

Abstract
The activation loop (A-loop) plays a key role in regulating the catalytic activity of protein kinases. Phosphorylation in this region enhances the phosphoryl transfer rate of the kinase domain and increases its affinity for ATP. Furthermore, the A-loop possesses autoinhibitory functions in some kinases, where it collapses onto the protein surface and blocks substrate binding when unphosphorylated. Due to its flexible nature, the A-loop is usually disordered and untraceable in kinase domain crystal structures. The resulting lack of structural information is regrettable as it impedes the design of drug A-loop contacts, which have proven favourable in multiple cases. Here, we characterize the binding with A-loop engagement between type 1.5 kinase inhibitor ‘example 172’ (EX172) and Mer tyrosine kinase (MerTK). With the help of crystal structures and binding kinetics, we portray how the recruitment of the A-loop elicits a two-step binding mechanism which results in a drug-target complex characterized by high affinity and long residence time. In addition, the type 1.5 compound possesses excellent kinome selectivity and a remarkable preference for the phosphorylated over the dephosphorylated form of MerTK. We discuss these unique characteristics in the context of known type 1 and type 2 inhibitors and highlight opportunities for future kinase inhibitor design.
Keywords: activation loop, activation segment, MerTK, TAM
Introduction
Mer tyrosine kinase (MerTK) is a member of the TAM family (Tyro3, Axl and MerTK) of receptor tyrosine kinases (RTKs), which act as sensors for apoptotic phosphatidylserine. Once activated, TAM receptors dimerize in the membrane, which enables autophosphorylation of the intracellular kinase domains and triggers downstream signalling events. TAM RTKs are ubiquitously expressed but most abundant in the nervous, vascular, reproductive and immune systems [1]. In line with the broad distribution of the receptors, TAM signalling has pleiotropic functions, with dampening inflammation being the most prominent role [2]. Accordingly, mutant mice lacking the TAM receptors develop autoimmune diseases [3]. Notably, TAM RTKs also exert their immune-inhibitory function in the tumour microenvironment, where they prevent chronic activation of antigen-presenting cells (APCs) and boost apoptotic cell clearance in macrophages; a tumour-promoting process called efferocytosis [2]. On the tumour side, MerTK activity has been directly linked to the stimulation of growth and metastasis [4]. TAM RTKs have, therefore, attracted considerable attention as targets in the immuno-oncology space [5,6].
MerTK crystal structures published so far (as of 09-2020, PDB entries: 2p0c, 3bpr, 3brb, 3tcp, 4m3q, 4mh7, 4mha, 5k0k, 5k0x, 5tc0, 5td2, 5u6c and 6mep) capture the kinase domain in an inactive form with ‘helix-C out’ conformation (for kinase nomenclature consult [7]). In contrast with the active state, helix-C appears rotated and moved away from the active site DFG motif, thereby breaking the canonical salt bridge between catalytic Lys619 and conserved Glu637. The translocation of helix-C creates a pocket adjacent to the ATP-binding site, referred to as ‘allosteric back pocket’ [8], which can be occupied by type 1.5, type 2 and type 3 inhibitors. Binding to the back pocket allows for contacts between small molecule inhibitor and the kinase activation loop (A-loop). Such interactions have been described for several kinases, including c-Met, MEK1, RIPK1 and RIPK2 (reviewed by [9]), and can contribute to both binding potency and kinome selectivity.
Experimental
Protein expression and purification
Variations of human MerTK kinase constructs were co-expressed with protein tyrosine phosphatase PTPN1; in accordance with literature precedent [10].
For biochemical activity measurements, the full intracellular domain (R528-M999) with an N-terminal GST-TEV-6His tag and C-terminal Avi- and FLAG-tags was expressed in Spodoptera frugiperda. Protein was purified from cell lysate via immobilized metal affinity chromatography (IMAC) followed by proteolytic cleavage of the GST tag, additional de-phosphorylation with lambda phosphatase and final purification with anion exchange chromatography.
For use in SPR measurements, the above protocol was modified by additional co-expression of BirA and supplementation of the growth medium with 60 µM biotin, to afford biotinylation of the Avi-tag. Activated MerTK protein was prepared by in vitro autophosphorylation. The final buffer contained 20 mM Tris–HCl pH 8.0, 270 mM NaCl, 5% glycerol, 1 mM TCEP. Biotinylation and phosphorylation of all protein batches was verified by LC–MS.
For crystallisation, N-terminally His6-tagged constructs of the kinase domain only (E571-V864) were expressed in Escherichia coli; and purified by IMAC followed by proteolytic cleavage of the affinity tag and size-exclusion chromatography. Protein was concentrated to ∼10 mg/ml in a final buffer of 20 mM Tris–HCl pH 8.0, 500 mM NaCl, 5% glycerol, 1 mM TCEP.
