

Abstract
We report herein the design, synthesis, and pharmacological characterization of a library of novel aryl pyrazol-1-yl-propanamides as selective androgen receptor degraders (SARDs) and pan-antagonists that exert broad-scope AR antagonism. Pharmacological evaluation demonstrated that introducing a pyrazole moiety as the B-ring structural element in the common A-ring–linkage–B-ring nonsteroidal antiandrogens’ general pharmacophore allowed the development of a new scaffold of small molecules with unique SARD and pan-antagonist activities even compared to our recently published AF-1 binding SARDs such as UT-155 (9) and UT-34 (10). Novel B-ring pyrazoles exhibited potent AR antagonist activities, including promising distribution, metabolism, and pharmacokinetic properties, and broad-spectrum AR antagonist properties, including potent in vivo antitumor activity. 26a was able to induce an 80% tumor growth inhibition of xenografts derived from the enzalutamide-resistant (Enz-R) VCaP cell line. These results represent an advancement toward the development of novel AR antagonists for the treatment of Enz-R prostate cancer.
Graphical Abstract
1. INTRODUCTION
Prostate cancer (PC) is the second leading cause of cancer-related death, after lung cancer, in American men. Globally, both the number of PC cases and mortality have increased significantly.3,4 Longer life expectancy and increasing geriatric male population are some of the contributors to increasing PC incidence. PC depends on the activation of androgen receptor (AR) signaling for its development, progression, growth, and survival.5–7
Approximately 20–40% of PC patients treated with radiation and radical prostatectomy will experience tumor recurrence.4 Once the tumor recurs, androgen ablation therapy or androgen deprivation therapy (ADT) is the standard of care for most patients. ADT is achieved through surgical castration (orchi-ectomy) or chemical castration (injection of gonadotropin-releasing hormone agonist or antagonist), both of which cause a reduction in testosterone biosynthesis by testes. In addition to ADT, secondary hormonal suppression is provided by direct competitive ligand binding domain (LBD)-directed AR antagonists termed as antiandrogens such as flutamide (1),8 bicalutamide (2),9 nilutamide (3), enzalutamide (4),10 apalutamide (5),11,12 or darolutamide (6)13,14 or androgen synthesis inhibition such as abiraterone acetate (7) plus prednisone (Figure 1).15 Secondary hormonal suppression, that is, added to ADT, has been approved to treat castration-sensitive PC (CSPC) or castration-resistant prostate cancer (CRPC),16–18 with the approval trend toward their use earlier in the natural history of the disease in order to more effectively delay disease progression.19
Figure 1.
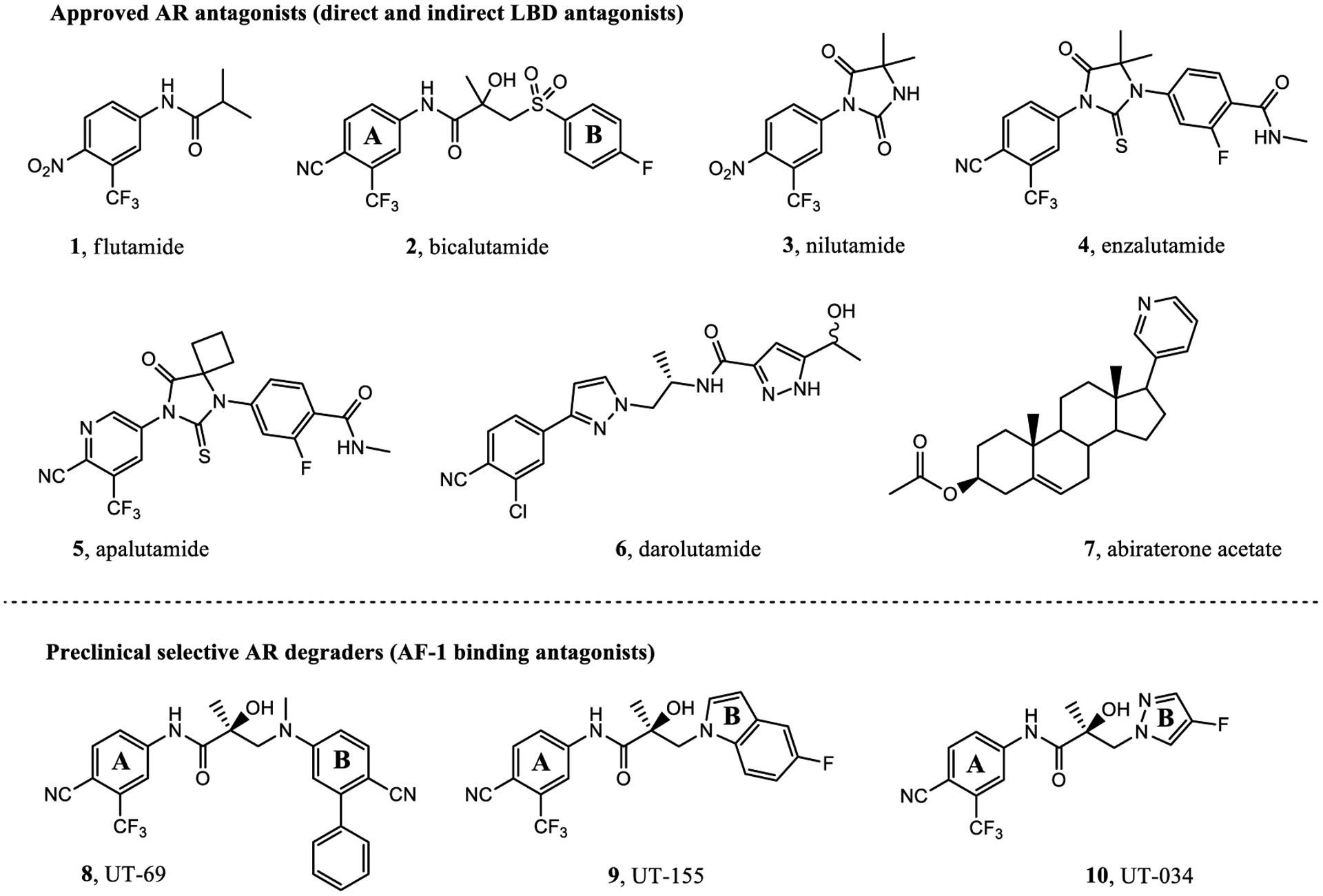
Overview of direct antiandrogens (1–6), an indirect LBD antagonist (7), and our preclinical SARDs (8–10). Clinically approved agents include first-generation (1–3) and second-generation (4–6) antiandrogens and an indirect androgen synthesis inhibitor (7). Also shown are our SARDs (8–10), which are pan-antagonists in preclinical development for the treatment of antiandrogen-resistant PC.
ADT is initially effective for advanced PCs; however, sustained ADT treatment, in combination with antiandrogens, often only stabilizes the disease for 2–3 years before PC becomes refractory, resulting in a more aggressive CRPC tumor phenotype where tumors become resistant to (ongoing ADT and) secondary hormonal therapies.20 Resistance to any one of 2, 4, 5, or 7 can emerge just months after initiation and studies suggest that 6 (darolutamide) may behave similarly in the CPRC population (6 approved for mCSPC).14 Despite resistance to secondary hormonal therapies in CRPC whether direct (1–6) or indirect (7), AR signaling continues to be fundamental for tumor growth and disease progression. Correspondingly, novel mechanisms to inhibit the AR axis are needed in hormone-resistant PCs.21
Although the exact mechanisms of CRPC progression are not always known clinically nor are they mutually exclusive, preclinical and clinical research has demonstrated numerous contributing factors to the emergence of CRPC that include (1) compensatory production of intratumoral androgens (e.g., DHT synthesized from adrenal precursors),5,22,23 (2) AR gene amplifications and overexpression,24–26 (3) AR LBD point mutations,25,27,28 (4) alterations in the expression of coregula-tory proteins,29,30 (5) ligand-independent activation of AR,31–35 (6) constitutively active truncated AR splice variants (AR SVs),36 and (7) induction of intracrine androgen metabolic enzymes.20,37,38 Direct and indirect antiandrogen therapies all target AR at the LBD and eventually fail because of the resistance mechanisms mentioned above.39 The development of AR antagonists for CRPC with novel mechanisms of action that are capable of durably treating patients with resistance to 2, 4, 5, and 7 (cross-resistance of 7 to 4 and 5 is common; 1 and 3 are rarely used) or darolutamide (6) (approved in 2019; patterns of resistance to 6 are still emerging) is an urgent need.
To provide clinical benefit for CRPC or to circumvent the emergence of CRPC, the next generation of AR-targeted therapeutics ideally should be able to bind to novel and/or multiple domains of the AR and inhibit a broad scope of AR functions across the broad scope of AR sequences present and emerging in the heavily pretreated CPRC population.40,41 Such novel antagonists ideally will maintain activity in wild-type (wt), point mutant, AR SVs, and/or AR overexpressing pathogenic states with sufficient potency to maintain suppression of the AR axis as PC becomes progressively more refractory to treatment.
Binding to a non-LBD site and degradation of the AR protein are promising preclinical approaches to rationally target CRPC; however, a clinical proof of concept is still needed.42–46 Degradation of AR can be achieved by genetic knockdown technologies, such as antisense oligonucleotides, RNA interference, and DNA editing. Despite genetic approaches having great therapeutic potential, it remains clinically challenging because of technical difficulties in delivering oligonucleotides (polyanionic macromolecules) to the prostate and metastatic tumors. Furthermore, oligonucleotide uptake into the tumor cells is poor.47,48 Alternatively, targeted destruction of the AR by protein knockdown technologies which degrade AR via the ubiquitin proteasome system (UPS) remains a promising group of options yet to be tested definitively in the clinical setting.2,44,49,50
In recent antitumor studies, our laboratory has reported the discovery and characterization of UT-69 (8) and UT-155 (9) as first-in-class AR antagonists that selectively inhibit tumor growth and degrade the AR (full-length) and AR SVs (truncated) within these tumors.1 We also reported a novel series of aryl indol-1-yl propanamides and aryl indolin-1-yl propanamides as selective androgen receptor degraders (SARDs).51 These SARD activities were mediated through the UPS as determined by UPS inhibitor studies.1,2 Among these SARDs, 8 and 9 bind both to the N-terminal domain (NTD) at the transcriptional activation units (Tau)-1 and −5 (Tau-1 and Tau-5) of the AF-1 domain, which has not been targeted for degradation previously, and additionally, these SARDs competitively bind the LBD. Recently, an aryl pyrazol-1-yl propanamide (10; termed UT-34 therein) was demonstrated to bind the same Tau-1 and Tau-5 NTD sites but have improved pharmacokinetic (PK) properties and was characterized to have unprecedented xenograft efficacy in the models of enzalutamide (4) resistance (Enz-R).2
All our preclinical SARDs reported so far have degraded AR and inhibited AR function, and our leads from each scaffold exhibited in vitro inhibitory potency in screening assays (e.g., LBD binding, transcriptional inhibition, AR degradation, and antiproliferative assays) and greater in vivo efficacy (Hershberger assay and various AR-dependent CPRC xenografts) than the approved AR antagonists. To discover preclinical leads to advance to clinical testing, herein, we explored the structure- activity relationships (SARs) within the pyrazol-1-yl series with the goal of improving upon the unprecedented activities of 10.
2. RESULTS AND DISCUSSION
2.1. Chemistry.
We designed and synthesized a series of pyrazol-1-yl-propanamide compounds similar to 10 with varying mono-substituents of the pyrazole B-ring (Series I),2 variations of the aromatic A-ring (Series II), varying the disubstituents of the pyrazole B-ring (Series III), or modifications of the linkage moiety (Series IV), as shown in Table 1.
Table 1.
Structures of Pyrazol-1-yl-propanamide AR Antagonists
| Series I. Monosubstitution of the Pyrazole Moiety (B-ring) | |||
| ID | Structure | ID | |
| 16a | 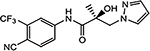 |
10 (UT-034) |
 |
| 16b | 16c | 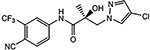 |
|
| 16d | 16e | ||
| 16f | 16g | ||
| 16h | 16i | 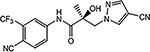 |
|
| 16j | 16k | ||
| 16l | 16m | ||
| 16n | 16o | ||
| 16p | 16q | ||
| 16r | 16s | ||
| 16t | 16u | ||
| 16v | 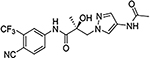 |
16w | 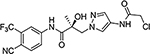 |
| 16x | 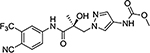 |
||
| Series II Variations of the Aromatic A-Ring | |||
| 21a | 21b | ||
| 21c | 21d | 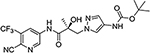 |
|
| 21e | 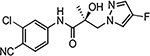 |
21f | 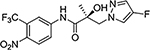 |
| 21g | 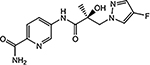 |
21h | 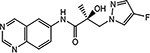 |
| 21i | 21J | ||
| Series III. Disubstitution of the Pyrazole B-ring | |||
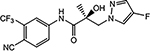
| 26a | 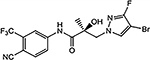 |
26b | |
| 26c | 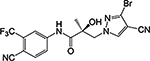 |
26d | 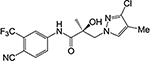 |
| 26e | 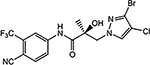 |
26f | 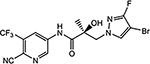 |
| 26g | 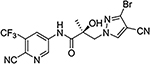 |
26h | 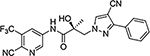 |
| Series IV. Modification of Linkage Moiety | |||
|
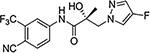
29a (R-isomer of 10) |
 |
29b | |
| 29c | 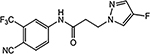 |
29d | 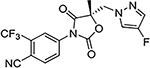 |
| 29e |  |
29f | |
2.1.1. Series I: Monosubstitutions of the Pyrazole Moiety (B-Ring).
The syntheses of 16a–16x were performed according to Scheme 1. Commercially available (R)-3-bromo-2-hydroxy-2-methylpropanoic acid (11) was treated with SOCl2 to transform acid 11 to acid chloride (R)-3-bromo-2-hydroxy-2-methylpropanoyl chloride (not shown), which reacted with aniline (12) to afford the bromide compound (13). Intermediate 13 has also been synthesized previously by Tucker (PMID: 3625091 and 3361581) and our lab (PMID: 8867996). Under basic conditions (e.g., K2CO3), 13 was transformed to a key oxirane intermediate (14). Alkylation of commercially available pyrazoles (15) by their reaction with 14 afforded the pyrazol-1-yl-propanamides 16a–16x, as shown in Scheme 1. Series I and all other compounds tested herein were screened in vitro for AR LBD binding (Ki), inhibition of transactivation (IC50), AR degradation (% degradation) of full-length (AR FL in LNCaP cells) and SV (AR SV in 22RV1 cells) ARs in PC cell lines, and degradation potency (DC50 values) in LNCaP cells (Table 2). Optimal SARDs and pan-antagonists are compounds that potently inhibit AR transactivation (IC50) and optionally degrade AR FL or AR SV and possess in vivo efficacy in models of antiandrogen-resistant CRPC of greater potency than 10.
Scheme 1. Synthesis of Pyrazol-1-yl-propanamides 16a–16xa.
