

Abstract
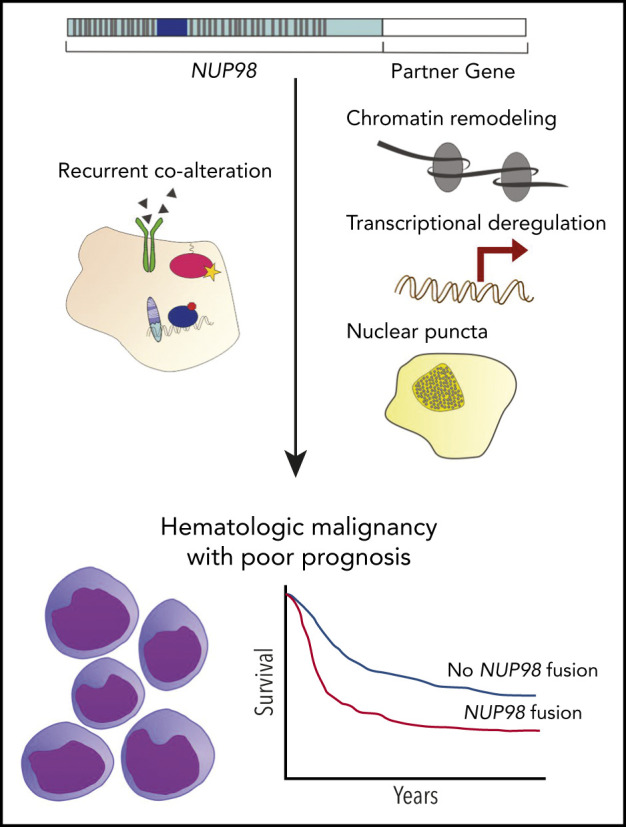
Nucleoporin 98 (NUP98) fusion oncoproteins are observed in a spectrum of hematologic malignancies, particularly pediatric leukemias with poor patient outcomes. Although wild-type full-length NUP98 is a member of the nuclear pore complex, the chromosomal translocations leading to NUP98 gene fusions involve the intrinsically disordered and N-terminal region of NUP98 with over 30 partner genes. Fusion partners include several genes bearing homeodomains or having known roles in transcriptional or epigenetic regulation. Based on data in both experimental models and patient samples, NUP98 fusion oncoprotein–driven leukemogenesis is mediated by changes in chromatin structure and gene expression. Multiple cofactors associate with NUP98 fusion oncoproteins to mediate transcriptional changes possibly via phase separation, in a manner likely dependent on the fusion partner. NUP98 gene fusions co-occur with a set of additional mutations, including FLT3–internal tandem duplication and other events contributing to increased proliferation. To improve the currently dire outcomes for patients with NUP98-rearranged malignancies, therapeutic strategies have been considered that target transcriptional and epigenetic machinery, cooperating alterations, and signaling or cell-cycle pathways. With the development of more faithful experimental systems and continued study, we anticipate great strides in our understanding of the molecular mechanisms and therapeutic vulnerabilities at play in NUP98-rearranged models. Taken together, these studies should lead to improved clinical outcomes for NUP98-rearranged leukemia.
Visual Abstract
Introduction
Chromosomal translocations represent critical genetic lesions in many cancer types, including childhood leukemias. The majority of pediatric leukemia patients display abnormal karyotypes, including ∼75% of children with acute myeloid leukemia (AML).1 Furthermore, with the advent of genome and transcriptome sequencing, cryptic events involving translocations not evident using conventional karyotyping have been identified.2 Chromosomal alterations commonly result in the aberrant fusion of 2 genes to generate a product that may possess oncogenic potential, in part through deregulation of the involved genes.
Although chromosomal translocations were among the first recognized alterations in human malignancies, studies detailing the molecular underpinnings of oncogenic fusion proteins continue to provide deeper insights into their biological consequences. The presence or absence of various gene fusions is now regularly used for risk stratification in clinical trials for leukemia patients.3-6 Furthermore, the expression of fusion oncoproteins specifically in malignant cells represents an important opportunity for therapeutic targeting; this principle was first successfully demonstrated in the development of imatinib for individuals with chronic myeloid leukemia (CML) and other malignancies expressing the BCR-ABL1 fusion oncoprotein.7,8
Structure and function of NUP98 in normal cells
A recurrent translocation of increasingly appreciated importance, especially in children with leukemia, involves the nucleoporin 98 (NUP98) gene at chromosome 11p15. NUP98 is a part of the nuclear pore complex, a multiprotein structure responsible for the transport of various macromolecules into and out of the nucleus. NUP98 resides at the center of the nuclear pore complex,9,10 and proper localization is dependent on autoproteolytic cleavage into the N-terminal 90-kDa and C-terminal 8-kDa polypeptides.11 Although NUP98 mediates selective transport of RNA molecules between the cytoplasm and nucleus, it also has functions in transcriptional regulation and mitotic progression.12-14
Structurally, the N terminus of NUP98 consists of a series of intrinsically disordered phenylalanine-glycine (FG)/glycine-leucine-phenylalanine-glycine (GLFG) repeats,15 which interact with cofactors including CREB-binding protein (CREBBP) and p300 (EP300), as well as export factors XPO1 (also known as CRM1) and TAP.16-18 The FG/GLFG repeat domain is split by a Gle2-binding-sequence (GLEBS) domain capable of binding RNA export factor RAE1.19 The C-terminal portion of NUP98 contains RNA-binding and autoproteolytic cleavage sites (Figure 1A).11
Figure 1.

Stucture of wild-type NUP98 and NUP98 fusions. (A) Structure of wild-type NUP98. (B) Structure of NUP98 fusions, including relevant functional domains and malignancies reported for each partner gene. The longest fusion gene is shown along with alternate breakpoints. Arrows denote breakpoints, and asterisk (*) denotes fusion partners for which multiple isoforms have been identified. CMML, chronic myelomonocytic leukemia; HMG, high-mobility group; IQ, isoleucine glutamine; JMML, juvenile myelomonocytic leukemia; MYST, MOZ, Ybf2, Sas2, and TIP60; PHD, plant homeodomain; SET, Su(var)3-9, enhancer-of-zeste and trithorax.
NUP98 fusions involve many partner genes with some overlapping features
A role for NUP98 in cancer was first described in 1996, when Nakamura et al and Borrow et al independently reported that NUP98 was fused to HOXA9 in AML cases bearing t(7;11)(p15;p15).20,21 Since this initial discovery, over 30 NUP98 fusion partners have been identified in patients with hematologic malignancies, including both de novo and therapy-related AML, myelodysplastic syndrome (MDS),22,23 CML,24,25 T-cell acute lymphoblastic leukemia (T-ALL),26,27 and mixed-phenotype acute leukemia (MPAL) (Table 1; Figure 1B).28,29 NUP98 fusions are present most frequently in childhood AML, representing ∼5% of patients.4,30-33
Table 1.
NUP98 fusion partner genes and characteristics
| Fusion partner | Chr. | HOX gene? | Transcription-related domain/function | Disease | Ref. |
|---|---|---|---|---|---|
| ADD3 | 10q25 | No | None identified | Therapy-related AML, T-ALL | 152 |
| ANKRD28 | 3p25 | No | None identified | MDS/AML | 153 |
| BPTF | 17q24 | No | PHD domain | T-ALL, AML | 45 |
| CCD28A (C6orf80) | 6q24 | No | None identified | AML, T-ALL | 154 |
| DDX10 | 11q22 | No | RNA helicase, involved in ribosome assembly | De novo or therapy-related MDS and AML, CML, CMML | 22 |
| GSX2 | 4q12 | Yes | HOX | AML | 40 |
| HHEX | 10q23 | Yes | HOX | AML | 39 |
| HMGB3 | Xq28 | No | Nucleosome remodeling (HMG box) | Therapy-related AML | 79 |
| HOXA9 | 7p15 | Yes | HOX | De novo or therapy-related AML, MDS, CML, CMML | 20,21 |
| HOXA11 | 7p15 | Yes | HOX | CML, JMML | 24 |
| HOXA13 | 7p15 | Yes | HOX | De novo AML, MDS, CML, CMML | 24 |
| HOXC11 | 12q13 | Yes | HOX | De novo or therapy-related AML | 35 |
| HOXC13 | 12q13 | Yes | HOX | De novo or therapy-related AML | 155 |
| HOXD11 | 2p31 | Yes | HOX | De novo AML | 37 |
| HOXD13 | 2p31 | Yes | HOX | De novo or therapy-related AML, CML | 38 |
| IQCG | 3q29 | No | None identified | MPAL | 29 |
| JADE2 | 5p31 | No | PHD domain | JMML | 46 |
| KAT7 (HBO1) | 17q21 | No | HAT | CMML | 52 |
| KDM5A (JARID1A) | 12p13 | No | PHD domain, H3K4 demethylation | AML | 47 |
| LNP1 | 3q12 | No | None identified | AML | 156 |
| KMT2A (MLL) | 11q23 | No | H3K4 methyltransferase | AML | 51 |
| MLLT10 (AF10) | 10p12 | No | OM-LZ domain, putative transcription factor | Atypical MDS resembling CMML | 40 |
| NSD1 | 5q35 | No | SET and PHD domains | MDS, AML, MPAL | 48 |
| NSD3 | 8p11 | No | SET and PHD domains | MDS, de novo or therapy-related AML | 49 |
| PHF23 | 17p13 | No | PHD domain | AML | 50 |
| PRRX1 | 1q24 | Yes | HOX | AML, CML, therapy-related MDS | 41 |
| PRRX2 | 9q34 | Yes | HOX | Therapy-related AML | 42 |
| POU1F1 | 3p11 | Yes | HOX | Therapy-related AML | 43 |
| PSIP1 (LEDGF) | 9p22 | No | Transcriptional coactivator | AML, CML, MDS | 53 |
| RAP1GDS1 | 4q23 | No | None identified–G-protein signaling | T-ALL, AML | 26 |
| RARA | 17q21 | No | DNA-binding domain | AML | 157 |
| RARG | 12q13 | No | DNA-binding domain | AML | 158 |
| SETBP1 | 18q12 | No | SET-binding protein | T-ALL | 27 |
| TOP1 | 20q12 | No | Topoisomerase | MDS, AML | 159 |
| TOP2B | 3p24 | No | Topoisomerase | De novo AML | 160 |
| VRK1 | 14q32 | No | None identified | T-ALL | 161 |
Chr., chromosome; CMML, chronic myelomonocytic leukemia; HAT, histone acetyltransferase; HMG, high-mobility group; JMML, juvenile myelomonocytic leukemia; MPN, myeloproliferative neoplasm; OM-LZ, octapeptide motif leucine zipper; Ref., reference.
