

Abstract
Background:
This report highlights a rapidly progressive case of Creutzfeldt-Jakob Disease (CJD) whose time from symptom onset to death spanned less than two months. We also explore the most recently available inpatient demographics data for discharges with CJD in the United States.
Methods:
We reviewed a CJD case and systematically analyzed a retrospective cohort of CJD discharges using the Healthcare Cost and Utilization Project (HCUP) to evaluate the existing national data on the status of CJD demographics and dispositions in the United States in 2016.
Results:
An estimated total of 710 hospital discharges with a diagnosis of CJD were seen across the United States in 2016. According to HCUP, the average age of patients was 66.15 ± 11.54 years with 48.6 % female. Average time to intubation from admission to hospital was 4.71 ± 7.32 days with a rate of intubation of 6.34 %. The mean hospital cost was $19,901.25 ± $18,743.48. The rate of in-hospital mortality was 8.45 %. No significant geographical differences were noted (p = 0.49). No significant differences were seen among incidence in specific ethnic groups (p = 0.33) or income quartiles (p = 0.90).
Conclusions:
Our data shows that the incidence of CJD in 2016 appears to be equally distributed among individuals in the United States by demographic categories. Additionally, our case-study from 2019 illustrates an important example for diagnosing a rapidly-progressing case of CJD.
Keywords: Creutzfeldt-Jacob Disease, Healthcare cost and utilization project, Demographics, Rapidly-progressing, Sporadic
1. Introduction
Creutzfeldt-Jakob Disease (CJD) is a rare insidious degenerative brain disorder first described in 1920 by Hans Gerhard Creutzfeldt in a report titled “a new and unusual type of neurological disease.” Creutzfeldt’s case report described a 22-year-old woman who presented with tremors, unsteady gait, involuntary eye movements, jerking limbs, and dementia who died one year after the onset of symptoms [1]. The following year, in 1921, physician Alfons Maria Jakob wrote four case reports on patients with similar symptoms, adding histopathological observations of nerve cell-loss and a characteristic “sponge-like” appearance of brain tissue [2]. It is from these early reports of rapidly-progressive dementia with specific pathological findings that the disease was eponymized with the names of the two physician scientists who first described it.
CJD results from the pathological effects of abnormally folded proteins called prions. In approximately 85 %, the causes are classified as sporadic with no clear definitive origin. The remaining 15 % can be attributed to a specific genetic mutation in the prion protein (PrP) gene and are often inherited in an autosomal dominant pattern. Of note, < 1% of CJD cases are acquired from iatrogenic or oral exposure to infected brain or nervous tissue [3,4]. Indeed, CJD and other prion diseases of the brain such as Kuru and Gertmann-Sträussler-Scheinker (GSS) disease typically present as rapidly progressive dementia involving memory loss, mood changes, problems with coordination, and visual disturbances (NINDS, 2020). Prion diseases are clinically and pathologically heterogeneous. Nevertheless, six prion isotype combinations predominate in the classification schema of sporadic CJD determined by the presence of methionine or valine at the polymorphic codon 129 in a patient’s prion protein gene. These include MM1, MM2, MV1, MV2, VV1, and VV2 [5].
As symptoms progress, the neurocognitive decline becomes more pronounced leading to myoclonus, blindness, extremity weakness, coma, and ultimately death. Manifesting within a year of symptom onset. CJD is generally preceded by a prodromal phase including the nonspecific findings of fatigue, sleep changes, and visual disturbances. As the disease progresses, patients may start to experience changes in behavior, emotional lability, extrapyramidal signs, myoclonus jerks, and severe rapid cognitive decline [6]. Diagnostic studies may reveal 3 Hz spike waves on EEG, 14-3-3 (or t-tau) protein identified on an otherwise nonspecific lumbar puncture, and/or cortical ribboning on a T2 fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) scan [7].
In the case of sporadic CJD, a comprehensive World Health Organization (WHO) diagnostic criterion can be appreciated in Table 1 [8,9]. Of note, RT-QuIC diagnostic accuracy is significantly superior previous methods, including 14-3-3, t-tau and EEG [10,11], with t-tau considered a superior CSF screening marker than 14-3-3 [11,12]. We herein present a case of sporadic CJD with rapid progression followed by a demographic assessment of patients diagnosed with CJD at the national level as reported in the Healthcare Cost and Utilization Project’s (HCUP) national database [13].
Table 1.
World Health Organization Diagnostic Criteria for Sporadic CJD.
| Definitive Diagnosis | Standard Neuropathological Techniques |
| AND/OR | • Immunohistochemically |
| AND/OR | • Western Blot Protease-Resistant PrP |
| AND/OR | • Presence of Scrapie-Associated Fibrils |
| Probable Diagnosis | Neuropsychiatric disorder plus positive RT-QuIC in cerebrospinal fluid (CSF) or other tissues |
| OR | Rapidly progressive dementia; and at least two out of the following four clinical features: |
| • Myoclonus | |
| • Visual or Cerebellar Signs | |
| • Pyramidal/Extrapyramidal Signs | |
| • Akinetic Mutism | |
| AND | Positive result on at least one of the following laboratory tests: |
| • Typical EEG (periodic sharp wave complexes) during an illness of any duration | |
| • Positive 14-3-3 CSF assay in patients with a disease duration of less than 2 years | |
| • High signal in caudate/putamen on magnetic resonance imaging (MRI) brain scan or at least two cortical regions (temporal, parietal, occipital) either on diffusion-weighted imaging (DWI) or fluid attenuated inversion recovery (FLAIR) | |
| AND | without routine investigations indicating an alternative diagnosis. |
2. Case study
In late fall of 2019, a 58-year-old right handed aspirin naïve male with past medical history of hypertension and gastroesophageal reflux disease presented to our emergency department with complaints of right-sided weakness, trouble walking, aphasia, and slurred speech. History was obtained from the patient’s wife, who stated that the patient had been having brief episodes of confusion and word-finding difficulty over the past month lasting approximately one minute. During this period, she also noted mood changes described as un-characteristically “grumpier,” and disproportionately frustrated with basic tasks. The patient initially attributed his cognitive symptoms to recent changes in his hypertension medications made by his primary care physician (PCP). Additionally, the patient’s wife began noticing that the patient ceased using his right (dominant) side for motor tasks. Videos obtained shortly before admission showed the patient dispaying brief episodes of rhythmic jerking of his right arm, intermittent intention tremor of his right hand, and fasciculations on his right leg while at rest.
