Dr. Lucyna Kramer and Dr. Izabela Miechowicz for excellent help with the statistical analysis and Dr. Ewa Ignatowicz for careful reading of the manuscript and language corrections.
Majchrzak‐Celińska A, Dybska E, Barciszewska A‐M. DNA methylation analysis with methylation‐sensitive high‐resolution melting (MS‐HRM) reveals gene panel for glioma characteristics. CNS Neurosci Ther. 2020;26:1303–1314. 10.1111/cns.13443
Majchrzak‐Celińska and Dybska are contributed equally to this work.
REFERENCES
- 1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803‐820. [DOI] [PubMed] [Google Scholar]
- 2. Capper D, Jones DTW, Sill M, et al. DNA methylation‐based classification of central nervous system tumours. Nature. 2018;555(7697):469‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ceccarelli M, Barthel F, Malta T, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hegi ME, Diserens A‐C, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997‐1003. [DOI] [PubMed] [Google Scholar]
- 5. Rivera AL, Pelloski CE, Gilbert MR, et al. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro‐Oncol. 2010;12(2):116‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shen D, Liu T, Lin Q, et al. MGMT promoter methylation correlates with an overall survival benefit in chinese high‐grade glioblastoma patients treated with radiotherapy and alkylating agent‐based chemotherapy: a single‐institution study. PLoS One. 2014;9(9):e107558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncol. 2009;10(5):459‐466. [DOI] [PubMed] [Google Scholar]
- 8. Wick W, Weller M, van den Bent M, et al. MGMT testing—the challenges for biomarker‐based glioma treatment. Nat Rev Neurol. 2014;10(7):372‐385. [DOI] [PubMed] [Google Scholar]
- 9. Kim H, Wang X, Jin P. Developing DNA methylation‐based diagnostic biomarkers. J Genet Genomics. 2018;45(2):87‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Šestáková Š, Šálek C, Remešová H. DNA methylation validation methods: a coherent review with practical comparison. Biol Proced Online. 2019;21(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hussmann D, Hansen LL. Methylation‐sensitive high resolution melting (MS‐HRM) In: Tost J, ed. DNA Methylation Protocols. Methods in Molecular Biology. New York, NY: Springer; 2018:551‐571. [DOI] [PubMed] [Google Scholar]
- 12. Switzeny OJ, Christmann M, Renovanz M, Giese A, Sommer C, Kaina B. MGMT promoter methylation determined by HRM in comparison to MSP and pyrosequencing for predicting high‐grade glioma response. Clin Epigenetics. 2016;8(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao F, Bapat B. The role of methylation‐specific PCR and associated techniques in clinical diagnostics In: Garcia‐Gimėnez JL, ed. Epigenetic Biomarkers and Diagnostics, 1st ed Academic Press, Elsevier Inc; 2016:155‐173. [Google Scholar]
- 14. Hattori N, Ushijima T. Analysis of gene‐specific DNA methylation In: Tollefsbol T, ed. Handbook of Epigenetics, 2nd ed San Diego, CA: Academic Press; 2017:113‐123. [Google Scholar]
- 15. Reich TR, Switzeny OJ, Renovanz M, et al. Epigenetic silencing of XAF1 in high‐grade gliomas is associated with IDH1 status and improved clinical outcome. Oncotarget. 2017;8(9):15071‐15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Binabaj MM, Bahrami A, ShahidSales S, et al. The prognostic value of MGMT promoter methylation in glioblastoma: a meta‐analysis of clinical trials. J Cell Physiol. 2018;233(1):378‐386. [DOI] [PubMed] [Google Scholar]
- 17. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646‐674. [DOI] [PubMed] [Google Scholar]
- 18. Chai R‐C, Chang Y‐Z, Wang Q‐W, et al. A novel DNA methylation‐based signature can predict the responses of MGMT promoter unmethylated glioblastomas to temozolomide. Front Genet. 2019;10:910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de Souza CF, Sabedot TS, Malta TM, et al. A distinct DNA methylation shift in a subset of glioma CpG island methylator phenotypes during tumor recurrence. Cell Rep. 2018;23(2):637‐651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klughammer J, Kiesel B, Roetzer T, et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat Med. 2018;24(10):1611‐1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baharudin R, Tieng FYF, Lee L‐H, Ab Mutalib NS. Epigenetics of SFRP1: the dual roles in human cancers. Cancers. 2020;12(2):445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu YU, Liu X, Zheng H, et al. Multiple roles of sFRP2 in cardiac development and cardiovascular disease. Int J Biol Sci. 2020;16(5):730‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu B, Han Y, Jiang LU, et al. Clinicopathological and prognostic significance of the RUNX3 expression in gastric cancer: a systematic review and meta‐analysis. Int J Surg. 2018;53:122‐128. [DOI] [PubMed] [Google Scholar]