NUP98 fusion oncoproteins universally involve the N-terminal portion of NUP98 (including the functional FG/GLFG repeat and GLEBS domains) and the C-terminal portion of the fusion partner. Confirming a conserved and critical role for these regions in leukemogenesis, the reciprocal fusion products are variably expressed.34 Although 3′ rapid amplification of complementary DNA ends, reverse transcription–polymerase chain reaction, and fluorescence in situ hybridization identified several of the first identified NUP98 fusions, many NUP98 fusions are cryptic on conventional cytogenetic analysis. Accordingly, only the recent development of next-generation sequencing technologies has revealed their high prevalence in pediatric leukemias.
NUP98 fusion partners can be separated into 2 categories: those with and without a homeodomain moiety (Table 1; Figure 1B). Seven clustered “class I” HOX genes (HOXA9,20,21 HOXA11,24 HOXA13,24 HOXC11,35 HOXC13,36 HOXD11,37 and HOXD1338) and 5 nonclustered “class II” HOX genes (HHEX,39 GSX2,40 PRRX1,41 PRRX2,42 and POU1F143) are among the partners with retained homeodomains. Of the NUP98-HOX gene fusions, NUP98-HOXA9 is the most common and best studied, and it has been used as an experimental model for the other NUP98-HOX gene fusions.
Non-HOX gene fusion partners are observed more frequently in hematologic malignancies. Most of the genes in this fusion-partner family share a coiled-coil domain.44 Because these domains exist in only 3% to 5% of all genes, this commonality supports an important role for this moiety in leukemogenesis, although the specific contribution still requires further investigation. Additional domains are observed in multiple non-HOX gene fusion partners, including plant homeodomain (PHD) domains (BPTF,45 JADE2,46 KDM5A [JARID1A],47 MLLT10,40 NSD1,48 NSD3,49 and PHF2350) and SET domains (KMT2A [MLL],51 NSD1,48 and NSD349) (Figure 1B). Furthermore, other fusion partners include domains with roles in transcriptional and/or epigenetic regulation: SETBP1 is a SET-binding protein,27 KAT7 (HBO1) is a histone acetyltransferase,52 and PSIP1 is a transcriptional coactivator.53
Some NUP98 fusion oncoproteins are more frequent in distinct disease subsets. NUP98 rearrangements with NSD1 are the most common and have been observed primarily in myelomonocytic leukemias (French-American-British [FAB] M4 and M5 AML).31,34 NUP98-KDM5A fusion oncoproteins are observed in monocytic, erythroid, and megakaryoblastic leukemia (FAB M5, M6, and M7).3,54,55 Finally, in T-ALL, RAP1GDS1 is the most common NUP98 fusion partner.56
NUP98 rearrangements predict poor prognosis
Early case reports noted unfavorable outcomes in NUP98-rearranged leukemia patients,53,57,58 and later studies including larger cohorts of patients have confirmed that the presence of NUP98 gene fusion defines a high-risk leukemia subset (Table 2).3,4,31-34,55,59-69 For example, a report by Marceau-Renaut et al including 10 NUP98 fusion-positive pediatric AML patients demonstrated that these children had high rates of induction failure, a trend that remained significant after multivariate analysis. This cohort also exhibited only 10% event-free survival 3 years following diagnosis.60 Other studies have reached similar conclusions regarding the poor outcome of children with NUP98 fusions and have extended these observations to include the contribution of co-occurring mutations, such as FLT3–internal tandem duplication (FLT3-ITD) and WT1.4,32,64,70 Notably, Ostronoff and coauthors70 observed that AML patients harboring NUP98 fusions with FLT3-ITD display rates of relapse and of death twice as high as rates in a cohort of similarly diagnosed patients without these genetic aberrations.33 In examining the results of multiple Children’s Oncology Group (COG) trials, Bolouri et al identified significantly lower 10-year overall survival rates in pediatric AML patients with FLT3-ITD mutation and WT1 alteration or NUP98-NSD1 gene fusion as compared with patients without FLT3-ITD mutation or FLT3-ITD mutation alone.4 Furthermore, in an additional study, nearly 50% of children with chemotherapy-resistant AML harbored NUP98 gene fusions.71
Table 2.
Outcomes of NUP98-rearranged hematological malignancy
| Tumor | Age | No. of patients | Fusion partner | Outcome | Reference | |
|---|---|---|---|---|---|---|
| Total | Fusion | |||||
| AML | Pediatric | 124 | 6 | NSD1 | 33.3% 4-y EFS and OS | 59 |
| AML | Pediatric | 246 | 15 | NSD1 | 13%, 52%, 81% 3-y EFS, OS, CIR, respectively | 32 |
| AML | Pediatric | 181 | 25 | NSD1 | 25% and 33% EFS with and without FLT3 alteration, respectively | 33 |
| AML | Pediatric | 98 | 18 | Mostly NSD1 (also KDM5A, TOP1) | 30% and 48% 5-y DFS and OS, respectively | 31 |
| AML | Pediatric | 385 | 10 | 9 NSD1, 1 KDM5A | 10% and 25% EFS and OS, respectively | 60 |
| AML | <3 y | 232 | 6 | KDM5A | 33.3%, 16.7%, 83.3% 3-y OS, EFS, CIR, respectively | 61 |
| AEL | Pediatric | 35 | 7 | 5 KDM5A, 2 NSD1 | ∼35% 5-y OS | 3 |
| AMKL | Pediatric | 87 | 5 | KDM5A | 35%, 25%, 62% 5-y OS, EFS, CIR, respectively | 62 |
| AMKL | Pediatric | 153 | 14 | KDM5A | 36%, 36%, 36% 4-y OS, EFS, CIR, respectively | 63 |
| FLT3-ITD+ AML | Pediatric | 44 | 8 | NSD1 | 4% complete response rate | 64 |
| AML | Pediatric | 238 | 12 | NSD1 | 31%, 8%, 83% 4-y OS, EFS, CIR, respectively | 34 |
| AML | Adult | 727 | 9 | NSD1 | 11%, 0%, 89% 4-y OS, EFS, CIR, respectively | 34 |
| AML | Pediatric | 2381 | 46 | KDM5A | 34%, 30%, 64% 4-y OS, EFS, RR, respectively | 55 |
| AML | Adult | 17 | 16 | HOXA9 | 4-mo median DFS, 8-mo median OS | 65 |
| AML | Adult | 493 | 11 | HOXA9 | 13.5-mo median OS, 6-mo median DFS | 66 |
| AML | Adult | 257 | 6 | NSD1 | 1.8-mo median EFS | 67 |
| AML | Younger adult, <60 y | 504 | 7 | NSD1 | 43% complete response rate | 68 |
AEL, acute erythroid leukemia; AMKL, acute megakaryoblastic leukemia; CIR, cumulative incidence of relapse; DFS, disease-free survival; EFS, event-free survival; OS, overall survival; RR, relapse rate.
Mechanisms of NUP98 fusion oncoprotein-mediated leukemogenesis
Mouse models of many NUP98 fusion oncoproteins have been developed using a variety of experimental approaches. These include (1) transplantation of mouse hematopoietic stem or progenitor cells (HSPCs) transduced with retroviral/lentiviral vectors expressing NUP98 fusion oncoproteins into irradiated recipients3,39,52,72-81; (2) transgenic or “knockin” genetically engineered mouse models (GEMMs)82-86; (3) transplantation of modified human CD34+ cells into immunocompromised animals87-89; or (4) establishment of patient-derived xenografts in immunocompromised mice33,90 (Table 3). Each approach has generated models that are capable of recapitulating various aspects of human disease, provided mechanistic insights, and/or validated important cofactors or concomitant genetic alterations. The most widely used mouse model, a transgenic model that expresses NUP98-HOXD13 controlled by the Vav promoter, was developed by Aplan and colleagues.83 It provides a faithful representation of MDS, which progresses to various acute leukemias in a subset of animals after long latency.83 Current mouse models (summarized in Table 3) are discussed further in relevant sections later in text.
Table 3.
Mouse models of NUP98 fusion oncoproteins
| Model type | Fusion | Disease and/or key findings | Co-lesion |
|---|---|---|---|
| HSPC transplant | NUP98-HOXA9 | MPD progressing to AML, first proved oncogenicity of NUP98 fusions in vivo72 |
Meis1 (A)72 BCR-ABL1(A)122 ETV6 (TEL)-PDGFRB (A)122 Kmt2a−/− (D)106,120 FLT3-ITD (A)120 |
| HSPC transplant | NUP98-HOXD13 | MPD progressing to leukemia73 | Meis1 (A)73 |
| HSPC transplant | NUP98-TOP1 | MPD progressing to leukemia74 | Meis174 |
| HSPC transplant | NUP98-HOXA10 | MPD75 | Meis1 (A)75 |
| HSPC transplant | NUP98-NSD1 | AML76 | FLT3-ITD (A)118 |
| HSPC transplant | NUP98-HHEX | AML39 | |
| HSPC transplant | NUP98-PRRX1 | MPD78 | Meis1 (A)78 |
| HSPC transplant | NUP98-KDM5A | AML3,77 | |
| HSPC transplant | NUP98-HMGB3 | MPD79 | |
| HSPC transplant | NUP98-CCDC28A | MPD with a few mice developing AML80 | |
| HSPC transplant | NUP98-IQCG | AML81 | |
| HSPC transplant | NUP98-KAT7 | CMML52 | |
| GEMM | NUP98-HOXA9 | MPD progressing to AML82 | |
| GEMM | NUP98-HOXD13 | MDS progressing to AML83 |
FLT3-ITD (A)119 Bcl2 (D)113 Cdkn2b (p15Ink4b)−/− (A)115 Bbc3 (Puma)−/− (D)114 Ep300 (p300)−/− (A)98 Crebbp (Cbp)−/− 98 |
| GEMM | NUP98-TOP1 | AML after very long latency84 | |
| GEMM | NUP98-RAP1GS1 | AML after very long latency84 | |
| GEMM | NUP98-PHF23 | Various acute leukemias85 | |
| GEMM | NUP98-KMT2A | MDS progressing to AML86 | |
| CD34+ | NUP98-HOXA9 | Engraftment and myelomonocytic blasts after 5-7 wk87 | |
| CD34+ | NUP98-HOXD13 | AML88 | MN1 (A)88 |
| CD34+ | NUP98-KDM5A | AML89 | |
| PDX | NUP98-KDM5A | Engraftment with primary and recurrence from same patient in all mice tested90 | |
| PDX | NUP98-NSD1 | Engraftment after 5-6 mo33 | |
| PDX | NUP98-NSD1 | Multiple distinct patient samples162 |
(A), accelerated disease with co-lesion; (D), delayed disease with co-lesion; MPD, myeloproliferative disease; PDX, patient-derived xenograft. See Table 1 for expansion of other abbreviations.