On the morning of admission, the family contacted the patient’s PCP, informing them that the patient had woken up “in a fog” with worsening symptoms and new-onset slurred speech with difficulty walking. The PCP recommended that the patient go to the emergency room immediately for a stroke work-up. At the time of presentation to the hospital, the patient exhibited apraxia on the right finger to nose test, mild aphasia, and slurring of speech. The patient was subsequently admitted to the stroke service for further workup. Throughout the day, the patient’s symptoms appeared to fluctuate considerably. Diffuse weighted imaging (DWI) and FLAIR-MRI scans were obtained on hospital day 2 and showed cortical ribboning in both occipital lobes, most pronounced on the right with some in parietal and frontal involvement (Fig. 1). CT angiography and magnetic resonance angiography also obtained at this time were negative for arterial stenosis or occlusions.
Fig. 1.
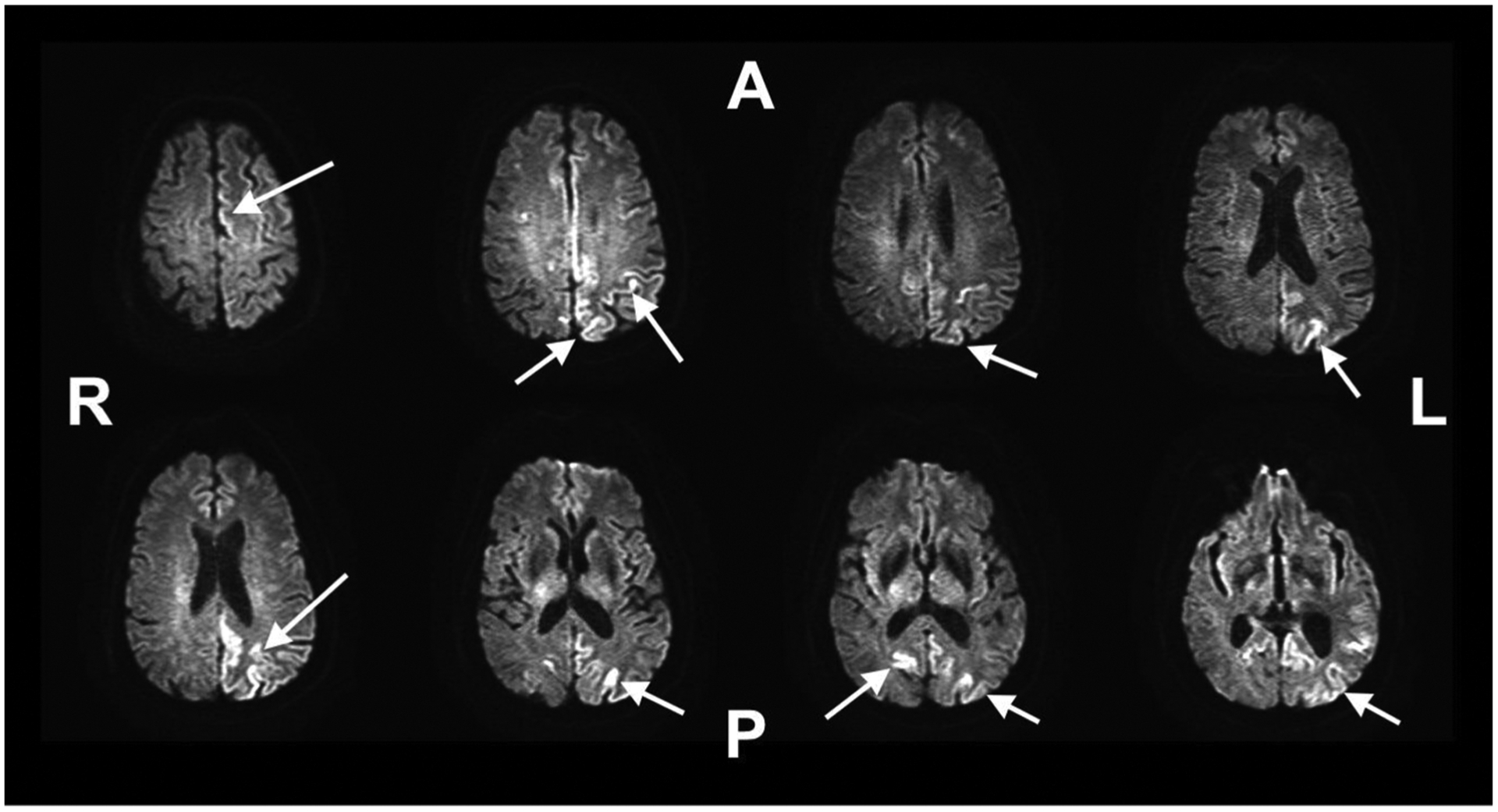
Cortical ribboning seen in patient’s T2 FLAIR MRI scan as indicated by arrows. Most cortical ribboning is localized to the left hemisphere. A = Anterior, P = Posterior, R = Right Hemisphere, L = Left Hemisphere.
