

Abstract
Cyclin-dependent kinase 2 (CDK2) is a potential therapeutic target for the treatment of cancer. Development of CDK2 inhibitors has been extremely challenging as its ATP-binding site shares high similarity with CDK1, a related kinase whose inhibition causes toxic effects. Here, we report the development of TMX-2172, a heterobifunctional CDK2 degrader with degradation selectivity for CDK2 and CDK5 over not only CDK1, but transcriptional CDKs (CDK7 and CDK9) and cell cycle CDKs (CDK4 and CDK6) as well. In addition, we demonstrate that antiproliferative activity in ovarian cancer cells (OVCAR8) depends on CDK2 degradation and correlates with high expression of cyclin E1 (CCNE1), which functions as a regulatory subunit of CDK2. Collectively, our work provides evidence that TMX-2172 represents a lead for further development and that CDK2 degradation is a potentially valuable therapeutic strategy in ovarian and other cancers that overexpress CCNE1.
Keywords: cancer, cell cycle, CDK2, CDK5, drug design, protein degradation
Graphical Abstract
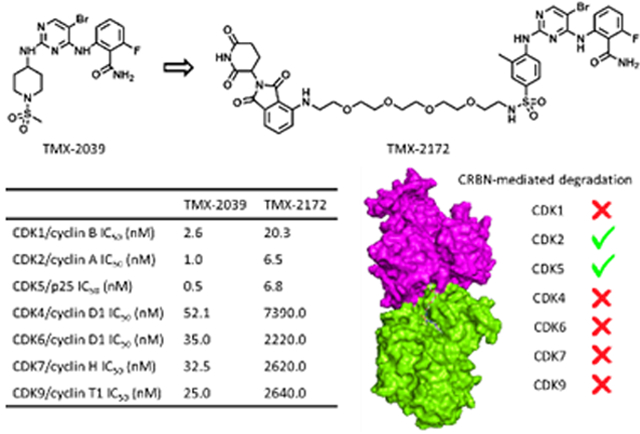
Selective targeting of closely related members of cyclin-dependent kinase family remains difficult. Here, a heterobifunctional degrader molecule, TMX-2172, is described. Dual degradation of CDK2 and CDK5 in OVCAR8 cells is achieved by reducing the binding affinity of the pyrimidine kinase inhibitor for CDK4, CDK6, CDK7, and CDK9, and by differential recognition between CDK2 and CDK1 mediated by the E3 ligase cereblon.
Under normal growth conditions, entry into the cell cycle is initiated by expression of cyclin D. Cyclin D activates the cyclin-dependent kinases CDK4 and CDK6, which in-turn induces expression of genes required for cell cycle progression.[1] Completion of the G1-phase and entry into the DNA-synthesis phase (S-Phase) of the cell cycle requires sequential activation of CDK2 through engagement of cyclin E (CCNE) and cyclin A.[2] Activation of CDK1 through cyclin A and B binding, as well as through additional post-translational events, is subsequently required for completion of the G2 phase and mitotic phases of the cell cycle.[2] Tissue homeostasis is orchestrated in part through extensive regulatory control over entry into and progression through the cell cycle and cancer can develop through multiple events that result in a loss of these regulatory controls. Ever since the initial discovery of CDKs as key controllers of yeast cell growth in the 1980s, there have been substantial efforts to develop CDK-targeting therapeutics. Early CDK inhibitors failed clinically due to unfavorable efficacy to toxicity profiles; however, more recently several dual CDK4 and CDK6 inhibitors have achieved remarkable success in the treatment of breast cancer.[3] The development of selective inhibitors of CDK1 and CDK2 have been stymied by the similarity of their ATP-binding pockets and the observation that potent CDK1 inhibition results in clinical toxicity.[4] These pharmacological findings are also consistent with recent genome-wide CRISPR/Cas9 screens that demonstrate that while CDK1 is a critical gene for proliferation of all cells, deletion of CDK2 results in more selective antiproliferative effects.[5] Although CDK2 seems dispensable in vivo,[6] recent studies have suggested that CDK2 has many cell cycle independent activities which cannot be compensated for by CDK1.[7] For example, an elevated level of CDK2, but not CDK1, is responsible for differentiation escape in AML.[8] CDK2 is a positive regulator of the DNA damage response including homologous recombination, non-homologous end-joining, and tumor suppressor p53.[9] Cancer cells with CCNE amplification, such as high grade serous ovarian cancers (HGSOC), exhibit selective vulnerability to CDK2 inhibition.[10] Taken together, these findings indicate that a selective CDK2 inhibitor might have a therapeutic benefit.
