

Abstract
The maintenance of T cell tolerance in the periphery proceeds through several mechanisms, including anergy, immuno-regulation, and deletion via apoptosis. We examined the mechanism underlying the induction of CD8 T cell peripheral tolerance to a self-Ag expressed on pancreatic islet β-cells. Following adoptive transfer, Ag-specific clone 4 T cells underwent deletion independently of extrinsic death receptors, including Fas, TNFR1, or TNFR2. Additional experiments revealed that the induction of clone 4 T cell apoptosis during peripheral tolerance occurred via an intrinsic death pathway that could be inhibited by overexpression of Bcl-2 or targeted deletion of the proapoptotic molecule, Bim, thereby resulting in accumulation of activated clone 4 T cells. Over-expression of Bcl-2 in clone 4 T cells promoted the development of effector function and insulitis whereas Bim−/− clone 4 cells were not autoaggressive. Examination of the upstream molecular mechanisms contributing to clone 4 T cell apoptosis revealed that it proceeded in a p53, E2F1, and E2F2-independent manner. Taken together, these data reveal that initiation of clone 4 T cell apoptosis during the induction of peripheral tolerance to a cross-presented self-Ag occurs through a Bcl-2-sensitive and at least partially Bim-dependent mechanism.
The majority of T cells with high avidity for self-peptide-MHC complexes are eliminated during a process known as central (thymic) tolerance (1, 2). Despite the efficiency of thymic deletion in eliminating self-reactive T cells, not all self-Ags are expressed in the thymus at a sufficient level to induce central tolerance. Therefore, additional mechanisms, including the induction of T cell anergy, immuno-regulation, and deletion, exist to promote T cell tolerance in the peripheral lymphoid organs (3, 4). Recent studies from a number of groups have demonstrated that potentially self-reactive naive CD8 T cells can be deleted in the periphery as a consequence of recognizing self-Ags that are cross-presented by APCs in secondary lymphoid tissues (5–8), a process referred to as cross-tolerance. Dendritic cells have been implicated as key regulators of this process as they possess the unique ability to capture both foreign and self-Ag from the parenchyma and present it to naive T cells circulating through secondary lymphoid tissues (9, 10). Under noninflammatory conditions, such recognition of Ag results in an abortive form of T cell activation that is followed by apoptotic death.
Using the clone 4 TCR transgenic (Tg) line that is specific for an H-2 Kd-restricted peptide epitope of the influenza hemagglutinin (HA), we have previously demonstrated that adoptive transfer of clone 4 CD8 T cells into InsHA mice, which express the viral HA on their pancreatic islet β cells (11), results in T cell activation by cross-presenting APCs in the pancreatic lymph nodes (LN) (6, 8). Clone 4 T cells activated in this manner are deleted after undergoing several rounds of division (6, 8), but the mechanism of CD8 T cell death used during this process remains unclear.
Apoptosis of mature T cells can be initiated through two distinct pathways: via death ligands extrinsic to the cell, such as FasL or TNF, or through an intrinsic pathway that can be initiated through a variety of stress-related factors, including cytokine withdrawal. The latter mechanism can be inhibited by the prosurvival molecule Bcl-2 (12, 13). Several studies have shown that signaling through Fas and TNFR1/2 promotes the apoptosis of activated T cells, a process referred to as activation-induced cell death (AICD) (14–18). Recent studies, however, have also implicated an intrinsic death pathway in several types of T cell tolerance, including thymic and peripheral deletion (19–21). In particular, several groups have demonstrated that Bcl-2 and Bim (Bcl-2-interacting mediator of death) can modulate both central and peripheral T cell tolerance (19, 21), although the up-stream molecular signals linking TCR activation to the initiation of an intrinsic apoptotic mechanism are still unknown.
Candidate molecules include the tumor suppressor protein, p53, which has been shown to initiate Fas-independent apoptosis that can be inhibited by Bcl-2 (22, 23), although AICD of T cells appears to occur independently of p53 in vivo (24, 25). Other candidate molecules include a member of the E2F family of transcription factors, such as E2F1, which was shown to regulate TCR-induced AICD (26). The specificity of E2F1 in apoptosis is still unclear, as it has been implicated in the regulation of Fas-dependent death in T cells (27), but also in the up-regulation of proapoptotic Bcl-2 family members, including Bim (28). Interestingly, E2F1-deficient mice develop autoimmunity, suggesting a role for E2F1 in the maintenance of tolerance (29).
In this report, we sought to examine the mechanism of clone 4 T cell apoptosis during peripheral tolerance to cross-presented self-Ag in InsHA mice. Our results indicate that peripheral deletion of naive CD8 T cells occurs independently of extrinsic death ligands and instead, involves a p53-, E2F1-, and E2F2-independent intrinsic apoptotic mechanism that can be inhibited by over-expression of Bcl-2. Abrogation of peripheral tolerance through the over-expression of Bcl-2 led to the acquisition of CD8 T cell effector function that ultimately led to the induction of pancreatic islet insulitis. In contrast, Bim−/− clone 4 T cells did not exhibit enhanced cytokine production and only initiated minimal islet peri-insulitis. These data suggest that defects in CD8 T cell apoptosis may contribute to the onset of organ-specific autoimmunity.
Materials and Methods
Mice
B10.D2 and BALB/c mice were purchased from the breeding colony of The Scripps Research Institute. B10.D2, B10.D2 Thy1.1, and BALB/c InsHA Tg mice, homozygous for the HA gene, and clone 4 TCR Tg mice were generated and characterized as previously described (6, 11, 30). Clone 4 mice were backcrossed with BALB/c mice for at least ten generations and were then crossed with BALB/c Thy1.1 mice for two generations to obtain clone 4 TCR mice homozygous for Thy1.1. C57BL/6 TNFR1−/−, TNFR2−/−, and p53−/− mice were obtained from The Jackson Laboratory. BALB/c lpr/lpr and C57BL/6 Bcl-2 Tg mice (31) were kindly provided by Dr. Frank Chisari and Dr. Charlie Surh, respectively (The Scripps Research Institute, La Jolla, CA). BALB/c E2F1−/− and E2F2−/− mice (29, 32) were provided by Dr. James DeGregori (University of Colorado Health Science Center, Denver, CO). C57BL/6 Bim−/− mice were kindly provided by Dr. Douglas Green (La Jolla Institute for Allergy and Immunology, La Jolla, CA). All mice obtained on a C57BL/6 background were backcrossed onto the B10.D2 background for more than ten generations before use. All mice were bred and maintained under specific pathogen-free conditions in The Scripps Research Institute’s animal facility. Experimental procedures were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Purification of naive TCR Tg T cells
Single-cell suspensions were prepared from the LNs of clone 4 (B10.D2) or clone 4 Thy1.1 (BALB/c) TCR Tg mice. Cell suspensions were depleted of CD4+, CD11b+, CD45R+, DX5+, and Ter-119+ cells using the MACS CD8α+ T cell isolation kit (Miltenyi Biotech). Clone 4 T cells were purified by negative selection per the manufacturer’s instructions and had a naive phenotype (CD44low, CD62Lhigh, and CD69-negative) as indicated by flow cytometry using FITC-conjugated Abs (Pharmingen) (data not shown). Purified clone 4 T cells were washed once in cold HBSS and then re-suspended at 5 × 107 cells/ml in HBSS.
CFSE labeling of naive clone 4 T cells
A total of 2 μl of a 5 mM solution of CFSE (Molecular Probes) in DMSO (Sigma-Aldrich) was added per 5 × 107 cells/ml in HBSS and incubated for 10 min at 37°C. Cells were then washed once with ice-cold HBSS. A total of 3–5 × 106 CFSE-labeled clone 4 T cells were injected i.v. in 200 μl of HBSS.
FTY720 treatment
The sphingosine-1-phosphate receptor-1 agonist (FTY720) was generously provided by Dr. Hugh Rosen (The Scripps Research Institute) (33–36). B10.D2 InsHA recipients were treated with FTY720 (3 mg/kg) dissolved in 200 μl HBSS i.p. on days −1, 1, 3, and 5 (clone 4 T cells were adoptively transferred on day 0).