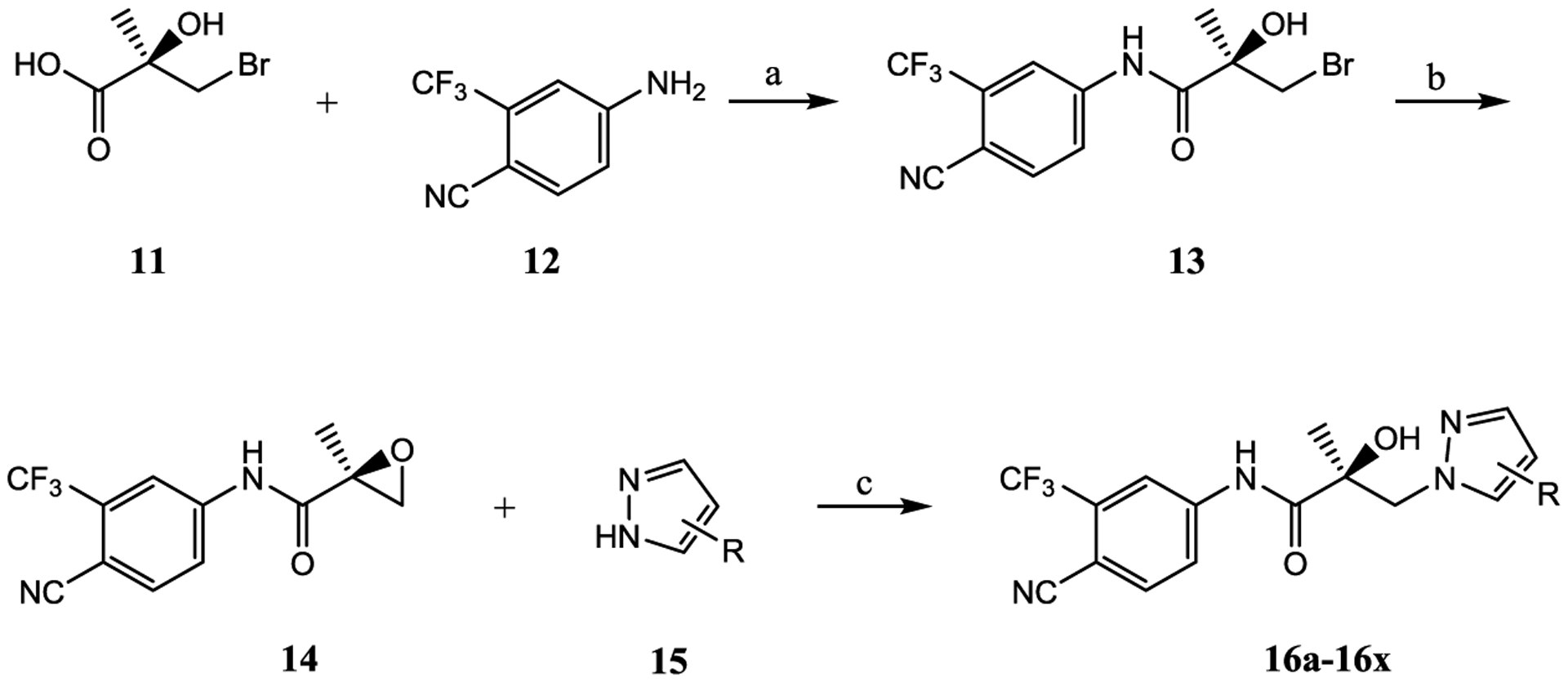
aReagents and conditions: (a) 1. SOCl2 in THF, −10 to 0 °C. 2. Et3N in THF, −10 to 0 °C, and then to 50 °C, 2–3 h; (b) 2-butanone, K2CO3, reflux; and (c) NaH in THF, 0 °C to rt.
Table 2.
In Vitro AR Activity of 16a–16x (Series I) and Approved Antiandrogens
 | |||||
|---|---|---|---|---|---|
| binding (Ki)/transactivation (IC50) (μM) |
SARD activity (% degradation) | ||||
| compound ID (R-group) | Ki (DHT = 1 nM)a | IC50b | full length (LNCaP) at 1 μM | splice variantc (22RV1) at 10 μM | F.L. DC50 (μM) |
| 16a (4-H) | 7.398 | 1.442 | 0 | 0 | |
| 10 (4-F)d | >10 | 0.199 | 100 | 100 | 0.74 |
| 16b (3-F) | 0.821 | 0.220 | 82 | 73 | 0.47 |
| 16c (4-CI) | >10 | 0.136 | 71 | 34 | 0.97 |
| 16d (4-Br) | >10 | 0.427 | 42 | 0 | |
| 16e (4-I) | N.A.e | 2.038 | N.A.e | N.A.e | |
| 16f (4-COCH3) | 1.056 | 0.758 | 30 | N.A. | |
| 16g (4-CF3) | 0.898 | 0.071 | 80 | 100 | 0.29 |
| 16h (3-CF3) | 0.512 | 0.205 | 67 | 54 | 0.73 |
| 16i (4-CN) | 1.499 | 0.045 | 90 | 100 | 0.32 |
| 16j (4-NO2) | 2.225 | 0.036 | 20 | N.A. | |
| 16k (4-OCH3) | N.A. | no effect | 0 | 0 | |
| 16l (4-CH3) | 1.552 | 8.087 | N.A. | N.A. | |
| 16m (4-phenyl) | >10 | 1.152 | 0 | 0 | |
| 16n (3-phenyl) | 3.660 | 4.770 | 0 | 0 | |
| 16o [4-(4-fluorophenyl)] | 0.612 | 0.969 | 72 | 0 | 0.86 |
| 16p [3-(4-fluorophenyl)] | >10 | 1.069 | 54 | 81 | 0.99 |
| 16q (4-ethynyl) | N.A. | 0.276 | 7, 28f | N.A. | |
| 16r [4-(4-OH-but-1-yn-1-yl)] | >10 | no effect | 51, 80f | N.A. | 0.97 |
| 16s (4-carbamoyl) | N.A. | no effect | N.A. | N.A. | |
| 16t (4-NH2) | 0.223 | agonist | N.A. | ||
| 16u (4-NHCOOtBu) | 1.382 | 1.153 | 20 | 0 | |
| 16v (4-NHCOCH3) | >10 | no effect | N.A. | ||
| 16w (4-NHCOCH2Cl) | >10 | no effect | N.A. | N.A. | |
| 16x (4-NHCOOCH3) | >10 | 0.827 | N.A. | N.A. | |
| 2 (R-bicalutamide)g | 0.509 | 0.248 | |||
| 4 (enzalutamide)g | 3.641 | 0.216 | |||
| 5 (apalutamide)g | 1.452 | 0.160 | |||
| 6 (darolutamide) | 0.011h | 0.065h | N.A. | N.A. | |
AR binding was determined by competitive binding of 1 nM [3H] MIB to recombinant LBD of wild-type AR (wtAR). DHT was used in each experiment as a standard agent and the values are normalized to DHT, with the IC50 of DHT taken as 1 nM.
Inhibition of transactivation was determined by transfecting HEK-293 cells with full-length wtAR, GRE-LUC, and CMV-renilla luciferase for transfection control. Cells were treated 24 h after transfection with a dose response of compounds (1 pM to 10 μM) in the presence of 0.1 nM R1881 (antagonist mode) or in the absence of R1881 (agonist mode). Luciferase assay was performed 24 h after treatment using a dual-luciferase (firefly and Renilla) assay kit (Promega, Madison, WI).
SARD activity was assayed by treating LNCaP or 22RV1 cells for determining FL AR (at 1 μM of antagonist) or SV AR (at 10 μM of antagonist) protein levels, respectively. Cells were maintained in charcoal-stripped, serum-containing medium for 48 h and treated with the indicated doses of antagonist for 24 h in the presence of 0.1 nM R1881 (agonist). Cells were harvested and Western blot for AR was performed using AR-N20 or PG-21 antibody that is directed toward the NTD of AR and actin (internal control for protein loading). The AR FL and AR SV bands were quantified and normalized to actin bands and represented as percent inhibition from vehicle-treated cells.
Result was reported in the literature in the same assay as described here.2
N.A. indicates data not available.
The two values indicate SARD assays run with 1 and 10 μM of antagonist.
Transcriptional activation was performed in the same assay in antagonist mode and the IC50 values are reported.56
Binding affinity and wtAR inhibition of transactivation were reported in the literature for the mixture of diastereomers of darolutamide.57
Compound 16a, which has no substitution on the pyrazole ring, possessed weak AR inhibitory activity with an IC50 value of 1.442 μM. AR inhibition in vitro is defined as the ability to inhibit R1881-induced wtAR transcriptional activity as measured by the luciferase assay [see values in the transactivation (IC50) column of Table 2], referred to as in vitro AR inhibition herein. Introducing a halogen on the pyrazole significantly increased the AR inhibitory activity, except for the 4-iodo compound 16e. The order of AR inhibitory potency with halogen substitution was 16c (4-Cl, 0.136 μM) > 10 (4-F, 0.199 μM) > 16b (3-F, 0.220 μM) > 16d (4-Br, 0.427 μM) > 16a (4-H, 1.442 μM) > 16e (4-I, 2.038 μM). Compounds with 4-substitution exhibited more potent AR inhibitory activity than that of their 3-substitution counterparts. For example, compare 10 (4-F) to 16b (3-F), 16g (4-CF3) to 16h (3-CF3), 16m (4-phenyl) to 16n (3-phenyl), and 16o [4-(4-fluorophenyl)] to 16p [3-(4-fluorophenyl)], respectively.
In general, the stronger the electron-withdrawing group (EWGs) is on the pyrazole ring, the more potent is the AR inhibitory activity, with the potency order of 16j (4-NO2, 0.036 μM) > 16i (4-CN, 0.045 μM) > 16g (4-CF3, 0.071 μM) > 16h (3-CF3, 0.205 μM) > 16q (4-ethynyl, 0.276 μM) > 16f (4-COCH3, 0.758 μM). Compounds bearing an electron-donating group on the pyrazole ring showed low-potency AR inhibitory activity (16l, 16u, and 16x), no AR inhibitory activity (16k, 16s, 16v, and 16w), or even AR agonist activity (16t which is 4-NH2). Interestingly, 16r bearing [4-(4-OH-but-1-yn-1-yl)] on the pyrazole ring exhibited no AR inhibitory activity but showed 51% AR full-length protein degradation activity.
With regard to SARD activity, substitution of the pyrazole ring seems to be necessary (16a; 0%/0% in % degradation) but some electron-donating groups such as 4-OCH3 of 16k and 4-phenyl or 3-phenyl in 16m and 16n are inactive. Like AR inhibitory potency (discussed above), the strength of the EWGs and 4-substitution seem to contribute favorably as seen in 10 (4-F; 100%/100% for AR FL and AR SV efficacies), 16g (4-CF3; 80%/100% efficacy), and 16i (4-CN; 90%/100% efficacy), whereas 3-substituted EWGs possessed slightly lower SARD activity as can be seen in 16b (3-F; 82%/73% efficacy) and 16h (3-CF3; 67%/54% efficacy). However, 16p (3-(4-fluorophenyl)) is superior to its 4-position isomer 16o (4-(4-fluorophenyl)) with 54%/81% versus 72%/0% degradation efficacies.
As seen previously,51 inhibitory potency (IC50) does not always correlate with % degradation. For example, the most potent inhibitor 16j (4-NO2; 0.036 μM) was a poor degrader, also the most potent Series I halogen 16c (4-Cl; 0.136 μM) demonstrated only moderate SARD activity (71%/34%). Also as seen previously,51 LBD binding (Ki) does not correlate with AR inhibitory potency (IC50) or SARD activity. For example, nonbinders 10 and 16c (Ki values >10 μM) inhibited and degraded, whereas nonbinder 16r degraded but was not an inhibitor. At this point, it is not clear how to differentiate the SARs of the AR inhibition and the SARD activity seen with these noncanonical ligands. Further complicating the SAR of these compounds is that the structural information for the AF-1 region has not been elucidated, and AR antagonism observed may be NTD-mediated for some compounds, while other compounds may have contributions from both NTD and LBD. We are in the process of learning about how our molecules interact with AR and the importance of each binding site.
The SARs of % efficacy of SARD activity (considering AR FL SARD and AR SV SARD activity in aggregate because of their limitation as semiquantitative values) seems to correlate with the AR inhibitory potency to some degree and LBD Ki to a [much] lesser degree. As discussed previously,51 the screening profile is intended to allow us to maximize FL and SV SARD efficacies and AR inhibitory potency to provide the most potent and broad-scope antagonists for testing in the models of antiandrogen-resistant CRPC. In some cases, such as 16g, 16i, and 26a, high-efficacy SARDs (>70% for both AR FL and AR SV) are potent AR inhibitors (<0.100 μM IC50); and moderate efficacy SARDs were moderate potency inhibitors such as 16b, 16c, 16h, and 26c. However, as discussed herein, % SARD efficacy does not always correlate well with in vitro inhibitory potency or LBD binding. As mentioned above, 16j was a poor degrader, possessing only 20% AR FL efficacy (N.A. for SV), but very potent inhibition (0.036 μM) compared to LBD binding (2.225 μM), and 26f, which possessed only 8, 15% AR FL efficacy at 1 and 10 μM but extremely potent inhibition (0.035 μM), which is >10-fold more potent than LBD binding (0.567 μM). Therefore, we have reconsidered placing primary emphasis of these molecules as AR degraders (i.e., SARDs) instead of placing emphasis of their ability to inhibit, and in most cases degrade, all AR forms tested to date. In an effort to determine the contribution of AR SARD activity to the AR antagonism observed, the degradation potency values (DC50 values) in LNCaP cells have been reported here for the first time. These values were approximately 4-to 10-fold greater than IC50 values (Tables 2–5), suggesting that SARD activity alone may not explain the potent AR pan-antagonism of these compounds. Hence, these broad-scope and potent noncanonical AR antagonists are discussed as SARDs and pan-antagonists herein.
Table 5.
In Vitro AR Activity of 29a–29f (Series IV)
| ID (R1) | Linker | Binding (Ki) / Transactivation (IC50) (μM) | SARD Activity (% degradation) | F.L. DC50 (μM) | ||
|---|---|---|---|---|---|---|
| Ki (DHT = 1 nM)a | IC50a | Full Lengtha (LNCaP) at 1 μM, 10 μM | Splice Varianta (22RV1) at 10 μM | |||
| 29a (4-F) (R-isomer) |  |
>10 | 0.192 | 84 | N.A.b | - |
| 29b (4-F) | 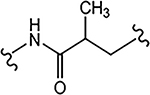 |
>10 | 0.462 | 60 | 70 | 0.74 |
| 29c (4-F) | 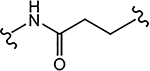 |
>10 | 2.124 | 35 | 40 | - |
| 29d (4-F) | 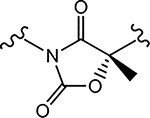 |
N.A.b | 1.131 | 18, 50c | N.A.b | - |
| 29e (4-NH2) | 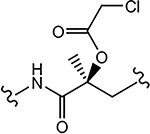 |
>10 | 0.901 | N.A. | N.A. | - |
AR binding, transactivation, and degradation assays were performed and values are reported as described in Table 2.
N.A. indicates data not available.
The two values indicate SARD assays run with 1 and 10 μM of antagonist.
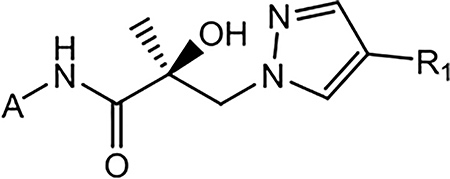
2.1.2. Series II: Modifications of Aromatic A-Ring.
Subsequent synthetic modifications were aimed at the replacement of the A-ring of 10 to explore the effect of different A-rings or A-ring substitutions on AR inhibition and SARD activity. 21a–21j were prepared by the route shown in Scheme 2. Treatment of acid 11 with SOCl2 provided the acid chloride (R)-3-bromo-2-hydroxy-2-methylpropanoyl chloride (not shown), which was reacted with various amines (17) under basic Et3N conditions to furnish bromoamides 18 with different A-rings. Basic conditions (e.g., K2CO3) transformed bromoamides 18 to the oxirane intermediates 19, followed by coupling with various pyrazoles 20 under the sodium hydride basic conditions to produce the target compounds 21a–21j, as shown in Scheme 2. The compounds were tested in vitro as discussed above for AR activity (Table 3).