Surface plasmon resonance
Dephosphorylated or autophosphorylated biotinylated MerTK tyrosine kinase (R528-M999) were used for surface plasmon resonance (SPR) experiments. A series S streptavidin Biacore chip (#BR100531, Cytiva) was docked onto either a T200 or 8 K Biacore instrument (Cytiva). Buffer consisting of 20 mM Tris (pH 7.7), 150 mM NaCl, 10 mM MgCl2, 0.5 mM TCEP (tris(2-carboxyethyl)phosphine), 0.005% (v/v) Tween-20 (polyoxyethylene(20)sorbitan monolaurate) and 1% (v/v) DMSO (dimethyl sulfoxide) was used for immobilization and binding experiments. Prior to running each experiment, a desorb method was run and the instrument was primed three times with buffer. All immobilization and binding experiments were equilibrated to 25°C. MerTK protein was immobilized at a density of between 2000 and 7000 RU. Unbound streptavidin sites were blocked with 50 μM amino-PEG biotin (#21346, ThermoFisher). Compounds were prepared in assay buffer in a 384-well polypropylene microplate (#781201, Greiner) and care was taken to ensure a final concentration of 1% (v/v) DMSO. The ‘Single Cycle Kinetic’ method was used to generate binding sensorgams and a 50% DMSO wash was used between samples. All data were double referenced and solvent (DMSO) corrected. Data were fitted to either a 1 : 1 or two-state binding model using the Biacore Insight software (Cytiva). Binding parameters were extracted from the Insight analysis software with the exception of the dissociation rate constant for the two-state model which was defined as koff = (k2k4/(k2 + k3 + k4)).
MerTK biochemical activity
The ability of inhibitors to cause inhibition of MerTK (6His_R528-M999_Thrombin-Avi-Flag, dephosphorylated) in vitro was assessed using Rapidfire LCMS method that quantified Axltide (CKKSRGDYMTMQIG-acid, Cambridge Research Biochemicals) peptide phosphorylation levels. MerTK (1 nM final) in assay buffer (20 mM HEPES pH 7.5, 0.006% Brij-35, 0.5 mM TCEP, 10 mM, 10 mM Mg(OAc)2, 0.02% Pluoronic-F127) was added to compound plates using a liquid dispenser; Multidrop Combi. Assay plates were equilibrated for 30 min before initiating the reaction by addition of Axltide (10 μM final) and ATP (at Kmapp; 80 μM final) substrates in assay buffer. The reaction was allowed to progress for 120 min before being stopped with 0.1% formic acid in water. The Axltide peptide substrate and the phosphorylated Axltide phosphopeptide product levels were determined by mass spectrometry (Rapidfire RF360 and Agilent triple quad mass spectrometer system) using aqueous phase; 0.1% formic acid in water and organic phase; 0.1% formic acid in 90% methanol on type E (C8 rapidfire cartridge) to elute.
Ratios were plotted to generate concentration-response profiles and the dose-response curves were fit to the data using the non-linear regression analysis; four parameter logistic smart fit method in the Assay analyzer and Condoseo applications of the Genedata® Screener software (Genedata, Inc., Basel, Switzerland).
Protein crystallography and structural biology
MerTK protein with surface entropy reduction mutations, MerTK (GSHM_E571-V864, K591R/K693R/K702R/K856R), was crystallized by vapour diffusion at 20°C in a condition of 4.3 M NaCl, 0.1 M Tris pH 8.5. The reproducibility of crystallisation was substantially improved by micro-seeding. For soaking procedures, crystals were transferred into 1.9 M NaCl, 0.1 M Tris pH 8.5, 20% DMSO, containing 10–20 mM of the compound. Soaking was carried out over 1–24 h; the crystals subsequently flash frozen in the soaking solution. The apo crystal was cryo-protected with 1.9 M NaCl, 0.1 M Tris pH 8.5, 30% ethylene glycol, instead.
LDC1267, merestinib and EX172 were co-crystallized with MerTK kinase domain. In each case 1 mM compound was added to the protein from 100 mM DMSO stocks. The complexes were briefly incubated on ice, then subjected to sparse matrix crystallization screens at 20°C. LDC1267 was co-crystallized with MerTK (GSHM_E571-V864, K591R/K693R/K702R/K856R) in 4.4 M NaCl, 0.05 M Tris pH 7.5 and the crystals flash frozen in 2.5 M NaCl, 0.05 M Tris pH 8.5, 20% glycerol. Crystals of merestinib-MerTK complex (GSHM_E571-V864, ΔM659-Q662) grew in 23% PEG 6000, 0.75 M LiCl, 0.1 M MES pH 6.2 and were cryo-cooled in reservoir solution supplemented with 20% butane-2,3-diole. MerTK (GS_E571-V864) in complex with EX172 crystallized in 0.1 M HEPES pH 6.6, 3.2 M NaCl. The crystals were cryo-cooled in reservoir solution supplemented with 25% ethylene glycol.