Flow cytometry
Pancreatic and peripheral LN, including inguinal, axillary, cervical, and mandibular, were harvested and processed to obtain single-cell suspensions. All mAbs and secondary reagents used for FACS analysis were purchased from Pharmingen. Cells were then incubated for 30 min on ice with anti-Thy1.1-PE or anti-Thy1.1-PE, and anti-CD8α-PerCP. After washing three times with HBSS containing 0.1% w/v BSA (Sigma-Aldrich) and 0.02% w/v sodium azide, cells were analyzed with a FACSCalibur or LSR II flow cytometer and CellQuest (BD Biosciences) or FlowJo software (Tree Star).
To measure CD8 T cell apoptosis in vivo, single-cell suspensions were prepared from the peripheral and pancreatic LN and Annexin V staining was measured by flow cytometry using Annexin V-allophycocyanin staining Ab (BD Biosciences) according to the manufacturer’s instructions. Briefly, cells were stained with Pacific Blue-conjugated anti-CD8 mAb (Caltag Laboratories) and PE-conjugated anti-Thy1.1 mAb (BD Pharmingen) followed by washing twice with cold PBS and then re-suspended in 1× binding buffer at a concentration of 1 × 106 cells/tube. Next, cells were incubated with Annexin V-allophycocyanin for 15 min at room temperature in the dark and the % Annexin V+ clone 4 T cells was determined by flow cytometry.
To measure Ag-specific production of IFN-γ, LN cells or splenocytes were incubated in RPMI 1640 10% FCS with 1 μg/ml the Kd HA peptide and 1 μl/ml brefeldin A containing Golgi-Plug solution (Pharmingen) for 6 h at 37°C. After washing, cells were stained to detect CD8 and Thy1.2 or Thy1.1 as described above. Cells were then permeabilized and stained to detect intracellular IFN-γ with IFN-γ-allophycocyanin mAb using the Cytofix/Cytoperm Plus kit (Pharmingen) according to the manufacturer’s instructions.
Blood glucose analysis
Blood samples were obtained by retro-orbital bleeding, and blood glucose levels were determined weekly using an Accu-Check Advantage glucose meter (Roche). Mice were considered diabetic when blood glucose levels were >300 mg/dl.
Histology
Pancreata were excised and fixed overnight in 10% (v/v) formalin solution (Sigma-Aldrich) and processed for paraffin embedding. Paraffin-embedded tissue was cut using a microtome and sections were placed onto saline-coated Superfrost slides for processing (Fisher Scientific). After de-paraffinizing tissue, serial sections were stained with eosin and then counterstained with Mayer’s hematoxylin (Sigma-Aldrich).
Results
CD8 T cell peripheral tolerance occurs independently of Fas, TNFR1, or TNFR2
To elucidate the mechanism responsible for CD8 T cell apoptosis during peripheral tolerance, we first addressed the potential contribution of extrinsic death signals to this process. Clone 4 TCR Tg mice were backcrossed to mice deficient in Fas (lpr/lpr mice, which have a natural mutation in the Fas receptor), TNFR1, or TNFR2. Naive clone 4 T cells derived from each of these mice were labeled with CFSE and transferred into InsHA recipients. Eight days later, peripheral and pancreatic LN were harvested and the donor cells were analyzed by flow cytometry. As expected from previous results (5, 6, 8), clone 4 T cells underwent proliferation in the pancreatic LN (Fig. 1), but not in peripheral LN or in mice that did not express HA (6). lpr/lpr, TNFR1−/−, or TNFR2−/− clone 4 T cells underwent a similar level of activation in the pancreatic LN of InsHA recipients (Fig. 1). Furthermore, there were no statistically significant differences in either the percentage or total number of activated (dividing) wild-type vs lpr/lpr, TNFR1−/−, or TNFR2−/− clone 4 T cells accumulating in the peripheral or pancreatic LN of InsHA mice 8 days after transfer (Fig. 1 and data not shown). The lack of accumulation of the activated clone 4 T cells suggested that these particular genes did not contribute to the induction of CD8 T cell peripheral tolerance in InsHA mice.
FIGURE 1.
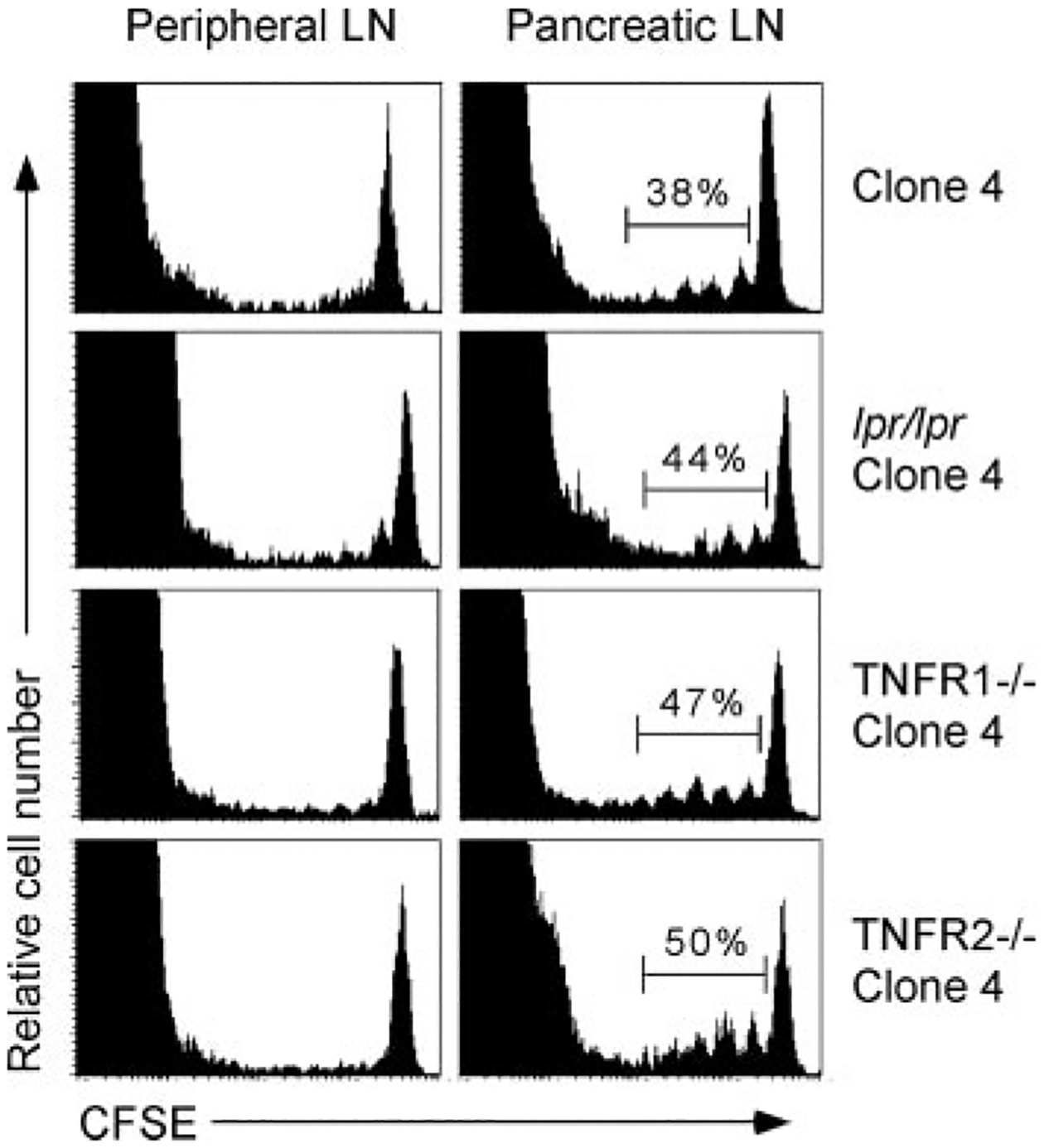
Peripheral deletion of clone 4 T cells occurs independently of death ligands. Clone 4, lpr/lpr clone 4, TNFR1−/− clone 4, or TNFR2−/− clone 4 T cells were CFSE labeled and transferred into InsHA recipients. Eight days later, peripheral and pancreatic LNs were harvested and donor CFSE-labeled CD8 T cells were analyzed by flow cytometry. Data are representative of three to four mice per group from one of two independent experiments with similar results.