The patient also underwent an epilepsy work-up with a 24 -h EEG ordered on hospital day 2. The EEG described a posterior dominant rhythm of up to 8 Hz, better developed on the right during arousals with an admixed theta in the anterior background and slower frequencies on the left. Occasional generalized triphasic waves were also noted. This was initially interpreted as possible left hemispheric periodic epileptiform discharges. Discharges that could be interpreted as either interictal manifestations of high degree epileptogenic potentials or alternatively, as representing acute injury. In certain instances, after prolonged status epilepticus, such findings can be interpreted as an end-stage of seizure activity. Mild generalized slowing is also indicative of generalized disturbance of cerebral function. Moreover, left hemispheric focal slowing indicates a focal disturbance of cerebral function secondary to a structural or physiologic etiology (Fig. 2).
Fig. 2.
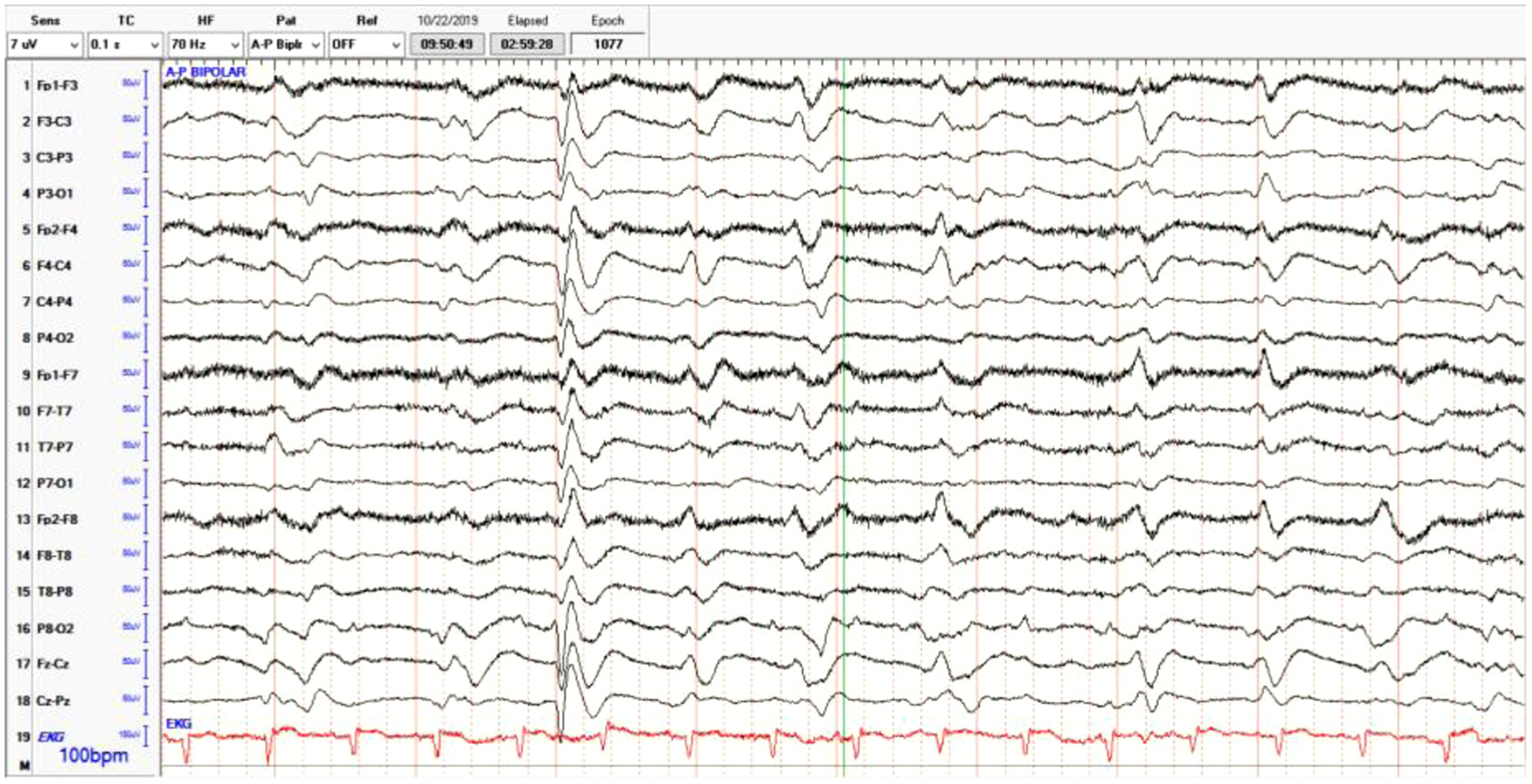
Electroencephalogram (EEG) findings indicating poorly-formed triphasic discharges interspersed with intermittent complexes.
On hospital day 3, the patient was loaded with Keppra and Vimpat and transferred to the general neurology service for further workup. Lumbar puncture obtained by the general neurology service at this time showed < 5 WBC, 1 RBC, glucose 61, protein 46. A meningoencephalitis panel of the patient’s CSF was negative. Additional tests, including an autoimmune panel, real-time quaking-induced conversion (RT-QuIC), and protein 14-3-3, were sent to offsite laboratories for processing. The patient was subsequently given five days of solumedrol with apparent improvement of symptoms. On hospital day 8 the patient developed labored breathing and acute worsening of mental status and was transferred to the Neurologic Intensive Care Unit (ICU) for emergent intubation due to inability to protect his airway.