Developing selective agents for other CDKs has also been challenging. CDK5 is an atypical CDK that is activated by non-cyclin proteins, p35, p39 and their respective truncated products p25 and p29.[11] Although traditionally viewed as a neuro-specific kinase,[12] CDK5 has recently emerged as having understudied roles in cancer.[11] However, the lack of efforts to develop CDK5-selective research tool compounds has impeded the target validation of CDK5.
There have been several previous efforts to develop CDK2 selective inhibitors using non-covalent, covalent, and allosteric binding strategies.[13] For example, NU6300 is a covalent CDK2 inhibitor that targets a lysine residue located proximal to the ATP-binding pocket.[14] However, the ability of this compound to target CDK1 and other targets has not been reported. Another study demonstrated that 8-anilino-1-naphthalene sulfonate (ANS; structure shown in Figure S1), a widely used fluorescent probe, is an allosteric CDK2 inhibitor.[15] A fragment library screening effort identified compound 1 (Figure S1) as an allosteric covalent CDK2 binder with reported selectivity over CDK1.[16] Recently, a virtual screening effort aimed at identifying compounds that disrupt protein-protein interaction between CDK2 and its negative regulatory protein CRIF1 identified F1142-3225 (Figure S1).[17] Although potentially promising, the full extent of potential off-target space of the allosteric compounds reported thus far is yet to be determined and the cellular activity and selectivity need further investigation and optimization.
Small molecule degraders, also known as PROteolysis TArgeting Chimeras (PROTACs) are a targeting modality of rapidly increasing importance.[18] PROTACs are heterobifunctional molecules that incorporate two chemical “arms”, one that binds to the protein target of interest, and the other that recruits an E3 ubiquitin ligase. The functional consequence of this PROTAC-mediated ternary complex formation is target poly-ubiquitination and proteasomal degradation. The majority of reported PROTAC molecules employ an existing inhibitor scaffold as the target-binding arm, and incorporate either cereblon (CRBN) or Von Hippel-Lindau (VHL) as the E3 ligase recruiting element. One of the key benefits of degrader molecules is their increased selectivity when compared to the parental inhibitor. For example, a highly selective CDK9 degrader Thal-SNS-032 was developed based on a pan-CDK inhibitor SNS-032,[19] and a CDK6-selective degrader BSJ-03-123 was derived from the dual CDK4/6 inhibitor palbociclib.[20] Therefore, these previous results suggest that a degrader-based strategy may lead to selective agents for additional CDKs.
Here, we report the discovery of TMX-2172 that shows high degradation selectivity for CDK2 and CDK5 over other cell cycle CDKs (CDK1, CDK4, and CDK6) and transcriptional CDKs (CDK7 and CDK9). We demonstrate that TMX-2172 exhibits antiproliferative activity against OVCAR8, an ovarian cancer cell line that overexpresses CCNE1. We show that this activity depends on the ability of the compound to degrade CDK2 but not CDK5, suggesting that TMX-2172 may represent a useful lead in the context of cancers with CCNE1 overexpression.
During the preparation of this manuscript, a CDK2 targeting PROTAC molecule (A9, Figure S1) was disclosed; however, although A9 seems selective with respect to CDK5, its selectivity amongst other CDKs, especially over its close paralog CDK1, was not described.[21]
To identify an appropriate CDK2-targeting ligand for development of a degrader, we queried our in-house library of kinase inhibitors. Our selection criteria were: (1) single digit nanomolar enzymatic IC50 on CDK2; and (2) molecular weight below 500 Da. Based on this, we selected TMX-2039, a pan-CDK inhibitor with low nanomolar enzymatic IC50s for both cell cycle CDKs (CDK1, CDK2, CDK4, and CDK6) and transcriptional CDKs (CDK7 and CDK9) (Figure 1). We prepared an initial set of degrader molecules by conjugating TMX-2039 through the solvent-facing sulfonamide position via a PEG or an alkyl linker to an imide moiety that recruits the CRL4CRBN ubiquitin ligase complex (Table 1). Biochemical assays using CDK2/cyclin A and CDK1/cyclin B confirmed that this linker attachment strategy retained the potency of the parental compound with IC50s below 30 nM (Table 1).
Figure 1.