CD8 T cell peripheral tolerance can be inhibited by over-expression of Bcl-2
Given that clone 4 T cell tolerance appeared to occur independently of these extrinsic death ligands, we next addressed the role of the intrinsic death pathway in CD8 T cell peripheral tolerance. Clone 4 TCR Tg mice were crossed with Bcl-2 Tg mice to obtain clone 4 T cells that constitutively express Bcl-2. CFSE-labeled clone 4 or Bcl-2 clone 4 T cells were transferred into InsHA mice and 4 or 8 days after transfer, peripheral and pancreatic LN were harvested and the donor cells were analyzed by flow cytometry. Four days after transfer, the activation profiles of clone 4 and Bcl-2 clone 4 T cells were similar (Fig. 2A). However, by 8 days after transfer, Bcl-2 clone 4 T cells exhibited a much greater degree of accumulation in the pancreatic LN as compared with wild-type clone 4 T cells (Fig. 2A). Quantitation of the total number of donor Bcl-2 clone 4 T cells revealed a significant accumulation of activated (dividing) cells as compared with wild-type clone 4 T cells in both the peripheral and pancreatic LN of InsHA mice (Fig. 2B). This process required the recognition of cognate Ag, as Bcl-2 clone 4 T cells transferred into HA-negative B10.D2 animals did not proliferate (data not shown). Importantly, the accumulation of Bcl-2 clone 4 T cells in the pancreatic LN of InsHA mice was associated with a significant reduction in Annexin V expression as compared with wild-type clone 4 T cells (Fig. 2C), providing direct evidence that the expression of Bcl-2 can prevent clone 4 T cells apoptosis during the induction of peripheral tolerance.
FIGURE 2.
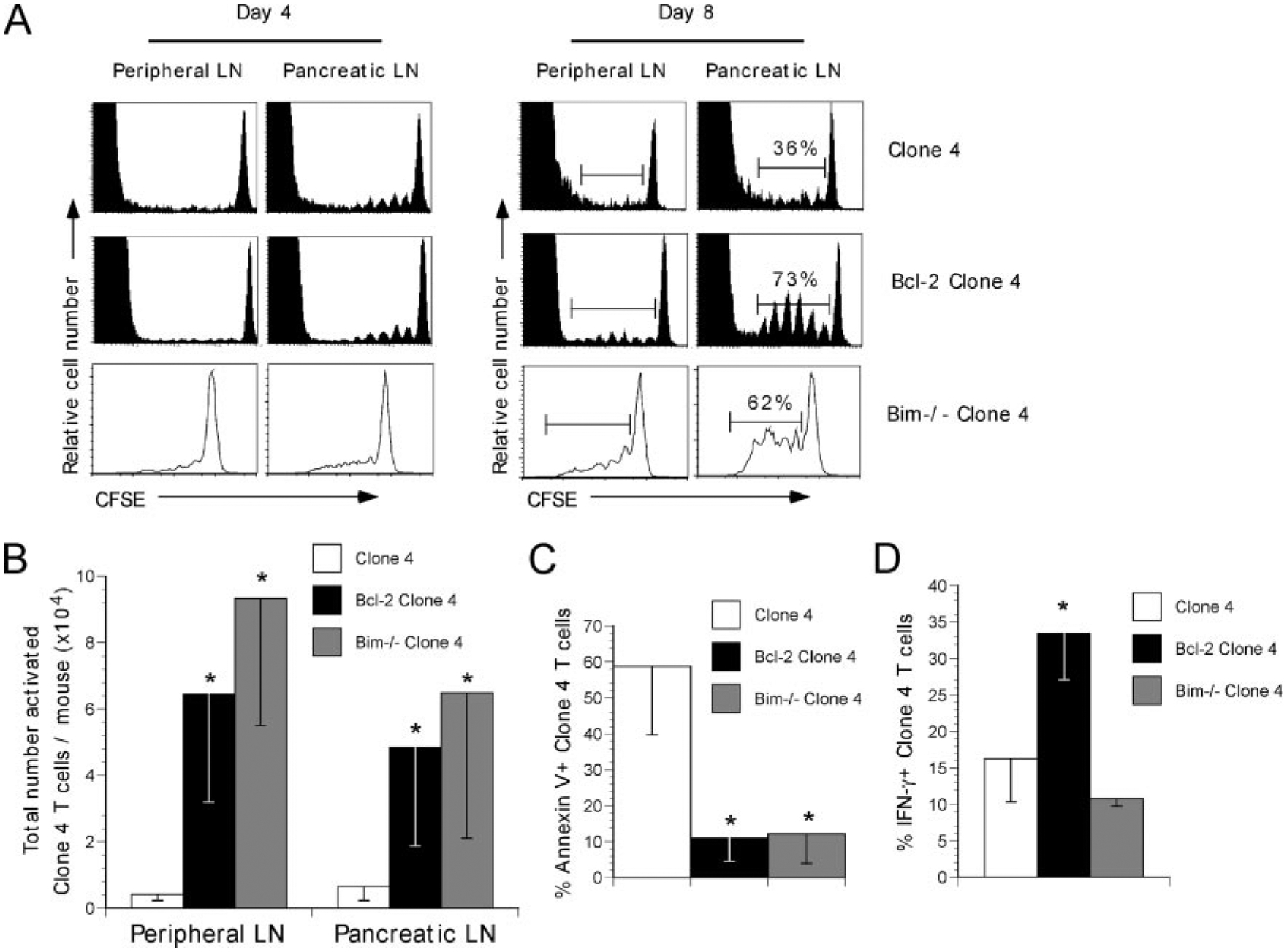
Bcl-2 clone 4 T cells are resistant to the induction of peripheral tolerance in InsHA mice. A, clone 4, Bcl-2 clone 4, or Bim−/− Thy1.2 clone 4 T cells were CFSE labeled and transferred into Thy1.2 B10.D2 (CL4, Bcl-2 CL4) or Thy1.1 B10.D2 (Bim−/− CL4) InsHA recipients. Four or eight days later, peripheral and pancreatic LNs were harvested and donor CFSE-labeled CD8 T cells were analyzed by flow cytometry. Graphs depict total Thy1.2 CD8 T cells (host and donor CL4 or Bcl-2 CL4) or donor Thy1.1 CD8 T cells (Bim−/− CL4). The percentage of activated clone 4 T cells is indicated. B, Total numbers of activated (dividing, based upon CFSE dilution, as gated in A) clone 4, Bcl-2 clone 4, or Bim−/−. Clone 4 T cells (from A) in the peripheral or pancreatic LNs of InsHA mice were determined by flow cytometry and calculated as: (percent activated clone 4 T cells per sample) × (total number of cells per animal). C, The extent of donor clone 4 T cell apoptosis in the pancreatic LNs was assessed by Annexin V expression and determined by flow cytometry. D, The extent of intracellular IFN-γ production was measured in activated clone 4 T cells from A following re-stimulation in vitro with Kd HA peptide for 6 h at 37°C. All graphs depict the mean ± SD of two to four mice per group. Data are representative of one of three independent experiments with similar results. (*, p < 0.05).
Given that the over-expression of Bcl-2 can enhance the survival of naive CD8 T cells, we sought to exclude the possibility that the increased accumulation of Bcl-2 clone 4 T cells was a by-product of the enhanced ability of the naive Bcl-2 clone 4 T cells to survive and subsequently traffic to the pancreatic LN of InsHA mice. To this end, large numbers of CFSE-labeled wild-type clone 4 T cells (up to 20 × 106/mouse) were adoptively transferred into InsHA mice. Eight days later, the peripheral and pancreatic LN were harvested and the proliferative profile of the donor clone 4 T cells was examined by flow cytometry. No difference was observed in clone 4 T cell proliferation or survival of the proliferating cells (data not shown). Taken together with the decrease extent of Bcl-2 clone 4 T cells apoptosis as compared with wild-type clone 4 T cells (Fig. 2C), these data suggest that the protective effects of Bcl-2 are due to a reduction in apoptosis of the activated cells, which is consistent with a role for a Bcl-2-sensitive mechanism in CD8 T cell tolerance (37).