At the time of arrival to the Neurological ICU, the patient presented with a GCS of 8, tachypnea, and gasping labored breathing with accessory muscle use. Following intubation, patient was started on plasma exchange therapy (PLEX) every other day for a total of five doses due to concerns of possible autoimmune encephalitis. Additionally, on hospital day 9 the patient underwent a repeat brain MRI as well as CT chest, abdomen, and pelvis to assess for a possible neoplastic source of the patient’s encephalopathy. A repeat brain MRI replicated the findings of cortical ribboning (Fig. 1), suggestive of possible prion disease. CT scans were unremarkable. An additional lumbar puncture was obtained Hospital day 11 to rule out cytologic, arbovirus and West Nile etiologies of the patient’s encephalitis. Patient’s mental status minimally improved during the duration of his PLEX therapy, with continued episodes of myoclonus-like activity.
On hospital day 15 the patient completed his final dose of PLEX with no improvement. A final 24 -h EEG study noted generalized slowing 1 hz periodic discharges with occasionally ceasing triphasic morphology and low voltage polymorphic activity in the background. These findings of generalized periodic discharges suggested bilateral structural damage, a notable change from unilateral damage in earlier EEGs. Generalized epileptiform discharges were noted with no discrete seizures indicating worsening diffuse cortical irritation.
A family meeting was held to discuss prognosis and further plans of care for the patient after which the patient’s code status was changed to “Do Not Resuscitate” with no escalation of care. The family wished to await the results of the RT QuIC and 14-3-3 before transferring the patient to comfort measures. On hospital day 22 the patient’s RT QuIC and 14-3-3 confirmed the diagnosis of CJD, and the patient’s family transitioned the patient to hospice. The patient died the following day on post-admission day 23.
The analysis from the autopsy tissue submitted to Case Western Reserve University’s National Prion Disease Pathology Surveillance Center completed three months after the patient’s death characterized the abnormal prion protein type by Western blot and carried out the histopathological and immunohistochemical examinations. The results confirmed the diagnosis of prion disease with the characteristics of sporadic Creutzfeldt-Jakob Disease MM1 according to the classification of sporadic prion disease proposed by Parchi et al. [5]. In addition, the report found that sequencing of the prion protein (PrP) gene with polymerase chain reaction (PCR), used to rule out the presence of a pathogenic mutation in the PrP coding region, ruled out a familial origin for our patient’s prion disease according to the latest criteria [14].
3. Materials and methods
We used the Healthcare Cost and Utilization Project (HCUP) national database, specifically the National Inpatient Sample (NIS), to perform a retrospective cohort study of the most recently available data on Creutzfeldt-Jakob Disease (CJD) in the United States (US). The NIS represents a 20 % random stratified sample of all discharges from US community hospitals. Importantly, the data excludes rehabilitation and long-term acute care hospitals centers. Individuals with a diagnosis upon discharge of CJD were identified within the HCUP NIS database by using the International Classification of Diseases 10th revision (ICD-10) codes A81.00, A81.01, and A81.09. We thus obtained data from of 142 confirmed cases CJD as reported on hospital discharge, from which we were able to extrapolate a total number of 710 reported cases in the US in 2016. Of note, the University of Texas Health Science Center at San Antonio was exempt in this analysis from full review by the Institutional Review Board. All statistical analyses were conducted using Stata (http://www.stata.com).
4. Results
During 2016, the Home Healthcare Costs and Utilization Project (HCUP) database reported 710 hospital discharges of CJD (48.6 % female). The average age of onset for patients with CJD was 66.15 ± 11.54 years. A Yates’ χ2 analysis comparing the age stratification between the CJD population and the general US population found that the age differences were significant with p-value of < 0.01, indicating that the increased incidence of CJD patients found in the decades between 50 and 70 is a true difference. Regionally, across the United States, 26.76 % of cases were localized to the Northeast, 26.76 % of cases to the Midwest, 27.46 % of cases in the South, and 17.61 % of cases in the west. A Yates’ χ2 analysis comparing the incidence of CJD cases across regions compared to population percentages of the general US population for these regions found no significant differences with a p-value of 0.49. Demographic data on ethnicity showed that 73.7 % of patients with CJD were white, 10.5 % were black, 12.0 % were Hispanic, 2.3 % were Asian, and 1.5 % were other. These demographic findings closely match the overall demographics in the United States based on 2015 estimates: 73 % white, 12.7 % black, 17.6 % Hispanic, 5.4 % Asian, 4.8 % other [15]. A Yates’ χ2 comparing the percentages of ethnic populations discharged with a CJD diagnosis to percent demographics of the US population found no significant differences with a p-value of 0.33. Similarly, no significant differences were found between CJD cases and the general population among individuals of in specific income quartiles. Individuals in the 1st income quartile, measured by zip code income averages and home values, accounted for 27.1 % of the reported cases, those in the 2nd income quartile accounted for 21.4 % of the cases, those in the 3rd income quartile accounted for 24.3 % of the cases, and those in the 4th income quartile accounted for 27.0 % of the cases. A Yates’ χ2 p-value of 0.90 indicated no significant statistical differences between income quartiles among CJD patients and the general population. Overall, 83.1 % of CJD cases were reported in urban teaching hospitals, likely due to the uncommon nature of the disease and need for specialized care. 4% of cases were reported in rural hospitals, and 14.1 % were reported in urban non-teaching hospitals. Based on the population of patients admitted to each hospital, a Yates’ χ2 p-value of < 0.01 establishes that the increased number of CJD discharges from urban teaching hospitals as compared to other hospitals among the general population of hospitalized patients is statistically different than the general patient population for all other diseases. The data also identified that the average time to intubation upon hospitalization was 4.71 ± 7.32 days in the 6.3 % of patients that required intubation. Finally, the average cost in US dollars for a CJD hospitalization was reported as $19,901.25 ± $18,743.48 with the average charge to patients and insurance providers totaling $82,446.24 ± $116,653.80 (Table 2).