NUP98 fusion oncoproteins exploit the functions of wild-type NUP98 and especially the transcriptional and/or chromatin-modifying activities of the partner genes to drive malignancy. Upregulated expression of Hoxa9, Hoxa7, and Meis1 were among the first transcriptional changes observed following NUP98-HOXA9 expression, and broader gene-expression analyses in myeloid cells confirmed the numerous transcriptional effects of the NUP98-HOXA9 fusion oncoprotein as compared with NUP98 or HOXA9 individually.91,92 Although upregulation of the Hoxa gene cluster has been observed in systems with NUP98 fused to various partners, exceptions do exist51,79 and NUP98-HOXA9 expression in marrow lacking Hoxa9 does not prevent immortalization.91 Furthermore, gene-expression profiling of NUP98-NSD1 or NUP98-KDM5A AML patients revealed transcriptional changes involving the upregulation of not only HOXA but also HOXB gene expression. This pattern differentiated NUP98-rearranged from KMT2A-rearranged samples, the latter of which display selective upregulation of the HOXA genes, and defines a unique transcriptional signature for AMLs with NUP98 rearrangements.34,54 Mouse models have demonstrated that additional conserved transcriptional targets, including CDK6, may be important in leukemogenesis.93
The molecular process mediating gene-expression changes in NUP98-rearranged cells is dependent on chromatin state, as reported by Wang and colleagues.76,77 In their studies, cells expressing NUP98-NSD1, NUP98-KDM5A, or NUP98-PHF23 fusion oncoprotein bound near the Hoxa gene loci along with transcriptional cofactors (described later in this section). Chromatin remodeling then allowed access to DNA regions that are otherwise tightly coiled during differentiation, which led to aberrant transcription of Hoxa and other important genes. This process was dependent on important functional domains in NUP98 fusion partners: the SET domain was required for an increase in transcription-activating H3K36 methylation, the PHD3 domain mediated transcription-activating H3K4 methylation, and either domain could decrease transcription-repressive H3K27 methylation patterns. Mutation or loss of the SET or PHD3 domain, respectively, significantly reduced proliferation rates in hematopoietic cells,76,77 illustrating the necessity of these mechanisms for cellular transformation.
Changes in chromatin remodeling and/or gene expression, however, require the temporal and spatial concentration of many pieces of cellular machinery. Recently, the formation of “transcription centers” has been demonstrated via the process of phase separation.94 Importantly, the intrinsically disordered FG/GLFG repeat domains in wild-type NUP98 mediate phase separation,95 and several NUP98 fusion oncoproteins have been shown to localize in nuclear puncta.34,96,97 An attractive hypothesis is that NUP98 fusion oncoproteins and other cofactors come together in membraneless organelles to deregulate transcription and drive leukemogenesis (Figure 2A).
Figure 2.

NUP98 regulates transcription and cell cycle progression through interactions with cofactors. (A) NUP98 fusion oncoproteins bind to various transcriptional cofactors (including XPO1, WDR-SET1-COMPASS, and/or KMT2A complexes) and remodel chromatin. This leads to expression of target genes, possibly through the formation-phase separatory transcription centers. (B) Wild-type NUP98 associates with the anaphase-promoting complex (APC/C), RAE1, and other cell-cycle proteins, contributing to regulation of the spindle-assembly checkpoint. (C) NUP98 fusion oncoproteins bind to components of the APC/C including CDC20, leading to the ubiquitination and premature degradation of securin. In turn, activation of separation causes premature spindle-assembly checkpoint. Ac, acetyl; Me, methyl; MOF, males absent on the first; NSL, nonspecific lethal; P, phospho; Ub, ubiquitin.
A number of additional proteins have indeed been shown to interact in complex with NUP98 fusions, and many of these cofactors contribute to the activation and repression of genes in the unique transcriptional signature described herein. CREBBP and EP300 were among the first such cofactors to be identified, and these transcriptional coactivators interact with the FG/GLFG repeat domains of NUP98.96 Expression of adenovirus E1A, which blocks the activity of CREBBP/EP300, specifically prevents the transcriptional activity of NUP98-HOXA9.96 CREBBP is likely less important than EP300 in this context, as loss of CREBBP in NUP98-HOXD13–transgenic mice does not affect disease development.98 Alternatively, EP300 binds the Hoxa gene locus in hematopoietic progenitors expressing NUP98-NSD1 or NUP98-KDM5A.76,77 Following expression of NUP98-HOXA9, EP300 also binds at loci for other upregulated genes, including Meis1 and Pbx3.99 To evaluate the requirement of EP300 in NUP98-rearranged leukemias, Yung and coauthors replaced the N-terminal NUP98 portion of a fusion gene with the EP300-interaction domain from E1a. This experiment showed that the histone acetyltransferase activity of EP300 alone is not responsible for the transformation of hematopoietic cells,100 suggesting that additional cofactors may play important, overlapping roles. Indeed, another critical cofactor in this system is the transcriptional repressor histone deacetylase 1 (HDAC1), which deacetylates genes that are downregulated upon NUP98-HOXA9 expression.99
The role of transcriptional cofactors capable of interacting with HOX genes has also been demonstrated in mouse models and human samples. Perhaps the most notable of these cofactors is MEIS1, as overexpression of Meis1 accelerates leukemogenesis in multiple NUP98 fusion mouse models.72,73,75,78,101 The interaction between MEIS1 and NUP98 fusion oncoproteins is likely indirect, as the MEIS1-binding site is lost in the N-terminal portion of HOX genes when fused with NUP98. Although this mechanism does not depend on the ability of MEIS1 to bind DNA,101 multiple functional domains in MEIS1 are required for its proleukemogenic effects in NUP98-HOXD13 bone marrow transplant recipients.102 Furthermore, Meis1 expression is not universally critical in NUP98-rearranged malignancies: latency in a NUP98-TOP1 bone marrow transplant model was not affected by coexpression of Meis1,74 and accumulating evidence suggests that other members of the MEIS family can contribute to leukemogenesis. For instance, expression of MEIS2 was upregulated in NUP98-rearranged patients from 2 large studies of FAB M6 or M7 AML3,62 and in a mouse model with CD34+ cells transduced with NUP98-HOXD13 and MN1.88,103 Interestingly, although MEIS2 expression has not been directly tested with in vivo models of NUP98 fusion oncoproteins, higher MEIS2 expression accelerates disease onset in other leukemia models.103,104 Available data suggest that there is a core subset of genes comprising the transcriptional signature of NUP98-rearranged leukemias, but with unique targets dysregulated in cases with specific genotypes; these targets may explain lineage specificity or other distinct phenotypes and require further study.
Members of other histone-modifying complexes also interact with NUP98 fusion proteins. Chromatin-immunoprecipitation studies show common DNA-binding sites for NUP98 fusions and KMT2A or WDR-SET1-COMPASS complexes.105,106 These multisubunit complexes contain proteins with SET methyltransferase domains that contribute to increased transcription. Their binding sites overlap not only with those of NUP98 fusion oncoproteins but also with histone H3 lysine 4 (H3K4) methylation and/or H4K16 acetylation marks of activated chromatin and lead to upregulated expression of Hoxa/b gene clusters.106 In vivo, Kmt2a (Mll) knockout prevents self-renewal and delays leukemia onset in NUP98-HOXA9 bone marrow recipients.106 Similarly, loss of the H4K16 acetyltransferase Kat8 (Mof) extends latency in a transplant model with coexpression of NUP98-HOXA9 and Meis1a.107 Although KMT2A and associated proteins seem to be common cofactors with multiple NUP98 fusion oncoproteins involving different fusion partners (including HOXA9, HOXD13, NSD1, PHF23, and TOP1),106 additional cofactors may vary by genotype; these interactions have not been fully characterized. Moreover, whether multiple complexes are capable of simultaneously binding any NUP98 fusion oncoprotein has yet to be determined.
Nuclear export protein XPO1 also has been identified in nuclear puncta, interacting with the FG/GLFG repeat domains of NUP98 in a Ran guanosine triphosphate–dependent manner.108 Furthermore, in mouse embryonic stem cells overexpressing NUP98-HOXA9, the Hox loci are DNA-binding sites for XPO1, RNA polymerase II, and fusion oncoproteins.109 Treatment with inhibitor of XPO1 cargo-binding leptomycin B or with HDAC inhibitor trichostatin A reduces DNA binding near Hox gene promoters and alters the localization of nuclear puncta.109 Taken together, these data suggest a link between XPO1, NUP98 fusions, and phase separation. Moreover, fusion protein expression leads to nuclear retention of XPO1 cargo transcription factors, including NF-κB and nuclear factor of activated T cells, perhaps further explaining changes in transcription following NUP98 rearrangement.108
Aside from their role in transcription, the NUP98 fusion oncoproteins alter formation of the mitotic spindle,110 consistent with the function of wild-type NUP98 in cell-cycle progression.13 Throughout mitosis, NUP98 fusion oncoproteins (unlike wild-type NUP98) interact with the CDC20 component of the anaphase-promoting complex/cyclosome (APC/C); this interaction mediates premature degradation of the anaphase-inhibitory protein securin111 and dysregulation of the mitotic checkpoint complex (Figure 2B-C).110 Together, these events may explain why up to 35% of cells expressing NUP98 fusion oncoproteins display abnormal mitoses.111
In vivo and ex vivo models offer additional insight regarding the contribution of cell-cycle dysregulation and aberrant DNA damage repair in NUP98 fusion–driven malignancy. In the NUP98-HOXD13–transgenic mouse model, impaired nonhomologous end joining may lead to G2/M cell-cycle arrest.112 Overexpression of BCL2 corrected these cell-cycle defects, prevented DNA damage, and prolonged survival.113 Similarly, loss of Bbc3 (Puma) in NUP98-HOXD13 mice extended survival with a significant reduction in DNA damage,114 whereas loss of tumor suppressor and cyclin-dependent kinase inhibitor p15Ink4b accelerated AML onset.115 Importantly, evidence of mitotic chromosomal breaks and DNA damage have been reported also in ex vivo blasts from a patient with NUP98-NSD1 AML,33 indicating the relevance of cell-cycle changes in human disease. Nevertheless, these effects are likely insufficient to mediate oncogenesis: loss of the NUP98 GLEBS domain, which binds to CDC20 and cell-cycle regulator RAE1, does not prevent leukemias in mouse bone marrow transplant models.100 It is, however, possible that mechanisms leading to increased DNA damage or altered cell-cycle progression may complement transcriptional changes to mediate leukemogenesis in some cases of NUP98 fusion.