Diffraction data were integrated with autoPROC/STARANISO [11–14] and structures solved by molecular replacement with AMoRe or Phaser from CCP4 [15–17]. Ligand co-ordinates plus restraints were generated with GRADE [18], structure co-ordinates modelled in COOT [19] and refinement carried out in BUSTER [18] and Refmac5 [20]. Structure figures were rendered with PyMOL.
Accession numbers
Coordinates and structure factors generated during this study are available at Protein Data Bank (PDB) under the following accession numbers: 7AB1, 7AB2, 7AAX, 7AAY, 7AAZ and 7AB0 (see Supplementary Table S1 for details).
Results
An improved MerTK crystal system
In the course of this work, we have optimized the crystallization of the MerTK kinase domain by introducing surface entropy reduction mutations, substituting four lysines with arginines [21]. The modified protein forms the same lattice as in the P21 literature precedent structures [10], however, with a centred orthorhombic unit cell (Supplementary Table S1). The novel C2221 crystal form allows for crystallization of apo-protein and subsequent compound soaking at high DMSO concentrations. Crystals generally result in well-resolved (<2 Å) structures, which contain one protein molecule per asymmetric unit.
Type 1 inhibitors
Gilteritinib is a dual Flt3/Axl inhibitor used for the treatment of acute myeloid leukaemia (AML) (Figure 1). The compound was reported to possess double-digit nanomolar off-target activity against MerTK [22], which confirmed in our biochemical and biophysical measurements (Table 1). UNC2025, also known as MRX-6313, was developed for the same indication, however, targets Flt3 and MerTK rather than Axl [23] (Figure 1). The structures of MerTK kinase domain complexed with gilteritinib and UNC2025 depict canonical type 1 binding: the molecules pack against the kinase hinge and gatekeeper Leu671, but do not enter the kinase back pocket or interfere with the conformation of the DFG motif (Figure 1). Each of the inhibitors place bulky, saturated rings in the ribose pocket and possess methylpiperazine solvent tails, which protrude out of the ATP-pocket along the kinase hinge. Since their binding does not entail rearrangements of the protein, type 1 inhibitors usually have faster kinetics than types 1.5 and 2. In good agreement with this trend gilteritinib demonstrated residence times below 1 min in our SPR measurements (Table 1). The biophysical characterization furthermore discovered a preference of gilteritinib for the active/phosphorylated form of MerTK, to which it binds with increased potency and extended residence time over the non-phosphorylated form (Table 1).
Figure 1. Comparison of unliganded MerTK with type 1 complexes.

(A) Superposition of apo-MerTK structure (PDB entry 7AB0, yellow), and complexes with gilteritinib (cyan, PDB entry 7AB1) and UNC2025 (red, PDB entry 7AB2). The compounds do not affect the overall conformation of the MerTK kinase domain or the position of the DFG motif. (B,C) Gilteritinib and UNC2025 bind to the kinase hinge without entering the back pocket. H-bonds are represented as yellow dashes with distances in Å indicated.
Table 1. Biochemical inhibition of MerTK kinase domain and biophysical binding by surface plasmon resonance (SPR).
| Biochem | SPR — dephosphorylated MerTK | SPR — phosphorylated MerTK | |||
|---|---|---|---|---|---|
| Compound | pIC501. | pKD2 | t1/2 (min) | pKD3 | t1/2 (min) |
 | |||||
Unless stated otherwise, all data reported are a mean of at least three independent measurements.
pIC50 SEM < 0.30.
pKd SEM < 0.21.
pKd SEM < 0.20.
*SPR data could not be reliably determined for UNC2025/MRX-6313.