Table 2.
Healthcare Cost and Utilization Project Breakdown of CJD cases in 2016 in the USA.
| Demographics | n (%)a | Expected % | Yates’ χ2 | Yates’ p-value | |
|---|---|---|---|---|---|
| Sample size, n | 710 | n/a | n/a | n/a | |
| Female | 345 (48.6) | 50a | 0.033 | 0.856 | |
| Age (mean + SD) | 66.15 ± 11.54 | n/a | n/a | n/a | |
| Decade | 0 to 9 | 5 (0.7) | 12.6 | 106.628 | < 0.001* |
| 10 to 19 | 0 (0.0) | 13.1 | |||
| 20 to 29 | 15(2.1) | 13.9 | |||
| 30 to 39 | 25 (3.5) | 13.1 | |||
| 40 to 49 | 125 (17.6) | 12.7 | |||
| 51 to 59 | 295 (41.6) | 13.7 | |||
| 61 to 69 | 155 (21.8) | 11.3 | |||
| 71 to 79 | 80 (11.3) | 6.2 | |||
| 81 to 89 | 10 (1.4) | 2.5 | |||
| Region | Northeast | 200(28.2) | 25a | 2.401 | 0.493 |
| Midwest | 190 (26.8) | 25a | |||
| South | 195 (27.5) | 25a | |||
| West | 125 (17.6) | 25a | |||
| Race | White | 490 (73.7) | 73b | 4.605 | 0.330 |
| Black | 70 (10.5) | 12.7b | |||
| Hispanic | 80 (12.0) | 17.6b | |||
| Asian | 15(2.3) | 5.4b | |||
| Native American | 0 (0) | 0.8b | |||
| Other | 10 (1.5) | 4.8b | |||
| Teaching Hospital/Location | Rural | 20 (2.8) | 9.1a | 13.656 | 0.001* |
| Urban non-teaching | 100 (14.1) | 25.5a | |||
| Urban teaching | 590 (83.1) | 65.4a | |||
| Income quartiles by Zip Code | 1 st | 190 (27.1) | 25b | 0.594 | 0.898 |
| 2nd | 150 (21.4) | 25b | |||
| 3rd | 170 (24.3) | 25b | |||
| 4th | 190 (27.1) | 25b | |||
| Average Time to Intubation (Days, mean + SD) | 4.71 ± 7.32 (6.3) | n/a | n/a | n/a | |
| Charge in US Dollars | $82446.24 ± 116653.8 | n/a | n/a | n/a | |
| Cost in US Dollars | $19901.25 ± 18743.48 | n/a | n/a | n/a |
Chi-square tests compare CJD population demographics based on the Healthcare Cost and Utilization Project (HCUP) to general US population statistics according to the
HCUP,
United States Census Bureau and
United States Center for Disease Control and Prevention.
Statistically significant difference from expected per population demographics (p < .05).
We were also interested in patient disposition. Since patients with CJD has a 100 % mortality rate, most commonly within a year of diagnosis [16], patient disposition after diagnosis is an important factor in understanding continuity of patient care. HCUP data reported that 16.9 % of CJD patients experienced a “routine” disposition, defined as being discharged to home or self-care or with a planned acute care hospital inpatient readmission; 4.9 % of patients had a disposition to a “short-term hospital,” defined as being transferred or discharged to a short-term inpatient hospital, federal health care facility, critical access hospital, or any of the above with a planned acute care hospital inpatient readmission; 42.3 % of patients had a disposition of being transferred to “another type of facility,” which include skilled nursing facilities, intermediate care facilities, hospice, long-term care facility, inpatient rehab facility, etc.; 26.8 % of patients experienced a “home health care” disposition, which involves a discharge to an organized home health service organization, or home hospice. Only 0.7 % left the hospital against medical advice, and a total of 8.5 % of patients died at the initial hospital of admission (Table 3). We further conducted two logistic regressions assessing the dispositions of “another type of facility” and “home health care” and HCUP demographic variables of age, sex, and length of stay. These were the two most common dispositions, with the largest number of patients capable of yielding significant results. The analyses found that older age was the most significant predictor discharge to “another type of facility” odds ratio = 1.048 (95 % confidence interval [1.012–1.085], and p = 0.008), with non-significant results in the remaining comparisons (Table 4).
Table 3.
Final Disposition of CJD Patients.
| Disposition of Patient | n | % |
|---|---|---|
| Routine | 120 | 16.9 |
| Short-term hospital | 35 | 4.9 |
| Another type of facility | 300 | 42.3 |
| Home Health Care (HHC) | 190 | 26.8 |
| Against medical advice (AMA) | 5 | 0.7 |
| Died at Initial Hospital | 60 | 8.5 |
| Total | 710 | 100 |
Table 4.