Recurrent cooperating events in NUP98-rearranged malignancies
Multiple genetic events are often required for full transition from a precancerous to a cancerous cell state, and such events often lead to phenotypes with increased proliferation or differentiation arrest.116 A host of evidence across various model systems suggests that NUP98 fusions prevent differentiation and promote self-renewal (Figure 3A). For example, HSPCs expressing NUP98-KDM5A exhibit sustained self-renewal,3 CD34+ cells expressing NUP98-HOXA9 show a block in erythroid differentiation,117 and NUP98-HOXD13–transgenic mice develop peripheral blood cytopenias with increased immature granulocytes compared with wild-type animals.83
Figure 3.

Co-altered genes and potential therapeutic approaches in NUP98-rearranged hematologic malignancies. (A) NUP98 fusion confers self-renewal and loss of differentiation. (B) Co-occurring alterations, including FLT3-ITD mutation, RAS mutation, and/or WT1 mutation may lead to a proliferative advantage and to hematologic malignancy. (C) Potential strategies for therapeutic targeting of hematologic malignancies with NUP98 gene fusions.
Relatively few genes are comutated with NUP98 rearrangement, consistent with the low mutational burden in pediatric AML.4 However, the observed mutations are strikingly recurrent, including, almost exclusively, alterations that mediate a proliferation advantage (Table 4; Figure 3B). Perhaps the most well characterized of these mutations is FLT3-ITD, which leads to autophosphorylation of the receptor and constitutive activation even in the absence of ligand. FLT3-ITD is a common event in AML and has been observed frequently with NUP98-NSD1.31,32,34,59,60,70 In mouse models, combining NUP98 fusions and FLT3-ITD alterations leads to more penetrant leukemia with a shorter latency.118-120 Consistent with this, NUP98 fusion and FLT3-ITD mutation also predict poorer outcomes in AML patient populations.32,34,64,70,121 Similarly, mutations in Wilms tumor 1 (WT1) are a common event in NUP98 fusion AMLs and have been noted in up to 55% of patients with NUP98-NSD1 fusions prior to therapy.60 Moreover, the co-occurrence of NUP98-NSD1 fusion and WT1 mutation predict poor clinical outcomes.32
Table 4.
Recurrent coalterations with NUP98 gene fusions
| Coalteration | Estimated frequency | Notes |
|---|---|---|
| FLT3-ITD mutation | 48%-92% NUP98-NSD131,34,59,60,70 7%-27% NUP98-HOXA965,66 |
Repeatedly observed in AML with many NUP98 fusion partners (rarely including NUP98-KDM5A3,31,58,62); predicts poor prognosis when observed with NUP98-NSD1 fusion and/or WT1 mutation32,34,60,70,129; association may be lost following treatment failure71 |
| WT1 mutation | 33%-55% NUP98-NSD159,60,131 44% NUP98-HOXA966 |
Observed in AML with various fusion partners3,31,32,34,59,60,66,71,129-131,163,164; predicts poor prognosis with NUP98-NSD1 fusion and/or FLT3-ITD mutation32,34,129 |
| NRAS mutation | 11%-29% NUP98-NSD159,60,131 22% NUP98-HOXA966 |
Observed in AML with various fusion partners3,33,34,59,60,66,129,130 |
| KRAS mutation | 11%-17% NUP98-NSD159,60 22% NUP98-HOXA966 |
Occurs with NUP98-HOXA9,66,129 -KDM5A,54 and -NSD133,59,60 |
| RB1 loss | 80%-100% NUP98-KDM5A3,62 | Observed in almost all NUP98-KDM5A cases3,62,125; not identified with other NUP98 fusion partners |
| BCR-ABL fusion | NUP98 fusion may drive CML transition into blast crisis or mediate resistance to imatinib25,165; not identified with NUP98-KDM5A or NUP98-NSD1 | |
| CEPBA mutation | Fairly infrequent with NUP98 fusion; generally only observed with NUP98 fusion as well as FLT3-ITD or WT1 mutation31,34 | |
| NOTCH1 mutation | Only identified in T-ALL with NUP98 fusion,123 including in some mouse models85,124 | |
| MYC mutation | Noted in NUP98-NSD1 cases; relatively infrequent71,131 | |
| KIT mutation | Identified with both NUP98-HOXA9 and NUP98-NSD1 fusions59,129 | |
| ASXL1 mutation | Likely results in epigenetic dysregulation, identified in few patients with NUP98 fusion62,166 | |
| Trisomy 8 | Observed in multiple cases with various fusion partners despite that NUP98 fusion is often noted as the only chromosomal rearrangement, especially in older patients25,31,34,65,166,167 |
For the most common coalterations, estimated frequencies were determined from available studies with >5 patients.
Whereas FLT3-ITD and WT1 alterations are observed with a wide array of NUP98 fusions across hematologic malignancies, some co-occurring genetic events are limited to specific patient populations. NUP98-HOXA9 fusions have been described as rare events in the blast crisis of CML and have been used to generate a mouse model of this disease.122 NOTCH1 alterations have been observed in NUP98-RAP1GDS1 T-ALL patients123 and in NUP98 fusion mouse models developing T-ALL.85,124 Finally, NUP98-KDM5A fusions present with a unique genomic profile, almost always involving the loss of RB1.3,62,125 Although KDM5A has been shown to promote cellular differentiation by binding to pRb,126-128 the molecular mechanisms underlying the co-occurrence of NUP98-KDM5A fusion and RB1 loss in leukemia are unknown and have not been modeled in vivo.
Other recurrent events observed with NUP98 rearrangements include NRAS, KRAS, KIT, and MYC mutations.3,33,34,54,59,66,71,129-131 Consistent with this, HSPCs from GEMMs bearing NUP98 fused to homeodomain and the NRAS G12D mutation showed higher levels of engraftment than cells from mice with single alterations when transplanted into lethally irradiated CD45.1 recipients.132 Furthermore, analysis of NUP98-HOXD13–transgenic mice that had progressed from MDS to AML identified that 9 of 32 samples (28%) also contained Ras mutations.133 Although common in AML, NPM1 and CEBPA mutations were not identified in leukemic NUP98-HOXD13 mice133 and are rare in NUP98 fusion-positive patients. Complex karyotypes are rare in many NUP98-rearranged leukemia patients, and many patients with cryptic fusions (including NUP98-NSD1) are cytogenetically normal. However, NUP98-KDM5A fusions were observed with chromosomal abnormalities in 10 of 10 patients (100%) with acute megakaryoblastic leukemia in 1 study.62 The roles of these chromosomal rearrangements and other recurrent genetic events, including ASXL1 mutations and trisomy 8, have yet to be elucidated.
Interestingly, recent data suggest that treatment may impact mutational and gene-expression profiles in NUP98-rearranged AML. McNeer et al showed that WT1 mutation (but not FLT3-ITD or RAS mutation) was observed in 4 of 6 NUP98-NSD1 patients (67%) who relapsed following chemotherapy treatment, and that gain of ELF1, gain of MYC, and/or loss of FRMD8 were frequent in this population.71 These findings, however, will require further validation and investigation in larger patient cohorts.
The challenge of therapeutic targeting of NUP98-rearranged leukemia
Despite our current knowledge of the molecular mechanisms and co-occurring genetic events in NUP98-rearranged malignancies, the ability to effectively treat patients bearing these translocations is very limited. Relapse is common, and during treatment follow-up the presence of NUP98 fusion may be assessed using next-generation sequencing or other methods as a sensitive indicator of minimal residual disease, as reported in previous case studies.67,134-136 Because NUP98 fusions are now recognized as a high-risk subtype of leukemia, current treatment paradigms often use chemotherapy followed by hematopoietic stem cell transplant (HSCT) during the first complete molecular remission.
Given the risks of HSCT, however, the development of an effective targeted therapy to treat patients with NUP98 fusion-positive malignancy is a valuable and exciting pursuit. Multiple approaches have been proposed to accomplish this goal (Figure 3C), which remains challenging given that proteins acting as transcription factors (like NUP98 fusions) have historically been considered “undruggable.” One potential strategy is to develop inhibitors against the PHD domain because several fusion partners require this moiety for sustained cell growth.77 Work by Gough et al demonstrated the validity of this approach by showing that PHD inhibitor disulfiram decreased the expression of Hoxa genes and led to apoptosis in cell lines derived from NUP98-PHF23–transgenic mice.85 Another possible approach is to inhibit cofactors necessary for the formation of nuclear puncta and/or transcriptional regulation. Here, inhibition of a cofactor, such as XPO1, might prevent access to the HOXA locus and other critical gene loci and thereby preclude expression of NUP98 fusion target genes. Selinexor is the most widely studied XPO1 inhibitor: this drug has been tested in a variety of cancer types, including pediatric AML, and is now approved for the treatment of relapsed/refractory multiple myeloma.137-139 A final strategy, considered recently and successfully in experimental models, involves inhibiting upregulated transcriptional targets, including CDK6.93
Furthermore, the role of chromatin remodeling in leukemogenesis driven by NUP98 fusion oncoproteins has prompted preclinical evaluation of epigenetic modulators. Both histone acetyltransferase and HDAC inhibitors display in vitro efficacy,140 demonstrating a delicate epigenetic balance in this system. Additionally, given the binding of fusion oncoproteins to KMT2A complexes, DOT1L and/or menin inhibitors may be valuable in this setting141; these compounds have been considered for KMT2A-rearranged leukemias and have shown reasonable toxicity profiles in phase 1 trials.142,143 However, loss of EP300 accelerated the transition from MDS to AML in the NUP98-HOXD13–transgenic mouse model,98 which indicates that blocking the activity of transcriptional or epigenetic regulators may have context-dependent effects and will need to be considered carefully prior to clinical evaluation.144
Efforts focused on targeting fusion partners or collaborating genetic events could also be considered. Selective lysine demethylase inhibitors, capable of blocking the activity of KDM5A have been identified,145 and similar compounds have shown the ability to overcome therapeutic resistance in other cancers.146 Additionally, inhibitors against the commonly coaltered FLT3 have gained US Food and Drug Administration (FDA) approval for AML147,148 and have demonstrated promising efficacy in NUP98-rearranged laboratory models, including mice with HSPCs bearing NUP98-HOXD13 and FLT3-ITD.118,119,149 FLT3-ITD, however, is a secondary event and is observed with extremely poor prognoses in NUP98 fusion patients.32,34,64,70,121 Because of this, FLT3 inhibitor combination regimens may be required for significant survival benefit.150
Finally, recent unbiased approaches have begun to suggest other clinically available drugs that may have selective efficacy in patients with NUP98 rearrangements. Gene set enrichment analysis and proteomic approaches suggested activation of JAK-STAT signaling in acute megakaryoblastic leukemia NUP98-KDM5A xenografts, and FDA-approved JAK inhibitors ruxolitinib and tofacitinib showed in vitro efficacy in NUP98 fusion models from this and another study.89,151 Small molecule profiling nominated aurora kinase A inhibition as a treatment of acute megakaryoblastic leukemia, and 2 aurora kinase A–targeting agents led to cell death in a NUP98-KDM5A model.90 Another recent report used high-throughput screening to identify the BCL2 inhibitor navitoclax and SRC/ABL inhibitor dasatinib for the treatment of NUP98-NSD1/FLT3-ITD–altered leukemias and found that the combination of these 2 agents was highly synergistic.149 Further genetic and pharmacologic exploration of therapeutic vulnerabilities in NUP98-rearranged leukemia is urgently needed to improve patient outcomes.