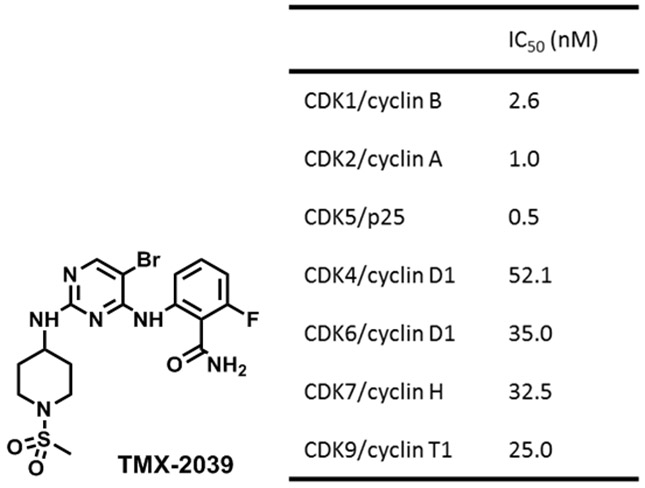
Chemical structure of TMX-2039 and its CDK inhibitory activities.
Table 1.
Biochemical IC50s of CDK1/2 for degraders.[a]
 | |||
| Compound | Linker | CDK2/cyclin A IC50 (nM) |
CDK1/cyclin B IC50 (nM) |
|---|---|---|---|
| TMX-1117 | 5.1 | 17.8 | |
| TMX-1128 | 4.9 | 11.6 | |
| TMX-1111 | 6.4 | 9.5 | |
| TMX-1146 | 7.3 | 15.5 | |
| TMX-1169 | 20.5 | 31.0 | |
| TMX-1162 | 17.1 | 8.5 | |
| TMX-1160 | 28.7 | 8.5 | |
All the IC50 values were tested in duplicate, value ranges are shown in Table S2 in the Supporting Information.
Next, we screened this series of compounds for CDK1 and CDK2 degradation in Jurkat cells at a concentration of 1 μM for a duration of 6 hours. Alkyl linked-PROTAC compounds TMX-1160 and TMX-1162 induced CDK2 degradation, while no CDK1 degradation was observed (Figure 2A), despite the potent inhibition in biochemical assays. We also observed that CDK7 and CDK9 were not degraded under these conditions, while CDK4 and CDK6, as well as CDK5, were potently degraded by TMX-1160 (Figure 2B). Although a different (or cell type-dependent) degradation profile was obtained in OVCAR8 cells, where CDK4 and CDK6 were not degraded (Figure S2), we decided to optimize TMX-1160 to achieve better degradation selectivity.
Figure 2.
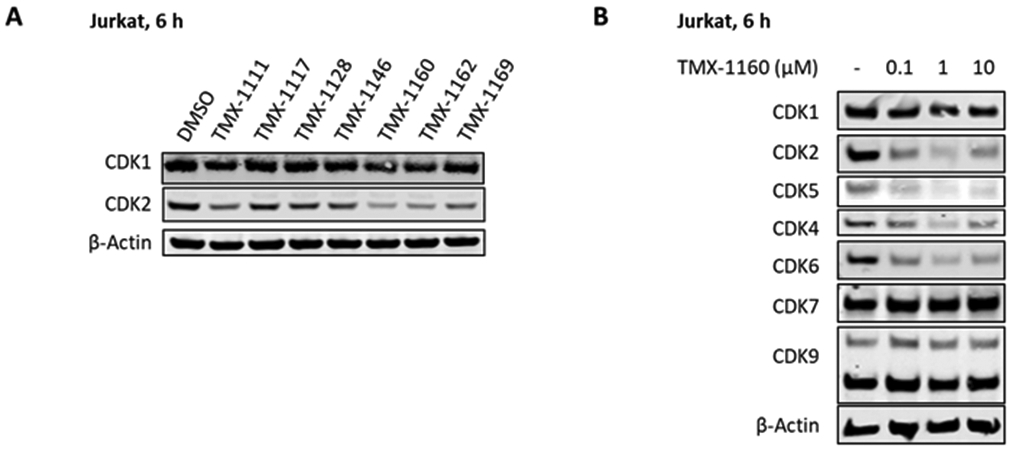
Immunoblot analysis of CDKs in Jurkat cells. (A) Immunoblot analysis of CDK1 and CDK2 treated with 250 nM of degraders for 6 hours. (B) Immunoblot analysis of CDKs treated with the indicated concentration of TMX-1160 for 6 hours.