- 24. Lu D, Ma Y, Zhu A, Han Y. An early biomarker and potential therapeutic target of RUNX 3 hypermethylation in breast cancer, a system review and meta‐analysis. Oncotarget. 2016;8(13):22166‐22174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen F, Liu X, Bai J, Pei D, Zheng J. The emerging role of RUNX3 in cancer metastasis (Review). Oncol Rep. 2016;35(3):1227‐1236. [DOI] [PubMed] [Google Scholar]
- 26. Majchrzak‐Celińska A, Słocińska M, Barciszewska A‐M, Nowak S, Baer‐Dubowska W. Wnt pathway antagonists, SFRP1, SFRP2, SOX17, and PPP2R2B, are methylated in gliomas and SFRP1 methylation predicts shorter survival. J Appl Genet. 2016;57:189‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Majchrzak‐Celińska A, Paluszczak J, Szalata M, Barciszewska A‐M, Nowak S, Baer‐Dubowska W. DNA methylation analysis of benign and atypical meningiomas: correlation between RUNX3 methylation and WHO grade. J Cancer Res Clin Oncol. 2015;141:1593‐1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Majchrzak‐Celińska A, Paluszczak J, Szalata M, et al. The methylation of a panel of genes differentiates low‐grade from high‐grade gliomas. Tumor Biol. 2015;36(5):3831‐3841. [DOI] [PubMed] [Google Scholar]
- 29. Yuan A, Rao MV, Veeranna, Nixon RA. Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb Perspect Biol. 2017;9(4):a018309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suh JH, Park C‐K, Park S‐H. Alpha internexin expression related with molecular characteristics in adult glioblastoma and oligodendroglioma. J Korean Med Sci. 2013;28(4):593‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shinawi T, Hill VK, Krex D, et al. DNA methylation profiles of long‐ and short‐term glioblastoma survivors. Epigenetics. 2013;8(2):149‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seigneur E, Südhof TC. Genetic ablation of all cerebellins reveals synapse organizer functions in multiple regions throughout the brain. J Neurosci. 2018;38(20):4774‐4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987‐996. [DOI] [PubMed] [Google Scholar]
- 34. Malpeli G, Innamorati G, Decimo I, et al. Methylation dynamics of RASSF1A and its impact on cancer. Cancers. 2019;11(7):959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Majchrzak‐Celińska A, Paluszczak J, Kleszcz R, et al. Detection of MGMT, RASSF1A, p15INK4B, and p14ARF promoter methylation in circulating tumor‐derived DNA of central nervous system cancer patients. J Appl Genet. 2013;54(3):335‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amornpisutt R, Proungvitaya S, Jearanaikoon P, Limpaiboon T. DNA methylation level of OPCML and SFRP1: a potential diagnostic biomarker of cholangiocarcinoma. Tumor Biol. 2015;36(7):4973‐4978. [DOI] [PubMed] [Google Scholar]
- 37. Li X, Hu F, Wang Y, et al. CpG island methylator phenotype and prognosis of colorectal cancer in northeast China. BioMed Res Int. 2014;2014:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stuopelytė K, Daniūnaitė K, Laurinavičienė A, Ostapenko V, Jarmalaitė S. High‐resolution melting‐based quantitative analysis of RASSF1 methylation in breast cancer. Medicina. 2013;49(2):78‐83. [PubMed] [Google Scholar]
- 39. Wojdacz TK, Dobrovic A. Methylation‐sensitive high resolution melting (MS‐HRM): a new approach for sensitive and high‐throughput assessment of methylation. Nucleic Acids Res. 2007;35(6):e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiao Z, Li B, Wang G, et al. Validation of methylation‐sensitive high‐resolution melting (MS‐HRM) for the detection of stool DNA methylation in colorectal neoplasms. Clin Chim Acta. 2014;431:154‐163. [DOI] [PubMed] [Google Scholar]
- 41. Methylation Analysis Using Methylation‐Sensitive HRM and DNA Sequencing. http://tools.thermofisher.com/content/sfs/brochures/cms_070933.pdf. Accessed March 10, 2019
