

Patient-derived xenografts (PDX) represent invaluable tools to study the biology of human cancers in vivo. The severely immuno-compromised NOD Prkdcscid Il2rg−/− (NSG) mouse strain is the most commonly used strain, in particular for hematologic malignancies. However, the lack of genetically modified (GM) NSG mice has greatly hampered functional studies in PDX models, in particular those that address the role of the microenvironment. The tumor microenvironment is a key player in cancer progression and a major contributor to therapeutic resistance.
A handful of studies have reported the generation of GM NSG mice through a time-consuming backcross of GM C57BL/6 mice onto an NSG background.1, 2, 3, 4 Here we describe a strategy that uses CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas9 (CRISPR associated protein 9) technology to efficiently generate GM NSG mice to specifically interrogate the role of niche-derived SPARC (Secreted Protein Acidic Rich in Cysteine) in vivo in different human leukemic contexts.
SPARC is a multifaceted matricellular protein that regulates key physiological processes by modulating cell–cell and cell–matrix interactions. Moreover, SPARC is shown to be differentially expressed in various tumors and proposed to modulate cancer cell activity.5 In leukemic contexts, SPARC is proposed to exert opposing effects depending on the leukemia type or even the specific subtype under study.6, 7 However, no study has so far investigated the role of niche-derived SPARC in human leukemia, in an in vivo setting.
In recent years, the use of targeted nucleases has revolutionized the field of genome engineering. In particular, CRISPRs and the Cas9 endonuclease system has proven to be by far the most versatile system. Cas9-induced double-strand breaks (DSBs) are rapidly resolved by DNA-dependent protein kinase (DNA-PK)-dependent non-homologous end joining (NHEJ), a repair mechanism that generates InDels and often results in functional inactivation of the targeted gene. Alternatively, precise editing can be achieved through the use of a donor template DNA homologous to the sequences flanking the DSB through a homology-directed repair (HDR) mechanism that is independent of DNA-PK.8 In wild-type cells, HDR is largely outcompeted by the fast acting NHEJ. However, because of the Prkdcscid mutation, NSG mice do not express DNA-PK and as such are impaired in their ability to efficiently resolve DSBs by NHEJ. We hypothesized that this deficiency would allow for efficient HDR, a fact that could be exploited to achieve precise genome editing in NSG background.
NSG zygotes were microinjected in the cytoplasm with Cas9, guide RNA (gRNA) and a template single-stranded DNA (ssDNA) (Supplementary Figure 1A and Supplementary Methods online), as previously described.9 Guide RNAs targeting exon 4 (gRNA#1) or exon 2 (gRNA#2) (Supplementary Figure1B) were initially tested for their ability to induce on target editing by transient transfection in NHEJ competent MS5 murine stromal cells. Using the surveyor assay, we observed efficient genome editing with both gRNAs. Although gRNA#2 was more efficient (Supplementary Figure 1C), we elected to use gRNA#1 because of the reduced number of predicted off-targets (Supplementary Table 1). To achieve a functional knockout in NSG mice, we used gRNA1 that targets an early-translated exon of the Sparc gene and a ssDNA template containing an in-frame STOP codon (Supplementary Figure 1B and Supplementary Methods online). This approach should halt translation while concomitantly inducing nonsense-mediated mRNA decay (NMD) of the edited transcript. Introduction of the STOP codon was also designed to generate a new restriction site (PleI) for subsequent evaluation of HDR editing by restriction fragment length polymorphism (RFLP) assay. Finally, the PAM sequence, a recognition motif required for Cas9 cleavage, was mutated in the template DNA to avoid repetitive cleavage of productively edited loci. The mutation of the PAM sequence was also designed to generate a second in-frame STOP codon to further ensure termination of translation (Supplementary Figure 1B and Supplementary Methods online). To further evaluate the need for PAM mutagenesis, ssDNA templates with or without PAM mutation were used at equal ratios in all described experiments.
HDR efficiency was subsequently evaluated in ex vivo cultured NSG embryos that were microinjected with gRNA#1, ssDNA and Cas9 at the zygote stage. A total of 13/65 (20%) embryos displayed productive repair by HDR as demonstrated by RFLP assay using PleI (Figure 1a and Supplementary Figure 2). Moreover, T7 endonuclease assay, which reflects overall editing by detecting DNA mismatches, revealed that most embryos were edited in this context (Figure 1a, bottom panel). This suggests that, in addition to HDR, other repair mechanisms are likely at play. DNA-PK independent alternative end-joining (Alt-EJ) mechanisms that rely on short homologous sequences at the junctions have been described. This type of Alt-EJ often yields products with deletions (for example, #E7, Figure 1a). This is in line with a recent report demonstrating the occurrence of microhomology-mediated end joining in NSG mice.10
Figure 1.