Multivariate logistic regression: Odds of final disposition for most prevalent CJD patient dispositions.
| Regression Variables | OR | p-value | 95% CI |
|---|---|---|---|
| Another Type of Facility Disposition (n = 300) | |||
| Age | 1.048 | .008* | 1.012, 1085 |
| Female | 0.927 | .835 | 0.454, 1.891 |
| Length of Stay (Days) | 1.020 | .246 | 0.987, 1.054 |
| Home Health Care Disposition (n = 190) | |||
| Age | 1.024 | .200 | 0.988, 1.061 |
| Female | 0.914 | .821 | 0.421, 1.987 |
| Length of Stay (Days) | 0.947 | .097 | 0.889, 1.010 |
Abbreviations: OR, odds ratio; CI, confidence intervals.
Statistically significant correlations between regression variables and disposition (p < 0.05).
In the context of our case study and our demographic analysis, we find that there are no stereotypical demographics for patients with CJD other than age. No statistically significant differences were seen among affected individuals and their relationship to demographic factors such as sex, race/ethnicity, geography, or SES based on normal demographic distributions in the US. Only age, with a mean of 66 years (± 11.5 years) and admission to an urban teaching hospital were found to correlate significantly with CJD as compared to the general population. Furthermore, analysis of disposition indicated that the majority of CJD patients get discharged to “another type of facility” comprising long-term and skilled nursing facilities and “home health care” comprising home hospice and other skilled care at home. Age was the only significant predictor of being discharged to “another type of facility,” with all other predictors and dispositions being non-significant or having a sample size too small for logistic regression. Although CJD has a 100 % mortality, our study showed that in-hospital mortality is only 8.5 %. This would indicate that most patients with CJD will die in care facilities such as hospices and skilled nurse facilities that are not the initial hospital of admission.
5. Discussion
Creutzfeldt-Jakob Disease and other prion diseases are exceedingly rare, with an estimated 459 CJD cases reported across the United States in 2016 according to CDC data and 710 discharges according to Healthcare Cost and Utilization Project (HCUP) data. The discrepancy between the total number of cases and discharges is primarily due to patients being discharged and re-admitted to hospital settings on more than one occasion. Indeed, making a successful diagnosis of CJD early in the disease course can be a difficult task, especially in rapidly-progressing cases [17,18]. More specific data highlighting the methodologies used to report CJD diagnoses within the HCUP database would be ideal, however since the HCUP database is not recording these variables, we cannot comment on these and thus this is a limitation of this study.
Common things being common, CJD is often treated as a disease of exclusion. Unsurprisingly, more prevalent diseases with similar presenting symptoms to CJD are given priority when developing a differential diagnosis. For example, neuropathological disorders such as stroke [19], epilepsy, meningitis, Alzheimer’s disease, paraneoplastic syndrome, autoimmune encephalitis, and other diseases are often placed higher in the differential and tested before considering a prion disease diagnosis [20]. Presently, screening approaches with 14-3-3 protein followed by a confirmatory RT-QuIC test are considered the optimal diagnostic approaches for the discrimination between CJD and other rapidly-progressive dementias [11]. Newer, less-invasive diagnostic methods involving magnetic resonance and diffusion weighted imaging are showing promising diagnostic performances comparable to the current RT-QuIC gold standard [21].
Though CJD has a near 100 % mortality, it is crucial to have an accurate and timely diagnosis. It has been shown that as more physicians become aware of CJD, patients can often be misdiagnosed as having CJD when they actually had a treatable disease. This can be seen in individuals showing positive results for 14-3-3 cerebrospinal fluid (CSF) protein, but without any other distinguishing CJD features. Between 2006 and 2009, the National Prion Disease Pathology Surveillance Center analyzed 1106 brains of patients submitted to them with symptoms of rapidly-progressive dementia, only 68 % of them appeared as true cases of prion disease. The 32 % of cases that were prion negative, Alzheimer’s disease and vascular dementia were the most commonly misdiagnosed disorders (54 % of the prion negative brains). The study also found that a total of 71 patients had potentially treatable diseases [20]. Another study of confirmed CJD patients sought to identify the most common misdiagnoses of CJD. This study of 163 patients with confirmed CJD on autopsy collected over a 5.5-year period found that the most commonly misdiagnosed diseases for CJD patients were neurodegenerative, autoimmune, infections (mainly viral), toxic/metabolic, and unspecified dementia. Furthermore, the study also reported that the mean time from disease onset to diagnosis was 7.9 months [22].
The most recent United States Center for Disease Control (CDC) criteria for CJD diagnoses (Table 1) stipulates that the 14-3-3 protein assay of CSF is not as specific as the more novel real-time quaking-induced conversion (RT-QuIC) [23,24]. RT-QuIC and autopsy findings in our patient established a definitive diagnosis of sporadic CJD by characterizing an abnormal prion protein type in the patient’s tissue sample using Western blot and histopathology and immunohistochemistry. It further ruled out the presence of the PrP gene using polymerase chain reaction (PCR) signifying that our patient’s disease was not familial in origin. Importantly, though very rare, PCR does not constitute a definitive diagnostic test or a precise test for the genetic forms of prion-associated diseases. Such sources of diagnostic errors can be attributed to sample mix-ups, and genotypic errors that can result from trace contamination of PCR reactions from other sources. Further, it is for this reason that these assays should be used in conjunction with other clinical, pathological, and laboratory findings, as previously described.