Scheme 2. Synthesis of Pyrazol-1-yl-propanamides 21a–21ja.
aReagents and conditions: (a) 1. SOCl2 in THF, −10 to 0 °C. 2. Et3N in THF, −10 to 0 °C and then heat to 50 °C, 2–3 h; (b) 2-butanone, K2CO3, reflux; and (c) NaH in THF, 0 °C to rt.
Table 3.
In Vitro AR Activity of 21a–21j (Series II)
| ID (R1) | Structure of the A-ring (A) |
Binding (Ki) / Transactivation (IC50) (μM) | SARD Activity (% degradation) | F.L. DC50 (μM) | ||
|---|---|---|---|---|---|---|
| Ki (DHT = 1 nM)a | IC50a | Full Lengtha (LNCaP) at 1 μM | Splice Varianta (22RV1) at 10 μM | |||
| 21a (4-F) | 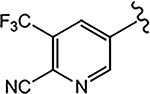 |
>10 | 0.062 | 54 | 81 | 0.88 |
| 21b (4-CF3) | 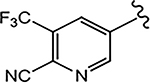 |
2.286 | 0.208 | 10 | N.A.b | - |
| 21c (4-CN) | 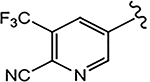 |
0.089 | 0.059 | 10 | N.A.b | - |
| 21d (4-NHCOOtBu) | 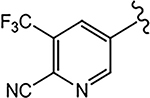 |
>10 | 6.108 | - | N.A.b | - |
| 21e (4-F) | 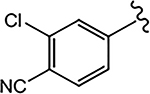 |
>10 | 0.427 | 42 | 0 | - |
| 21f (4-F) | 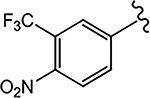 |
N.A.b | Partial Agonist | N.A.b | N.A.b | - |
| 21g (4-F) | 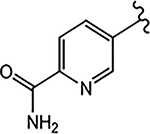 |
>10 | No effect | 0 | N.A.b | |
| 21h (4-F) | 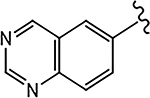 |
>10 | No effect | N.A. | N.A. | - |
| 21i (4-F) | 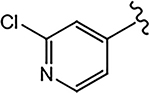 |
N.A. | 2.470 | 75 | N.A. | N.A. |
| 21j (4-F) | 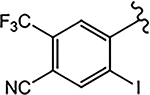 |
N.A. | 5.450 | N.A.b | N.A. | - |
AR binding, transactivation, and degradation assays were performed and values are reported as described in Table 2.
N.A. indicates data not available.
For the 4-F pyrazole, replacing a carbon (CH) with a nitrogen (N) at the 3′-position of the A-ring, that is, 3′-pyridino derivative of 10, delivered a more potent compound (21a) with an AR inhibitory IC50 value of 0.062 μM compared to its counterpart 10 (IC50 = 0.199 μM). However, in other instances, 3′-pyridino derivatives were equipotent or less potent than their phenyl A-ring analogues. The 3′-pyridino 21c (4-CN; 0.059 μM) showed almost equally potent AR inhibitory activity compared to its A-ring phenyl analogue 16i (IC50 = 0.045 μM). However, 3′-pyridino compounds 21b (4-CF3) and 21d (4-NHCOOtBu) showed lower activity (IC50 values of 0.208 and 6.108 μM, respectively) than their phenyl A-ring counterparts 16g and 16u. Other A-ring modifications of 10 decreased the AR inhibitory activity and % degradation when compared to 10 (0.199 μM; 100%/100%), including replacing the 3′-CF3 with a 3′-Cl (21e; 0.427 μM; 42%/0% degradation), replacing the 4′-CN with a 4′-NO2 (21f; partial agonist; N.A. % degradation), and other modifications as in 21g–21j. Interestingly, unlike other pyrazole propanamides, which show low or no AR LBD binding affinity (Ki), we found that the combination of 4-CN substituent in pyrazole and 3′-pyridino A-ring promoted the tight LBD binding seen for 21c (Ki = 0.089 μM) but relatively poor SARD activity (15%/N.A.).52
2.1.3. Series III: Disubstitution of the Pyrazole B-Ring.
Disubstitutions on pyrazole ring system might enhance inhibitory activity or improve metabolic stability and provide a better understanding of the SAR of the pyrazole ring. Series III compounds 26a–26h were designed and synthesized utilizing similar synthetic methods as in Schemes 1 and 2, as depicted in Scheme 3, and tested for AR activity (Table 4).
Scheme 3. Synthesis of Pyrazol-1-yl-propanamides 26a–26ha.
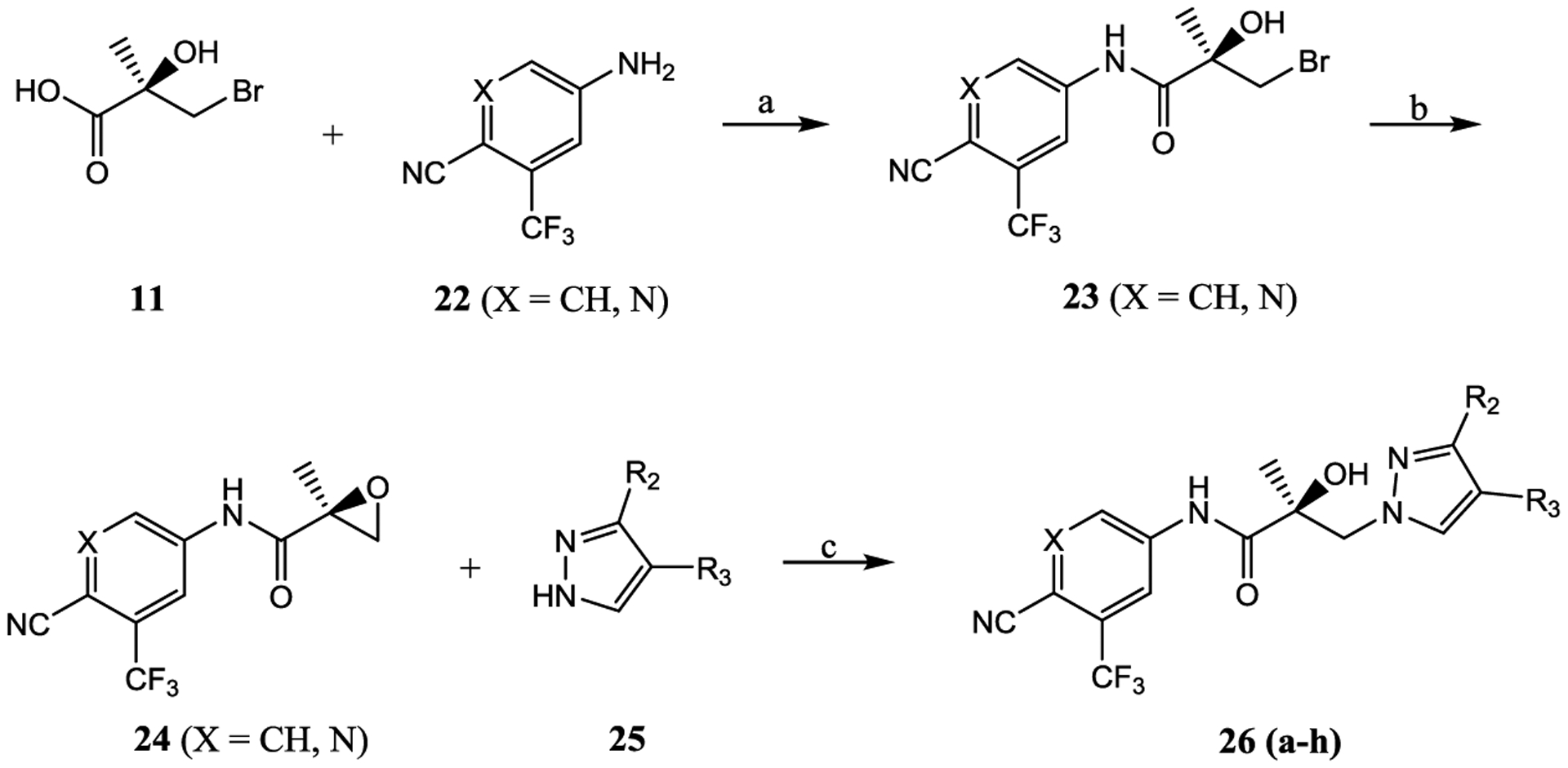
aReagents and conditions: (a) 1. SOCl2 in THF, −10 °C to 0 °C. 2. Et3N in THF, −10 to 0 °C and then heated to 50 °C, 2–3 h; (b) 2-butanone, K2CO3, and reflux; and (c) NaH in THF, 0 °C to rt.
Table 4.
In Vitro AR Activity of 26a–26h (Series III)
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| binding (Ki)/transactivation (IC50) (μM) | SARD activity (% degradation) | |||||||
| ID | X | R2 | R3 | Ki (DHT = 1 nM)a | IC50a | full lengtha (LNCaP) at 1 μM, 10 μM | splice varianta (22RV1) at 10 μM | F.L. DC50 (μM) |
| 26a | CH | F | Br | 0.607 | 0.084 | 70 | 80 | 0.86 |
| 26b | CH | F | 4-F-phenyl | 0.601 | 0.285 | N.A.c | toxic | |
| 26c | CH | Br | CN | 0.202 | 0.181 | 41, 23b | 32 | |
| 26d | CH | Cl | CH3 | 1.345 | 0.332 | 41, 83b | N.A.c | |
| 26e | CH | Br | Cl | 4.935 | 0.138 | N.A.c | N.A.c | |
| 26f | N | F | Br | 0.567 | 0.035 | 8, 15b | N.A.c | |
| 26g | N | Br | CN | N.A.c | 5.481 | 40, 80b | N.A.c | N.A. |
| 26h | N | phenyl | CN | N.A.c | 0.579 | 9, 55b | N.A.c | |
AR binding, transactivation, and degradation assays were performed and values are reported as described in Table 2.
The two values indicate the SARD assays run with 1 and 10 μM of antagonist.
N.A. indicates data not available.
Given our difficulty in improving the activity via A-ring modification [see Series II (Table 3); all changes except for 21a and 21c were not favorable], we expanded our attempts to optimize the pyrazole ring system for inhibitory activity by 3,4-disubstitution. 26a possessed two EWGs (3-F and 4-Br) on the pyrazole ring and exhibited potent inhibitory activity (IC50 value of 0.084 μM) and moderate- to high-efficacy AR FL and AR SV degradation (70–80% degradation). Compound 26a improved the AR inhibitory potency by 3–4-fold over the 3-F (16b; 0.220 μM; 82%/73%) and 4-Br (16d; 0.427 μM; 42%/0%) monosubstituted analogues and retained or improved upon degradation properties, supportive of further exploration of disubstitution. Replacing a carbon (CH) with a nitrogen (N) in the A-ring of 26a delivered the 3′-pyridino 26f, which was a very potent AR inhibitor with an IC50 value of 0.035 μM but poor SARD activity (8, 15%/N.A. for FL/SV) (Table 4). Compounds 26b–26e with disubstituents on pyrazole showed inhibitory activity comparable to 10 (0.199 μM) with AR inhibitory IC50 values in the order of 26e (3-Br, 4-Cl; 0.138 μM) > 26c (3-Br, 4-CN; 0.202 μM) > 26b (3-Br, 4-(4-fluorophenyl); 0.285 μM) > 26d (3-Cl, 4-methyl); 0.332 μM). Compounds 26e (0.138 μM) and 26c (0.202 μM) did not improve upon their monosubstituted analogues 16c (4-Cl; 0.136 μM) and 16i (4-CN; 0.045 μM). However, addition of halogens to 4-EDG pyrazoles at least partially rescued the activity, for example, compare to 26b [3-Br, 4-(4-fluorophenyl); 0.285 μM] and 16o [4-(4-fluorophenyl); 0.969 μM] and 26d [(3-Cl, 4-methyl); 0.332 μM] and 16l (4-methyl; 8.087 μM). Again, these results suggest that the EWG strength of the pyrazole substituents contribute favorably to inhibitory activity. Interestingly, the 3′-pyridino A-ring version of 26c afforded a >10-fold less potent inhibitor 26g with an AR inhibitory IC50 value of 5.481 μM despite 80% SARD efficacy in AR FL (but no efficacy in AR SV). Introducing an extra bromo (26g) or a phenyl (26h) group on the 3-position of the pyrazole greatly decreased the inhibitory activity to 5.481 or 0.579 μM, respectively, compared to 21c (4-CN; 0.059 μM). We found that 3-Br and 4-CN substituents on pyrazole promoted a tighter LBD binding (Ki = 0.202 μM) for 26c, and 3-F and 4-Br substituents on pyrazole (26a) delivered a potent inhibitor (IC50 = 0.084 μM) while retaining the SARD activity in AR FL and AR SV (70%/80%). Correspondingly, 26a and 26c were selected for further study.
2.1.4. Series IV: Modification of the Linkage Moiety.
Subsequent modifications were aimed at introducing different linkage/spacer groups between the aromatic A-ring and pyrazole B-ring to explore the importance of the structure of the linking element to improve the inhibitory potency and SARD activity. Series IV compounds 29a–29f were designed and synthesized utilizing similar synthetic methods as in Schemes 1–3, as depicted in Scheme 4, and 29a–29f were tested for their AR activity (Table 5 and Table 1).
Scheme 4. Synthesis of Pyrazol-1-yl-propanamides 29a–29fa.