Summary and future directions
Since the discovery of NUP98 as the key partner in 11p15 translocations, a large number of fusion partners have since been identified in hematological malignancies, most notably AML and T-ALL. Research over the last 25 years has provided many useful insights and valuable models for the study of NUP98-rearranged diseases; although many important questions remain unanswered, new technologies now provide the resources and approaches to address these areas.
The impact of NUP98 fusion oncoprotein expression on transcriptional activity and epigenetic states will likely require recently developed sequencing methods to study limited numbers of cells or even single cells. Inventive protocols will allow future research to interrogate gene expression, DNA binding, and active chromatin in precious patient samples and at higher resolution than ever before. These approaches will allow detailed analysis of individual samples as well as broad comparisons of the landscape of NUP98 translocations with various fusion partners and cooperating alterations in different diseases. Additionally, evidence of a distinct nuclear localization pattern in samples bearing NUP98 fusion oncoproteins suggests that phase separation may mediate transcription signatures, and studies are needed to better understand the structural properties and associated cofactors of NUP98 fusion oncoproteins in phase-separated bodies. Together, this work will deepen our understanding of the mechanisms of NUP98 fusion oncoproteins in altered hematopoiesis.
Furthermore, additional in vivo models are needed to more faithfully recapitulate human disease. One approach to accomplish this might use mice bearing cells from healthy cord blood donors or patient samples, and transgenic strains with the machinery for humanized hematopoiesis may enable these efforts. Moreover, to enhance our understanding of the acquisition and maintenance of NUP98 fusion–driven malignancies, mouse modeling should address the successive gain of NUP98 fusion and coalteration(s) at varying stages of HSPC maturation. Ultimately, these efforts and others will reveal common mediators of NUP98 fusion oncoprotein-driven leukemia as well as lineage- or genotype-specific mechanisms important in the development and progression of disease. Coupling approaches to better understand leukemia biology with innovative campaigns to identify effective therapeutic targets offer hope toward improved patient prognoses in NUP98-rearranged malignancies.
Acknowledgments
This work was supported by National Institutes of Health, National Cancer Institute grants U54 CA243124 (C.G.M. and J.M.K.) and R35CA197695 (C.G.M.).
Authorship
Contribution: N.L.M., J.M.K., and C.G.M. wrote the manuscript
Conflict-of-interest disclosure: C.G.M. has received research fuding from Loxo Oncology, Pfizer and Illumina; speaking fees from Amgen, served on an advisory board for Illumina, and holds stock in Amgen. The remaining authors declare no competing financial interests.
Correspondence: Charles G. Mullighan, St. Jude Children’s Research Hospital, 262 Danny Thomas Pl, Mail Stop 342, Memphis, TN 38105; e-mail: charles.mullighan@stjude.org; and Jeffery M. Klco, St. Jude Children’s Research Hospital, 262 Danny Thomas Pl, Mail Stop 342, Memphis, TN 38105; e-mail: jeffery.klco@stjude.org.
REFERENCES
- 1.Harrison CJ, Hills RK, Moorman AV, et al. Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J Clin Oncol. 2010;28(16):2674-2681. [DOI] [PubMed] [Google Scholar]
- 2.Kim JC, Zuzarte PC, Murphy T, et al. Cryptic genomic lesions in adverse-risk acute myeloid leukemia identified by integrated whole genome and transcriptome sequencing. Leukemia. 2020;34(1):306-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iacobucci I, Wen J, Meggendorfer M, et al. Genomic subtyping and therapeutic targeting of acute erythroleukemia. Nat Genet. 2019;51(4):694-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolouri H, Farrar JE, Triche T Jr., et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions [published corrections appear in Nat Med. 2018;24(4):526 and Nat Med. 2019;25(3):530]. Nat Med. 2018;24(1):103-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu Z, Churchman M, Roberts K, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun. 2016;7:13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol. 2015;12(6):344-357. [DOI] [PubMed] [Google Scholar]
- 7.Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344(14):1038-1042. [DOI] [PubMed] [Google Scholar]
- 8.Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031-1037. [DOI] [PubMed] [Google Scholar]
- 9.Krull S, Thyberg J, Björkroth B, Rackwitz HR, Cordes VC. Nucleoporins as components of the nuclear pore complex core structure and Tpr as the architectural element of the nuclear basket. Mol Biol Cell. 2004;15(9):4261-4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatel G, Desai SH, Mattheyses AL, Powers MA, Fahrenkrog B. Domain topology of nucleoporin Nup98 within the nuclear pore complex. J Struct Biol. 2012;177(1):81-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenblum JS, Blobel G. Autoproteolysis in nucleoporin biogenesis. Proc Natl Acad Sci USA. 1999;96(20):11370-11375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capelson M, Liang Y, Schulte R, Mair W, Wagner U, Hetzer MW. Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell. 2010;140(3):372-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laurell E, Beck K, Krupina K, et al. Phosphorylation of Nup98 by multiple kinases is crucial for NPC disassembly during mitotic entry. Cell. 2011;144(4):539-550. [DOI] [PubMed] [Google Scholar]
- 14.Kalverda B, Pickersgill H, Shloma VV, Fornerod M. Nucleoporins directly stimulate expression of developmental and cell-cycle genes inside the nucleoplasm. Cell. 2010;140(3):360-371. [DOI] [PubMed] [Google Scholar]
- 15.Radu A, Moore MS, Blobel G. The peptide repeat domain of nucleoporin Nup98 functions as a docking site in transport across the nuclear pore complex. Cell. 1995;81(2):215-222. [DOI] [PubMed] [Google Scholar]
- 16.Griffis ER, Altan N, Lippincott-Schwartz J, Powers MA. Nup98 is a mobile nucleoporin with transcription-dependent dynamics. Mol Biol Cell. 2002;13(4):1282-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oka M, Asally M, Yasuda Y, Ogawa Y, Tachibana T, Yoneda Y. The mobile FG nucleoporin Nup98 is a cofactor for Crm1-dependent protein export. Mol Biol Cell. 2010;21(11):1885-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blevins MB, Smith AM, Phillips EM, Powers MA. Complex formation among the RNA export proteins Nup98, Rae1/Gle2, and TAP. J Biol Chem. 2003;278(23):20979-20988. [DOI] [PubMed] [Google Scholar]
- 19.Pritchard CE, Fornerod M, Kasper LH, van Deursen JM. RAE1 is a shuttling mRNA export factor that binds to a GLEBS-like NUP98 motif at the nuclear pore complex through multiple domains. J Cell Biol. 1999;145(2):237-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamura T, Largaespada DA, Lee MP, et al. Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nat Genet. 1996;12(2):154-158. [DOI] [PubMed] [Google Scholar]
- 21.Borrow J, Shearman AM, Stanton VP Jr., et al. The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat Genet. 1996;12(2):159-167. [DOI] [PubMed] [Google Scholar]
- 22.Arai Y, Hosoda F, Kobayashi H, et al. The inv(11)(p15q22) chromosome translocation of de novo and therapy-related myeloid malignancies results in fusion of the nucleoporin gene, NUP98, with the putative RNA helicase gene, DDX10. Blood. 1997;89(11):3936-3944. [PubMed] [Google Scholar]
- 23.Nishiyama M, Arai Y, Tsunematsu Y, et al. 11p15 translocations involving the NUP98 gene in childhood therapy-related acute myeloid leukemia/myelodysplastic syndrome. Genes Chromosomes Cancer. 1999;26(3):215-220. [PubMed] [Google Scholar]
- 24.Fujino T, Suzuki A, Ito Y, et al. Single-translocation and double-chimeric transcripts: detection of NUP98-HOXA9 in myeloid leukemias with HOXA11 or HOXA13 breaks of the chromosomal translocation t(7;11)(p15;p15). Blood. 2002;99(4):1428-1433. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto K, Nakamura Y, Saito K, Furusawa S. Expression of the NUP98/HOXA9 fusion transcript in the blast crisis of Philadelphia chromosome-positive chronic myelogenous leukaemia with t(7;11)(p15;p15). Br J Haematol. 2000;109(2):423-426. [DOI] [PubMed] [Google Scholar]
- 26.Hussey DJ, Nicola M, Moore S, Peters GB, Dobrovic A. The (4;11)(q21;p15) translocation fuses the NUP98 and RAP1GDS1 genes and is recurrent in T-cell acute lymphocytic leukemia. Blood. 1999;94(6):2072-2079. [PubMed] [Google Scholar]
- 27.Panagopoulos I, Kerndrup G, Carlsen N, Strömbeck B, Isaksson M, Johansson B. Fusion of NUP98 and the SET binding protein 1 (SETBP1) gene in a paediatric acute T cell lymphoblastic leukaemia with t(11;18)(p15;q12). Br J Haematol. 2007;136(2):294-296. [DOI] [PubMed] [Google Scholar]
- 28.Romana SP, Radford-Weiss I, Ben Abdelali R, et al. ; Groupe Francophone de Cytogénétique Hématologique . NUP98 rearrangements in hematopoietic malignancies: a study of the Groupe Francophone de Cytogénétique Hématologique. Leukemia. 2006;20(4):696-706. [DOI] [PubMed] [Google Scholar]
- 29.Pan Q, Zhu YJ, Gu BW, et al. A new fusion gene NUP98-IQCG identified in an acute T-lymphoid/myeloid leukemia with a t(3;11)(q29q13;p15)del(3)(q29) translocation. Oncogene. 2008;27(24):3414-3423. [DOI] [PubMed] [Google Scholar]
- 30.Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555(7696):371-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Struski S, Lagarde S, Bories P, et al. NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia. 2017;31(3):565-572. [DOI] [PubMed] [Google Scholar]