Currently there is no treatment for Creutzfeldt-Jakob Disease. There are numerous aspects of CJD that make treatment-focused clinical trials difficult; namely, the rarity of the disease, the disease’s high mortality, and the aforementioned difficulty in obtaining a diagnosis early enough in the course of the disease to refer patients to centers conducting such trials [25]. Though limited in number, clinical trials focusing on improving survival time and limiting cognitive decline have been performed with new experimental guidelines for genetic prion disease clinical trials being recently proposed [26]. Two such studies from the University of California San Francisco and the United Kingdom’s PRION-1 Study tested possible benefits from the anti-malarial drug quinacrine, but neither study demonstrated an improvement in survival time [27,28]. One double-blind placebo-controlled trial using the drug Flupirtine showed some positive influence on slowing cognitive-decline [29]. Still other studies performed in Italy, Germany, and France looked at whether doxycycline could provide benefits in survival time. Indeed, some studies did show improved survival time on the scale of months with doxycycline, however others showed no benefit [30]. Other studies have hypothesized that polyanions like petnsan polysulfate and antibody therapies could theoretically provide benefit to patients suffering with CJD [31]. Unfortunately, as of the writing of this paper there are no gold standard or FDA-recommended therapies yet for improving outcome measures in CJD patients.
6. Conclusion
Creutzfeldt-Jakob Disease (CJD) is a rare, but devastating neuro-degenerative prion disease that resulted a total of 710 hospital discharges in 2016, as well as our case study patient in 2019. There are no differences between the patients from the standpoint of sex, demographics, geography, or socio-economic status based on normal demographic distributions in the US. The majority of CJD patients get discharged to “another type of facility” comprising long-term and skilled nursing facilities and “home health care” comprising home hospice and other skilled care at home, where most of them will die. Indeed, much remains to be learned about this disease, particularly when it comes to accurate and timely diagnoses, potentials for therapy, referral for involvement in clinical trials, and disposition.
Acknowledgments
We want to expressly thank the patient’s family for providing crucial information and for their unwavering support in the preparation of this report. It is their hope as well as ours that this publication may help shed light on this devastating disease and contribute to the growing knowledge that may eventually lead to more efficient diagnoses and future therapies. We also want to thank the South Texas MSTP program for their academic support. Educational funding for this study was supported by funds from the National Institutes of Health (NIH South Texas Medical Scientist Training Program: T32GM113898 and NCATS Translational Scientist Training Program: TL1TR002647-01). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- [1].Wolf JH, Foley P, Hans Gerhard Creutzfeldt (1885–1964): a life in neuro-pathology, J. Neural Transm 112 (8) (2005), 10.1007/s00702-005-0288-2 I–XCVII. [DOI] [PubMed] [Google Scholar]
- [2].Triarhou LC, Alfons Maria Jakob (1884–1931), Neuropathologist par Excellence, Eur. Neurol 61 (1) (2009) 52–58, 10.1159/000175123. [DOI] [PubMed] [Google Scholar]
- [3].Will RG, Alperovitch A, Poser S, Pocchiari M, Hofman A, Mitrova E, et al. , Descriptive epidemiology of Creutzfeldt-Jakob disease in six European countries, 1993–1995, Ann. Neurol 43 (6) (1998) 763–767. [DOI] [PubMed] [Google Scholar]
- [4].Bonda DJ, Manjila S, Mehndiratta P, Khan F, Miller BR, Onwuzulike K, Puoti G, Cohen ML, Schonberger LB, Cali I, Human prion diseases: surgical lessons learned from iatrogenic prion transmission, Neurosurg. Focus 41 (1) (2016) E10, 10.3171/2016.5.FOCUS15126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, et al. , Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects, Ann. Neurol 46 (2) (1999) 224–233. [PubMed] [Google Scholar]
- [6].Karch A, Raddatz LM, Ponto C, Hermann P, Summers D, Zerr I, Diagnostic profiles of patients with late-onset Creutzfeldt–Jakob disease differ from those of younger Creutzfeldt–Jakob patients: a historical cohort study using data from the German National Reference Center, J. Neurol 261 (5) (2014) 877–883, 10.1007/s00415-014-7283-1. [DOI] [PubMed] [Google Scholar]
- [7].Abdulmassih R, Min Z, An ominous radiographic feature: cortical ribbon sign, Intern. Emerg. Med 11 (2) (2015) 281–283, 10.1007/s11739-015-1287-4. [DOI] [PubMed] [Google Scholar]
- [8].Knight RSG, Surveillance of CJD in the UK, Retrieved on March 15, 2020 from Department of Health and Scottish Government Policy Research Programme, 3, 2017, pp. 1–85 https://www.cjd.ed.ac.uk/sites/default/files/NCJDRSU%20surveillance%20protocol-january%202017.pdf. [Google Scholar]
- [9].Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. , Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease, Brain 132 (10) (2009) 2659–2668, 10.1093/brain/awp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lattanzio F, Abu-Rumeileh S, Franceschini A, Kai H, Amore G, Poggiolini I, Rossi M, Baiardi S, McGuire L, Ladogana A, Pocchiari M, Green A, Capellari S, Parchi P, Prion-specific and surrogate CSF biomarkers in Creutzfeldt-Jakob disease: diagnostic accuracy in relation to molecular subtypes and analysis of neuropathological correlates of p-tau and Aβ42 levels, Acta Neuropathol. 133 (4) (2017) 559–578, 10.1007/s00401-017-1683-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Abu-Rumeileh S, Baiardi S, Polischi B, et al. , Diagnostic value of surrogate CSF biomarkers for Creutzfeldt-Jakob disease in the era of RT-QuIC, J. Neurol 266 (12) (2019) 3136–3143, 10.1007/s00415-019-09537-0. [DOI] [PubMed] [Google Scholar]
- [12].Hamlin C, Puoti G, Berri S, et al. , A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt-Jakob disease, Neurology 79 (6) (2012) 547–552, 10.1212/WNL.0b013e318263565f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].HCUP Home. Healthcare Cost and Utilization Project (HCUP). Agency for Healthcare Research and Quality, Rockville, MD: Retrieved November 4, 2019 www.hcup-us.ahrq.gov/home.jsp. [Google Scholar]
- [14].Kong Q, Surewicz WK, Petersen RB, Zou W, Chen SG, et al. , Inherited prion diseases, in: Prusiner SB (Ed.), Prion Biology and Diseases, 2nd edition, Cold Spring Harbor Laboratory Press, New York, 2004, pp. 673–775. [Google Scholar]
- [15].United States Census Bureau, U.S. Census Bureau, 2011–2015 American Community Survey 5-Year Estimates, July 1, Retrieved April 14, 2020, from (2015) https://archive.vn/Pm2oY.