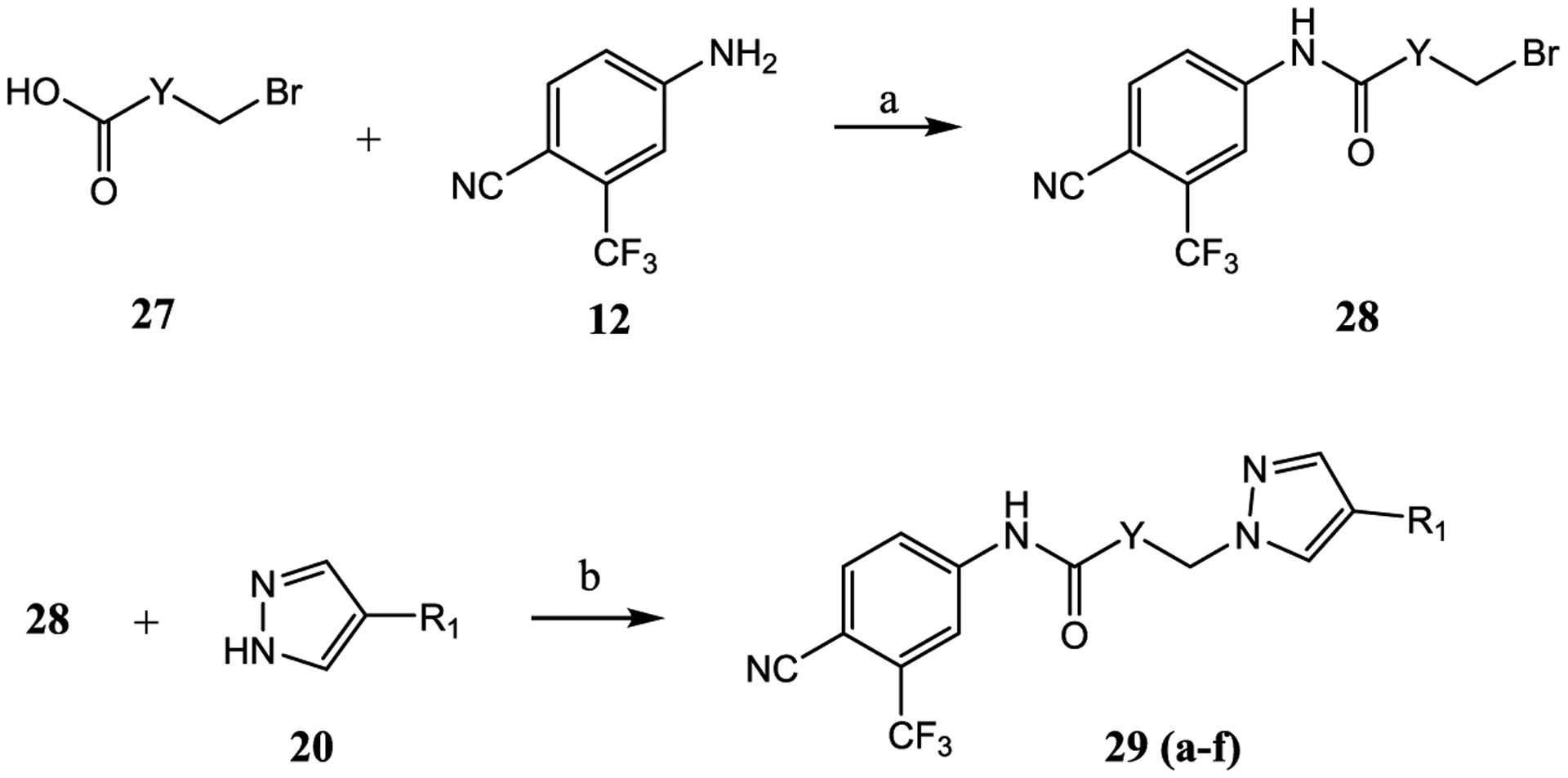
aReagents and conditions: (a) 1. SOCl2 in THF, −10 to 0 °C. 2. Et3N in THF, −10 to 0 °C and then heat to 50 °C, 2–3 h, and (b) NaH in THF, 0 °C to rt.
Similar to the previously reported R-isomer of 9,1 switching the chirality of 10 (the S-isomer) afforded the almost equipotent 29a (the R-isomer) with an AR inhibitory IC50 value of 0.192 μM and slightly decreased to 84% degradation compared to 10 (100%). Removal of the 2-hydroxyl moiety from the 2-hydroxy-2-methyl propanamide linker of 10 produced 29b with reduced AR inhibitory activity (0.462 μM) and 60%/70% FL/SV SARD activity compared to 10 (0.199 μM; 100%/100%). Removal of 2-methyl and 2-hydroxy moieties from linkage of 10 to produce the linear propanamide 29c further decreased the AR inhibitory and SARD activities. As an oxazolidin-2,4-dione linker variant of 10, 29d possessed groups similar to the amide and hydroxyl (as the oxygen in the carbamate) groups of the linker. 29d still showed activity but with substantially decreased AR inhibitory (1.131 μM) and SARD (18, 50%/N.A.) activities compared to 10 (0.199 μM; 100%/100%). Acylation of the 2-hydroxy AR agonist 16t (4-NH2) produced 29e, which recovered some antagonist activity with an AR inhibitory IC50 value of 0.901 μM, whereas introducing a second amide into the linker and varying the pyrazole attachment position as in 29f (see Table 1 for structure) produced an agonist. Although the linker element was not optimized in this initial SAR of Series IV, tolerance to chiral center inversion was again observed (unlike structurally similar propanamide SARMs), and it was established that there is no absolute requirement for the 2-hydroxy-2-methylpropamide linker for inhibitory and SARD activities.
The AR LBD affinity (for some compounds) and in vitro antagonist properties of Series I–III ranged from comparable to favorable relative to the known standard AR antagonists currently employed clinically for the treatment of PC. For example, 2, 4, 5, and 6 had LBD binding affinities of 0.509, 3.641, 1.452, and 0.011 μM (values for 2, 4, and 5 are internally determined vs for 6 is from the literature), and in vitro inhibition of 0.248, 0.216, 0.160, and 0.065 μM (values for 2, 4, and 5 are internally determined vs for 6 is from the literature); compared to 10 binding of >10 μM and antagonism of 0.199 μM. We found that compounds 16b, 16c, 16g, 16h, 16i, and 16m from Series I; 21a from Series II; 26a and 26c from Series III; and 29a from Series IV exhibited relatively potent AR inhibitory IC50 values in the range from 0.041 to 0.220 μM but, unlike 2, 4–6, were SARDs with degradation activity values in the range from 100 to 45%. Because these compounds with the exception of 16m were comparable to improved inhibitors relative to known LBD-targeted antiandrogens but possessed novel pan-antagonism and SARD activities, they were selected for further in vitro and in vivo study to help optimize PK properties and explore the pharmacodynamic (PD) potential of these NTD binding noncanonical antagonists. A particular emphasis was placed on improving upon pyrazole 10, which was the early lead in this chemical class of compounds.
2.2. In Vitro Metabolic Stability in Mouse, Rat, and Human Liver Microsomes.
Compounds with potent inhibitory activity of each series were selected to be further evaluated for in vitro metabolic stability in mouse liver microsomes (MLM) with cofactors for enzymes of both phase I and phase II metabolism. The half-life (T1/2) and intrinsic clearance (CLint) values were calculated as a predictor of the metabolism and PK properties of these compounds (Table 6). In overview, the CLint of these compounds was slower than previous generations of SARDs, producing relatively stable T1/2 values that range from 48.45 to >360 min for these pyrazol-1-yl-propanamides (16b, 16g, 16h, 16i, 16m, 21a, 26a, and 29a) with six of the nine tested pyrazoles being stable for >330 min in MLM. This is a vast improvement when compared to previous SARD templates such as 1.15 min for the tertiary amine 8, 12.11 min for lead indole 9,1 and 9–36 min for a variety of indole and indoline B-ring compounds previously published in the same in vitro assay51 and an improvement over 10 (T1/2 of 77.96 min).
Table 6.
In Vitro Metabolic Stability for Selected Compounds in MLMs
| MLMa | ||
|---|---|---|
| compound ID | T1/2 (min) | CLint (mL/min/mg) |
| 10 (4-F)b | 77.96 | 0.89 |
| 16b (3-F) | 64.07 | 1.02 |
| 16g (4-CF3) | >360 | 0 |
| 16h (3-CF3) | 330 | 0 |
| 16i (4-CN) | >360 | 0 |
| 16m (4-phenyl) | 48.45 | 14.31 |
| 21a (4-F) | >360 | 0 |
| 26a (3-F, 4-Br) | >360 | 0 |
| 29b (4-F) | >360 | 0 |
| 8b/9b | 1.15/12.11 | 208.8/57.26 |
| 4 (enzalutamide) | 10.04 hc | 86.3d |
Compounds were incubated together with MLMs with cofactors for phases I and II provided, as described in the Experimental Section.
Reported previously in using the same method as in the Experimental Section.1,2
T1/2 (h) after oral administration in humans as previously reported in ref. 58
CL (mL/h/kg) after oral administration in humans as previously reported.58
The likely metabolic liability in aryl bicycles such as indoles and indolines may be aryl hydroxylation of the B-ring. The A-ring and propanamide portions have been incorporated into many bioavailable compounds such as 2 (N-[4-cyano-3-(trifluoromethyl)phenyl]-3-(4-fluorophenyl)sulfonyl-2-hydroxy-2-methylpropanamide) and enobosarm ((2S)-3-(4-cya-nophenoxy)-N-[4-cyano-3-(trifluoromethyl)phenyl]-2-hydroxy-2-methylpropanamide), leaving the B-ring as the likely metabolically labile site. A possible rationale for improved PK properties with pyrazoles is the elimination of some of the possible aryl hydroxylation sites on the B-ring. Also, it is possible that the increased positive charged on the 2-position nitrogen atom of the pyrazole makes the compounds poor substrates for metabolic enzymes and/or improves biological partitioning. Further, the design principle of 3,4-dihalogenated pyrazoles like 26a was an attempt to more effectively block the metabolism of the B-ring, thereby decreasing hepatic CL.
Four further compounds were also characterized in rat liver microsomes (RLM) and human liver microsomes (HLM) as these readouts are relevant to suggest the stability of compounds for in vivo testing in PD models such as the rat Hershberger assay and xenograft in SRG rats (see infra) and ultimately in the clinic (Table 7). Series I compounds 16c and 16g were stable in RLM (T1/2 of >120 min) but 16c was less stable in HLM (T1/2 of 102 min). 21a (3′-pyridino, 4-F) and 26a (3-F, 4-Br) were stable (T1/2 of >120 min) in both RLM and HLM, which was similar to previously published data for 10 in RLM (181 min) and HLM (274 min) (cite ref. 2). The stability in RLM and HLM is consistent with the possibility of oral bioavailability of these pyrazoles, as previously seen with 10. However, 21a and 26a have improved in vitro efficacy relative to 16c and 10. Correspondingly, if sufficiently high blood levels of 21a and 26a are attained and the compounds are distributed to the site of action, that is, the tumor(s) throughout the body, it may be possible to improve the efficacy to treat antiandrogen-resistant CRPC compared to 10. Correspondingly, a few pyrazoles (16i, 21a, and 26a) with a variety of activity profiles were advanced to testing in the models of CRPC including resistance to 4 (MR49F cells harboring F876L AR point mutant) and 1 (LNCaP cells harboring the T877A).
Table 7.
In Vitro Metabolic Stability for Selective Compounds in RLM and HLM
| RLMa | HLMb | |||
|---|---|---|---|---|
| compound ID | T1/2 (min) | CLint (mL/min/mg) | T1/2 (min) | CLint (mL/min/mg) |
| 16c (4-Cl) | >120 | 0 | 102 | 6.78 |
| 16g (4-CF3) | >120 | 0 | >120 | 0 |
| 21a (4-F) | >120 | 0 | >120 | 0 |
| 26a (3-F, 4-Br) | >120 | 0 | >120 | 0 |
Compounds were incubated together with RLM with cofactors for phases I and II provided, as described in the Experimental Section.
Compounds were incubated together with HLM with cofactors for phases I and II provided, as described in the Experimental Section.
2.3. In Vitro PD in Models of Castration-Resistant PC.
As discussed above, compounds were screened in vitro in a competitive LBD binding assay (Ki), inhibitory AR transactivation assay (IC50), and AR FL (in LNCaP cells) and AR SV (in 22RV1 cells) degradation assays (% degradation) (Tables 2–5 above). Once strong in vitro screening profiles were accomplished for single molecule(s), in vitro metabolic stability criteria were also considered in the selection of compounds to be tested further in vitro (Tables 6 and 7 above). In order to improve the efficacy in in vivo testing, compounds were sought with superior in vitro screening profiles compared to 10 and tested further for transactivation selectivity between AR and PR, AR target gene expression in LNCaP cells, and proliferation studies in Enz-R PC cells (MR49F LNCaP cells).
2.3.1. Mutant AR and wtPR Antagonist Effects.
The selected compounds 16i (4-CN), 21a (3′-pyridino, 4-F), 26a (3-F, 4-Br), and 10 (4-F) were tested for their ability to antagonize a LBD point mutant AR, which confers an enzalutamide (4)-resistant (Enz-R) phenotype to PC cells. This F876L-mutant AR or wild-type PR (wtPR) was transfected into COS cells, a non-PC cell line, and quantified by luciferase assay (Figure 2). The compounds 16i, 21a, 26a, and 10 robustly inhibited the F876L-mutant AR with IC50 values of 0.043, 0.063, 0.084, and 0.219 μM (Figure 2) that are comparable to wtAR IC50 values of 0.045, 0.062, 0.084, and 0.199 μM (Tables 2–4). The ability to equipotently inhibit F876L and wtAR indicates that these SARDs exhibit pan-antagonism in a model of Enz-R. Further, this pan-antagonism cannot be explained by the AR LBD Ki values of 1.499, >10, 0.607, and >10 μM for 16i, 21a, 26a, and 10. Moreover, the increased potency of 16i, 21a, and 26a relative to 10 in wtAR inhibitory potency translated into this model of Enz-R.
Figure 2.
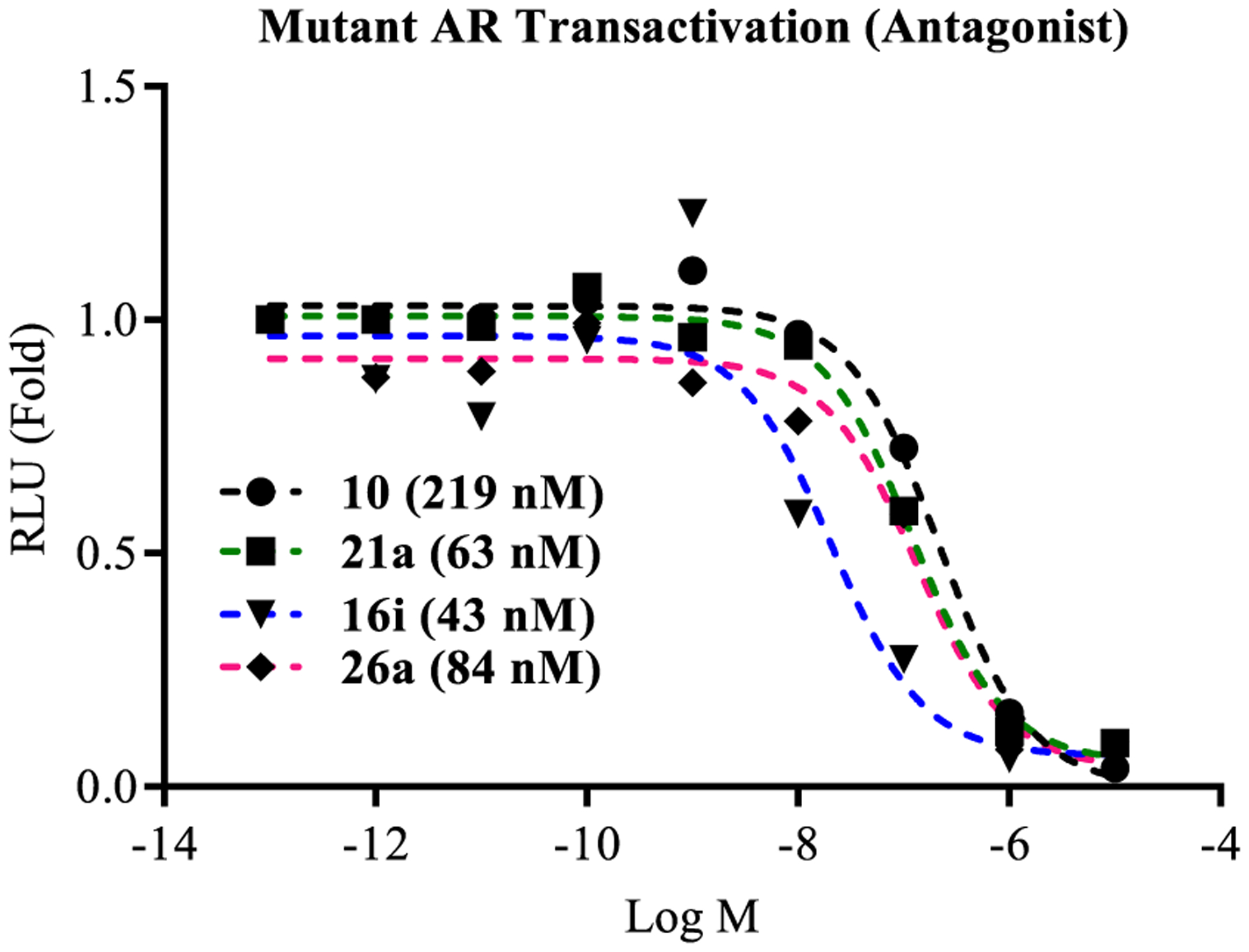
Antagonism of F876L-mutant AR transactivation. AR with phenylalanine 876 mutated to leucine (F876L), GRE-LUC, and CMV-renilla LUC were transfected in COS cells. Cells were treated 24 h after transfection with 0.1 nM R1881 (agonist) and a dose response of antagonists. Luciferase assay was performed 48 h after transfection. The effect of each compound was conducted in antagonistic mode (in the presence of 0.1 nM R1881). IC50 values were calculated and are provided in the figure.