Since activated Bcl-2 clone 4 T cells accumulated in large numbers, we next examined whether these cells were able to acquire effector functions that may promote their ability to induce autoimmunity. Clone 4 or Bcl-2 clone 4 T cells were transferred into InsHA mice and eight days later pancreatic LN were harvested. Next, donor clone 4 T cells were re-stimulated with HA peptide in vitro and the amount of intracellular IFN-γ production was assessed by flow cytometry. A larger proportion of activated Bcl-2 clone 4 T cells recovered from the pancreatic LN of InsHA mice exhibited IFN-γ production as compared with wild-type clone 4 T cells (Fig. 2D). These data indicate that expression of Bcl-2 not only inhibited peripheral deletion of clone 4 T cells, it also promoted the acquisition of effector function.
Bim−/− clone 4 T cells are resistant to the induction of apoptosis to a cross-presented self-Ag
Recent studies have shown that CD8 T cell deletion during peripheral tolerance to a cross-presented self-Ag is mediated through the proapoptotic Bcl-2 family member, Bim (21). To examine the role of Bim in clone 4 T cell peripheral tolerance, CFSE-labeled Bim-deficient clone 4 T cells were transferred into InsHA recipients, and 4 or 8 days after transfer, the peripheral and pancreatic LN were harvested and the donor cells were analyzed by flow cytometry. Consistent with the results obtained using Bcl-2 clone 4 T cells, Bim−/− clone 4 T cells were able to survive and accumulate in the peripheral and pancreatic LN of InsHA mice (Fig. 2B) and also exhibited a reduction in apoptosis as measured by Annexin V staining (Fig. 2C), suggesting that clone 4 T cell peripheral deletion occurred through a Bim-dependent mechanism. Interestingly, Bim−/− clone 4 T cells recovered from the pancreatic LN of InsHA mice 8 days after transfer did not up-regulate IFN-γ production as compared with wild-type clone 4 T cells (Fig. 2D). These data indicate that although Bim−/− clone 4 T cells were protected from apoptosis, in contrast to what was observed with Bcl-2 clone 4 T cells, the lack of Bim expression was not sufficient to promote the acquisition of clone 4 T cell effector function.
Adoptively transferred Bcl-2 clone 4 T cells can infiltrate islet β-cells
To assess whether Bcl-2 clone 4 T cells were able to promote islet destruction and the onset of autoimmune diabetes, naive clone 4 or Bcl-2 clone 4 T cells were transferred into InsHA recipients. Blood glucose levels were measured weekly and the pancreas was taken for histology 21 days after adoptive transfer. Histological analysis revealed that animals receiving Bcl-2 clone 4 T cells had evidence of lymphocyte infiltrates in the pancreatic islets (Fig. 3). Indeed, ~50% of the pancreatic islets examined exhibited either peri- or full insulitis, whereas Bim−/− clone 4 T cells only induced minimal islet peri-insulitis following adoptive transfer into InsHA mice (Fig. 3 and Table I). In contrast, none of the animals receiving wild-type clone 4 T cells, or clone 4 T cells deficient in Fas, TNFR1, or TNFR2, became diabetic or had signs of significant islet infiltrates (Table I and data not shown). It should be noted that although Bcl-2 clone 4 T cells induced a significant amount of islet damage, adoptive transfer of these cells did not induce overt diabetes in InsHA mice (data not shown) because destruction of greater than 80% of islets is required to result in overt diabetes.
FIGURE 3.
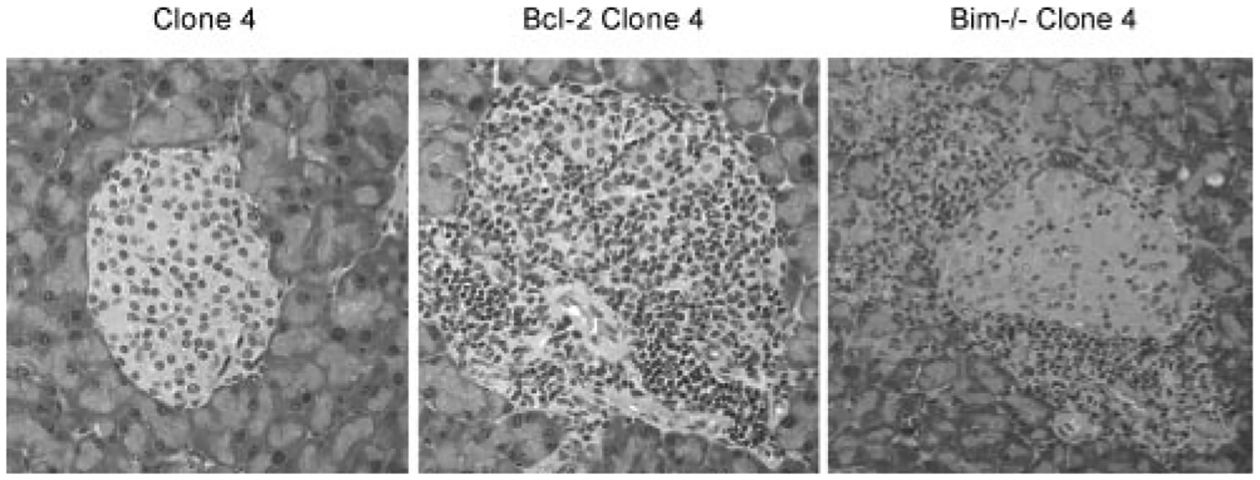
Bcl-2 clone 4 T cells are able to infiltrate pancreatic islet β-cells in InsHA mice. Clone 4, Bcl-2 clone 4, or Bim−/− clone 4 T cells were transferred into InsHA recipients. After 14–21 days, mice were sacrificed and the pancreas taken for histology. Representative pancreatic islet from mice receiving clone 4 T cells had no evidence of lymphocytic infiltrates or islet β-cell damage. Islets from mice receiving Bcl-2 clone 4 T cells exhibited a significant amount of islet β-cell infiltrates, whereas the adoptive transfer of Bim−/− clone 4 T cells only led to a minimal amount of islet β-cell peri-insulitis (see Table I). Representative islets demonstrating the most extensive level of lymphocyte infiltrates are shown. Original magnification: ×200.
Table I.
Adoptive transfer of Bcl-2 clone 4 T cells promotes insulitis in InsHA recipients (% of total islets)a
| Cells Transferred | Insulitis | Peri-Insulitis | No Infiltrates | Total Islets Scored |
|---|---|---|---|---|
| clone 4 | 0 | 0 | 100 | 21 |
| Ipr/lpr clone 4 | 0 | 0 | 100 | 39 |
| TNFR1−/− clone 4 | 0 | 0 | 100 | 33 |
| TNFR2−/− clone 4 | 0 | 0 | 100 | 28 |
| p53−/− clone 4 | 0 | 0 | 100 | 32 |
| Bim−/− clone 4 | 0 | 8.3 | 91.7 | 48 |
| Bcl-2 clone 4 | 27.5 | 25.5 | 47 | 51 |
The indicated clone 4 T cells were adoptively transferred into InsHA recipients. Three weeks later, the pancreas was taken and the extent of lymphocyte infiltrates was determined by histological analysis. Data are representative of three to four mice per group from two independent experiments.