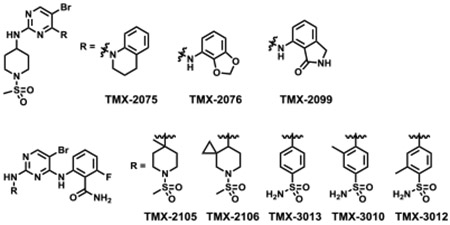
In general, based on previous work in this area, selectivity of a PROTAC molecule could be improved through either modifications of the target binding ligand and/or optimization of the linker.[22] In contrast to linker modifications that can potentially affect both target binding and ternary complex formation, modifications of the target-recruiting ligand primarily affect target binding. Therefore, we chose to focus on optimizing the binding selectivity of the TMX-2039 scaffold. First, bicyclic groups were introduced to replace 5-amino-2-fluorobenzamide to obtain TMX-2075, TMX-2076, and TMX-2099, which failed to improve selectivity. Installing a methyl and a spiro-cyclopropane group at the 4- or 3-position of the piperidine ring (TMX-2105 and TMX-2106) resulted in a significant activity loss on all CDKs. Finally, replacing the piperidine ring with a phenyl ring resulted in compounds that retained activity (TMX-3013 and TMX-3012), and eventually led to TMX-3010 that exhibited a hundred-fold discrimination for inhibiting CDK1, CDK2, and CDK5 over CDK4 and CDK6, as benchmarked to its CDK2 IC50 value (Table 2 and Table S3).
Table 2.
Biochemical IC50s on CDKs of TMX-2039 analogs.
 | |||||
|---|---|---|---|---|---|
| Compound | CDK1[a] IC50 (nM) |
CDK2[b] IC50 (nM) |
CDK5[c] IC50 (nM) |
CDK4[d] IC50 (nM) |
CDK6[d] IC50 (nM) |
| TMX-2075 | 365.0 | 75.3 | 154.0 | 124 | 127.0 |
| TMX-2076 | 105.0 | 17.8 | 32.4 | 981 | 487.0 |
| TMX-2099 | 3.7 | 0.6 | <0.5 | 10.2 | 4.9 |
| TMX-2105 | 1600.0 | 530.0 | 530.0 | 140.0 | 5100.0 |
| TMX-2106 | 369.0 | 33.2 | 57.0 | 33.4 | 546.0 |
| TMX-3013 | 0.9 | <0.5 | 0.5 | 24.5 | 15.6 |
| TMX-3010 | 1.2 | 0.7 | 0.9 | 135.0 | 84.1 |
| TMX-3012 | 0.8 | 0.7 | 0.6 | 79.6 | 24.0 |
w/cyclin B.
w/cyclin A.
w/p25.
w/cyclin D1. All the IC50 values were tested in duplicate, value ranges are shown in Table S2 in the Supporting Information.
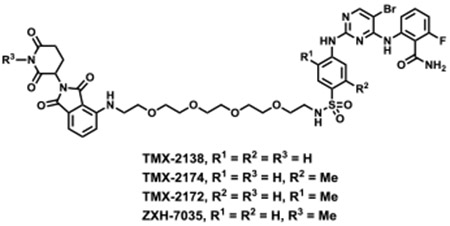
We used TMX-3013, TMX-3012, and TMX-3010 as starting points for additional PROTACs featuring a PEG linker and thalidomide as the CRBN-recruiting arm, resulting in TMX-2138, TMX-2174, and TMX-2172, respectively. As shown in Table 3, the linker moiety significantly impaired the inhibitory activity on CDK4 and CDK6, while single digit nanomolar enzymatic IC50s on CDK2 and CDK5 were maintained. TMX-2172 was shown to be the most selective against transcriptional CDKs. In a cellular CRBN engagement assay, TMX-2172 exhibited an IC50 of 46.9 nM, indicating its good cell permeability (Figure S5).[22] In Jurkat cells, TMX-2172 induced CDK2 degradation at a concentration of 250 nM, whereas other cell cycle CDKs (CDK1, CDK4, and CDK6) as well as transcriptional CDKs (CDK7 and CDK9) were not affected (Figure 3A). To confirm that CDK2 degradation was CRBN-dependent, we used CRBN-null Jurkat cells as well as a negative control compound ZXH-7035, containing a methylated glutarimide ring, which is incapable of engaging CRBN. In both cases, no CDK2 degradation was detected, supporting a CRBN-dependent mechanism for CDK2 degradation (Figure 3B and 3C). However, as shown in Figure 3A, TMX-2172 retained the ability to degrade CDK5.
Table 3.