Similar to our previously reported propanamide SARDs, these molecules also inhibited wtPR activity with IC50 values of 3.540, 0.235, 1.101, and 0.403 μM for 16i, 21a, 26a, and 10 (Figure 3) versus 0.045, 0.062, 0.084, and 0.199 μM for wtAR inhibition (Tables 2–5). Though wtPR inhibition was conserved, selectivity ratios ([PR IC50]/[AR IC50]) varied with values of 79-, 3.8-, 13.1-, and 2.0-fold for the compounds selected for testing, suggesting that AR selectivity could also be optimized with further testing. Importantly, none of these molecules had any effect on GR, MR, or ER transactivation (data not shown).
Figure 3.
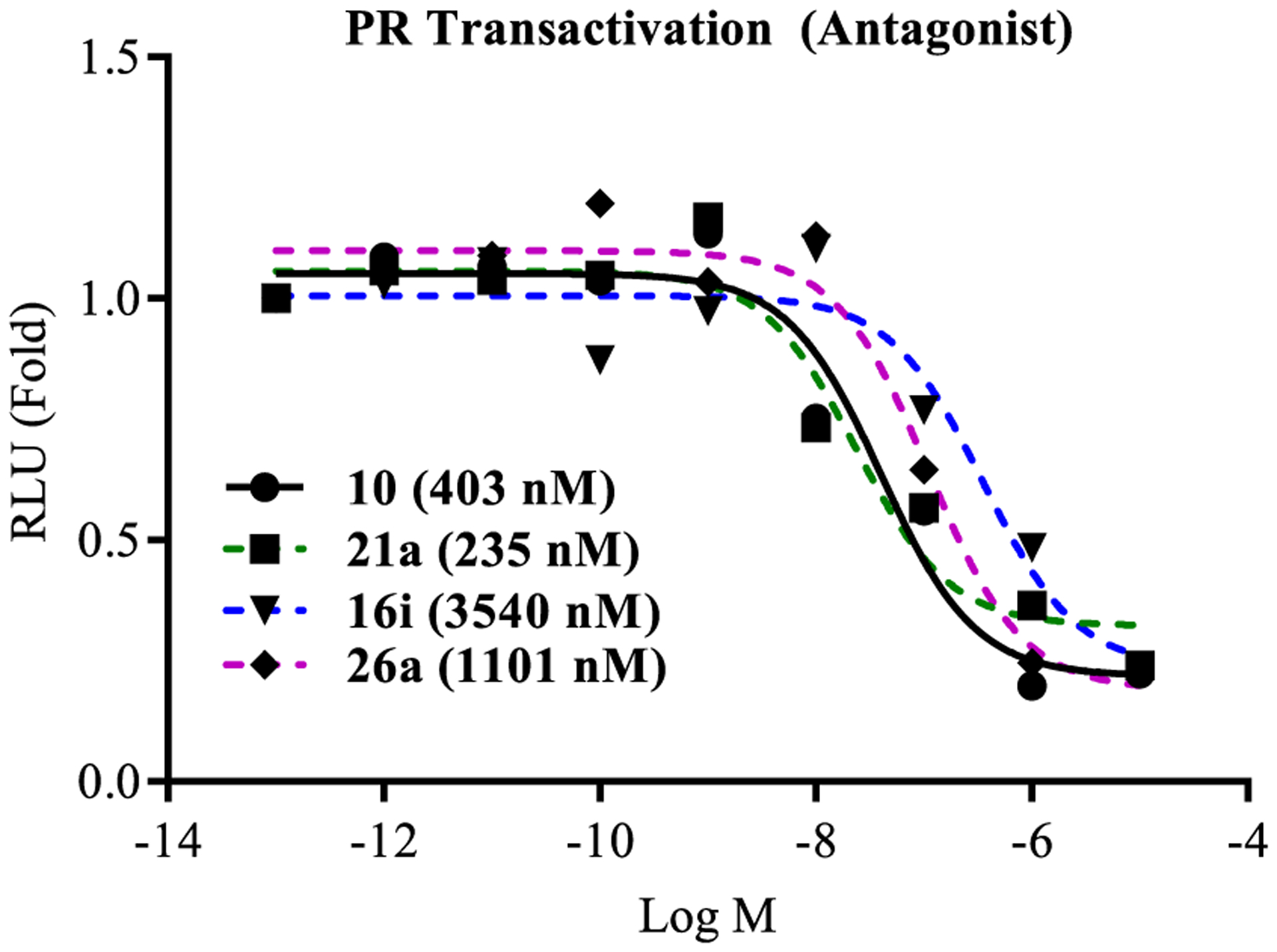
Antagonism of wtPR transactivation. COS cells were transfected with wtPR and a transactivation study was performed as indicated in the legend for Figure 2. IC50 values were calculated and are provided in the figure.
2.3.2. AR Target Gene Expression in CRPC Cells.
As 10 was reported to be effective in inhibiting the expression of FKBP5 in MR49F cells,2 we performed an AR target gene inhibitory experiment to determine the effect of lead pyrazole 26a on R1881-induced AR target gene expression in LNCaP cells (Figure 4). The LNCaP cell line is a very well-characterized model of CRPC that expresses the T877A point mutation of AR that confers resistance to 1. Compound 26a was chosen as the lead pyrazole as 26a possessed a balance of high-potency inhibition (0.084 μM) and high-efficacy degradation (70–80% for both AR FL and AR SV) with 3,4-disubstitution that blocked metabolism relative to 10 [T1/2 > 360 min vs 77.96 min in MLM (Table 6)] and 26a is also stable in RLM and HLM (>120 min). Consistent with the nM inhibition of wtAR (0.084 μM; Table 4) and F876L AR (0.084 μM; Figure 2) transactivation, FKBP5 gene expression in LNCaP cells was robustly inhibited by 26a at concentrations as low as 0.1 μM, indicating that the antiandrogenic effects include inhibition of endogenous gene expression (Figure 4) in another model of antiandrogen-resistant CRPC without loss of potency. As expected, the antiandrogen 4 also inhibited expression of the FKBP5 but at a slightly lower potency. The same results were observed with other AR target genes such as PSA and TMPRSS2 (data not shown). Cumulatively, the above data support that 26a has pan-antagonist effects in at least wtAR (Table 4), F876L (Figure 2), T877A (Figure 4), and AR SV (Table 4).
Figure 4.
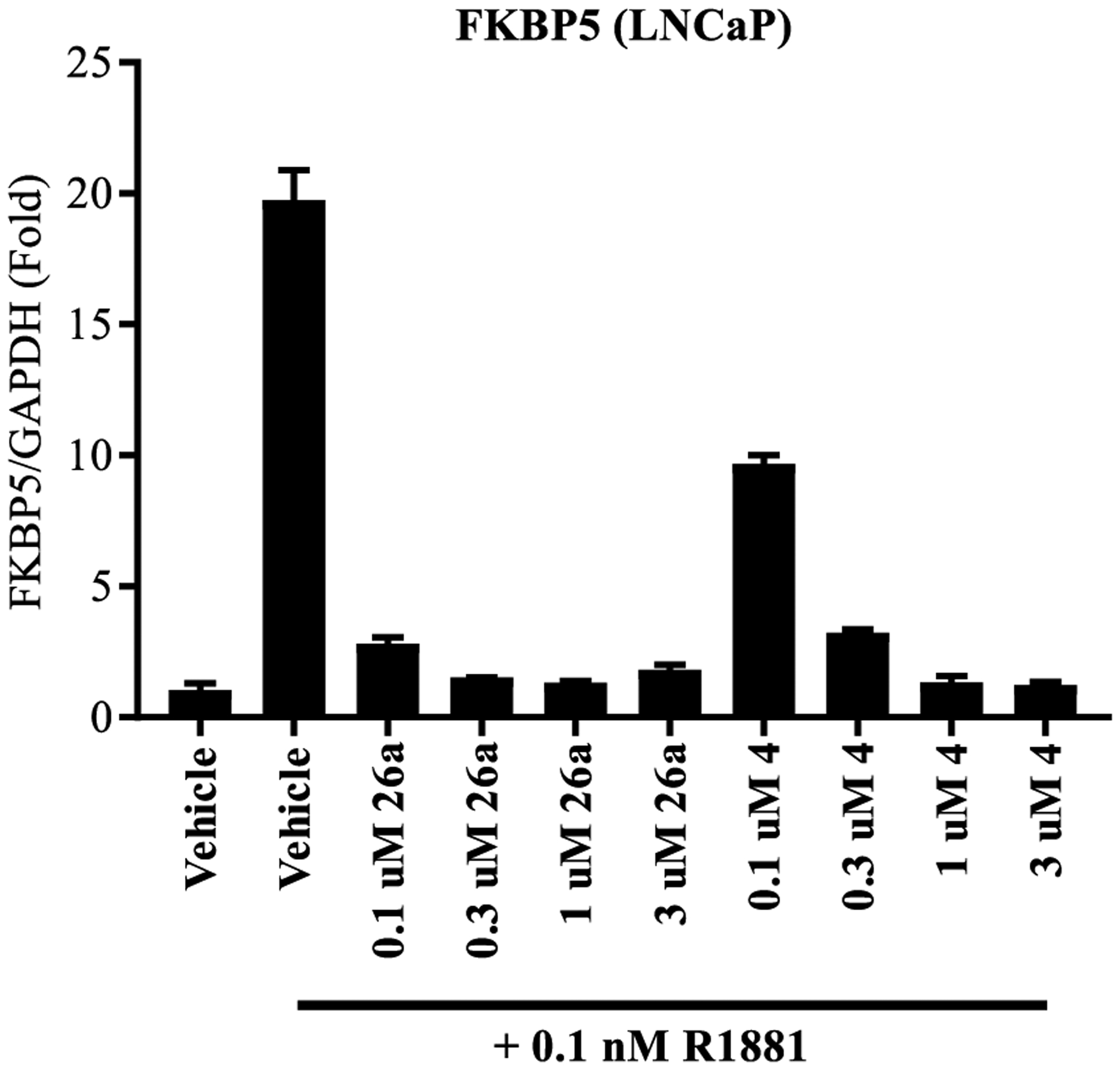
SARDs antagonize AR function in PC cell, LNCaP. LNCaP cells were maintained for 2 d in charcoal-stripped, serum-containing medium. The cells were treated with antagonist as indicated in the figure for 20–24 h, RNA was isolated, and expression of AR target gene, FKBP5, was measured and normalized to GAPDH using real-time PCR.
2.3.3. Proliferation Studies in Enz-R LNCaP Cells.
Proliferation studies were conducted with 26a to confirm that potent inhibition of AR-dependent gene expression in a model of CRPC harboring the T877A antiandrogen-resistant mutation (i.e., LNCaP cells) translated into antiproliferation in an even more refractory model of CRPC, that is, MR49F LNCaP cells harboring F876L and T877A point mutations of AR. As mentioned above, the F876L mutation confers enzalutamide (4) resistance (Enz-R) to MR49F cells; however, MR49F cells remain dependent on the AR for growth. MR49F cells were tested in the presence of a titrated dose of 26a or 4 as shown in Figure 5. Compound 26a demonstrated dose-responsive antiproliferation that showed potent, but partial, efficacy (~50–60% reduction from vehicle) at doses as low as 0.1 μM. The Enz-R of the MR49F model was demonstrated as the antiproliferation of 4 was ~100-fold less potent. For example 10 μM of 4 produced effects comparable to 0.1 μM of 26a, which was weak at ~20% efficacy and not significantly different from vehicle. Assuming that 26a can reach the tumors, this potent antiproliferation suggests that 26a may perform well in in vivo models of Enz-R CRPC.
Figure 5.
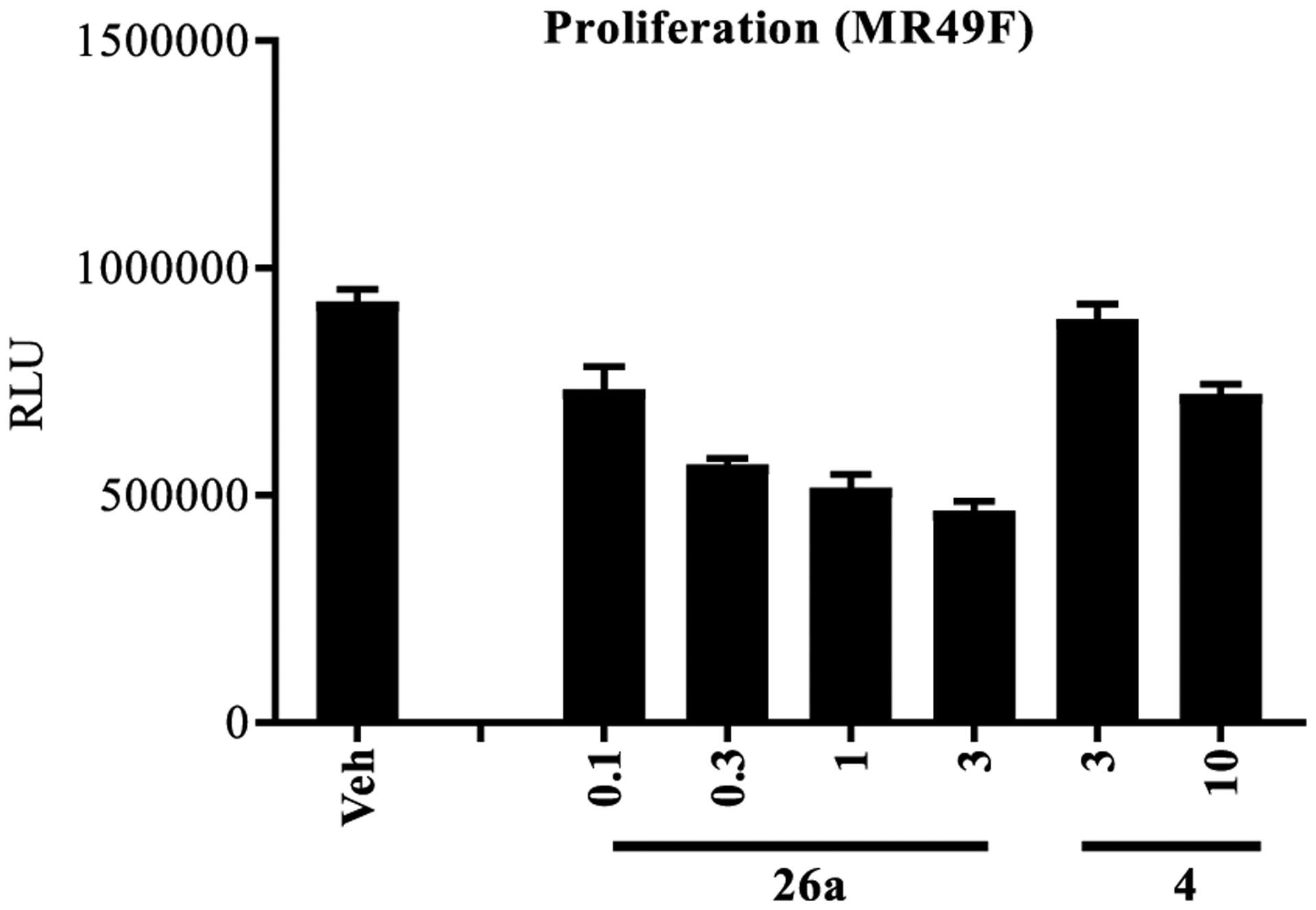
Enzalutamide-resistant LNCaP (MR49F) cellular antiproliferation. Enzalutamide (4)-resistant (Enz-R) LNCaP (MR49F) cells were plated in 1% charcoal-stripped, serum-containing medium and treated with 0.1 nM R1881 and a titration of antagonist as indicated in the figure. Cells were retreated 3 d after the first treatment and the number of viable cells was measured by the CellTiter-Glo assay (Promega, Madison, WI). N = 3.