Type 2 inhibitors
Merestinib is an inhibitor of c-Met and several other receptor tyrosine oncokinases, including MerTK [24], while LDC1267 was developed as a selective pan-TAM kinase inhibitor [25] (Table 1). The compounds occupy the ATP-pocket and cross the DFG motif to displace the sidechain of Phe742; a typical type 2 binding mode with ‘DFG-out’ conformation [7] (Figure 2). Despite the chemical similarity between merestinib and LDC1267, with a common F-aryl amide core, the MerTK kinase domain assumes surprisingly different conformations in the related crystal structures. While the LDC1267-complex formed the same crystals as the unliganded protein (structure 7AB0, Supplementary Table S1), merestinib co-crystallized with MerTK in ‘helix-C in’ conformation. Helix-C moved ∼3.4 Å toward the ATP-binding site when measured on the backbone carbon of Ser627. The new position enables the formation of the canonical salt bridge between Lys619 and conserved Glu637 on helix-C (Figure 2); one of the requisite interactions leading to a catalytically active kinase conformation. Along with this relocation goes a 50° rotation of the helical N-terminus (helix-A), which packs alongside helix-C now (Figure 2). This is the first report of a structure depicting MerTK in this active-like conformation. However, it is unclear whether the rearrangement of helices A and C happened as a result of merestinib binding, protein engineering or a combination of both. The protein construct co-crystallized with merestinib features a loop-truncation within the N-lobe (Supplementary Table S1), which could have contributed to the movement observed in the structure. The truncation melts the β-sheet formed by strands four and five and thereby makes room for the repositioning of helix-C (Figure 2). Type 2 binding describes compounds that displace the DFG motif phenylalanine (Phe742 in case of MerTK) from its native pocket to replace it with another hydrophobic group (Figure 2 and Figure 1). This design feature often leads to elevated compound hydrophobicity which, combined with the induced-fit binding mode, usually makes for slower kinetics than seen with type 1 compounds. In line with this general observation, merestinib remains bound to non-phosphorylated MerTK with a residence time of over 120 min and LDC1267 ∼13 min, significantly longer than type 1 compound gilteritinib (Table 1). Despite inducing the ‘DFG-out’ conformation upon binding, the type 2 molecules demonstrate single step kinetics in SPR experiments (Supplementary Figure S2). However, in contrast with gilteritinib, LDC1267 and merestinib exhibit a drop in potency and shortened residence times for the active/phosphorylated form of MerTK compared with the non-phosphorylated form (Table 1).
Figure 2. MerTK complexes with type 2 kinase inhibitors.

LDC1267-complex 7AAX is rendered in magenta, merestinib-complex 7AAY in purple. Alignment of the whole structures, using the CE algorithm (‘cealign’ in PyMOL). (A) Top and side view on the kinase domains. Alpha helices A and C (αA, αC) and beta strands 1–5 (β1–β5) of the kinase N-lobe are labelled. The merestinib structure depicts MerTK in ‘helix-C in’ conformation. Helix-C is repositioned 3.4 Å inward the ATP-pocket (measured on atom CA of Ser627 at the extremity of the helix), thereby enabling the formation of the canonical salt bridge between Lys619 and Glu637 (yellow dashes, distance in Å indicated). (B) Typical for type 2 compounds, both merestinib and LDC1267 displace Phe742 of the DFG motif.
EX172 — overall binding
Example 172 (EX172) is a is a dual MerTK/Axl inhibitor described in patent application US20170066742 (Table 1). The compound was selected from the 458 molecules exemplified in US20170066742 using ‘Frequency of Group’ (FOG) analysis [26]; based on the abundance of chemical moieties, the potency data provided and predicted physical properties (LogD, solubility).
Structure 7AAZ depicts how EX172 spans from the hinge region in the ATP-pocket to helix-C in the kinase back pocket, while maintaining a ‘DFG-in’ conformation (Figure 3). This binding mode renders EX172 a type 1.5 kinase inhibitor. The compound fills the space otherwise occupied by Phe742 when in ‘DFG-out’ with its cyclopentyl group and further rigidifies the DFG by placing its biphenyl moiety adjacent to it (Figure 3). EX172 features a terminal benzylpiperazine which contacts helix-C; packing against Phe634 and making H-bonds with conserved Glu637 (Figure 2); and interacts closely with the A-loop; packing against Tyr753 and making water-mediated H-bonds with Tyr754 and Gly743 (Figure 3). EX172 could not be soaked into preformed crystals. Instead it co-crystallized with MerTK in a unique conformation where the activation segment (A-loop) and the loop linking β-strand 3 and helix-C (helix-C loop) are stabilized by the compound (residues 744–767 and 621–630, respectively; Figure 3). The A-loop is recruited into forming a binding pocket around EX172 and itself acts as a scaffold for the helix-C loop. Neither of the two loops are usually resolved in MerTK structures due to flexibility and their conformation in EX172-complex 7AAZ is likely stabilized by interactions with the ligand. The terminal benzylpiperazine of EX172 appears crucial for reshaping the protein this way (Figure 3). A detailed analysis of A-loop flexibility in MerTK crystal structures can be found in Supplementary Figure S3.