Chemical structures and CDKs inhibitory activities of degraders.
 | ||||
|---|---|---|---|---|
| Compound | TMX-2138 | TMX-2172 | ZXH-7035 | TMX-2174 |
| CDK1/cyclin B IC50 (nM) |
8.7 | 20.3 | 8.9 | 8.9 |
| CDK2/cyclin A IC50 (nM) |
10.9 | 6.5 | 2.2 | 8.1 |
| CDK5/p25 IC50 (nM) |
7.0 | 6.8 | 2.6 | 7.1 |
| CDK4/cyclin D1 IC50 (nM) |
901.0 | 7390.0 | ND[a] | 2880.0 |
| CDK6/cyclin D1 IC50 (nM) |
465.0 | 2220.0 | ND | 1130.0 |
| CDK7/cyclin H IC50 (nM) |
286.0 | 2620.0 | ND | 343.0 |
| CDK9/cyclin T1 IC50 (nM) |
25.7 | 2640.0 | ND | 116.0 |
Not determined. All the IC50 values were tested in duplicate, value ranges are shown in Table S2 in the Supporting Information.
Figure 3.
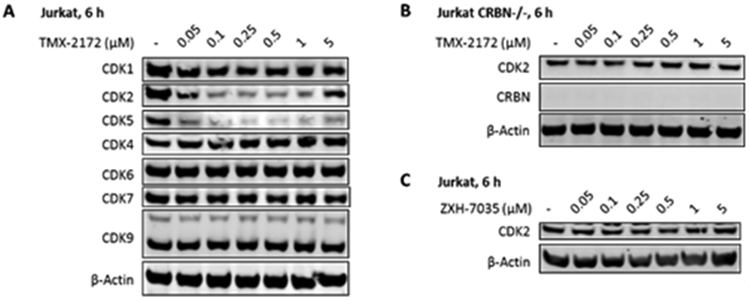
Immunoblot analysis of CDKs following treatment of Jurkat cells with TMX-2172 and its negative control. (A) Immunoblot analysis of CDKs in Jurkat cells treated with TMX-2172 for 6 hours. (B) Immunoblot analysis of CDK2 in CRBN-null Jurkat cells treated with TMX-2172 for 6 hours. (C) Immunoblot analysis of CDK2 in Jurkat cells treated with negative control ZXH-7035 for 6 hours.
To more fully characterize the selectivity profile of TMX-2172, we performed KINOMEscan, which profiles more than 400 kinases.[23] As shown in Data S1, 14 kinases exhibited >99% inhibition by 1 μM TMX-2172 compared to DMSO. CDK2 was the only CDK that showed potent inhibition (CDK1 is not included in KINOMEscan panel).
Next, we used a pulse-chase SILAC quantitative mass spectrometry (MS)-based proteomic method[24] to determine the range of targets degraded by TMX-2172 in OVCAR8 cells, a HGSOC cell line. As shown in Figure 4A, three kinases CDK2, CDK5, and Aurora A, the top hits from KINOMEscan, were found to be effectively degraded, while the degree of degradation of the other three kinases RSK1 (RPS6KA1), JNK2 (MAPK9), and STK33 was relatively weak. As shown in Figure 4B, both CDK2 and CDK5 were degraded in a dose dependent manner in OVCAR8 cells, while other CDKs, including CDK1, remained intact. A follow-up biochemical activity assay revealed an IC50 of 205.0 nM for Aurora A, and a higher concentration of TMX-2172 (5 μM) was needed to achieve observable degradation under the same conditions (Figure S3). Given that Aurora A levels are often reduced due to cell cycle arrest induced by other targeted kinase inhibitors,[25] we cannot rule out that the degradation of Aurora A is caused by secondary effects rather than direct degradation.
Figure 4.
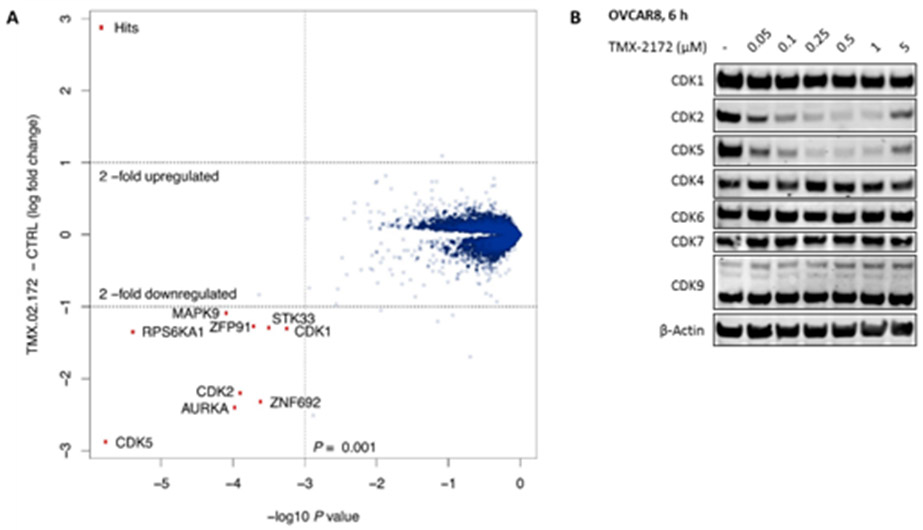
(A) Proteome-wide degradation selectivity of TMX-2172 at a dose of 250 nM in OVCAR8 after 6 hours treatment. (B) Immunoblot analysis of CDKs in OVCAR8 cells treated with TMX-2172 for 6 hours.