2.3.4. AR FL (F876L) and AR SV (AR-V7) Degradation in Models of CRPC.
FL AR degradation studies in MR49F cells were performed in order to confirm that the robust in vitro AR antagonism profiles of 16i (0.045 μM, 90, and 100% in wtAR inhibition, AR FL and AR SV degradation assays) and 26a (0.084 μM, 70, 80%) predicted SARD activity in this model of highly refractory CRPC. Compound 26a possessed the ability to suppress AR-dependent gene expression in LNCaP cells and suppress proliferation in MR49F cells as described above and was able to also degrade the FL AR (Figure 6 upper panel) in the Enz-R CRPC setting. Western blotting is not a quantitative method and it can be difficult to compare the AR levels between compounds based on relative band densities. Accordingly, GAPDH was also included as a protein loading control in each lane. The levels of AR are normalized to the level of GADPH in that lane. The western blots were quantified densitometrically and the AR/GADPH values are represented as fold change (under blots in Figure 6) or percent change from vehicle-treated cells (Tables 2–5).
Figure 6.
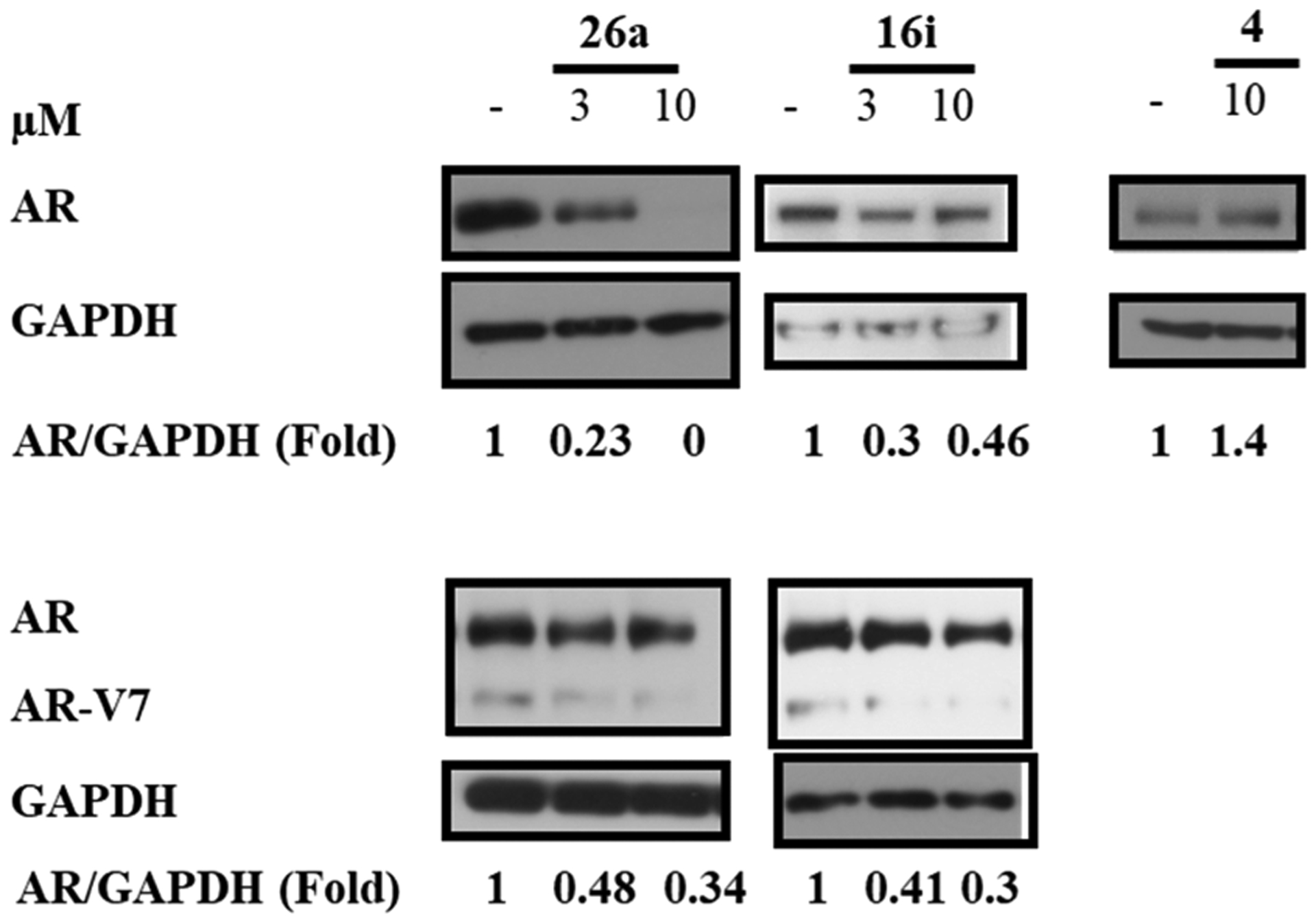
SARDs degrade enzalutamide resistance conferring escape mutant AR. Enzalutamide (4)-resistant (Enz-R) LNCaP cells (MR49F) (top panel) or 22RV1 cells (bottom panel) were maintained in charcoal-stripped, serum containing medium for 2 d and treated with 0.1 nM R1881 (agonist) and a titration of the SARD or 4 as indicated in the figure. Twenty-four hours after treatment, the cells were harvested and protein-extracted, and the proteins were blotted with AR-N20 antibody. Blots were stripped and reprobed with a GAPDH antibody. The ratio of AR to GAPDH or each lane is given under each blot.
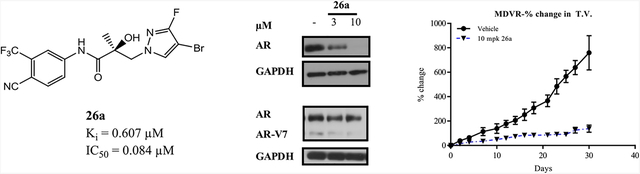
High-efficacy SARD activity was observed with 26a at 3 μM and complete degradation at 10 μM (Figure 6, top panel), indicating that this mutant AR FL that confers Enz-R in MR49F LNCaP cells is susceptible to destruction by 26a. 16i also demonstrated SARD activity but not full efficacy, whereas 4 produced no AR degradation in MR49F cells.
The lower panel demonstrates that the SARD activity is not just present for T877A (LNCaP; Tables 2–5) and F876L/T877A (MR49F LNCaP cells; Figure 6 upper panel) AR FL with point mutations in the LBD but also can degrade AR SVs such as the AR-V7 that lack the expression of the LBD (22RV1 cells; lower panel of Figure 6). As reported in Tables 4 and 2 (see AR SV degradation column), 26a and 16i were able to reduce AR-V7 levels in 22RV1 cells at 10 μM. Figure 6 confirmed the AR-V7 SARD activity at 3 and 10 μM, but % degradation was not complete for either SARD in this particular experiment. Lower % degradation for AR SV than AR FL is consistent with earlier reports and Tables 2–5, which revealed that AR SV degradation can be complete but generally at higher treatment concentrations (screened at 10 μM) than for AR FL (screening at 1 μM).51 PCs expressing AR SVs possess no binding site for traditional (or canonical) antiandrogens to bind AR, are associated with poor prognosis, and are believed to be pan-resistant to approved therapies including 1–7.53 Accordingly, the pyrazole SARDs and pan-antagonists such as 10,1,2 21a, and 26a (discussed infra) that possess PK properties compatible with oral administration at low dose afford a very broad scope of AR antagonistic abilities in at least:
T877A (LNCaP AR FL degradation in Tables 2–5 and inhibition of AR-dependent gene expression in Figure 4),
F876L (inhibition in COS cells in Figure 2),
F876L/T877A comutant (proliferation in MR49F cells in Figure 5),
AR-V7 (degradation of AR SV in 22RV1 cells in Tables 2–5 and Figure 6), and
AR amplification/overexpression (see VCaP data reported infra).
The broad-scope AR antagonism across various resistance-conferring AR mutants helps to ensure that treated tumors that are evolving to contain these and/or other AR mutations will remain sensitive to our SARDs and pan-antagonists. Further, our SARDs and pan-antagonists performed well in models of AR overexpression and/or AR gene recombination such as present in VCaP cells, suggesting that these PCs will not be able to resist this treatment either. In view of the fact that SARD activity may not be necessary for these activities, we have added discussion of our agents as AR pan-antagonists in this publication. Compound 26a was tested as a lead SARD and pan-antagonist in vitro (supra) and subjected to a series of in vivo tests to describe its PK (Section 2.4) and PD (Section 2.5) profiles in healthy rats and models of antiandrogen-resistant PC in rats.
2.4. In Vivo Rat PK.
The overall goal of this initial pyrazole SAR study was to improve upon 10 as a pyrazole lead. Assuming comparable oral bioavailability, Sections 2.1 and 2.3 demonstrate improved potency compared to 10 and a broad spectrum of in vitro activities for 26a, suggesting the possibility of improved in vivo AR-dependent tumor growth inhibition (TGI) over 10 including antiandrogen-resistant and/or CRPC tumors.2 Rat PK studies were conducted to confirm that pyrazole 26a, like 10,2 possessed improved PK properties compared to previous generations of our SARDs such as tertiary amine 8 and indole 9. Further, these studies allow more informed comparisons between the pyrazoles 26a and 10 in view of their PK criteria. Optimized PK properties within the pyrazole template provide the best chance to reveal optimized in vivo PD profiles for our molecules with their unique AR mechanism of action in in vivo models of advanced PCs.2
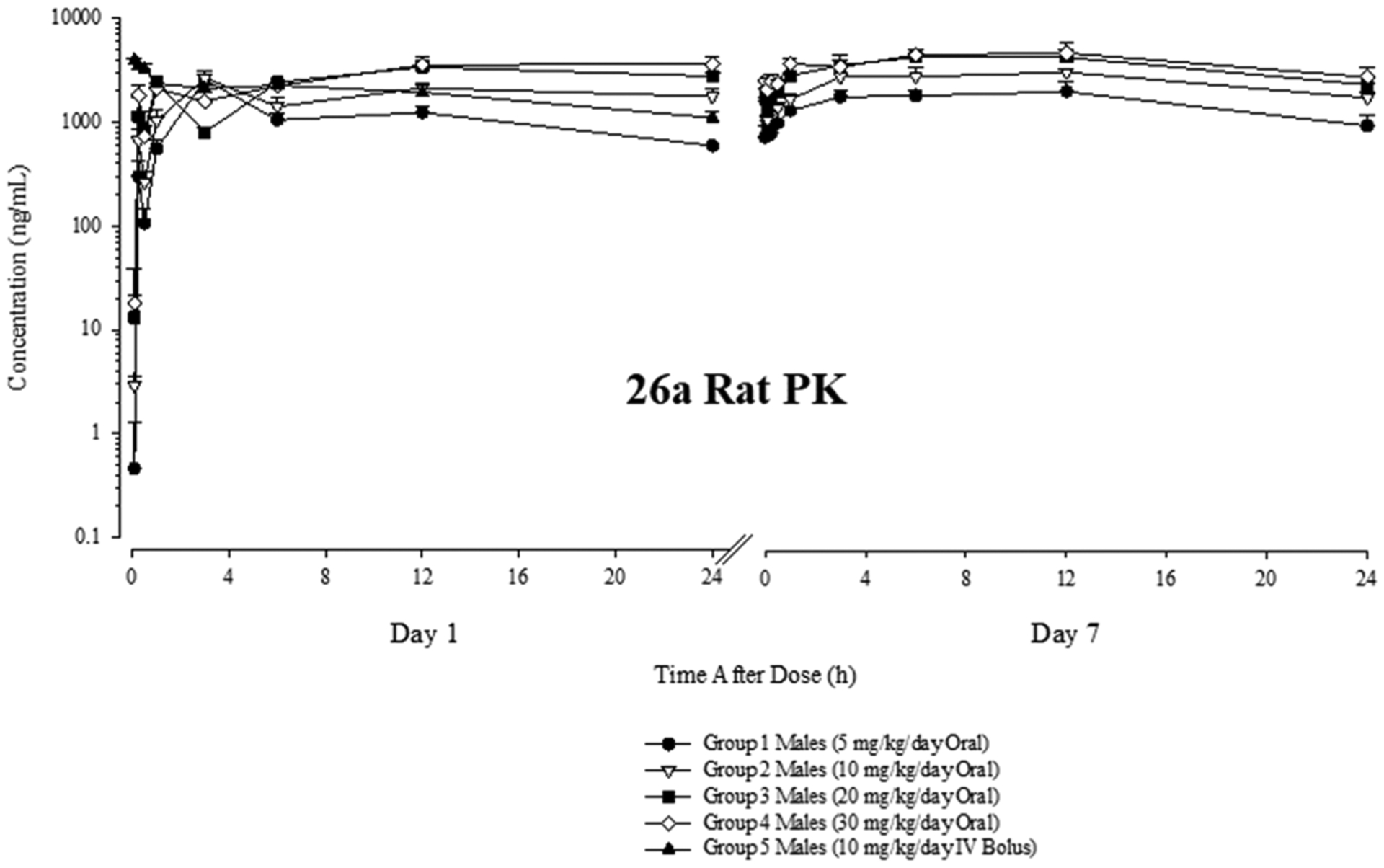
Male Sprague Dawley rats were given a single oral (po) daily dose on seven consecutive days or a single intravenous (iv) dose on day 1, and blood was sampled periodically at 0.083, 0.25, 0.5, 1, 3, 6, 12, and 24 h post dose. The doses of 5, 10, 20, and 30 mg/kg po (groups 1–4) and 10 mg/kg iv (group 5) were selected based on in vivo efficacies seen in a series of pilot experiments, which were similar to the Hershberger study discussed in detail in this section. Concentration-time curves were plotted from this data for 26a (Figure 7), and the PK parameters were calculated for 26a from this data (Table 8).
Figure 7.
Concentration–time plots in rats for 26a. Twelve week old male Sprague Dawley rats were dosed in five groups of five animals each (N = 5) at the doses shown. Blood samples were drawn at the shown time points and analyte concentrations were determined by MS/MS. The concentration- time plots for all dose groups are shown for day 1 (left) and day 7 (right).
Table 8.
Summary of 26a PK Parameters in Rata
| dose group | dose Level (mg/kg/day) | C0 (ng/mL) | Cmax (ng/mL) | DN Cmax (ng/mL)/(mg/kg/day) | Tmax (h) | AUC0–24 (h*ng/mL) | DN AUC0–24 (h*ng/mL)/(mg/kg/day) |
|---|---|---|---|---|---|---|---|
| l | 5 | NA | 2570 | 515 | 3.00 | 26,800 | 5350 |
| 2 | 10 | NA | 2680 | 268 | 3.00 | 44,600 | 4460 |
| 3 | 20 | NA | 3420 | 171 | 12.0 | 64,100 | 3200 |
| 4 | 30 | NA | 3650 | 122 | 24.0 | 71,500 | 2380 |
| 5 | 10 (iv) | 4200 | 3940 | 394 | 0.083 | 45,500 | 4550 |
C0—back-extrapolated concentration at time 0 (group 5 only). Cmax—maximum observed concentration. DN Cmax—dose normalized Cmax, calculated as Cmax/dose level. Tmax—time of maximum observed concentration. AUC0–24—area under the concentration–time curve from time 0 to 24 h, estimated by linear trapezoidal rule. DN AUC0–24—dose normalized AUC0–24, calculated as AUC0–24/dose level.