- [16].Belay ED, Schonberger LB, Variant Creutzfeldt-Jakob disease and bovine spongiform encephalopathy, Clin. Lab. Med 22 (4) (2002), 10.1016/s0272-2712(02)00024-0 849–vi. [DOI] [PubMed] [Google Scholar]
- [17].Parchi P, Strammiello R, Giese A, Kretzschmar H, Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future, Acta Neuropathol. 121 (1) (2011) 91–112, 10.1007/s00401-010-0779-6. [DOI] [PubMed] [Google Scholar]
- [18].Iwasaki Y, Creutzfeldt-Jakob disease, Neuropathology 37 (2) (2017) 174–188, 10.1111/neup.12355. [DOI] [PubMed] [Google Scholar]
- [19].Okamoto K, Abe T, Itoh Y, A case of creutzfeldt-jakob disease with stroke-like onset, J. Stroke Cerebrovasc. Dis (2020) 104788, , 10.1016/j.jstrokecerebrovasdis.2020.104788. [DOI] [PubMed] [Google Scholar]
- [20].Chitravas N, Jung RS, Kofskey DM, Blevins JE, Gambetti P, Leigh RJ, Cohen ML, Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease, Ann. Neurol 70 (3) (2011) 437–444, 10.1002/ana.22454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bizzi A, Pascuzzo R, Blevins J, Grisoli M, Lodi R, Moscatelli M, Castelli G, Cohen ML, Schonberger LB, Foutz A, Safar JG, Appleby BS, Gambetti P, Evaluation of a new criterion for detecting prion disease with diffusion magnetic resonance imaging, JAMA Neurol. (2020) e201319, , 10.1001/jamaneurol.2020.1319 Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Paterson RW, Torres-Chae CC, Kuo AL, Ando T, Nguyen EA, Wong K, et al. , Differential diagnosis of Jakob-Creutzfeldt disease, Arch. Neurol 69 (12) (2012), 10.1001/2013.jamaneurol.79 1578–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Atarashi R, Sano K, Satoh K, Nishida N, Real-time quaking-induced conversion, Prion 5 (3) (2014) 150–153, 10.4161/pri.5.3.16893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rhoads DD, Wrona A, Foutz A, Blevins J, Glisic K, Person M, Maddox RA, Belay ED, Schonberger LB, Tatsuoka C, Cohen ML, Appleby BS, Diagnosis of prion diseases by RT-QuIC results in improved surveillance, Neurology (2020), 10.1212/WNL.0000000000010086. [DOI] [PubMed] [Google Scholar]
- [25].Appleby BS, Many challenges of conducting clinical trials in CJD, J. Neurol. Neurosurg. Psychiatr 88 (2) (2016) 98, 10.1136/jnnp-2016-314578. [DOI] [PubMed] [Google Scholar]
- [26].Vallabh SM, Minikel EV, Schreiber SL, Lander ES, Towards a treatment for genetic prion disease: trials and biomarkers, Lancet Neurol. 19 (4) (2020) 361–368, 10.1016/S1474-4422(19)30403-X. [DOI] [PubMed] [Google Scholar]
- [27].Geschwind MD, Kuo AL, Wong KS, Haman A, Devereux G, Raudabaugh BJ, Johnson DY, Torres-Chae CC, Finley R, Garcia P, Thai JN, Cheng HQ, Neuhaus JM, Forner SA, Duncan JL, Possin KL, Dearmond SJ, Prusiner SB, Miller BL, Quinacrine treatment trial for sporadic Creutzfeldt-Jakob disease, Neurology 81 (23) (2013) 2015–2023, 10.1212/WNL.0b013e3182a9f3b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Collinge J, Gorham M, Hudson F, Kennedy A, Keogh G, Pal S, Rossor M, Rudge P, Siddique D, Spyer M, Thomas D, Walker S, Webb T, Wroe S, Darbyshire J, Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial, Lancet Neurol. 8 (4) (2009) 334–344, 10.1016/s1474-4422(09)70049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Otto M, Cepek L, Ratzka P, Doehlinger S, Boekhoff I, Wiltfang J, Irle E, Pergande G, Ellers-Lenz B, Windl O, Kretzschmar HA, Poser S, Prange H, Efficacy of flupirtine on cognitive function in patients with CJD: a double-blind study, Neurology 62 (5) (2004) 714–718, 10.1212/01.wnl.0000113764.35026.ef. [DOI] [PubMed] [Google Scholar]
- [30].Varges D, Manthey H, Heinemann U, Ponto C, Schmitz M, Schulz-Schaeffer WJ, et al. , Doxycycline in early CJD: a double-blinded randomised phase II and observational study, J. Neurol. Neurosurg. Psychiatr 88 (2) (2016) 119–125, 10.1136/jnnp-2016-313541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Korth C, Peters PJ, Emerging pharmacotherapies for creutzfeldt-jakob disease, Arch. Neurol 63 (4) (2006) 497, 10.1001/archneur.63.4.497. [DOI] [PubMed] [Google Scholar]