HGSOC often displays high CCNE1 expression and a dependency on CDK2 activity for proliferation. OVCAR8 cells were found to express high levels of CDK2 relative to other ovarian cancer cell lines (Figure S6) and were found to be dependent on CDK2 in a CRISPR screen.[5] We therefore screened our first generation degrader, TMX-1160, across 18 ovarian cancer cell lines and found that OVCAR8 was amongst the most sensitive to growth rate inhibition (Table S1). We next determined that the growth rate inhibitory effect of the lead degrader TMX-2172 on OVCAR8 cells was approximately 8.5-fold more potent than the non-degrading control ZXH-7035, demonstrating that degradation is an important component of the pharmacology of this inhibitor (Figure 5). Although CDK5 is also degraded by TMX-2172, genome-wide CRISPR screening suggests that CDK5 deletion does not cause growth defects in OVCAR8 cells.[26] We confirmed independently that CDK5 depletion by shRNA-knockdown does not impede OVCAR8 cell growth suggesting that the ability of TMX-2712 to degrade CDK5 is not a major contributor to its antiproliferative activity in OVCAR8 cells (Figure S4). Taken together, this suggests that TMX-2172-mediated degradation of CDK2 is the major mechanism by which TMX-2172 exerts antiproliferative effects in OVCAR8 cells.
Figure 5.

Growth rate inhibition in OVCAR8 cells treated with TMX-2172 and ZXH-7035 for 72 hours.
Selective targeting of CDK isoforms has been a long-standing challenge in drug development, and a major limitation for clinical development of CDK inhibitors. PROTACs have recently emerged as a new strategy that can improve targeting selectivity of previously promiscuous compounds.[19,27] This is, to a large extent, due to the requirement that PROTACs engage not only the target, but also the participant E3 ligase, in a way that results in a productive, ubiquitination-competent ternary complex. Thus, intrinsic differences in the target specific ubiquitination efficacy, and protein-protein interactions with E3 ligase result in more complex selectivity filters. For example, we and others recently employed the PROTAC strategy to develop selective degraders for CDK4 and CDK6, two closely related CDKs for which no isoform selective inhibitors exist.[20,28] One of the key obstacles to clinical targeting of CDK2 stems from off-target effects on CDK1, which results in dose-limiting toxicity. Here, we report development and characterization of TMX-2172, a CRBN-recruiting PROTAC that selectively degrades CDK2 and CDK5 while having no detectable degradation effect on CDK1. Moreover, we demonstrate that TMX-2172 does not degrade CDK4, CDK6, CDK7, and CDK9 by removing the ability of the pyrimidine kinase inhibitor to bind to these targets.
Our results using the HGSOC cell line OVCAR8, which harbors high levels of CCNE1 (a well-established activator of CDK2), demonstrate that TMX-2172 had strong growth rate inhibitory effects. To deconvolute CDK2- from CDK5-based effects in this cell line, we examined the effects of shRNA mediated depletion of CDK5 and observed that CDK5 knockdown had no effect on growth in OVCAR8 cells. These results suggest that the phenotypic effects of TMX-2172 in OVCAR8 cells are mainly driven by the loss of CDK2 rather than CDK5. This is further supported by the results we obtained when using a negative control compound, ZXH-7035, that inhibits CDK2 but does not lead to its degradation due to the loss of CRBN binding. Taken together, we consider TMX-2172 and ZXH-7035 to be an important addition to the current toolset for studying CDK2 function especially in model systems that have elevated CCNE1 levels. Moreover, our results suggest that CDK2 degradation may have therapeutic benefits in ovarian cancer, although further efforts to improve selectivity and drug-like properties will be needed to enable pharmacological studies of CDK2 degraders in cancers.