- 42. Smith E, Jones ME, Drew PA. Quantitation of DNA methylation by melt curve analysis. BMC Cancer. 2009;9(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wojdacz TK, Borgbo T, Hansen LL. Primer design versus PCR bias in methylation independent PCR amplifications. Epigenetics. 2009;4(4):231‐234. 10.4161/epi.9020 [DOI] [PubMed] [Google Scholar]
- 44. Wojdacz TK, Hansen LL, Dobrovic A. A new approach to primer design for the control of PCR bias in methylation studies. BMC Res Notes. 2008;1(1):1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Suvà ML. Genetics and epigenetics of gliomas. Swiss Med Wkly. 2014;144(4344):14018. [DOI] [PubMed] [Google Scholar]
- 46. Lai RK, Chen Y, Guan X, et al. Genome‐wide methylation analyses in glioblastoma multiforme. PLoS One. 2014;9(2):e89376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zukiel R, Nowak S, Barciszewska A‐M, Gawronska I, Keith G, Barciszewska MZ. A simple epigenetic method for the diagnosis and classification of brain tumors. Mol Cancer Res. 2004;2:196‐202. [PubMed] [Google Scholar]
- 48. Barciszewska A‐M, Giel‐Pietraszuk M, Perrigue PM, Naskręt‐Barciszewska M. Total DNA methylation changes reflect random oxidative DNA damage in gliomas. Cells. 2019;8(9):1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gao Y, Guan M, Su B, Liu W, Xu M, Lu Y. Hypermethylation of the RASSF1A gene in gliomas. Clin Chim Acta. 2004;349(1):173‐179. [DOI] [PubMed] [Google Scholar]
- 50. Muñoz J, Inda MDM, Lázcoz P, et al. Promoter methylation of RASSF1A associates to adult secondary glioblastomas and pediatric glioblastomas. ISRN Neurol. 2012;2012:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. BLUEPRINT consortium . Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat Biotechnol. 2016;34(7):726‐737. [DOI] [PubMed] [Google Scholar]
- 52. Akika R, Awada Z, Mogharbil N, Zgheib N. Region of interest methylation analysis: a comparison of MSP with MS‐HRM and direct BSP. Mol Biol Rep. 2017;44(3):295‐305. [DOI] [PubMed] [Google Scholar]
- 53. Migheli F, Stoccoro A, Coppedè F, et al. Comparison study of MS‐HRM and pyrosequencing techniques for quantification of APC and CDKN2A gene methylation. PLoS One. 2013;8(1):e52501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu F, Zhang H, Lu S, et al. Quantitative assessment of gene promoter methylation in non‐small cell lung cancer using methylation‐sensitive high‐resolution melting. Oncol Lett. 2018;15(5):7639‐7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. https://www.ncbi.nlm.nih.gov/gene/140689. Accessed March 14, 2019.
- 56. Ostrom QT, Cioffi G, Gittleman H, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro‐Oncol. 2019;21(Supplement_5):v1‐v100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chang L, Lei X, Qin YU, et al. Expression and prognostic value of SFRP1 and β‐catenin in patients with glioblastoma. Oncol Lett. 2016;11(1):69‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kafka A, Karin‐Kujundžić V, Šerman L, et al. Hypermethylation of Secreted Frizzled Related Protein 1 gene promoter in different astrocytoma grades. Croat Med J. 2018;59(5):213‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bell EH, Won M, Chang SM, et al. MGMT promoter methylation status independently predicts overall survival in anaplastic astrocytoma in NRG Oncology/RTOG 9813: a phase 3 trial of radiation plus nitrosourea versus radiation plus temozolomide. Int J Radiat Oncol Biol Phys. 2017;99(2):S99‐S100. [Google Scholar]
- 60. Franceschi E, Tosoni A, Minichillo S, et al. The prognostic roles of gender and O6‐Methylguanine‐DNA methyltransferase methylation status in glioblastoma patients: the female power. World Neurosurg. 2018;112:e342‐e347. [DOI] [PubMed] [Google Scholar]
- 61. Tian M, Ma W, Chen Y, et al. Impact of gender on the survival of patients with glioblastoma. Biosci Rep. 2018;38(6):BSR20180752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Reszec J, Szkudlarek M, Hermanowicz A, Bernaczyk PS, Mariak Z, Chyczewski L. N‐cadherin, beta‐catenin and connexin 43 expression in astrocytic tumours of various grades. Histol Histopathol. 2015;30(3):361‐371. [DOI] [PubMed] [Google Scholar]
- 63. Szopa W, Burley TA, Kramer‐Marek G, Kaspera W. Diagnostic and therapeutic biomarkers in glioblastoma: current status and future perspectives. Biomed Res Int. 2017;2017:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Fig S10
Table S1
Table S2
Table S3