Figure 3. MerTK in complex with EX172 (PDB entry 7AAZ).

The kinase domain N-lobe is rendered in white, the C-lobe in grey and the A-loop in orange. (A) Structure overview. EX172 packs its methylpiperazine against helix-C and stabilizes the A-loop. (B) The binding pocket in detail. Residues within 5 Å of the ligand are rendered as wires and the cavity they define as surface. H-bonds are depicted as yellow dashes with distances in Å indicated. (C) Model of the structure with A-loop tyrosine 754 phosphorylated. A phosphate would replace a water molecule which is found between tyrosine hydroxyl and EX172 piperazine in the original structure 7AAZ. A phosphate would directly contact the compound, an adjacent water and Arg758.
EX172 binds MerTK with a two-step mechanism
Characterization of the interaction between EX172 and the MerTK kinase domain by SPR results in biphasic sensorgrams composed of a rapid initial binding step, followed by a slower second process (Supplementary Figure S2). The observation that complex formation between MerTK and EX172 takes place in sequential events is in good agreement with the induced-fit conformation of MerTK seen in EXP172-complex structure 7AAZ (Figure 3). With the exception of 7AAZ and merestinib-complex 7AAY, all other MerTK structures published appear largely invariant and depict the same overall conformation of the kinase domain, including the position of helix-C (Supplementary Figure S3). In contrast with merestinib-complex 7AAY, which shows an inward movement of helix-C (Figure 2), EX172-complex 7AAZ depicts the helix sitting 1.9 Å further out when compared with the ‘default’ MerTK conformation (measured on the backbone carbon of Ile631 on aligned structures 7AAZ and 7AB0). This movement makes way for the bulky phenylpiperazine of EX172 (Figure 3), which clashes with apo-MerTK structure 7AB0 when superimposed. However, apo-MerTK can fit EX172 without structural changes when the benzylpiperazine is slightly rotated outward. Under the assumption that the ‘default’ MerTK conformation seen in more than ten crystal structures is the ground state, we propose a model where the placement of EX172 in the ATP- and back pocket of MerTK is the fast, initial binding event seen by SPR and the rearrangement of A-loop, helix-C loop and helix-C is the second. Induced-fit binding mechanisms have been reported to augment drug-target residence time in cases where the final complex represents a low-energy state [27]. That such effects can be used for protein kinases has recently been demonstrated in a case study on FAK and PYK2 [28]. The work describes how slow complex dissociation going along with a binding-induced conformational rearrangement of the DFG motif enable to design target selectivity into ATP-competitive inhibitors. It is likely that the conformational rearrangement of the MerTK kinase domain imposes structural barriers which limit compound dissociation and contribute to the comparatively long residence times determined for EX172 (Table 1 and Supplementary Figure S2).
EX172 binds to MerTK with phospho-selectivity
The engagement of the A-loop adds a second particular aspect to the binding between EX172 and MerTK, in addition to the two-step mechanism discussed above. Two of the A-loop residues which shape a pocket around the compound, Tyr753 and Tyr754 (Figure 3), have been reported as activity-modulating autophosphorylation sites [29]. Interestingly, phosphorylation of these residues does not impede the binding of EX172, but instead increases the compounds binding affinity ∼16-fold (Table 1). Since the structural characterization of phosphorylated MerTK protein failed, phosphorylation of Tyr753 and Tyr754 was simulated in a model (Figure 3). In brief: with the help of CCG MOE phosphate groups were added and the water molecule adjacent to Tyr754 removed; the structure was then minimized in an Amber force-field (resulting co-ordinates available in the Supplementary information). The modifications were accommodated in the model without significant structural rearrangements when compared with the input co-ordinates. Tyr753 places its phosphate at the centre of the helix-C loop (Figure 3) without forming H-bonds, while phospho-Tyr754 engages into numerous polar contacts with the piperazine of EX172, an adjacent water molecule and the sidechain of Arg758 (Figure 3). The additional polar contacts rationalize the gain in the potency of EX172 upon the phosphorylation of Mer. Hence, the structural model gives some confidence that the induced-fit conformation of non-phosphorylated MerTK seen in EX172-complex 7AAZ is also relevant in the context of phosphorylation on tyrosines 753 and 754 (Figure 3).