Additionally, although it would require further validation, we believe TMX-2172 may be a useful preliminary chemical tool to investigate CDK5 dependent biology in contexts where CDK2 is unlikely to play a role, such as in CDK2 deleted backgrounds, or in neurons post-mitosis, where CDK5 is known to play a critical role while other CDKs are not active or expressed.[29] This is of special importance given that CDK5-selective research tool compounds and drug leads are not currently available. Several investigations have noted cell-type specific degradation profiles for degrader molecules[30] and therefore it will be critical to re-examine the degradation selectivity profile of TMX-2172 in specific cell types of future interest.
Supplementary Material
Acknowledgements
We thank Dr. Milka Kostic (Twitter: @MilkaKostic) for her advice and support during the preparation of this manuscript. This work was supported in part by the National Institutes of Health (R01CA218278 and P01CA154303 awarded to N.S.G. and U54HL127365 awarded to P.K.S.).
Footnotes
Conflict of interest
N.S.G. is a founder, science advisory board member (SAB) and equity holder in Gatekeeper, Syros, Petra, C4, B2S, Aduro and Soltego. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Her2llc, Epiphanes, Deerfield and Sanofi. M.T., J.J., T.H.Z., and N.S.G. are inventors on CDK2/5 degrader patents. E.S.F. is a founder, SAB member and equity holder in Civetta Therapeutics, and an equity holder and SAB member in C4 Therapeutics. The Fischer lab receives or has received research funding from Astellas, Novartis, Voronoi and Deerfield. P.K.S. is a member of the SAB or Board of Directors of Merrimack Pharmaceutical, Glencoe Software, Applied Biomath and RareCyte Inc. and has equity in these companies.
References
- [1].Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC, Cell 1999, 98, 859–869. [DOI] [PubMed] [Google Scholar]
- [2].a) Edgar BA, Lehner CF, Science 1996, 274, 1646–1652; [DOI] [PubMed] [Google Scholar]; b) Hochegger H, Takeda S, Hunt T, Nat. Rev. Mol. Cell. Biol 2008, 9, 910–916. [DOI] [PubMed] [Google Scholar]
- [3].a) Spring LM, Wander SA, Andre F, Moy B, Turner NC, Bardia A, Lancet 2020, 395, 817–827; [DOI] [PubMed] [Google Scholar]; b) Sherr CJ, Beach D, Shapiro GI, Cancer Discov. 2016, 6, 353–367; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Whittaker SR, Mallinger A, Workman P, Clarke PA, Pharmaco.l Ther 2017, 173, 83–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Guha M, Nat. Rev. Drug. Discov 2012, 11, 892–894; [DOI] [PubMed] [Google Scholar]; b) Wood DJ, Korolchuk S, TatμM NJ, Wang LZ, Endicott JA, Noble MEM, Martin MP, Cell Chem. Biol 2019, 26, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, Meyers RM, Ali L, Goodale A, Lee Y, Jiang G, Hsiao J, Gerath WFJ, Howell S, Merkel E, Ghandi M, Garraway LA, Root DE, Golub TR, Boehm JS, Hahn WC, Cell 2017, 170, 564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Tetsu O, McCormick F, Cancer Cell 2003, 3, 233–245; [DOI] [PubMed] [Google Scholar]; b) Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P, Curr. Biol 2003, 13, 1775–1785; [DOI] [PubMed] [Google Scholar]; c) Enders GH, Genes Cancer 2012, 3, 614–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tadesse S, Anshabo AT, Portman N, Lim E, Tilley W, Caldon CE, Wang S, Drug Discov. Today 2020, 25, 406–413. [DOI] [PubMed] [Google Scholar]
- [8].Ying M, Shao X, Jing H, Liu Y, Qi X, Cao J, Chen Y, Xiang S, Song H, Hu R, Wei G, Yang B, He Q, Blood 2018, 131, 2698–2711. [DOI] [PubMed] [Google Scholar]