Accumulation of Bcl-2 clone 4 T cells in InsHA mice is not due to islet-specific damage
The accumulation of Bcl-2 clone 4 T cells in the pancreatic LN of InsHA mice could have been the result of several factors, such as the inhibition of apoptosis, proinflammatory cytokine milieu, or increased pancreatic islet β-cell destruction. The latter could lead to an enhanced antigenic load and thus a greater level of T cell priming. To exclude this possibility, InsHA mice were treated with a sphingosine-1-phosphate receptor 1 agonist (FTY720) that has been shown to block lymphocyte egress from the LN into peripheral tissues (36). Although drug-treated naive T cells can still respond to antigenic stimulation, they are unable to migrate into tissues, such as the pancreas. InsHA mice were treated were FTY720 on day −1, 1, 3, and 5 and then naive Bcl-2 clone 4 T cells were adoptively transferred on day 0. Seven days later, pooled peripheral or pancreatic LNs were harvested and donor cells examined by flow cytometry. As shown in Fig. 4A, the prevention of Bcl-2 clone 4 egress from the pancreatic LNs did not affect their initial activation and proliferation as similar CFSE-profiles were obtained from both treated and control groups. Quantitative analysis indicated that similar numbers of control or FTY720-treated Bcl-2 clone 4 T cells accumulated in the pancreatic LNs of InsHA recipient mice (Fig. 4B). The effectiveness of the drug-treatment was confirmed via histological analysis of the FTY720-treated InsHA recipients, which revealed no lymphocytic infiltrates in the pancreatic islets (data not shown). These data suggest the accumulation of Bcl-2 clone 4 T cells was not the result of destruction of islet β-cells.
FIGURE 4.
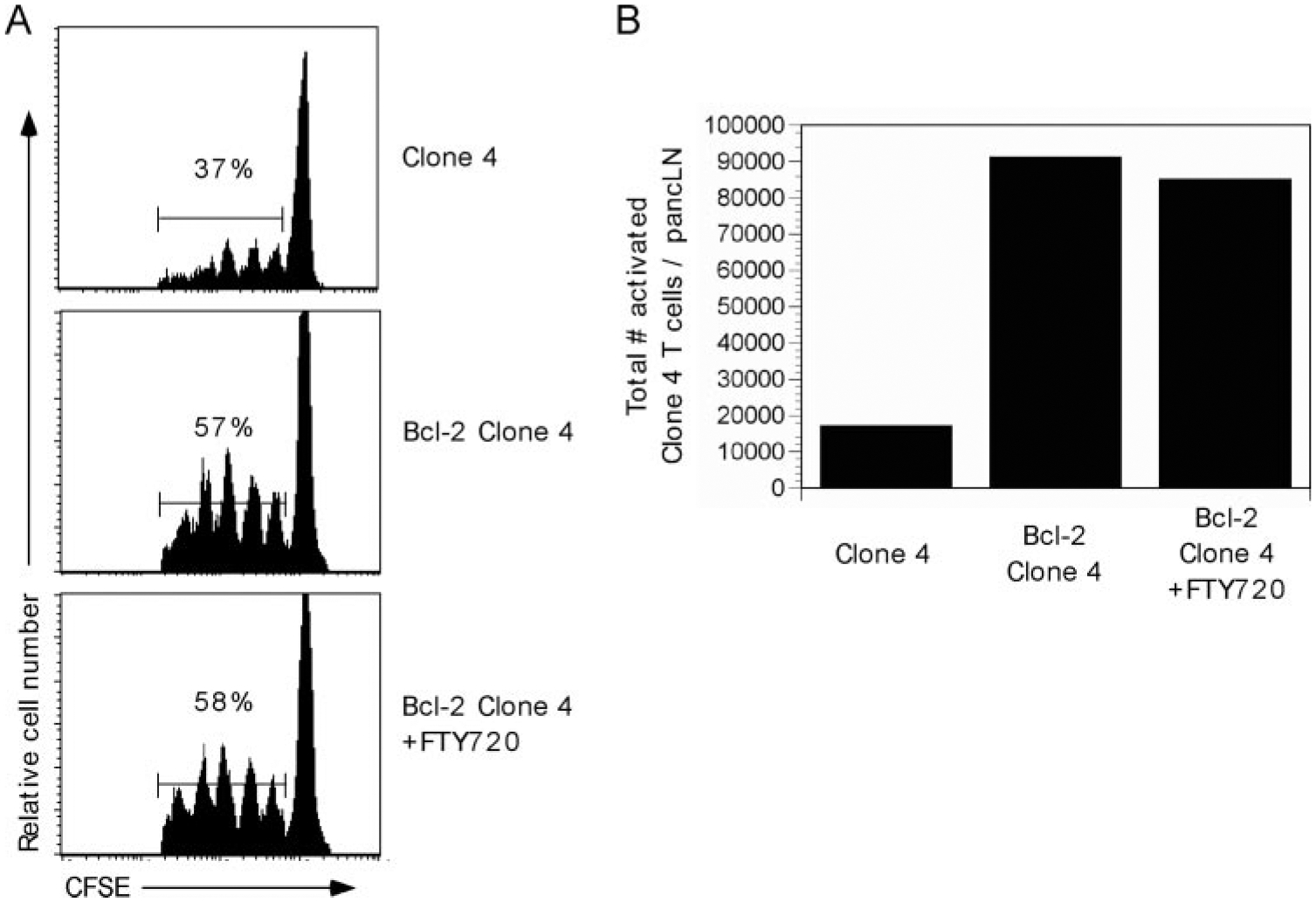
Accumulation of Bcl-2 clone 4 T cells in InsHA mice is due to their resistance to apoptosis rather than increased islet damage. InsHA mice were treated with FTY720 on day −1, 1, 3, and 5 i.p. On day 0, clone 4 or Bcl-2 clone 4 T cells were adoptively transferred into InsHA mice. A, Seven days later, peripheral and pancreatic LNs were harvested and donor CFSE-labeled clone 4 T cells were analyzed by flow cytometry. The percentage of activated clone 4 T cells is indicated. B, Total numbers of clone 4 or Bcl-2 clone 4 T cells (from A) were quantitated by flow cytometry. Graphs represent the mean of activated (based on CFSE dilution) clone 4 or Bcl-2 clone 4 T cells in the pancreatic LNs of InsHA mice. Data are representative of three to four mice per group from one of two independent experiments with similar results.
CD8 T cell peripheral tolerance occurs via a p53-, E2F1-, and E2F2-independent mechanism
Because the over-expression of Bcl-2 was able to prevent the apoptosis of clone 4 T cells during peripheral tolerance, we next sought to determine the up-stream signals that might mediate this process. Previous studies have been shown that p53-mediated apoptosis can be inhibited by up-regulated expression of Bcl-2 (23). Therefore, we investigated whether p53 was the up-stream molecule promoting the death of activated clone 4 T cells. Naive CD8 T cells from clone 4 or p53−/− clone 4 mice were CFSE-labeled and transferred into InsHA recipient mice. Eight days after transfer, peripheral and pancreatic LNs were harvested and the extent of proliferation monitored by flow cytometry. There was no significant difference in the activation or accumulation of p53−/− or wild-type clone 4 T cells in the pancreatic LNs of InsHA mice (Fig. 5), nor did p53−/− clone 4 T cells initiate pancreatic insulitis (Table I), suggesting that p53 was not involved in the induction of clone 4 T cell peripheral tolerance.
FIGURE 5.