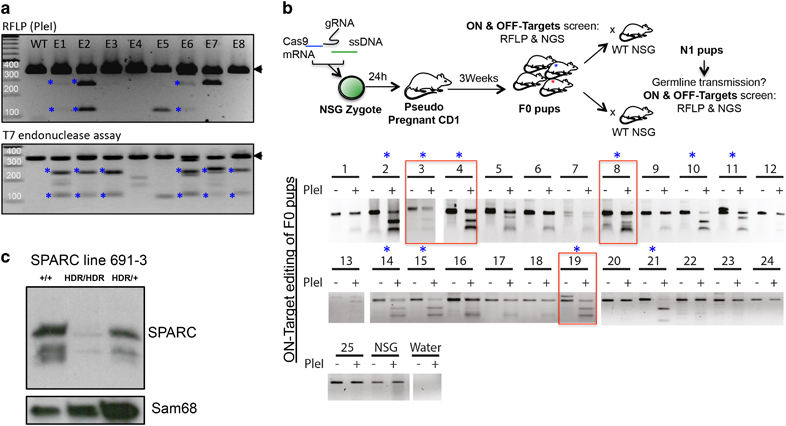
CRISPR/Cas9 allows for the fast and efficient generation of SPARC deficient NSG mice. (a) RFLP (PleI digest) and T7 endonuclease analysis of day 4 NSG embryos microinjected with gRNA#1, Cas9 and ssDNA template. Asterisks mark productively edited embryos. (b) Schematic view of the workflow to generate NSG-edited line (top) and RFLP analysis of tail DNA from chimeric F0 mice (bottom). Asterisks mark HDR-edited mice. A subset of F0 mice (red squares) were backcrossed to WT NSG mice to obtain N1 progeny and evaluate germline transmission of the HDR+ allele. (c) Western blot on bone marrow cells shows complete loss of SPARC protein in SPARCHDR/HDR animals, whereas a significant reduction is also observed in heterozygous SPARCHDR/+ mice. Sam68 is used as loading control.
Importantly, when microinjected NSG zygotes were implanted in pseudo-pregnant CD1 females, they gave rise to edited F0 progeny. RFLP analysis demonstrated that 40% of the mice (10/25) underwent productive HDR (Figure 1b). Importantly, this level of HDR editing in NSG zygotes is not restricted to the SPARC locus, as we observed comparable efficiencies when we targeted, using the same workflow, two other loci located on chromosome 5 (21.4% HDR+ F0 pups) and chromosome 8 (23.5% HDR+ F0 pups (Nevmerzhitskaya A and Medyouf H, manuscript in preparation). Of note, editing of the SPARC locus using the same workflow in C57BL/6 zygotes gave rise to a comparable frequency of 29% HDR-edited embryos (n=7/24, Supplementary Figure 3). Taken together, these data demonstrate that HDR is highly active in NSG mice and can be exploited for the rapid generation of knock-in and knockout mice.
To precisely define and quantify the editing, we amplified the predicted on- and off-target sites from all HDR+ F0 pups (Figure 1b, asterisk) and incorporated mouse-specific barcodes to be used as sample identifiers on the Illumina (San Diego, CA, USA) MiSeq platform (Supplementary Figure 4A and Supplementary Methods). F0 editing was highly efficient (mean edited variant allele frequency=75%) and mediated through either HDR (mean HDR+ variant allele frequency=35%) or Alt-EJ (mean variant allele frequency=30%) (Supplementary Figures 4B and C). Of note, edited sequences with and without PAM mismatch were observed at comparable frequencies in the F0 pups, thereby suggesting that PAM motif conservation in the ssDNA template does not decrease the frequency of productive HDR editing (Supplementary Figure 4B). Importantly, no editing was observed at predicted off-target sites in any of the analyzed mice. This was further confirmed by high coverage sequencing in the N1 progeny obtained from four of the F0 founders (#3, #4, #8 and #19; data not shown). We speculate that this lack of off-target editing might be contributed to by the use of a highly selective gRNA with limited predicted off-targets, as well as the inability of this strain to carry out error-prone NHEJ.