Kinome selectivity of EX172
EX172 was screened biochemically against a large set of human kinases to establish selectivity against the human kinome (ThermoFisher SelectScreen Kinase Profiling Services) (Figure 4, Supplementary Table S1). At a concentration of 0.1 μM the compound inhibited only three protein kinases to more than 35%; MerTK (99.5% inhibition), Axl (97.0%) and Chk2 (55.2%). Follow up dose-response assays gave IC50 (nM) values at 0.87 (Mer), 4.3 (Axl) and 102 (Chk2) showing that EX172 was 117-fold selective for MerTK and 24-fold selective for Axl compared with the next most inhibited kinase Chk2. Noteworthy, the third TAM family member Tyro3 was inhibited to 31.4% by 0.1 μM EX172, with a concentration-response assay resulting an IC50 of 532 nM. This result renders EX172 a dual MerTK/Axl inhibitor with a particularly clean kinome profile. The similar potency of EX172 for MerTK and Axl is in good agreement with the importance of A-loop interactions discussed above. The kinase domains of MerTK (E571-V864) and Axl (E520-A813) share 69% sequence identity overall and differ only in one sidechain on the A-loop; S750 in MerTK which corresponds to N699 in Axl (Supplementary Figure S1).
Figure 4. EX172 is a selective MerTK/Axl inhibitor.
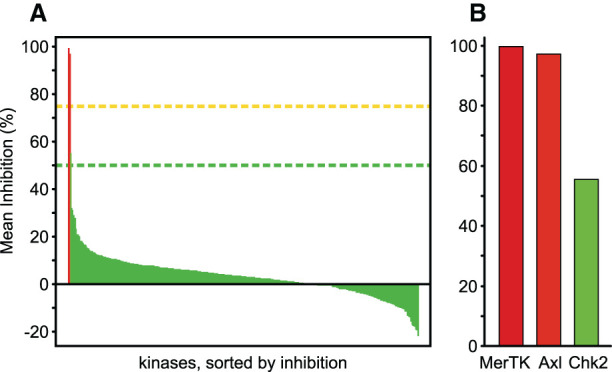
The compound was tested at a concentration of 0.1 μM in a biochemical screen against a panel of 395 human kinases (ThermoFisher SelectScreen Kinase Profiling Services). (A) Inhibition of all kinases tested. (B) Kinases with >35% inhibition in detail. The raw data are listed in Supplementary Table S2.
Discussion
A-loop interactions remain a curiosity among kinase inhibitors but have been reported in some cases and the potential for target selectivity discussed [9]. Here, we demonstrate that stabilization of A-loop interactions may give rise to both exquisite selectivity, as well as a specific kinetic profile. Furthermore, it could be envisioned that specific compounds enable selectivity for a particular phosphorylation state of the target, when interacting favourably with a phosphorylated residue. The lock-in binding mechanism seen between the A-loop of MerTK and EX172 results in residence times which compare well with the type 2 molecules tested. With slow enough dissociation times, the lifetime of the drug-target complex might surpass the compound's pharmacokinetic lifetime in circulation and positively affect pharmacodynamics, along the lines seen for covalent binders [30].
A-loop interactions benefit kinome selectivity
The A-loop is part of the kinase domain peptide-binding cleft, a region of lower sequence conservation, adjacent to the ATP-binding site [31]. Engaging into contacts with the loop, therefore, represents an opportunity to augment the target selectivity of ATP-pocket binders, when desired. The feasibility of this approach has been demonstrated with Necrostatins directed against RIPK1 [32]. This family of type 3 inhibitors binds in the kinase back pocket, stabilizing an inactive ‘helix-C out’ and ‘DLG-out’ conformation (RIPK1 belongs to a subset of kinases where the phenylalanine of the DFG motif is substituted by a leucine). Much like compound EX172 characterized here, Necrostatin analogue Nec-1 recruits the A-loop into forming a binding pocket and forms an H-bond with one of the A-loop phospho-acceptor residues, Ser161 [33]. The contact is productive as Nec-1 shows reduced binding to Ser161Ala substituted protein [33]. Nec-1 displayed exclusive selectivity towards RIPK1 in a biochemical screen of over 400 human kinases [34], a similar degree of selectivity as EX172 has demonstrated here (Figure 4). It is reasonable to assume that this selectivity is partially attributed to the sequence-specific interaction between the compounds and the kinase A-loop.
A-loop interactions enable phospho-selectivity
A key observation is that EX172 shows selectivity for the active/phosphorylated state over the unphosphorylated form of the protein (Table 1). The other examples of A-loop interacting compounds discussed here show the opposite effect. Type 2 compounds LDC1267 and merestinib bind the active MerTK kinase domain with reduced affinity and faster kinetics (Table 1), and the affinity of Nec-1 for RIPK1 drops ∼10-fold upon phosphorylation of Ser161, mimicked by a glutamate residue [33]. In contrast with the aforementioned inhibitors, the binding mode of EX172 can accommodate phosphorylation of the MerTK A-loop and benefits from additional polar contacts, resulting in higher potency than the other compounds (Figure 3, Table 1). This increased binding affinity of EX172 for active MerTK measured by SPR agrees well with the IC50 determined in a kinase activity assay (Table 1). The data on EX172 demonstrates that it is feasible to develop selective kinase inhibitors, tuned to favour a defined phosphorylation state of a kinase. This opens opportunities to modulate the drug phenotype, or simply to avoid large in vitro to in vivo potency drops by addressing the most biologically relevant form.