Like 10, 26a demonstrated a robust PK profile in rats characterized by micromolar blood levels and a long terminal elimination half-life (t1/2) (Table 8) consistent with daily oral dosing. An advantage of 26a over 10 is its relatively long t1/2, which is in excess of 24 versus 2.6 h (calculated based on the 7 day rat PK data reported in Ponnusamy’s paper for 10).2 The exact t1/2 value of 26a could not be calculated as the t1/2 was longer than the 24 h dosing interval (Figure 7). 26a had decreasing oral bioavailability at higher doses as revealed by the decreasing dose-normalized area under the concentration–time curve from 0 to 24 h (DN AUC0–24) values and increasing time of maximum concentration (Tmax) values for groups 1–4 with increasing 26a dose (Table 8). The calculated oral bioavailabilities for 5, 10, 20, and 30 mg/kg doses of 26a were 1.18, 0.982, 0.705, and 0.524. Nonetheless, the longer t1/2 of 26a relative to 10 at least partially offset the decreasing oral bioavailability at high doses and 26a attained marginally increased absolute exposures compared to 10. For example, the AUC0–24 values for 30 mg/kg po 26a and 10 were 71,500 and 62,000 h*ng/mL, respectively. The latter value, again, is calculated from the 7 day rat PK data presented in Ponnusamy’s paper.2
Correspondingly, 26a exhibits a PK profile sufficiently robust to maintain high blood levels in vivo via oral daily dosing in rats. Also shown is preliminary rat PK data for 30 mg po 21a (Figure S1). The concentration versus time plot demonstrated reduced in vivo stability, with the vast majority of 21a eliminated by 24 h, which is in sharp contrast to 30 mg po 26a where blood levels at 24 h were barely reduced from their Cmax (Figure 7). Nonetheless, 21a at 30 mg po demonstrated sufficiently low CL to allow observation of its PD character in rats. [Unfortunately, despite an interesting and potent in vitro panel of activities, 16i demonstrated lethality at 5 mg/kg in vivo and was not evaluated further]. Correspondingly, 21a and 26a were studied in rat Hershberger assays, but 26a was chosen as the lead for the xenograft studies reported infra.
The micromolar Cmax blood levels and long t1/2 observed for 26a suggested PK properties in rats consistent with revealing any high efficacy AR antagonism of 26a in vivo that was engendered by the data in Sections 2.1 and 2.3. Sections 2.1 and 2.3 demonstrated that 26a inhibited a broad spectrum of antiandrogen activities in vitro with increased potency compared to 10, including in the models of antiandrogen-resistant CRPC. Oral daily dosing in rats with 26a should be able to maintain blood levels above the IC50 value of AR antagonism (Table 4) and inhibitory effects on AR-dependent transcription (Figure 4) and proliferation (Figure 5), as would be necessary to suppress the AR axis in AR-dependent xenografts. Further, the low micromolar drug levels seen for 21a (Figure S1), 26a (Figure 7 and Table 8), and 102 were in excess of DC50 values for 21a (880 nM), 26a (860 nM), and 10 (740 nM) (see Tables 2–4), suggesting that SARD activity may contribute to in vivo AR antagonism, as seen previously with 10 where intratumoral degradation was observed.2 Correspondingly, we expected to observe TGI in models of CRPC and/or antiandrogen resistance, similar to 10.2 In summary, the more potent in vitro profile of 26a (Sections 2.1 and 2.3) relative to 10 and the more stable μM range plasma concentrations of 26a with daily dosing (Figure 7) relative to 10 or 21a suggest that a robust in vivo PD profile will be observed with 26a in models of CRPC and antiandrogen resistance.
2.5. In Vivo AR Antagonist Activity.
2.5.1. Hershberger Assays.
In order to prove that these compounds with robust PK properties have clinically meaningful SARD and pan-antagonist activity in vivo, we performed Hershberger assays in intact rats for 21a and 26a which, as shown above, demonstrated oral bioavailability in rats (Figures S1 and 7). The Hershberger assay has been used to demonstrate anabolic selectivity of androgens for decades.54 Rat ventral prostate (VP), seminal vesicle (SV), and levator ani (LA) muscle are AR-dependent tissues whose size (reflected by their weight) responds rapidly to castration. Upon castration, these organs atrophy within 3–7 days to organ weights that are approximately 85% (VP), 90% (SV), and 50% (LA) reduced compared to their intact organ weights. Traditionally, agonists are dosed to prevent (whereby agonist is given upon castration) or restore (agonist is given after tissue atrophies) anabolic tissue weights [LA or other skeletal muscles and bone (the latter takes months not days to atrophy and restore)] to intact levels or greater, without increasing androgenic tissue (SV or VP) weights back to intact levels. As employed herein, that is, antagonist mode, young intact animals were used wherein the endogenous androgen milieu provided AR-mediated support for the VP, SV, and LA weights, as reflected by the 0% change for the vehicle columns in Figure 8.
Figure 8.
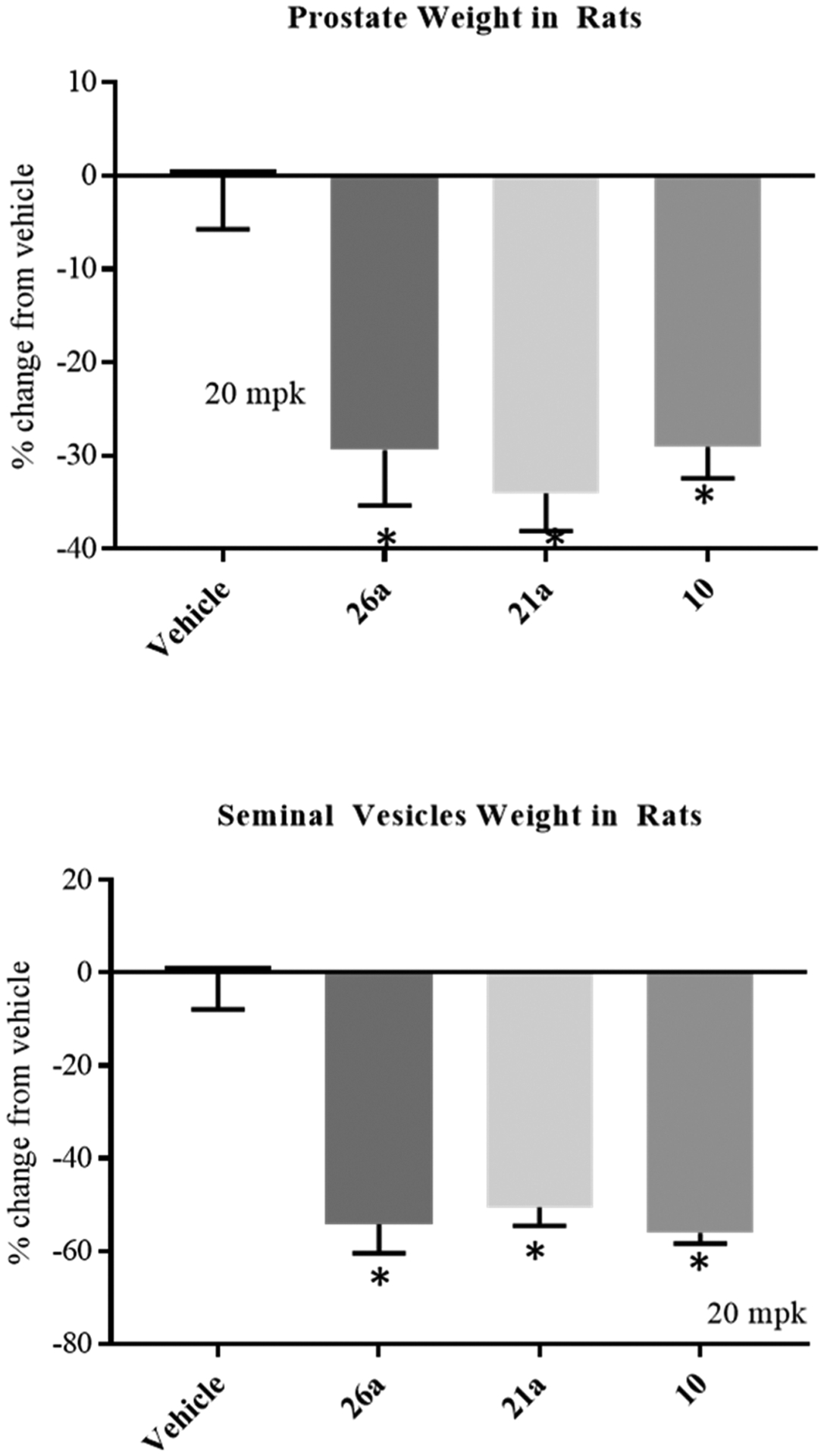
SARDs and pan-antagonists inhibit androgen-dependent organs in rats. The top and bottom panels show the reduction of VP and SV weights following the treatment of intact rats with 20 mg/kg (mpk) po daily of antagonist or vehicle for 14 days (n = 5/group). Rats were sacrificed at the end of the treatment period and weights of prostate and SVs were measured and normalized to body weight.
Exogenous antagonists 21a and 26a, with potent in vitro inhibition [0.062 and 0.084 μM (Tables 3 and 4)], were dosed to observe their AR antagonism in vivo. Previously, we demonstrated that 10 (4-F pyrazole; 0.199 μM inhibition in vitro) was able to reduce the VP weight by ~70–80% at 60 mg/kg po [Figure S4C, middle panel in Ponnusamy’s paper] versus ~40% for 30 mg/kg po of 4, both in rats.2 Improved potency of in vivo AR antagonism was seen for derivatives of 10 with (1) the addition of the 3′-pyridino N to 10 as in 21a or (2) an additional halogen on the pyrazole such as 3-F or 4-Br in 26a. At 20 mg/kg of 21a and 26a, that is, one-third of the dose of 10 mentioned above, VP weights were reduced by approximately 35 and 30% (Figure 8, top panel), demonstrating that the in vivo PD properties intrinsic to 21a and 26a are observable at lower doses relative to 10. In SV at this dose, approximately 45–50% reductions were provided by 21a and 26a. Addition of the 3′-pyridino (21a) or dihalogenation of pyrazole (26a) of 10 seemed favorable for in vivo antagonism, suggesting that xenograft potencies should also be improved. Consistent with Section 2.4, these results confirm that orally administered 26a was absorbed and distributed to the site of action in AR target organs and suggest that these compounds should also distribute to tumors in xenograft models and exert antitumor effects in sensitive models.
2.5.2. Enz-R (MDVR) VCaP Xenografts in Rats.
The VCaP cell line is derived from a vertebral bone metastasis from a patient with hormone refractory PC (https://atcc.org/Products/ all/CRL-2876.aspx; accessed January 20, 2020).55 VCaP is commonly used as a model of CRPC, which expresses both AR SV (AR-V7) and overexpression of AR FL (TMPRSS2-ERG gene fusion). VCaP, the parental cell line for the MDVR VCaP used in the experiments below, is a model of highly advanced PC where multiple mechanisms of hormone resistance have emerged in response to androgen ablation in a single AR axis-driven cell line. The parental VCaP cells are nonetheless sensitive to enzalutamide (4); however, MDVR VCaP cells possess acquired Enz-R in addition to the resistance mechanisms in the parental cell line. Previously, we demonstrated that VCaP are partially sensitive to 4, whereas MDVR VCaP are not sensitive.2 Furthermore, 60 mg/kg po of 10 regressed MDVR VCaP xenografts and degraded AR and AR-V7 intratumorally,2 whereas 10 mg/kg po of 10 only produced about 50% TGI [Figure 6E left panel of ref. 2].
Following the demonstration with 26a of an in vitro screening panel that was superior to 10, in vitro activity in MR49F (an Enz-R LNCaP cell line) and in vivo antagonism in Hershberger assays, there was confidence in our ability to demonstrate activity in Enz-R MDVR VCaP xenografts. In order to allow direct comparison of 26a to 10, the MDVR VCaP xenografts were performed as previously published for 10.2 For 10, castration was not necessary to demonstrate the efficacy in this model (unlike all previous AR antagonists to our knowledge); however, 10 was not stable in mice; therefore, intact SRG rats were used as the host for MDVR VCaP xenograft experiments. Treatment of intact SRG rats (studies performed at HERA Biolabs, Lexington KY) with 10 mg/kg po daily of 26a produced comparable efficacy of up to 83% TGI (Figure 9, top panel) versus 10 required 20–30 mg/kg to achieve similar results [see Figure 6E, left panel of ref. 2], whereas 4 failed to durably achieve any effect (not shown; previously published) [see Figure 6, panel C].2 Tumor weights measured at the end of the study also demonstrated a significant inhibition (lower panel of Figure 9).
Figure 9.
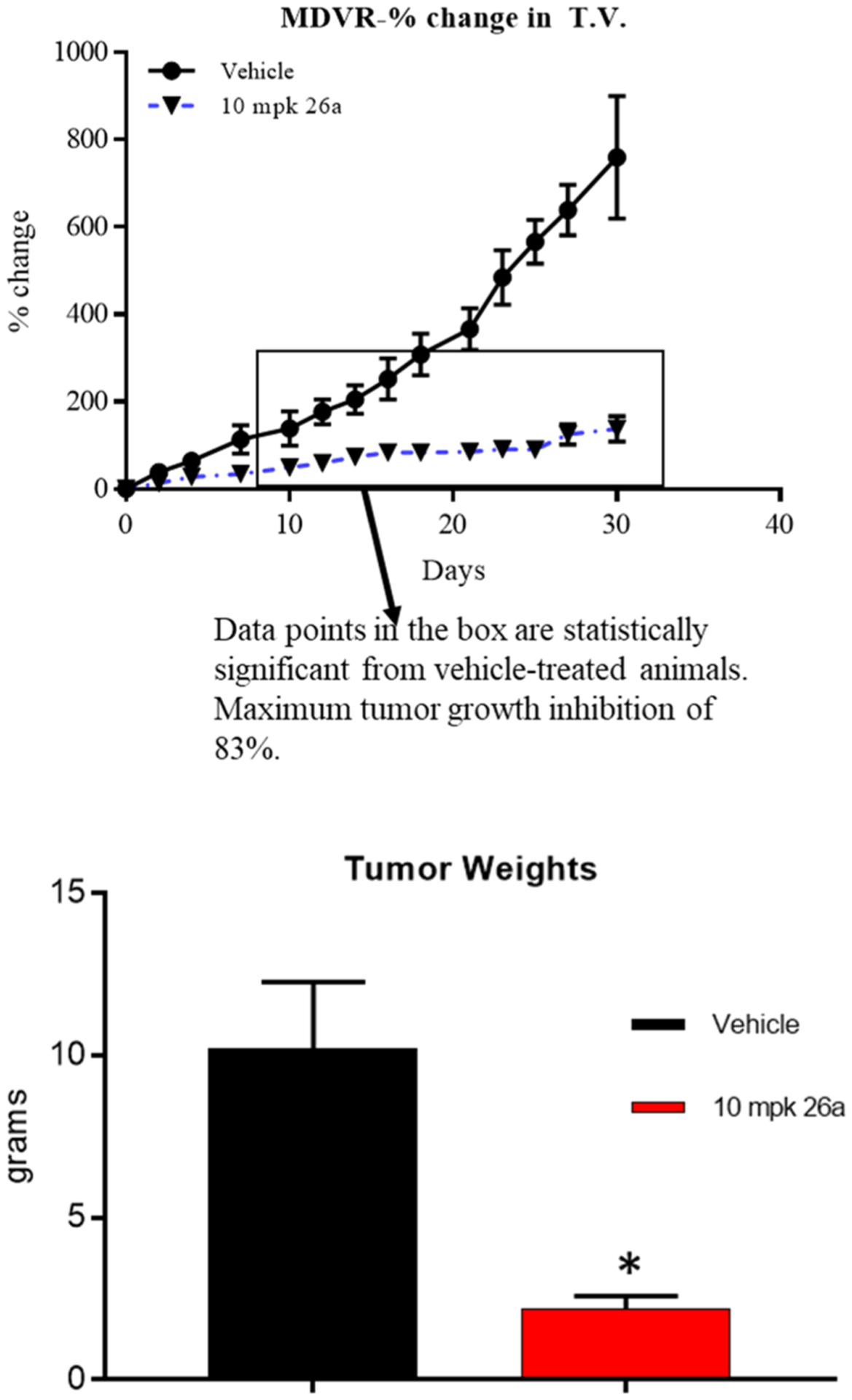
SARDs and pan-antagonists inhibit growth of Enz-R PC. Enz-R MDVR cells (10 × 106 cells/rat) were implanted subcutaneously in male SRG (Sprague Dawley-Rag2: IL2rg KO) rats. When the tumors reached 1000–3000 mm3, the animals were randomized and treated (intact). Once the tumors attain 2000–3000 mm3, the animals were treated orally with vehicle (DMSO/PEG-300 15:85) or 10 mg/kg/day of 26a. Tumor volume (T.V.) was measured twice weekly and represented as a percent change (upper panel) or weight at sacrifice (lower panel).