Notably, we used RFLP analysis to show that the HDR+ allele exhibited efficient germline transmission to the N1 progeny (Supplementary Figure 5A) in all four F0 founders tested (line 691, F0 founders #3, #4, #8, #19). To establish a SPARC line, we intercrossed the progeny of 691#3 carrying the HDR allele after HDR-mediated editing was further confirmed by standard Sanger sequencing (data not shown). Heterozygous mating demonstrated Mendelian transmission (Supplementary Figure 5B). Molecular analysis showed that both SPARC RNA and protein were absent from homozygous SPARCHDR/HDR (referred to as SPARC-deficient) mice, indicating that the mRNA produced from the HDR-edited allele is likely degraded through NMD in vivo, a well known translation-coupled mechanism that eliminates mRNAs containing premature translation-termination codons (Figure 1c and Supplementary Figure 5C). Finally, similar to their C57BL/6 counterpart, NSG SPARC-deficient mice appear normal until ∼4–6 months of age, when they develop severe cataract formation (Supplementary Figure 6).11
Because mouse and human SPARC have a 92% sequence identity at the protein level, we used our NSG SPARC-deficient model to interrogate in vivo the specific effect of niche-derived SPARC in human leukemia. Initially, we focused on acute myeloid leukemia (AML), where seemingly conflicting results have been reported. Indeed, high SPARC expression has been shown to associate with adverse outcome in cytogenetically normal AMLs,6 whereas SPARC appears to rather reduce the growth of low SPARC expressing MLL-rearranged (MLLR) AML blasts ex vivo.7 However, thus far, no study has investigated the role of niche-derived SPARC in the pathogenesis of human AML in vivo. When compared with wild-type mice, NSG SPARCHDR/HDR animals injected with THP-1, a SPARClow MLL-AF9+ line,7 exhibited a dramatically increased leukemic burden both at an early time point post transplant (Figure 2b) and in terminally ill animals (Supplementary Figure 7). Conversely, xenotransplantation of a cytogenetically normal primary AML case in our model rather showed a tendency toward lower burden in SPARC-deficient animals (Figure 2b).
Figure 2.
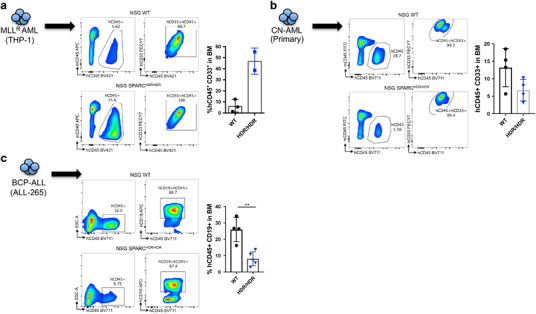
NSG SPARC mice reveal opposing effects of niche-derived SPARC in human leukemia. Percentage and immunophenotyping of hCD45+ cells in the bone marrow of WT and SPARCHDR/HDR mice engrafted with 106 MLL-AF9-expressing THP-1 cells (WT NSG n=3; SPARC KO n=2) analyzed 5 weeks post transplant (a), 106 cells from a cytogenetically normal (CN) primary AML case (WT NSG n=4; SPARC KO n=4) analyzed 11 weeks post transplant (b) or 105 cells from a BCP-ALL relapsed case (ALL-265; WT NSG n=4; SPARC KO n=5) analyzed 5 weeks post transplant (c). The data show a significantly increased leukemic burden in SPARCHDR/HDR mice receiving THP-1 cells (a). In stark contrast, leukemic burden is dramatically decreased in SPARCHDR/HDR mice receiving the primary CN-AML case or the relapsed BCP-ALL case (ALL-265) (b, c). Of note, additional THP-1 and BCP-ALL cases (ALL-199) are depicted in supplementary Figures 7 and 8. Each dot represents an independent mouse. Unpaired Student’s t-test, **P<0.01.
Similarly, divergent roles for SPARC have been proposed in lymphoid malignancies, where absence of stromal SPARC predicts poor prognosis in diffuse large B-cell lymphoma,12 whereas its leukemia-specific upregulation is a recurrent event in relapsed pediatric B-cell acute lymphoblastic leukemia (B-ALL).13 We therefore used PDX to evaluate the impact of exogenous SPARC in relapsed pediatric B-cell precursor ALL cases (BCP-ALL; ALL-265 and ALL-199).14 In stark contrast to the THP-1 model, SPARC deficiency dramatically decreased the BCP-ALL burden in mice (Figure 2c and Supplementary Figure 8), thereby suggesting that tumor microenvironment-derived SPARC can elicit downstream signals that effectively promote the growth of an aggressive form of BCP-ALL.
Taken together, these data clearly highlight in vivo that niche-derived SPARC modulates leukemic cell behavior. Most importantly, this effect appears to be highly specific to the cellular context.
This work describes a new strategy to generate genetically modified NSG mice that are of great value to interrogate the functional relevance of key niche factors in PDX models in vivo. In addition to this technical aspect, the approach was also successfully used to demonstrate, for the first time, the divergent functions of niche-derived SPARC in human leukemia in an in vivo setting. Future studies will explore the disease-specific programs that mediate SPARC downstream effects in vivo. Identifying such programs might pave the way to the design of new therapeutic strategies, in particular for patients in urgent need for alternative therapies.