The first structure of MerTK in ‘helix-C in’ conformation
It is not clear whether the ‘helix-C out’ conformation is a common feature all kinases share. However, structural characterization of the kinome suggests that kinases have varying propensities to adopt the conformation (Figure 2). The back pocket ligand binding space made available by ‘helix-C out’ can, therefore, be utilized to design target selectivity into kinase inhibitors. In case of MerTK, it appears that the kinase domain easily assumes or even favours the ‘helix-C out’ conformation. Regardless, with merestinib-complex 7AAY we present a first structure depicting MerTK in an active-like form with ‘helix-C in’. Where desired, the structure can be utilized to design type 2 inhibitors for MerTK which are agnostic to the conformational state of helix-C.
Acknowledgements
We thank Dr. Jason Breed for crystallographic data collection, and the beamline scientists at DLS I02, I03 and I04 for technical assistance.
Abbreviations
- A-loop
activation loop/activation segment
- EX172
compound example 172
- helix-C loop
loop linking β-strand 3 and helix-C
- IMAC
immobilized metal affinity chromatography
- MerTK
Mer tyrosine kinase
- RTK
receptor tyrosine kinase
- SEM
standard error of the mean
Competing Interests
All authors are currently or have been (R.C.O and J.W.) employees and shareholders of AstraZeneca at the time of the study.
Funding
The study was funded solely by AstraZeneca.
CRediT Contribution
Alexander Pflug: Investigation, writing — original draft, writing — review and editing. Marianne Schimpl: Investigation, writing — review and editing. J. Willem M. Nissink: Investigation, writing — review and editing. Ross C. Overman: Investigation, writing — review and editing. Philip B. Rawlins: Investigation, writing — review and editing. Caroline Truman: Investigation, writing — review and editing. Elizabeth Underwood: Investigation, writing — review and editing. Juli Warwicker: Investigation, writing — review and editing. Jon J. Winter-Holt: Investigation, writing — review and editing. William McCoull: Investigation, writing — review and editing.
Supplementary Material
References
- 1.Lemke G. and Rothlin C.V. (2008) Immunobiology of the TAM receptors. Nat. Rev. Immunol. 8, 327–336 10.1038/nri2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paolino M. and Penninger J.M. (2016) The role of TAM family receptors in immune cell function: implications for cancer therapy. Cancers (Basel) 8, 97 10.3390/cancers8100097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu Q. and Lemke G. (2001) Homeostatic regulation of the immune system by receptor tyrosine kinases of the tyro 3 family. Science 293, 306–311 10.1126/science.1061663 [DOI] [PubMed] [Google Scholar]
- 4.Cook R.S., Jacobsen K.M., Wofford A.M., DeRyckere D., Stanford J., Prieto A.L., et al. (2013) MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Invest. 123, 3231–3242 10.1172/JCI67655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myers K.V., Amend S.R. and Pienta K.J. (2019) Targeting Tyro3, Axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Mol. Cancer 18, 94 10.1186/s12943-019-1022-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huelse J.M., Fridlyand D.M., Earp S., DeRyckere D. and Graham D.K. (2020) MERTK in cancer therapy: targeting the receptor tyrosine kinase in tumor cells and the immune system. Pharmacol. Ther. 213, 107577 10.1016/j.pharmthera.2020.107577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Modi V. and Dunbrack R.L. (2019) Defining a new nomenclature for the structures of active and inactive kinases. Proc. Natl. Acad. Sci. U.S.A. 116, 6818–6827 10.1073/pnas.1814279116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fabbro D., Cowan-Jacob S.W. and Moebitz H. (2015) Ten things you should know about protein kinases: IUPHAR review 14. Br. J. Pharmacol. 172, 2675–2700 10.1111/bph.13096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suebsuwong C., Pinkas D.M., Ray S.S., Bufton J.C., Dai B., Bullock A.N. et al. (2018) Activation loop targeting strategy for design of receptor-interacting protein kinase 2 (RIPK2) inhibitors. Bioorg. Med. Chem. Lett. 28, 577–583 10.1016/j.bmcl.2018.01.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang X., Finerty P., Walker J.R., Butler-Cole C., Vedadi M., Schapira M. et al. (2009) Structural insights into the inhibited states of the Mer receptor tyrosine kinase. J. Struct. Biol. 165, 88–96 10.1016/j.jsb.2008.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tickle I.J., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C. et al. (2018) STARANISO, Global Phasing Ltd, Cambridge, U.K [Google Scholar]