- 32.Niktoreh N, Walter C, Zimmermann M, et al. Mutated WT1, FLT3-ITD, and NUP98-NSD1 fusion in various combinations define a poor prognostic group in pediatric acute myeloid leukemia. J Oncol. 2019;2019:1609128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bisio V, Zampini M, Tregnago C, et al. NUP98-fusion transcripts characterize different biological entities within acute myeloid leukemia: a report from the AIEOP-AML group. Leukemia. 2017;31(4):974-977. [DOI] [PubMed] [Google Scholar]
- 34.Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood. 2011;118(13):3645-3656. [DOI] [PubMed] [Google Scholar]
- 35.Taketani T, Taki T, Shibuya N, Kikuchi A, Hanada R, Hayashi Y. Novel NUP98-HOXC11 fusion gene resulted from a chromosomal break within exon 1 of HOXC11 in acute myeloid leukemia with t(11;12)(p15;q13). Cancer Res. 2002;62(16):4571-4574. [PubMed] [Google Scholar]
- 36.La Starza R, Trubia M, Crescenzi B, et al. Human homeobox gene HOXC13 is the partner of NUP98 in adult acute myeloid leukemia with t(11;12)(p15;q13). Genes Chromosomes Cancer. 2003;36(4):420-423. [DOI] [PubMed] [Google Scholar]
- 37.Taketani T, Taki T, Shibuya N, et al. The HOXD11 gene is fused to the NUP98 gene in acute myeloid leukemia with t(2;11)(q31;p15). Cancer Res. 2002;62(1):33-37. [PubMed] [Google Scholar]
- 38.Raza-Egilmez SZ, Jani-Sait SN, Grossi M, Higgins MJ, Shows TB, Aplan PD. NUP98-HOXD13 gene fusion in therapy-related acute myelogenous leukemia. Cancer Res. 1998;58(19):4269-4273. [PubMed] [Google Scholar]
- 39.Jankovic D, Gorello P, Liu T, et al. Leukemogenic mechanisms and targets of a NUP98/HHEX fusion in acute myeloid leukemia. Blood. 2008;111(12):5672-5682. [DOI] [PubMed] [Google Scholar]
- 40.Soler G, Kaltenbach S, Dobbelstein S, et al. Identification of GSX2 and AF10 as NUP98 partner genes in myeloid malignancies [letter]. Blood Cancer J. 2013;3(7):e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakamura T, Yamazaki Y, Hatano Y, Miura I. NUP98 is fused to PMX1 homeobox gene in human acute myelogenous leukemia with chromosome translocation t(1;11)(q23;p15). Blood. 1999;94(2):741-747. [PubMed] [Google Scholar]
- 42.Gervais C, Mauvieux L, Perrusson N, et al. A new translocation t(9;11)(q34;p15) fuses NUP98 to a novel homeobox partner gene, PRRX2, in a therapy-related acute myeloid leukemia. Leukemia. 2005;19(1):145-148. [DOI] [PubMed] [Google Scholar]
- 43.Lisboa S, Cerveira N, Bizarro S, et al. POU1F1 is a novel fusion partner of NUP98 in acute myeloid leukemia with t(3;11)(p11;p15). Mol Cancer. 2013;12:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hussey DJ, Dobrovic A. Recurrent coiled-coil motifs in NUP98 fusion partners provide a clue to leukemogenesis. Blood. 2002;99(3):1097-1098. [DOI] [PubMed] [Google Scholar]
- 45.Roussy M, Bilodeau M, Jouan L, et al. NUP98-BPTF gene fusion identified in primary refractory acute megakaryoblastic leukemia of infancy. Genes Chromosomes Cancer. 2018;57(6):311-319. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz JR, Ma J, Lamprecht T, et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun. 2017;8:1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Zutven LJ, Onen E, Velthuizen SC, et al. Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes Chromosomes Cancer. 2006;45(5):437-446. [DOI] [PubMed] [Google Scholar]
- 48.Jaju RJ, Fidler C, Haas OA, et al. A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood. 2001;98(4):1264-1267. [DOI] [PubMed] [Google Scholar]
- 49.Rosati R, La Starza R, Veronese A, et al. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood. 2002;99(10):3857-3860. [DOI] [PubMed] [Google Scholar]
- 50.Reader JC, Meekins JS, Gojo I, Ning Y. A novel NUP98-PHF23 fusion resulting from a cryptic translocation t(11;17)(p15;p13) in acute myeloid leukemia. Leukemia. 2007;21(4):842-844. [DOI] [PubMed] [Google Scholar]
- 51.Kaltenbach S, Soler G, Barin C, et al. NUP98-MLL fusion in human acute myeloblastic leukemia. Blood. 2010;116(13):2332-2335. [DOI] [PubMed] [Google Scholar]
- 52.Hayashi Y, Harada Y, Kagiyama Y, et al. NUP98-HBO1-fusion generates phenotypically and genetically relevant chronic myelomonocytic leukemia pathogenesis. Blood Adv. 2019;3(7):1047-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahuja HG, Hong J, Aplan PD, Tcheurekdjian L, Forman SJ, Slovak ML. t(9;11)(p22;p15) in acute myeloid leukemia results in a fusion between NUP98 and the gene encoding transcriptional coactivators p52 and p75-lens epithelium-derived growth factor (LEDGF). Cancer Res. 2000;60(22):6227-6229. [PubMed] [Google Scholar]
- 54.de Rooij JD, Hollink IH, Arentsen-Peters ST, et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia. 2013;27(12):2280-2288. [DOI] [PubMed] [Google Scholar]
- 55.Noort S, Wander P, Alonzo TA, et al. The clinical and biological characteristics of NUP98-KDM5A in pediatric acute myeloid leukemia [published online ahead of print 7 May 2020]. Haematologica. doi:10.3324/haematol.2019.236745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cimino G, Sprovieri T, Rapanotti MC, Foà R, Mecucci C, Mandelli F. Molecular evaluation of the NUP98/RAP1GDS1 gene frequency in adults with T-acute lymphoblastic leukemia. Haematologica. 2001;86(4):436-437. [PubMed] [Google Scholar]
- 57.Panarello C, Rosanda C, Morerio C. Cryptic translocation t(5;11)(q35;p15.5) with involvement of the NSD1 and NUP98 genes without 5q deletion in childhood acute myeloid leukemia. Genes Chromosomes Cancer. 2002;35(3):277-281. [DOI] [PubMed] [Google Scholar]
- 58.Tosić N, Stojiljković M, Colović N, Colović M, Pavlović S. Acute myeloid leukemia with NUP98-HOXC13 fusion and FLT3 internal tandem duplication mutation: case report and literature review. Cancer Genet Cytogenet. 2009;193(2):98-103. [DOI] [PubMed] [Google Scholar]
- 59.Shiba N, Ichikawa H, Taki T, et al. NUP98-NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes Chromosomes Cancer. 2013;52(7):683-693. [DOI] [PubMed] [Google Scholar]
- 60.Marceau-Renaut A, Duployez N, Ducourneau B, et al. Molecular profiling defines distinct prognostic subgroups in childhood AML: a report from the French ELAM02 Study Group. HemaSphere. 2018;2(1):e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hara Y, Shiba N, Yamato G, et al. Patients aged less than 3 years with acute myeloid leukaemia characterize a molecularly and clinically distinct subgroup. Br J Haematol. 2020;188(4):528-539. [DOI] [PubMed] [Google Scholar]
- 62.de Rooij JD, Branstetter C, Ma J, et al. Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet. 2017;49(3):451-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Rooij JD, Masetti R, van den Heuvel-Eibrink MM, et al. Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood. 2016;127(26):3424-3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shimada A, Iijima-Yamashita Y, Tawa A, et al. Risk-stratified therapy for children with FLT3-ITD-positive acute myeloid leukemia: results from the JPLSG AML-05 study. Int J Hematol. 2018;107(5):586-595. [DOI] [PubMed] [Google Scholar]
- 65.Wei S, Wang S, Qiu S, et al. Clinical and laboratory studies of 17 patients with acute myeloid leukemia harboring t(7;11)(p15;p15) translocation. Leuk Res. 2013;37(9):1010-1015. [DOI] [PubMed] [Google Scholar]
- 66.Chou WC, Chen CY, Hou HA, et al. Acute myeloid leukemia bearing t(7;11)(p15;p15) is a distinct cytogenetic entity with poor outcome and a distinct mutation profile: comparative analysis of 493 adult patients. Leukemia. 2009;23(7):1303-1310. [DOI] [PubMed] [Google Scholar]
- 67.Fasan A, Haferlach C, Alpermann T, Kern W, Haferlach T, Schnittger S. A rare but specific subset of adult AML patients can be defined by the cytogenetically cryptic NUP98-NSD1 fusion gene. Leukemia. 2013;27(1):245-248. [DOI] [PubMed] [Google Scholar]
- 68.Thol F, Kölking B, Hollink IH, et al. Analysis of NUP98/NSD1 translocations in adult AML and MDS patients. Leukemia. 2013;27(3):750-754. [DOI] [PubMed] [Google Scholar]
- 69.Miyamura T, Moritake H, Nakayama H, et al. Clinical and biological features of paediatric acute myeloid leukaemia (AML) with primary induction failure in the Japanese Paediatric Leukaemia/Lymphoma Study Group AML-05 study. Br J Haematol. 2019;185(2):284-288. [DOI] [PubMed] [Google Scholar]
- 70.Ostronoff F, Othus M, Gerbing RB, et al. NUP98/NSD1 and FLT3/ITD coexpression is more prevalent in younger AML patients and leads to induction failure: a COG and SWOG report. Blood. 2014;124(15):2400-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McNeer NA, Philip J, Geiger H, et al. Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia. 2019;33(8):1934-1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kroon E, Thorsteinsdottir U, Mayotte N, Nakamura T, Sauvageau G. NUP98-HOXA9 expression in hemopoietic stem cells induces chronic and acute myeloid leukemias in mice. EMBO J. 2001;20(3):350-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pineault N, Buske C, Feuring-Buske M, et al. Induction of acute myeloid leukemia in mice by the human leukemia-specific fusion gene NUP98-HOXD13 in concert with Meis1. Blood. 2003;101(11):4529-4538. [DOI] [PubMed] [Google Scholar]
- 74.Gurevich RM, Aplan PD, Humphries RK. NUP98-topoisomerase I acute myeloid leukemia-associated fusion gene has potent leukemogenic activities independent of an engineered catalytic site mutation. Blood. 2004;104(4):1127-1136. [DOI] [PubMed] [Google Scholar]
- 75.Pineault N, Abramovich C, Ohta H, Humphries RK. Differential and common leukemogenic potentials of multiple NUP98-Hox fusion proteins alone or with Meis1. Mol Cell Biol. 2004;24(5):1907-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang GG, Cai L, Pasillas MP, Kamps MP. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol. 2007;9(7):804-812. [DOI] [PubMed] [Google Scholar]