Supplementary information
Acknowledgements
We thank Dr Henner Farin and Dr Michael Milsom for helpful discussions, Maresa Weitmann for technical help and Dr Michaela Socher, Dr Boris Brill and all members of the DKFZ and GSH Laboratory Animal Core Facility for excellent animal welfare and husbandry. We thank Dr Sebastian Wagner and Dr Khalil Abou Elaradat from the Frankfurt DKTK sequencing facility for the MiSeq runs and Dr Stefan Stein for the THP-1 line. We thank H Altmann and C Röllig from the SAL biobank (Dresden) and Dr Vick Binje from the Helmholtz Center (Munich) for providing samples. HM is supported by the European Research Council (ERC Grant No. 639795) and the German José Carreras Leukemia Foundation (Award No. DJCLS A 14/01).
Author contributions
IT-G, AN, EC, DS, EB, AC and HM performed experiments. AM performed the analysis of NGS data. UK and FVdH isolated the NSG zygotes and performed the cytoplasmic microinjections. IJ, JPB and UP contributed reagents and discussed results. HM designed and supervised the study. HM and ITG wrote the manuscript.
Competing interests
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on the Leukemia website
The original version of this article was revised due to a retrospective Open Access order.
I Tirado-Gonzalez, E Czlonka and A Nevmerzhitskaya: These three authors contributed equally to this work.
Change history
12/1/2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
- 1.Cosgun KN, Rahmig S, Mende N, Reinke S, Hauber I, Schafer C. Kit regulates HSC engraftment across the human-mouse species barrier. Cell Stem Cell. 2014;15:227–238. doi: 10.1016/j.stem.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins ED, Duarte D, Akinduro O, Khorshed RA, Passaro D, Nowicka M. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature. 2016;538:518–522. doi: 10.1038/nature19801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McIntosh BE, Brown ME, Duffin BM, Maufort JP, Vereide DT, Slukvin II. Nonirradiated NOD,B6.SCID Il2rgamma-/- Kit(W41/W41) (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Rep. 2015;4:171–180. doi: 10.1016/j.stemcr.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Passaro D, Di Tullio A, Abarrategi A, Rouault-Pierre K, Foster K, Ariza-McNaughton L. Increased vascular permeability in the bone marrow microenvironment contributes to disease progression and drug response in acute myeloid leukemia. Cancer Cell. 2017;32:324–341 e326. doi: 10.1016/j.ccell.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tai IT, Tang MJ. SPARC in cancer biology: its role in cancer progression and potential for therapy. Drug Resist Updat. 2008;11:231–246. doi: 10.1016/j.drup.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Alachkar H, Santhanam R, Maharry K, Metzeler KH, Huang X, Kohlschmidt J. SPARC promotes leukemic cell growth and predicts acute myeloid leukemia outcome. J Clin Invest. 2014;124:1512–1524. doi: 10.1172/JCI70921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DiMartino JF, Lacayo NJ, Varadi M, Li L, Saraiya C, Ravindranath Y. Low or absent SPARC expression in acute myeloid leukemia with MLL rearrangements is associated with sensitivity to growth inhibition by exogenous SPARC protein. Leukemia. 2006;20:426–432. doi: 10.1038/sj.leu.2404102. [DOI] [PubMed] [Google Scholar]
- 8.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sweeney CL, Choi U, Liu C, Koontz S, Ha SK, Malech HL. CRISPR-mediated knockout of Cybb in NSG mice establishes a model of chronic granulomatous disease for human stem-cell gene therapy transplants. Hum Gene Ther. 2017;28:565–575. doi: 10.1089/hum.2017.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilmour DT, Lyon GJ, Carlton MB, Sanes JR, Cunningham JM, Anderson JR. Mice deficient for the secreted glycoprotein SPARC/osteonectin/BM40 develop normally but show severe age-onset cataract formation and disruption of the lens. EMBO J. 1998;17:1860–1870. doi: 10.1093/emboj/17.7.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer PN, Fu K, Greiner T, Smith L, Delabie J, Gascoyne R. The stromal cell marker SPARC predicts for survival in patients with diffuse large B-cell lymphoma treated with rituximab. Am J Clin Pathol. 2011;135:54–61. doi: 10.1309/AJCPJX4BJV9NLQHY. [DOI] [PubMed] [Google Scholar]
- 13.Chow YP, Alias H, Jamal R. Meta-analysis of gene expression in relapsed childhood B-acute lymphoblastic leukemia. BMC Cancer. 2017;17:120. doi: 10.1186/s12885-017-3103-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebinger S, Ozdemir EZ, Ziegenhain C, Tiedt S, Castro Alves C, Grunert M. Characterization of rare, dormant, and therapy-resistant cells in acute lymphoblastic leukemia. Cancer Cell. 2016;30:849–862. doi: 10.1016/j.ccell.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.