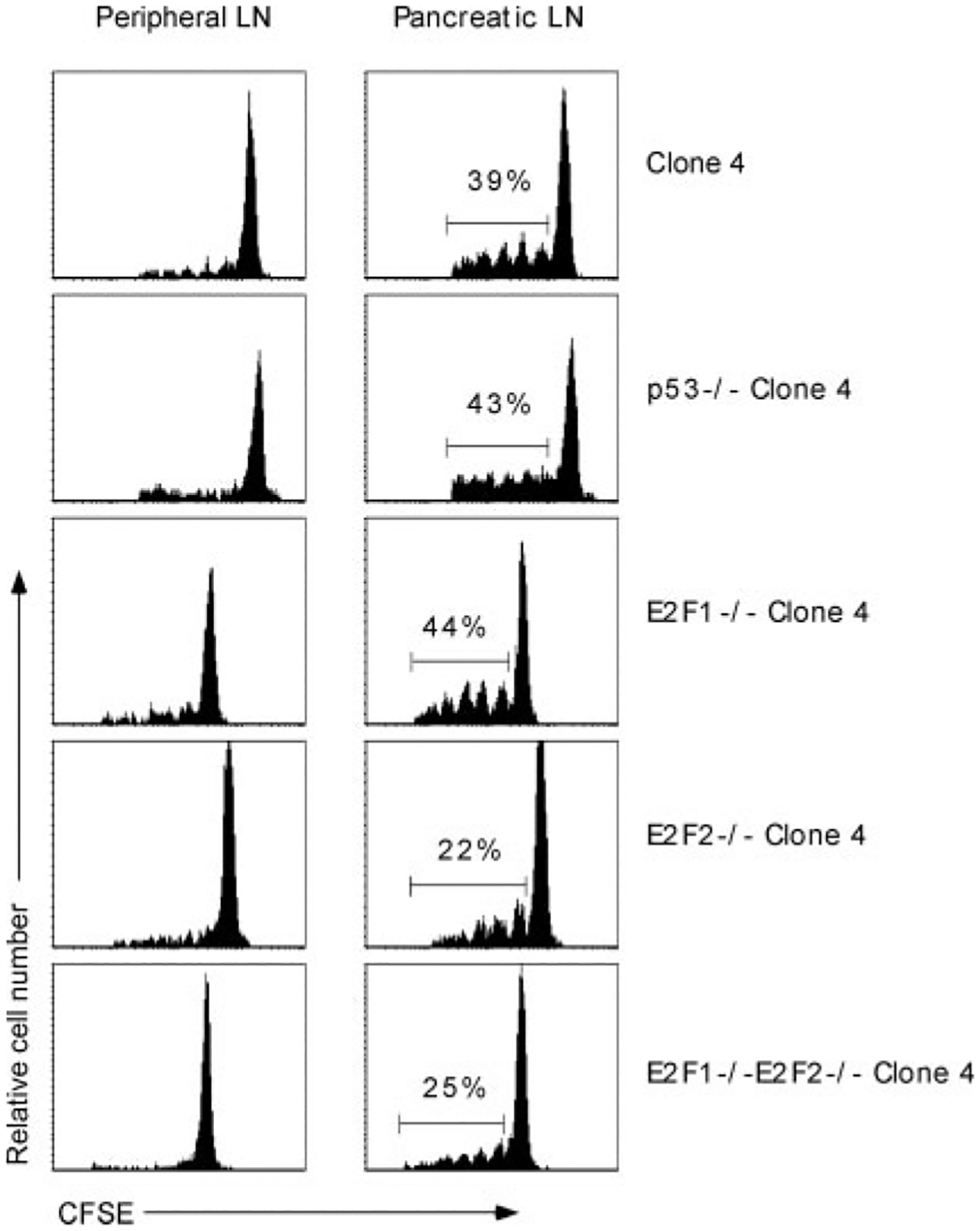
Peripheral deletion of clone 4 T cell in InsHA mice occurs independently of p53, E2F1, and E2F2. p53−/−, E2F1−/−, E2F2−/−, or wild-type clone 4 T cells were CFSE labeled and transferred into InsHA recipients. Eight days later, peripheral and pancreatic LNs were harvested and donor CFSE-labeled CD8 T cells were analyzed by flow cytometry. The percentage of activated clone 4 T cells is indicated. Data are representative of three to four mice per group from one of two to three independent experiments with similar results.
Several recent studies have demonstrated that the transcription factors E2F1 and E2F2 can also mediate the induction of T cell apoptosis (26, 32). In addition, E2F1 has been shown to directly affect the activation of several molecules involved in the intrinsic apoptotic pathway, including Bim and Apaf-1 (28, 38). Therefore, given our data indicating that clone 4 T cell apoptosis occurred via an intrinsic (Bcl-2-sensitive) apoptotic mechanism, we next investigated the potential contribution of E2F1 and E2F2 to CD8 T cell peripheral tolerance. Naive CFSE-labeled E2F1−/− or E2F2−/− clone 4 T cells were adoptively transferred into InsHA mice. Eight days later, peripheral or pancreatic LNs were harvested from the recipient mice and the proliferation and accumulation of the donor cells was examined by flow cytometry. No statistically significant difference was observed in the activation or accumulation of E2F1−/− or E2F2−/− clone 4 T cells as compared with wild-type clone 4 T cells (Fig. 5). Furthermore, there was no increase in intracellular IFN-γ production by the E2F1- or E2F2-deficient clone 4 T cells (data not shown). Although other groups have reported that the combined deficiency in E2F1/E2F2 can lower the threshold of TCR-induced activation in CD4 T cells (39), double-deficient clone 4 T cells underwent normal activation and deletion following adoptive transfer into InsHA recipients (Fig. 5). Importantly, no statistically significant differences were detected among the total number of activated wild-type vs p53−/−, E2F1−/−, E2F2−/−, or E2F1/E2F2−/− clone 4 T cells accumulating in the pancreatic LNs of InsHA mice 8 days after transfer (data not shown). These data suggest that the induction of clone 4 T cell peripheral tolerance in InsHA mice occurs via a p53-, E2F1-, and E2F2-independent apoptotic mechanism.
Discussion
In this study, we set out to determine the apoptotic mechanism used during the induction of clone 4 T cell peripheral tolerance to a cross-presented self-Ag. As there have been numerous reports implicating members of the TNF-family of death receptors as the initiators of T cell apoptosis during peripheral tolerance, we first examined the role of Fas, TNFR1, and TNFR2. No role was found for these molecules in the induction of CD8 T cell peripheral tolerance (Fig. 1). Additionally, clone 4 T cells deficient in any one of these molecules did not initiate autoimmunity in InsHA mice (Table I), which was consistent with the inability of these molecules to alter the proliferative profile and survival of activated clone 4 T cells.
Considering the lack of evidence for a role of an extrinsic death ligand in CD8 T cell peripheral tolerance, we next addressed the possibility that clone 4 T cell apoptosis occurred via a TCR-mediated intrinsic death pathway. To test this hypothesis, we examined the effect of the anti-apoptotic molecule, Bcl-2, on clone 4 T cell peripheral tolerance. Following adoptive transfer, Bcl-2 clone 4 T cells accumulated in the peripheral and pancreatic LNs of InsHA mice, which correlated with a reduced apoptosis of the Bcl-2 clone 4 T cells (Fig. 2C). In addition, Bcl-2 clone 4 T cells spontaneously acquired effector functions and were able to promote insulitis (Figs. 2 and 3 and Table I). The ability of donor Bcl-2 clone 4 T cells to infiltrate the pancreatic islets led us to question whether the accumulation of Bcl-2 clone 4 T cells was due to increased islet damage and thus, the increased availability of Ag, or other factors such as the inhibition of apoptosis or increased cytokine production. To prevent the initiation of Bcl-2 clone 4 T cell-mediated islet damage, InsHA recipient mice were first treated with an S1P receptor-1 agonist drug (FTY720) that has been shown to prevent lymphocyte egress from the LNs (33, 34, 36). Treatment with FTY720 had no effect on the activation, proliferation, or accumulation of Bcl-2 clone 4 T cells (Fig. 4), suggesting that Bcl-2 was primarily inhibiting clone 4 T cell apoptosis. However, we cannot exclude the possibility that the accumulation of Bcl-2 clone 4 T cells in the pancreatic LNs was also influenced by the presence of proinflammatory cytokines, such as IFN-γ (Fig. 2D). It is possible that, in addition to the anti-apoptotic effects of Bcl-2, the inflammatory milieu generated in the pancreatic LNs also promoted the recruitment and/or survival of Bcl-2 clone 4 T cells.
Given the ability of Bcl-2 to inhibit the peripheral deletion of clone 4 T cells in vivo, we next sought to determine the molecular mechanisms linking TCR signaling to the induction of the intrinsic apoptotic pathway. Recent studies have implicated the proapoptotic Bcl-2 family member, Bim, in the deletion of autoreactive thymocytes, mature T cells undergoing superantigen-induced deletion, and CD8 T cells recognizing peripherally expressed self-Ag in RIP-mOVA Tg mice (19–21). Consistent with the results obtained using Bcl-2 clone 4 T cells, Bim−/− clone 4 T cells accumulated in the peripheral and pancreatic LNs of InsHA mice (Fig. 2B) and exhibited a reduction in apoptosis (Fig. 2C). However, in contrast with results obtained using Bcl-2 clone 4 T cells, the adoptive transfer of Bim−/− clone 4 T cells into InsHA mice did not promote their ability to produce IFN-γ and only led to a minimal amount of islet peri-insulitis (Figs. 2D and 3 and Table I).