- 12.Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W. et al. (2011) Data processing and analysis with the autoPROC toolbox. Acta Cryst. D 67, 293–302 10.1107/S0907444911007773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans P.R. and Murshudov G.N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 10.1107/S0907444913000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C. and Read R.J. (2007) Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Navaza J. (1994) AMore: an automated package for molecular replacement. Acta Cryst. A 50, 157–163 10.1107/S0108767393007597 [DOI] [Google Scholar]
- 17.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Cryst. D 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., et al. (2017) BUSTER Version 2.11.7, Global Phasing Ltd., Cambridge, U.K [Google Scholar]
- 19.Emsley P., Lohkamp B., Scott W.G. and Cowtan K. (2010) Features and development of Coot. Acta Cryst. D 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murshudov G.N., Vagin A.A. and Dodson E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. D 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 21.Derewenda Z.S. (2004) Rational protein crystallization by mutational surface engineering. Structure 12, 529–535 10.1016/j.str.2004.03.008 [DOI] [PubMed] [Google Scholar]
- 22.Mori M., Kaneko N., Ueno Y., Yamada M., Tanaka R., Saito R. et al. (2017) Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Invest. New Drugs 35, 556–565 10.1007/s10637-017-0470-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang W., DeRyckere D., Hunter D., Liu J., Stashko M.A., Minson K.A., et al. (2014) UNC2025, a potent and orally bioavailable MER/FLT3 dual inhibitor. J. Med. Chem. 57, 7031–7041 10.1021/jm500749d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan S.B., Peek V.L., Ajamie R., Buchanan S.G., Graff J.R., Heidler S.A., et al. (2013) LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Invest. New Drugs 31, 833–844 10.1007/s10637-012-9912-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paolino M., Choidas A., Wallner S., Pranjic B., Uribesalgo I., Loeser S., et al. (2014) The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 507, 508–512 10.1038/nature12998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tyrchan C., Boström J., Giordanetto F., Winter J. and Muresan S. (2012) Exploiting structural information in patent specifications for key compound prediction. J. Chem. Inf. Model. 52, 1480–1489 10.1021/ci3001293 [DOI] [PubMed] [Google Scholar]
- 27.Copeland R.A. (2016) The drug–target residence time model: a 10-year retrospective. Nat. Rev. Drug Discov. 15, 87–95 10.1038/nrd.2015.18 [DOI] [PubMed] [Google Scholar]
- 28.Berger B.-T., Amaral M., Kokh D.B., Nunes-Alves A., Musil D., Heinrich T., et al. (2020) Structure-Kinetic-Relationship Reveals the Mechanism of Selectivity of FAK Inhibitors Over PYK2, SSRN Scholarly Paper, Social Science Research Network, Rochester, NY
- 29.Ling L., Templeton D. and Kung H.J. (1996) Identification of the major autophosphorylation sites of Nyk/Mer, an NCAM-related receptor tyrosine kinase. J. Biol. Chem. 271, 18355–18362 10.1074/jbc.271.31.18355 [DOI] [PubMed] [Google Scholar]
- 30.Bradshaw J.M., McFarland J.M., Paavilainen V.O., Bisconte A., Tam D., Phan V.T., et al. (2015) Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 11, 525–531 10.1038/nchembio.1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knight J.D.R., Qian B., Baker D. and Kothary R. (2007) Conservation, variability and the modeling of active protein kinases. PLoS One 2, e982 10.1371/journal.pone.0000982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Degterev A., Hitomi J., Germscheid M., Ch'en I.L., Korkina O., Teng X., et al. (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 10.1038/nchembio.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie T., Peng W., Liu Y., Yan C., Maki J., Degterev A. et al. (2013) Structural basis of RIP1 inhibition by necrostatins. Structure 21, 493–499 10.1016/j.str.2013.01.016 [DOI] [PubMed] [Google Scholar]
- 34.Christofferson D.E., Li Y., Hitomi J., Zhou W., Upperman C., Zhu H. et al. (2012) A novel role for RIP1 kinase in mediating TNF α production. Cell Death Dis. 3, e320 10.1038/cddis.2012.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bond C.S. and Schüttelkopf A.W. (2009) ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Cryst. D 65, 510–512 10.1107/S0907444909007835 [DOI] [PubMed] [Google Scholar]
- 36.Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., et al. (2018) Molprobity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 10.1002/pro.3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.