- 77.Wang GG, Song J, Wang Z, et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature. 2009;459(7248):847-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hirose K, Abramovich C, Argiropoulos B, Humphries RK. Leukemogenic properties of NUP98-PMX1 are linked to NUP98 and homeodomain sequence functions but not to binding properties of PMX1 to serum response factor. Oncogene. 2008;27(46):6056-6067. [DOI] [PubMed] [Google Scholar]
- 79.Petit A, Ragu C, Della-Valle V, et al. NUP98-HMGB3: a novel oncogenic fusion. Leukemia. 2010;24(3):654-658. [DOI] [PubMed] [Google Scholar]
- 80.Petit A, Ragu C, Soler G, et al. Functional analysis of the NUP98-CCDC28A fusion protein. Haematologica. 2012;97(3):379-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pan MM, Zhang QY, Wang YY, et al. Human NUP98-IQCG fusion protein induces acute myelomonocytic leukemia in mice by dysregulating the Hox/Pbx3 pathway. Leukemia. 2016;30(7):1590-1593. [DOI] [PubMed] [Google Scholar]
- 82.Iwasaki M, Kuwata T, Yamazaki Y, et al. Identification of cooperative genes for NUP98-HOXA9 in myeloid leukemogenesis using a mouse model. Blood. 2005;105(2):784-793. [DOI] [PubMed] [Google Scholar]
- 83.Lin YW, Slape C, Zhang Z, Aplan PD. NUP98-HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood. 2005;106(1):287-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Saw J, Curtis DJ, Hussey DJ, Dobrovic A, Aplan PD, Slape CI. The fusion partner specifies the oncogenic potential of NUP98 fusion proteins. Leuk Res. 2013;37(12):1668-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gough SM, Lee F, Yang F, et al. NUP98-PHF23 is a chromatin-modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD histone reader function. Cancer Discov. 2014;4(5):564-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fisher JN, Thanasopoulou A, Juge S, et al. Transforming activities of the NUP98-KMT2A fusion gene associated with myelodysplasia and acute myeloid leukemia. Haematologica. 2020;105(7):1857-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chung KY, Morrone G, Schuringa JJ, et al. Enforced expression of NUP98-HOXA9 in human CD34(+) cells enhances stem cell proliferation. Cancer Res. 2006;66(24):11781-11791. [DOI] [PubMed] [Google Scholar]
- 88.Imren S, Heuser M, Gasparetto M, et al. Modeling de novo leukemogenesis from human cord blood with MN1 and NUP98HOXD13. Blood. 2014;124(24):3608-3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cardin S, Bilodeau M, Roussy M, et al. Human models of NUP98-KDM5A megakaryocytic leukemia in mice contribute to uncovering new biomarkers and therapeutic vulnerabilities. Blood Adv. 2019;3(21):3307-3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thiollier C, Lopez CK, Gerby B, et al. Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J Exp Med. 2012;209(11):2017-2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Calvo KR, Sykes DB, Pasillas MP, Kamps MP. Nup98-HoxA9 immortalizes myeloid progenitors, enforces expression of Hoxa9, Hoxa7 and Meis1, and alters cytokine-specific responses in a manner similar to that induced by retroviral co-expression of Hoxa9 and Meis1. Oncogene. 2002;21(27):4247-4256. [DOI] [PubMed] [Google Scholar]
- 92.Ghannam G, Takeda A, Camarata T, Moore MA, Viale A, Yaseen NR. The oncogene Nup98-HOXA9 induces gene transcription in myeloid cells. J Biol Chem. 2004;279(2):866-875. [DOI] [PubMed] [Google Scholar]
- 93.Schmoellerl J, Barbosa IAM, Eder T, et al. CDK6 is an essential direct target of NUP98 fusion proteins in acute myeloid leukemia. Blood. 2020;136(4):387-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Boija A, Klein IA, Sabari BR, et al. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell. 2018;175(7):1842-1855.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schmidt HB, Görlich D. Nup98 FG domains from diverse species spontaneously phase-separate into particles with nuclear pore-like permselectivity. eLife. 2015;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kasper LH, Brindle PK, Schnabel CA, Pritchard CE, Cleary ML, van Deursen JM. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol. 1999;19(1):764-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fahrenkrog B, Martinelli V, Nilles N, et al. Expression of leukemia-associated Nup98 fusion proteins generates an aberrant nuclear envelope phenotype. PLoS One. 2016;11(3):e0152321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cheng G, Liu F, Asai T, et al. Loss of p300 accelerates MDS-associated leukemogenesis. Leukemia. 2017;31(6):1382-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rio-Machin A, Gómez-López G, Muñoz J, et al. The molecular pathogenesis of the NUP98-HOXA9 fusion protein in acute myeloid leukemia. Leukemia. 2017;31(9):2000-2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yung E, Sekulovic S, Argiropoulos B, et al. Delineating domains and functions of NUP98 contributing to the leukemogenic activity of NUP98-HOX fusions. Leuk Res. 2011;35(4):545-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pineault N, Abramovich C, Humphries RK. Transplantable cell lines generated with NUP98-Hox fusion genes undergo leukemic progression by Meis1 independent of its binding to DNA. Leukemia. 2005;19(4):636-643. [DOI] [PubMed] [Google Scholar]
- 102.Argiropoulos B, Palmqvist L, Yung E, et al. Linkage of Meis1 leukemogenic activity to multiple downstream effectors including Trib2 and Ccl3. Exp Hematol. 2008;36(7):845-859. [DOI] [PubMed] [Google Scholar]
- 103.Lai CK, Norddahl GL, Maetzig T, et al. Meis2 as a critical player in MN1-induced leukemia. Blood Cancer J. 2017;7(9):e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vegi NM, Klappacher J, Oswald F, et al. MEIS2 is an oncogenic partner in AML1-ETO-positive AML. Cell Rep. 2016;16(2):498-507. [DOI] [PubMed] [Google Scholar]
- 105.Franks TM, McCloskey A, Shokirev MN, Benner C, Rathore A, Hetzer MW. Nup98 recruits the Wdr82-Set1A/COMPASS complex to promoters to regulate H3K4 trimethylation in hematopoietic progenitor cells. Genes Dev. 2017;31(22):2222-2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xu H, Valerio DG, Eisold ME, et al. NUP98 fusion proteins interact with the NSL and MLL1 complexes to drive leukemogenesis. Cancer Cell. 2016;30(6):863-878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Valerio DG, Xu H, Chen CW, et al. Histone acetyltransferase activity of MOF is required for MLL-AF9 leukemogenesis. Cancer Res. 2017;77(7):1753-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Takeda A, Sarma NJ, Abdul-Nabi AM, Yaseen NR. Inhibition of CRM1-mediated nuclear export of transcription factors by leukemogenic NUP98 fusion proteins. J Biol Chem. 2010;285(21):16248-16257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oka M, Mura S, Yamada K, et al. Chromatin-prebound Crm1 recruits Nup98-HoxA9 fusion to induce aberrant expression of Hox cluster genes. eLife. 2016;5:e09540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Salsi V, Fantini S, Zappavigna V. NUP98 fusion oncoproteins interact with the APC/C(Cdc20) as a pseudosubstrate and prevent mitotic checkpoint complex binding. Cell Cycle. 2016;15(17):2275-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Salsi V, Ferrari S, Gorello P, et al. NUP98 fusion oncoproteins promote aneuploidy by attenuating the mitotic spindle checkpoint. Cancer Res. 2014;74(4):1079-1090. [DOI] [PubMed] [Google Scholar]
- 112.Puthiyaveetil AG, Reilly CM, Pardee TS, Caudell DL. Non-homologous end joining mediated DNA repair is impaired in the NUP98-HOXD13 mouse model for myelodysplastic syndrome. Leuk Res. 2013;37(1):112-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Slape CI, Saw J, Jowett JB, et al. Inhibition of apoptosis by BCL2 prevents leukemic transformation of a murine myelodysplastic syndrome. Blood. 2012;120(12):2475-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guirguis AA, Slape CI, Failla LM, et al. PUMA promotes apoptosis of hematopoietic progenitors driving leukemic progression in a mouse model of myelodysplasia. Cell Death Differ. 2016;23(6):1049-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Humeniuk R, Koller R, Bies J, Aplan P, Wolff L. Brief report: Loss of p15Ink4b accelerates development of myeloid neoplasms in Nup98-HoxD13 transgenic mice. Stem Cells. 2014;32(5):1361-1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Grove CS, Vassiliou GS. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Dis Model Mech. 2014;7(8):941-951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Abdul-Nabi AM, Yassin ER, Varghese N, Deshmukh H, Yaseen NR. In vitro transformation of primary human CD34+ cells by AML fusion oncogenes: early gene expression profiling reveals possible drug target in AML. PLoS One. 2010;5(8):e12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Thanasopoulou A, Tzankov A, Schwaller J. Potent co-operation between the NUP98-NSD1 fusion and the FLT3-ITD mutation in acute myeloid leukemia induction. Haematologica. 2014;99(9):1465-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Greenblatt S, Li L, Slape C, et al. Knock-in of a FLT3/ITD mutation cooperates with a NUP98-HOXD13 fusion to generate acute myeloid leukemia in a mouse model. Blood. 2012;119(12):2883-2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shima Y, Yumoto M, Katsumoto T, Kitabayashi I. MLL is essential for NUP98-HOXA9-induced leukemia. Leukemia. 2017;31(10):2200-2210. [DOI] [PubMed] [Google Scholar]
- 121.Akiki S, Dyer SA, Grimwade D, et al. NUP98-NSD1 fusion in association with FLT3-ITD mutation identifies a prognostically relevant subgroup of pediatric acute myeloid leukemia patients suitable for monitoring by real time quantitative PCR. Genes Chromosomes Cancer. 2013;52(11):1053-1064. [DOI] [PubMed] [Google Scholar]