The reasons underlying the differences observed between Bcl-2 Tg and Bim−/− clone 4 T cells are still unclear, but may reflect differences in the extent to which each apoptotic pathway is used during tolerance induction. Indeed, the induction of T cell apoptosis through both Bim-dependent and -independent pathways can be inhibited by over-expression of Bcl-2 (40), suggesting that Bcl-2 has a more global protective effect. Interestingly, previous studies have demonstrated that Bcl-2 Tg or Bim−/− OT-I T cells accumulated to a similar degree during the induction of peripheral tolerance to a cross-presented self-Ag in RIP-mOVA mice (21). One critical difference between these two model systems is that when high numbers of naive OT-I T cells are adoptively transferred into RIP-mOVA mice, these cells spontaneously acquire effector function and can initiate diabetes (21). In contrast, naive clone 4 T cells do not spontaneously acquire effector functions or induce diabetes following their adoptive transfer into InsHA mice (Fig. 2D and Table I) (8, 41). This has allowed us to observe an effect of Bcl-2 over-expression in promoting acquisition of effector function.
Thus far, our data indicated that CD8 T cell deletion during peripheral tolerance in InsHA mice occurred through a Bim- and Bcl-2-sensitive mechanism. To explore the upstream molecular mechanisms required to induce clone 4 T cell apoptosis, we investigated the role of p53, a well-characterized proapoptotic molecule that has been shown to induce apoptosis in a Fas-independent manner that can be inhibited by Bcl-2 (22, 23, 42, 43). Although the induction of T cell apoptosis following viral immunization or stimulation with anti-CD3 mAb in vitro was shown to occur in a p53-independent manner (24, 25), the role of p53 in CD8 T cell peripheral tolerance is unknown. As shown in Fig. 5, p53−/− clone 4 T cells did not accumulate in the pancreatic LNs of InsHA mice, suggesting that this molecule was not involved in clone 4 T cell apoptosis during peripheral tolerance. Interestingly, recent work suggested that the p53 homologue, p73, may also promote the apoptosis of mature T cells (26, 44). Future experiments will be needed to address the contribution of p73 to clone 4 T cell peripheral tolerance.
Next, we assessed the contribution of another proapoptotic molecule, E2F1, in the deletion of clone 4 T cells. E2F1 was initially of interest as a candidate apoptosis-inducing molecule based its ability to directly up-regulate several down-stream proapoptotic molecules, including Bim and Apaf-1 (28, 38). Although E2F1 was recently reported to mediate T cell apoptosis and lower the threshold of TCR-induced activation (26, 29), we did not observe any statistically significant difference in the proliferation or accumulation of E2F1-deficient vs wild-type clone 4 T cells following adoptive transfer into InsHA mice (Fig. 5). Similar results were obtained with E2F2−/− or E2F1−/−E2F2−/− clone 4 T cells (Fig. 5), suggesting that neither of the molecules was required for the initiation of clone 4 T cell proliferation or apoptosis during peripheral tolerance to a cross-presented self-Ag. The role of E2F1 in CD8 T cell apoptosis may depend upon the stimulatory environment, such that strong TCR stimulation initiates a distinct signaling cascade. Indeed, one recent report suggests that E2F1 may regulate an extrinsic Fas-dependent death pathway in both CD4 and CD8 T cells following strong TCR ligation (27).
To further probe the downstream molecular mechanisms regulating clone 4 T cell apoptosis during peripheral tolerance, we have begun to examine the role of additional molecules including the proapoptotic Bcl-2 family members, Noxa and Puma (p53-up-regulated modulator of apoptosis) (45–47). Given that CD8 T cell peripheral tolerance appears to occur in a p53-independent manner and that Noxa is induced in a p53-dependent manner (45, 46), it is unlikely that Noxa plays a significant role in CD8 T cell apoptosis during peripheral deletion. In contrast, Puma has been shown to be regulated in both a p53-dependent and -independent manner (47, 48). Furthermore, recent studies have demonstrated that the apoptosis of activated T cells can occur through a Bim-independent pathway that involves up-regulation Puma (40, 49). Although we have observed an increase in Puma mRNA expression in activated clone 4 T cells recovered from the pancreatic LNs of InsHA mice (data not shown), additional experiments will be required to determine whether Puma is the key proapoptotic molecule that mediates Bim-independent clone 4 T cell apoptosis in vivo.
In conclusion, these results indicate that the peripheral deletion of clone 4 T cells in response to cross-presented self-Ag in the pancreatic LNs of InsHA mice occurs via a Bcl-2-sensitive and Bim-dependent intrinsic apoptotic mechanism that is not prevented by the absence of p53, E2F1, or E2F2. In addition, prevention of peripheral tolerance by over-expression of Bcl-2 can promote the onset of spontaneous autoimmunity. Although further studies will be needed to clarify the molecular mechanisms involved in peripheral deletion, these data indicate that defects in CD8 T cell apoptosis during peripheral tolerance can play an important role in the modulation of organ-specific autoimmunity.
Acknowledgments
We thank Judith Biggs, Kristi Marquardt, and Rebecca Trenney for excellent technical assistance and members of the Sherman laboratory for helpful discussions.
This work was supported by National Institutes of Health Grants R01 DK50824 and T32 AI07606 (to L.A.S.), an Achievement Awards for College Scientists Foundation Fellowship (to W.L.R.), and a Juvenile Diabetes Research Foundation Fellowship (to C.-H.W.).
6. Abbreviations used in this paper:
- Tg
transgenic
- HA
hemagglutinin
- AICD
activation-induced cell death
- LN
lymph node
Footnotes
Disclosures
The authors have no financial conflict of interest.
References
- 1.Kappler JW, Roehm N, and Marrack P. 1987. T cell tolerance by clonal elimination in the thymus. Cell 49: 273–280. [DOI] [PubMed] [Google Scholar]
- 2.Kishimoto H, and Sprent J. 2000. The thymus and central tolerance. Clin. Immunol 95: S3–S7. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, and Lenardo MJ. 1997. Molecules involved in cell death and peripheral tolerance. Curr. Opin. Immunol 9: 818–825. [DOI] [PubMed] [Google Scholar]
- 4.Walker LS, and Abbas AK. 2002. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol 2: 11–19. [DOI] [PubMed] [Google Scholar]
- 5.Morgan DJ, Kurts C, Kreuwel HT, Holst KL, Heath WR, and Sherman LA. 1999. Ontogeny of T cell tolerance to peripherally expressed antigens. Proc. Natl. Acad. Sci. USA 96: 3854–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan DJ, Kreuwel HT, and Sherman LA. 1999. Antigen concentration and precursor frequency determine the rate of CD8+ T cell tolerance to peripherally expressed antigens. J. Immunol 163: 723–727. [PubMed] [Google Scholar]
- 7.Miller JF, Kurts C, Allison J, Kosaka H, Carbone F, and Heath WR. 1998. Induction of peripheral CD8+ T-cell tolerance by cross-presentation of self antigens. Immunol. Rev 165: 267–277. [DOI] [PubMed] [Google Scholar]
- 8.Hernandez J, Aung S, Redmond WL, and Sherman LA. 2001. Phenotypic and functional analysis of CD8+ T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J. Exp. Med 194: 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sallusto F, and Lanzavecchia A. 1999. Mobilizing dendritic cells for tolerance, priming, and chronic inflammation. J. Exp. Med 189: 611–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banchereau J, and Steinman RM. 1998. Dendritic cells and the control of immunity. Nature 392: 245–252. [DOI] [PubMed] [Google Scholar]