- [9].a) Ruffner H, Jiang W, Craig AG, Hunter T, Verma IM, Mol. Cell Biol 1999, 19, 4843–4854; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC, Nature 2005, 434, 598–604; [DOI] [PubMed] [Google Scholar]; c) Diederichs S, BaμMer N, Ji P, Metzelder SK, Idos GE, Cauvet T, Wang W, Moller M, Pierschalski S, Gromoll J, Schrader MG, Koeffler HP, Berdel WE, Serve H, Muller-Tidow C, J. Biol. Chem 2004, 279, 33727–33741; [DOI] [PubMed] [Google Scholar]; d) Wang Y, Prives C, Nature 1995, 376, 88–91; [DOI] [PubMed] [Google Scholar]; e) Bačević K, Lossaint G, Achour TN, Georget V, Fisher D, Dulic V, Sci. Rep 2017, 7, 13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Yang L, Fang D, Chen H, Lu Y, Dong Z, Ding HF, Jing Q, Su SB, Huang S, Oncotarget 2015, 6, 20801–20812; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Au-Yeung G, Lang F, Azar WJ, Mitchell C, Jarman KE, Lackovic K, Aziz D, Cullinane C, Pearson RB, Mileshkin L, Rischin D, Karst AM, Drapkin R, Etemadmoghadam D, Bowtell DD, Clin. Cancer Res 2017, 23, 1862–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pozo K, Bibb JA, Trends Cancer 2016, 2, 606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Shah K, Lahiri DK, J. Cell Sci 2014, 127, 2391–2400; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cheung ZH, Ip NY, Trends Cell Biol. 2012, 22, 169–175. [DOI] [PubMed] [Google Scholar]
- [13].Tadesse S, Caldon EC, Tilley W, Wang S, J. Med. Chem 2019, 62, 4233–4251. [DOI] [PubMed] [Google Scholar]
- [14].Anscombe E, Meschini E, Mora-Vidal R, Martin MP, Staunton D, Geitmann M, Danielson UH, Stanley WA, Wang LZ, Reuillon T, Golding BT, Cano C, Newell DR, Noble ME, Wedge SR, Endicott JA, Griffin RJ, Chem. Biol 2015, 22, 1159–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Betzi S, Alam R, Martin M, Lubbers DJ, Han H, Jakkaraj SR, Georg GI, Schonbrunn E, ACS Chem. Biol 2011, 6, 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Craven GB, Affron DP, Allen CE, Matthies S, Greener JG, Morgan RML, Tate EW, Armstrong A, Mann DJ, Angew. Chem. Int. Ed. Engl 2018, 57, 5257–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ran Q, Xiang Y, Stephen P, Wu C, Li T, Lin SX, Li Z, J. Am. Chem. Soc 2019, 141, 1420–1424. [DOI] [PubMed] [Google Scholar]
- [18].a) Burslem GM, Crews CM, Cell 2020, 181, doi: 10.1016/j.cell.2019.11.031; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kostic M, Jones LH, Trends Pharmacol, Sci. 2020, 41, 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z, Zhang T, Kwiatkowski N, Boukhali M, Green JL, Haas W, Nomanbhoy T, Fischer ES, Young RA, Bradner JE, Winter GE, Gray NS, Nat. Chem. Biol 2018, 14, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jiang B, Wang ES, Donovan KA, Liang Y, Fischer ES, Zhang T, Gray NS, Angew. Chem. Int. Ed. Engl 2019, 58, 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhou F, Chen L, Cao C, Yu J, Luo X, Zhou P, Zhao L, Du W, Cheng J, Xie Y, Chen Y, Eur. J. Med. Chem 2020, 187, 111952. [DOI] [PubMed] [Google Scholar]
- [22].Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Safaee N, Jedrychowski MP, Ponthier CM, Ishoey M, Zhang T, Mancias JD, Gray NS, Bradner JE, Fischer ES, Nat. Chem. Biol 2018, 14, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP, Nat. Biotechnol 2011, 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- [24].An J, Ponthier CM, Sack R, Seebacher J, Stadler MB, Donovan KA, Fischer ES, Nat. Commun 2017, 8, 15398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nomanbhoy TK, Sharma G, Brown H, Wu J, Aban A, Vogeti S, Alemayehu S, Sykes M, RosenblμM JS, Kozarich JW, Biochemistry 2016, 55, 5434–5441. [DOI] [PubMed] [Google Scholar]
- [26].See: https://depmap.org/portal/gene/CDK5?tab=dependency&dependency=Sanger_CRISPR
- [27].Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, Wang J, Hamman BD, Ishchenko A, Crews CM, Cell Chem. Biol 2018, 25, 78–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rana S, Bendjennat M, Kour S, King HM, Kizhake S, Zahid M, Natarajan A, Bioorg. Med. Chem. Lett 2019, 29, 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dhavan R, Tsai LH, Nat. Rev. Mol. Cell Biol 2001, 2, 749–759. [DOI] [PubMed] [Google Scholar]
- [30].Huang HT, Dobrovolsky D, Paulk J, Yang G, Weisberg EL, Doctor ZM, Buckley DL, Cho JH, Ko E, Jang J, Shi K, Choi HG, Griffin JD, Li Y, Treon SP, Fischer ES, Bradner JE, Tan L, Gray NS, Cell Chem Biol 2018, 25, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.