- 122.Dash AB, Williams IR, Kutok JL, et al. A murine model of CML blast crisis induced by cooperation between BCR/ABL and NUP98/HOXA9. Proc Natl Acad Sci USA. 2002;99(11):7622-7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Crescenzi B, Nofrini V, Barba G, et al. NUP98/11p15 translocations affect CD34+ cells in myeloid and T lymphoid leukemias. Leuk Res. 2015;39(7):769-772. [DOI] [PubMed] [Google Scholar]
- 124.Lin YW, Nichols RA, Letterio JJ, Aplan PD. Notch1 mutations are important for leukemic transformation in murine models of precursor-T leukemia/lymphoma. Blood. 2006;107(6):2540-2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lopez CK, Malinge S, Gaudry M, Bernard OA, Mercher T. Pediatric acute megakaryoblastic leukemia: multitasking fusion proteins and oncogenic cooperations. Trends Cancer. 2017;3(9):631-642. [DOI] [PubMed] [Google Scholar]
- 126.Benevolenskaya EV, Murray HL, Branton P, Young RA, Kaelin WG Jr.. Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol Cell. 2005;18(6):623-635. [DOI] [PubMed] [Google Scholar]
- 127.Lin W, Cao J, Liu J, et al. Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc Natl Acad Sci USA. 2011;108(33):13379-13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Defeo-Jones D, Huang PS, Jones RE, et al. Cloning of cDNAs for cellular proteins that bind to the retinoblastoma gene product. Nature. 1991;352(6332):251-254. [DOI] [PubMed] [Google Scholar]
- 129.Taketani T, Taki T, Nakamura T, et al. High frequencies of simultaneous FLT3-ITD, WT1 and KIT mutations in hematological malignancies with NUP98-fusion genes. Leukemia. 2010;24(11):1975-1977. [DOI] [PubMed] [Google Scholar]
- 130.Yang J, Lyu X, Zhu X, et al. Chromosome t(7;11)(p15;p15) translocation in acute myeloid leukemia coexisting with multilineage dyspoiesis and mutations in NRAS and WT1: A case report and literature review. Oncol Lett. 2017;13(5):3066-3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lavallée VP, Lemieux S, Boucher G, et al. Identification of MYC mutations in acute myeloid leukemias with NUP98-NSD1 translocations. Leukemia. 2016;30(7):1621-1624. [DOI] [PubMed] [Google Scholar]
- 132.Dong Y, Xia C, Weng Q, et al. Synergy of NUP98-HOXA10 fusion gene and NrasG12D mutation preserves the stemness of hematopoietic stem cells on culture condition. Cells. 2019;8(9):E951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Slape C, Liu LY, Beachy S, Aplan PD. Leukemic transformation in mice expressing a NUP98-HOXD13 transgene is accompanied by spontaneous mutations in Nras, Kras, and Cbl. Blood. 2008;112(5):2017-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Terui K, Kitazawa J, Takahashi Y, et al. Successful treatment of acute myelomonocytic leukaemia with NUP98-HOXD11 fusion transcripts and monitoring of minimal residual disease. Br J Haematol. 2003;120(2):274-276. [DOI] [PubMed] [Google Scholar]
- 135.Yamamoto K, Nakamachi Y, Yakushijin K, et al. Expression of the novel NUP98/PSIP1 fusion transcripts in myelodysplastic syndrome with t(9;11)(p22;p15). Eur J Haematol. 2012;88(3):244-248. [DOI] [PubMed] [Google Scholar]
- 136.Au CH, Ho DN, Ip BBK, et al. Rapid detection of chromosomal translocation and precise breakpoint characterization in acute myeloid leukemia by nanopore long-read sequencing. Cancer Genet. 2019;239:22-25. [DOI] [PubMed] [Google Scholar]
- 137.Chari A, Vogl DT, Gavriatopoulou M, et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N Engl J Med. 2019;381(8):727-738. [DOI] [PubMed] [Google Scholar]
- 138.Alexander TB, Lacayo NJ, Choi JK, Ribeiro RC, Pui CH, Rubnitz JE. Phase I study of selinexor, a selective inhibitor of nuclear export, in combination with fludarabine and cytarabine, in pediatric relapsed or refractory acute leukemia. J Clin Oncol. 2016;34(34):4094-4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang AY, Liu H. The past, present, and future of CRM1/XPO1 inhibitors. Stem Cell Investig. 2019;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Giotopoulos G, Chan WI, Horton SJ, et al. The epigenetic regulators CBP and p300 facilitate leukemogenesis and represent therapeutic targets in acute myeloid leukemia. Oncogene. 2016;35(3):279-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Deshpande AJ, Deshpande A, Sinha AU, et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell. 2014;26(6):896-908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Krivtsov AV, Evans K, Gadrey JY, et al. A menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell. 2019;36(6):660-673.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Stein EM, Garcia-Manero G, Rizzieri DA, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood. 2018;131(24):2661-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Man N, Nimer SD. p300 suppresses leukemia development in NUP98-HOXD13 driven myelodysplastic syndrome. Oncotarget. 2018;9(42):26603-26604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gale M, Sayegh J, Cao J, et al. Screen-identified selective inhibitor of lysine demethylase 5A blocks cancer cell growth and drug resistance. Oncotarget. 2016;7(26):39931-39944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hinohara K, Wu HJ, Vigneau S, et al. KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance [published correction appears in Cancer Cell. 2019;35(2):330-332]. Cancer Cell. 2018;34(6):939-953.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381(18):1728-1740. [DOI] [PubMed] [Google Scholar]
- 149.Kivioja JL, Thanasopoulou A, Kumar A, et al. Dasatinib and navitoclax act synergistically to target NUP98-NSD1+/FLT3-ITD+ acute myeloid leukemia. Leukemia. 2019;33(6):1360-1372. [DOI] [PubMed] [Google Scholar]
- 150.Ofran Y. Concealed dagger in FLT3/ITD+ AML. Blood. 2014;124(15):2317-2319. [DOI] [PubMed] [Google Scholar]
- 151.Yin M, Chung YJ, Lindsley RC, et al. Engineered Bcor mutations lead to acute leukemia of progenitor B-1 lymphocyte origin in a sensitized background. Blood. 2019;133(24):2610-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lahortiga I, Vizmanos JL, Agirre X, et al. NUP98 is fused to adducin 3 in a patient with T-cell acute lymphoblastic leukemia and myeloid markers, with a new translocation t(10;11)(q25;p15). Cancer Res. 2003;63(12):3079-3083. [PubMed] [Google Scholar]
- 153.Ishikawa M, Yagasaki F, Okamura D, et al. A novel gene, ANKRD28 on 3p25, is fused with NUP98 on 11p15 in a cryptic 3-way translocation of t(3;5;11)(p25;q35;p15) in an adult patient with myelodysplastic syndrome/acute myelogenous leukemia. Int J Hematol. 2007;86(3):238-245. [DOI] [PubMed] [Google Scholar]
- 154.Tosi S, Ballabio E, Teigler-Schlegel A, Boultwood J, Bruch J, Harbott J. Characterization of 6q abnormalities in childhood acute myeloid leukemia and identification of a novel t(6;11)(q24.1;p15.5) resulting in a NUP98-C6orf80 fusion in a case of acute megakaryoblastic leukemia. Genes Chromosomes Cancer. 2005;44(3):225-232. [DOI] [PubMed] [Google Scholar]
- 155.Panagopoulos I, Isaksson M, Billström R, Strömbeck B, Mitelman F, Johansson B. Fusion of the NUP98 gene and the homeobox gene HOXC13 in acute myeloid leukemia with t(11;12)(p15;q13). Genes Chromosomes Cancer. 2003;36(1):107-112. [DOI] [PubMed] [Google Scholar]
- 156.Gorello P, Brandimarte L, La Starza R, et al. t(3;11)(q12;p15)/NUP98-LOC348801 fusion transcript in acute myeloid leukemia. Haematologica. 2008;93(9):1398-1401. [DOI] [PubMed] [Google Scholar]
- 157.Zhu HH, Yang MC, Wang F, et al. Identification of a novel NUP98-RARA fusion transcript as the 14th variant of acute promyelocytic leukemia. Am J Hematol. 2020;95(7):E184-E186. [DOI] [PubMed] [Google Scholar]
- 158.Such E, Cervera J, Valencia A, et al. A novel NUP98/RARG gene fusion in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood. 2011;117(1):242-245. [DOI] [PubMed] [Google Scholar]
- 159.Ahuja HG, Felix CA, Aplan PD. The t(11;20)(p15;q11) chromosomal translocation associated with therapy-related myelodysplastic syndrome results in an NUP98-TOP1 fusion. Blood. 1999;94(9):3258-3261. [PubMed] [Google Scholar]
- 160.Nebral K, Schmidt HH, Haas OA, Strehl S. NUP98 is fused to topoisomerase (DNA) IIbeta 180 kDa (TOP2B) in a patient with acute myeloid leukemia with a new t(3;11)(p24;p15). Clin Cancer Res. 2005;11(18):6489-6494. [DOI] [PubMed] [Google Scholar]
- 161.Chen B, Jiang L, Zhong ML, et al. Identification of fusion genes and characterization of transcriptome features in T-cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2018;115(2):373-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.St. Jude Children’s Research Hospital PROPEL: public resource of patient-derived and expanded leukemias. www.stjude.org/PROPEL. Accessed 20 August 2020.
- 163.Gallego Hernanz MP, Torregrosa Diaz JM, Sorel N, et al. Long-term molecular remission in a patient with acute myeloid leukemia harboring a new NUP98-LEDGF rearrangement. Cancer Med. 2019;8(4):1765-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Luo H, Zhang S, Li K, et al. A novel entity of acute myeloid leukaemia with recurrent RARG-rearrangement resembling acute promyelocytic leukaemia. Leuk Res. 2019;77:14-16. [DOI] [PubMed] [Google Scholar]
- 165.Yamamoto M, Kakihana K, Kurosu T, Murakami N, Miura O. Clonal evolution with inv(11)(p15q22) and NUP98/DDX10 fusion gene in imatinib-resistant chronic myelogenous leukemia. Cancer Genet Cytogenet. 2005;157(2):104-108. [DOI] [PubMed] [Google Scholar]
- 166.Cui J, Xie J, Qin L, Chen S, Zhao Y, Wu D. A unique acute myeloid leukemia patient with cryptic NUP98-NSD1 gene and ASXL1 mutation. Leuk Lymphoma. 2016;57(1):196-198. [DOI] [PubMed] [Google Scholar]
- 167.Mecucci C, La Starza R, Negrini M, et al. t(4;11)(q21;p15) translocation involving NUP98 and RAP1GDS1 genes: characterization of a new subset of T acute lymphoblastic leukaemia. Br J Haematol. 2000;109(4):788-793. [DOI] [PubMed] [Google Scholar]