- 11.Lo D, Freedman J, Hesse S, Palmiter RD, Brinster RL, and Sherman LA. 1992. Peripheral tolerance to an islet cell-specific hemagglutinin transgene affects both CD4+ and CD8+ T cells. Eur. J. Immunol 22: 1013–1022. [DOI] [PubMed] [Google Scholar]
- 12.Hildeman DA, Zhu Y, Mitchell TC, Kappler J, and Marrack P. 2002. Molecular mechanisms of activated T cell death in vivo. Curr. Opin. Immunol 14: 354–359. [DOI] [PubMed] [Google Scholar]
- 13.Strasser A, O’Connor L, and Dixit VM. 2000. Apoptosis signaling. Annu. Rev. Biochem 69: 217–245. [DOI] [PubMed] [Google Scholar]
- 14.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, and Lenardo MJ. 1995. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature 377: 348–351. [DOI] [PubMed] [Google Scholar]
- 15.Sytwu HK, Liblau RS, and McDevitt HO. 1996. The roles of Fas/APO-1 (CD95) and TNF in antigen-induced programmed cell death in T cell receptor transgenic mice. Immunity 5: 17–30. [DOI] [PubMed] [Google Scholar]
- 16.Van Parijs L, Biuckians A, and Abbas AK. 1998. Functional roles of Fas and Bcl-2-regulated apoptosis of T lymphocytes. J. Immunol 160: 2065–2071.9498742 [Google Scholar]
- 17.Refaeli Y, Van Parijs L, London CA, Tschopp J, and Abbas AK. 1998. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity 8: 615–623. [DOI] [PubMed] [Google Scholar]
- 18.Van Parijs L, Peterson DA, and Abbas AK. 1998. The Fas/Fas ligand pathway and Bcl-2 regulate T cell responses to model self and foreign antigens. Immunity 8: 265–274. [DOI] [PubMed] [Google Scholar]
- 19.Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, and Strasser A. 2002. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 415: 922–926. [DOI] [PubMed] [Google Scholar]
- 20.Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, and Marrack P. 2002. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity 16: 759–767. [DOI] [PubMed] [Google Scholar]
- 21.Davey GM, Kurts C, Miller JF, Bouillet P, Strasser A, Brooks AG, Carbone FR, and Heath WR. 2002. Peripheral deletion of autoreactive CD8 T cells by cross presentation of self-antigen occurs by a Bcl-2-inhibitable pathway mediated by Bim. J. Exp. Med 196: 947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Connor L, Harris AW, and Strasser A. 2000. CD95 (Fas/APO-1) and p53 signal apoptosis independently in diverse cell types. Cancer Res. 60: 1217–1220. [PubMed] [Google Scholar]
- 23.Chiou SK, Rao L, and White E. 1994. Bcl-2 blocks p53-dependent apoptosis. Mol. Cell. Biol 14: 2556–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grayson JM, Lanier JG, Altman JD, and Ahmed R. 2001. The role of p53 in regulating antiviral T cell responses. J. Immunol 167: 1333–1337. [DOI] [PubMed] [Google Scholar]
- 25.Boehme SA, and Lenardo MJ. 1996. TCR-mediated death of mature T lymphocytes occurs in the absence of p53. J. Immunol 156: 4075–4078. [PubMed] [Google Scholar]
- 26.Lissy NA, Davis PK, Irwin M, Kaelin WG, and Dowdy SF. 2000. A common E2F-1 and p73 pathway mediates cell death induced by TCR activation. Nature 407: 642–645. [DOI] [PubMed] [Google Scholar]
- 27.Cao Q, Xia Y, Azadniv M, and Crispe IN. 2004. The E2F-1 transcription factor promotes caspase-8 and bid expression, and enhances Fas signaling in T cells. J. Immunol 173: 1111–1117. [DOI] [PubMed] [Google Scholar]
- 28.Hershko T, and Ginsberg D. 2004. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J. Biol. Chem 279: 8627–8634. [DOI] [PubMed] [Google Scholar]
- 29.Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG Jr., Livingston DM, Orkin SH, and Greenberg ME. 1996. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell 85: 549–561. [DOI] [PubMed] [Google Scholar]
- 30.Morgan DJ, Liblau R, Scott B, Fleck S, McDevitt HO, Sarvetnick N, Lo D, and Sherman LA. 1996. CD8+ T cell-mediated spontaneous diabetes in neonatal mice. J. Immunol 157: 978–983. [PubMed] [Google Scholar]
- 31.Strasser A, Harris AW, and Cory S. 1991. bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell 67: 889–899. [DOI] [PubMed] [Google Scholar]
- 32.Murga M, Fernandez-Capetillo O, Field SJ, Moreno B, Borlado LR, Fujiwara Y, Balomenos D, Vicario A, Carrera AC, Orkin SH, et al. 2001. Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity 15: 959–970. [DOI] [PubMed] [Google Scholar]
- 33.Rosen H, Sanna G, and Alfonso C. 2003. Egress: a receptor-regulated step in lymphocyte trafficking. Immunol. Rev 195: 160–177. [DOI] [PubMed] [Google Scholar]
- 34.Xie JH, Nomura N, Koprak SL, Quackenbush EJ, Forrest MJ, and Rosen H. 2003. Sphingosine-1-phosphate receptor agonism impairs the efficiency of the local immune response by altering trafficking of naive and antigen-activated CD4+ T cells. J. Immunol 170: 3662–3670. [DOI] [PubMed] [Google Scholar]
- 35.Rosen H, Alfonso C, Surh CD, and McHeyzer-Williams MG. 2003. Rapid induction of medullary thymocyte phenotypic maturation and egress inhibition by nanomolar sphingosine 1-phosphate receptor agonist. Proc. Natl. Acad. Sci. USA 100: 10907–10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, et al. 2002. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296: 346–349. [DOI] [PubMed] [Google Scholar]
- 37.Petschner F, Zimmerman C, Strasser A, Grillot D, Nunez G, and Pircher H. 1998. Constitutive expression of Bcl-xL or Bcl-2 prevents peptide antigen-induced T cell deletion but does not influence T cell homeostasis after a viral infection. Eur. J. Immunol 28: 560–569. [DOI] [PubMed] [Google Scholar]
- 38.Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Muller H, and Helin K. 2001. Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell. Biol 3: 552–558. [DOI] [PubMed] [Google Scholar]
- 39.Zhu JW, Field SJ, Gore L, Thompson M, Yang H, Fujiwara Y, Cardiff RD, Greenberg M, Orkin SH, and DeGregori J. 2001. E2F1 and E2F2 determine thresholds for antigen-induced T-cell proliferation and suppress tumorigenesis. Mol. Cell. Biol 21: 8547–8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bauer A, Villunger A, Labi V, Fischer SF, Strasser A, Wagner H, Schmid RM, and Hacker G. 2006. The NF-κB regulator Bcl-3 and the BH3-only proteins Bim and Puma control the death of activated T cells. Proc. Natl. Acad. Sci. USA 103: 10979–10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Redmond WL, Hernandez J, and Sherman LA. 2003. Deletion of naive CD8 T cells requires persistent antigen and is not programmed by an initial signal from the tolerogenic APC. J. Immunol 171: 6349–6354. [DOI] [PubMed] [Google Scholar]
- 42.Marin MC, Hsu B, Meyn RE, Donehower LA, el-Naggar AK, and McDonnell TJ. 1994. Evidence that p53 and bcl-2 are regulators of a common cell death pathway important for in vivo lymphomagenesis. Oncogene 9: 3107–3112. [PubMed] [Google Scholar]
- 43.Wang Y, Szekely L, Okan I, Klein G, and Wiman KG. 1993. Wild-type p53-triggered apoptosis is inhibited by bcl-2 in a v-myc-induced T-cell lymphoma line. Oncogene 8: 3427–3431. [PubMed] [Google Scholar]
- 44.Wan YY, and DeGregori J. 2003. The survival of antigen-stimulated T cells requires NFκB-mediated inhibition of p73 expression. Immunity 18: 331–342. [DOI] [PubMed] [Google Scholar]
- 45.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, and Tanaka N. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053–1058. [DOI] [PubMed] [Google Scholar]
- 46.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, and Strasser A. 2003. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302: 1036–1038. [DOI] [PubMed] [Google Scholar]
- 47.Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al. 2003. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 4: 321–328. [DOI] [PubMed] [Google Scholar]
- 48.Nakano K, and Vousden KH. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7: 683–694. [DOI] [PubMed] [Google Scholar]
- 49.Erlacher M, Labi V, Manzl C, Bock G, Tzankov A, Hacker G, Michalak E, Strasser A, and Villunger A. 2006. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J. Exp. Med 203: 2939–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]