Consistent with the observed high-potency antitumor activity, 26a was observed in this study at an average concentration within the tumors of 881 nM, which is 10-fold higher than its IC50 value in wtAR or F876L (both 84 nM). Further, intratumoral levels were only slightly reduced from the 1319 nM average concentration of 26a in the blood of these animals (Table 9). This supports efficient distribution of 26a into tumors, in addition to VP and SV, and supports its use in advanced PC.
Table 9.
Serum and Tumor Drug Concentration of 26a
| 26a (3-F, 4-Br) | ||
|---|---|---|
| serum (nM)a | tumor (nM)a | |
| animal 1 | 1611.663 | 962.6859 |
| animal 2 | 1360.036 | 913.4912 |
| animal 3 | 1143.556 | 666.0278 |
| animal 4 | 1160.002 | 983.6887 |
| average | 1318.814 | 881.4484 |
| S. E. | 109.3146 | 73.29447 |
Twenty to 24 h after the last dose (day 28), the animals were sacrificed, and blood and tumors were collected for further analysis. The serum was separated from blood, and drug concentration in serum and tumor was measured using the LC–MS/MS method (n = 4).
The in vitro DC50 values (concentration of the 50th percentile of degradation efficacy) in LNCaP cells for 21a (880 nM) and 26a (860 nM) reported in Tables 3 and 4 were comparable to the intratumoral levels attained in the MDVR VCaP xenografts. Despite the different cell types between in vitro and in vivo studies, the data suggest the possibility of suboptimal exposures for full-efficacy SARD activity in the tumors of this experiment. Nonetheless, this presumed half-efficacy intratumoral SARD activity may contribute to the TGI. In overview, it may be possible to improve the antitumor activity with increased intratumoral levels, that is, at increased dose of 26a, or with improved degradation potency analogues.
The results clearly indicate that 26a was stable in rats (like 10) and was very potent and highly efficacious in this AR overexpressing and AR-V7 expressing model of Enz-R CRPC. The results further suggest that the improved PK and PD of 26a translated into more potent in vivo efficacies compared to 10, providing a dose-sparing SARD and pan-antagonist if, as of yet to be unobserved, toxicities become dose limiting. Further, the improved PK may translate into improved penetration throughout the cancer patient allowing better suppression of distant metastatic growth. All the above increase our chances of observing clinically significant reduction in disease burden when trialed in a human population (as supported by HLM studies in Table 7) expressing a broad spectrum of CRPC-resistant mechanisms. This population would still be sensitive even if expressing AR SVs (like AR-V7), AR gene amplications to overexpress AR (like TMPRSS2-ERG), or LBD-directed antiandrogen resistance (like Enz-R and/or darolutamide resistance observed in MR49F or MDV VCaP cells) or combinations thereof as in MDVR VCaP.
These results confirm that for 26a, our in vitro screening paradigm was successful in selecting an improved lead compound from our library of SARDs and pan-antagonists that was highly efficacious in an in vivo model of CRPC. Though full-efficacy in vitro SARD activity such as published for 10 is unique and should be beneficial in AR-dependent disease, it may not be necessary for efficacy in the clinic. This is supported by the more potent and comparable efficacy antitumor activity in vivo for 26a, which was not a full-efficacy SARD in vitro (70%/80%; Table 4), unlike 10 (100%/100%; Table 2). Although exact and incontrovertible mechanistic explanations of the high efficacy of 26a are not possible, its potent in vivo efficacy is also incontrovertible. The pyrazole template represents the optimal B-ring template presented to date, and 26a is an optimized lead from this template. 10 or 26a is believed to hold great potential for overcoming multiple mechanisms of CPRC present in the clinic.
3. CONCLUSIONS
Compounds 16c, 16g, 16i, and 16j from Series I; 21a and 21c from Series II; and 26a, 26c, 26e, and 2b6f from Series III exhibited potent inhibitory activity in vitro, while compounds 16b, 16c, 16bg, and 16bi from Series I; only 21a from Series II; and 26a and 26g from Series III possessed potent SARD activity in vitro (Tables 2–4). Compared to previous SARD templates such as 1.15 min for the tertiary amine 8 and 12.11 min for lead indole 9, these pyrazol-1-yl-propanamides, such as 16g, 16i, 21a, and 26a, significantly improved their stability in vitro in MLM (Table 6), and 21a and 26a were stable in RLM and HLM (Table 7). Compounds 16i, 21a, and 26a robustly inhibited the F876L-mutant AR with IC50 values of 0.043, 0.063, and 0.084 μM (Figure 2), as well as inhibited wt PR activity with IC50 values of 3.540, 0.235, and 1.101 μM (Figure 3). Compound 26a effectively inhibited the expression of FKBP5 in LNCaP cells at concentrations as low as 0.1 μM, indicating that the antiandrogenic effects include inhibition of endogenous gene expression (Figure 4), as well as demonstrated dose-responsive antiproliferation at doses as low as 0.1 μM (Figure 5). Compound 26a also produced superior in vivo rat PK and PD properties compared to 10 and 21a, with relatively long t1/2 values that were well in excess of 24 h (Figure 7) and AR antagonism in rat Hershberger assay, with approximately 30% (VP), and 50% (SV) reduced compared to their intact organ weights (Figure 8), which was comparable to 21a.
Enz-R (MDVR) VCaP xenograft experiments with 10 mg/kg po daily of 26a in an intact rat model demonstrated high drug levels intratumorally (881 nM) and producing an efficacy of 83% TGI (Figure 9, top panel), which was comparable to 10 at 20–30 mg/kg po. The results clearly indicate that 26a was very potent and highly efficacious in this AR overexpressing and AR-V7 expressing model of Enz-R CRPC and collectively satisfied all the criteria for a next-generation AR antagonist for Enz-R PC. Compound 26a is believed to hold great potential for overcoming multiple mechanisms of CPRC present in the clinic and is one of the several compounds being evaluated for their potential to advance to IND enabling studies.
4. EXPERIMENTAL SECTION
4.1. Chemistry.
4.1.1. General Procedures, Materials, and Information.
All solvents and chemicals were used as purchased without further purification. The progress of all reactions was monitored by thin-layer chromatography (TLC) analysis on silica gel 60 F254 plates (Merck). Column chromatography was performed with a silica gel column (Merck Kieselgel 60, 70–230 mesh, Merck).
General methods:
All nonaqueous reactions were performed in oven-dried glassware under an inert atmosphere of dry nitrogen. All the reagents and solvents were purchased from Aldrich (St. Louis, MO), Alfa-Aesar (Ward Hill, MA), Combi-Blocks (San Diego, CA), and Ark Pharm (Libertyville, IL) and used without further purification. Analytical TLC was performed on Silica Gel GHLF 10 × 20 cm Analtech TLC Uniplates (Analtech, Newark, DE) and was visualized by fluorescence quenching under UV light. The Biotage SP1 Flash Chromatography Purification System (Charlotte, NC) (Biotage SNAP Cartridge, silica, 50 & 100 g) was used to purify the compounds. 1H NMR and 13C NMR spectra were recorded on a Bruker Ascend 400 (400 MHz) (Billerica, MA) spectrometer. Chemical shifts for 1H NMR were reported in parts per million (ppm) downfield from tetramethylsilane (δ) as the internal standard in deuterated solvent and coupling constants (J) are in hertz (Hz). The following abbreviations are used for spin multiplicity: s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, dd = doublet of doublets, dt = doublet of triplets, qd = quartet of doublets, dquin = doublet of quintets, m = multiplet, and br s = broad singlet. Low-resolution mass spectra were acquired using a Brucker ESQUIRE electrospray/ion trap instrument in the positive and negative modes. High-resolution mass spectrometry (HRMS) data were acquired on a Waters Xevo G2-S QTOF (Milford, MA) system equipped with an Acquity I-class UPLC system. The purity of the final compounds was analyzed by an Agilent 1100 HPLC system (Santa Clara, CA). HPLC conditions: 45% acetonitrile at flow rate of 1.0 mL/min using a LUNA 5 μ C18 100A column (250 × 4.60 mm) purchased from Phenomenex (Torrance, CA) at ambient temperature. UV detection was set at 340 or 245 nm. Purities of the compounds were established by careful integration of areas for all peaks detected and determined as ≥95% for all compounds tested for biological study.
4.1.2. Synthesis of 13, 18, 23, and 28 Using 13 as an Example.
(R)-3-Bromo-2-hydroxy-2-methylpropanoic acid 11 (5.00 g, 27 mmol) was dissolved in tetrahydrofuran (THF) (27 mL, 5.4 vol) in an EasyMax 100 mL reactor. Agitation was set to 400 rpm and the solution was cooled to 2.5 °C. Thionyl chloride (2.39 mL, 1.20 equiv, 0.48 vol) was slowly added to the reaction mixture over 30 min while maintaining the reaction temperature below 12 °C. The reaction mixture was stirred for 1.5 h. The reaction was cooled to −5 °C. Triethylamine (5.0 mL, 1.30 equiv, 1 vol) was slowly added to the reaction mixture, keeping the temperature below 12 °C. 4-Amino-2-(trifluoromethyl)benzonitrile 12 (4.85 g, 0.95 equiv, 0.97 wt) and THF (3.37 mL, 0.67 vol) were then charged to the batch. The batch was then heated to 50 ± 5 °C and agitated for 2 h. The batch was then cooled to 20 ± 5 °C, followed by the addition of water (14.7 mL, 2.9 vol) and toluene (20.2 mL, 4.0 vol). After brief agitation, the layers were separated. The organic layer was then washed with water (14.7 mL, 2.9 vol). The batch was then concentrated to 5 ± 0.5 vol (4 ± 0.5 wt) while maintaining the batch temperature below 50 °C, followed by the addition of toluene (30 mL, 6 vol). The batch was then distilled to 5 ± 0.5 vol (4 ± 0.5 wt) and the batch temperature reduced to 2.5 ± 2.5 °C. The batch was then filtered and the filter cake was washed with toluene twice (8.5 mL each, 1.7 vol each). The batch was then dried under 25–30 in. vacuum to provide (R)-3-bromo-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide 13. Yield = 85%. 1H NMR (400 MHz, DMSO-d6): δ 10.55 (s, 1H, NH), 8.55 (s, 1H, ArH), 8.32 (d, J = 8.8 Hz, 1H, ArH), 8.12 (d, J = 8.8 Hz, 1H, ArH), 6.43 (s, 1H, OH), 3.84 (d, J = 10.4 Hz, 1H, CH), 3.59 (d, J = 10.4 Hz, 1H, CH), 1.48 (s, 3H, CH3). HRMS [C12H11BrF3N2O2+]: calcd, 350.9956; found, 350.9961.
4.1.3. Synthesis of 14, 19, and 24 Using 14 as an Example.
To a solution of (R)-3-bromo-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide 13 (5.00 g, 0.018504 mol) in 25 mL of 2-butanone was added potassium carbonate (3.836 g, 0.027756 mol). The resulting reaction mixture was heated at reflux for 2 h under argon atmosphere. After ending the reaction by establishing TLC, the reaction was cooled to room temperature (rt), filtered through a pad of Celite, and rinsed the pad of Celite with 15 mL of 2-butanone. The filtrate was concentrated under vacuum and dried under 25–30 in. vacuum to provide (S)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-methyloxirane-2-carboxamide 14. Yield = 99%. 1H NMR (400 MHz, DMSO-d6): δ 10.22 (s, 1H, NH), 8.41 (s, 1H, ArH), 8.22 (d, J = 8.8 Hz, 1H, ArH), 8.09 (d, J = 8.8 Hz, 1H, ArH), 3.09 (d, J = 4.8 Hz, 1H, CH), 3.02 (d, J = 4.8 Hz, 1H, CH), 1.55 (s, 3H, CH3). HRMS [C12H10F3N2O2+]: calcd, 271.0694; found, 271.0696.
4.1.4. General Procedure A for the Synthesis of 16(a–y), 21(a–j), 26(a–h), and 29(a–p) Using 10 (UT-34) as an Example.
To a solution of 4-fluoro-pyrazole (0.10 g, 0.00116 mol), or general pyrazole 15, in anhydrous THF (10 mL), which was cooled in an ice–water bath under an argon atmosphere, was added sodium hydride (60% dispersion in oil, 0.12 g, 0.00291 mol). After addition, the resulting mixture was stirred for 3 h. (R)-3-Bromo-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide 13 (0.41 g, 0.00116 mol) or (S)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-methyloxirane-2-carboxamide 14 (0.313 g, 0.00116 mol) was added to the above solution, and the resulting reaction mixture was allowed to stir overnight at rt under argon atmosphere. The reaction was quenched with water and extracted with ethyl acetate. The organic layer was washed with brine, dried with MgSO4, filtered, and concentrated under vacuum. The product was purified by a silica gel column using ethyl acetate and hexanes (1:1) as eluents to afford 0.13 g of 10 as a white solid.
4.1.4.1. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methyl-3-(1H-pyrazol-1-yl)propanamide (16a).
16a was prepared following general procedure A. The crude product was purified by a silica gel column using ethyl acetate and hexanes (2:1) as eluents to afford 0.52 g of the titled compound as a white solid. Yield = 52%. 1H NMR (400 MHz, DMSO-d6): δ 10.39 (s, 1H, NH), 8.48 (d, J = 2.0 Hz, 1H, ArH), 8.22 (dd, J = 8.2 Hz, J = 2.0 Hz, 1H, ArH), 8.08 (d, J = 8.2 Hz, 1H, ArH), 7.66–7.65 (m, 1H, pyrazole-H), 7.39–7.38 (m, 1H, pyrazole-H), 6.28 (s, 1H, OH), 6.25–6.23 (m, 1H, pyrazole-H), 4.50 (d, J = 13.6 Hz, 1H, CH), 4.29 (d, J = 13.6 Hz, 1H, CH), 1.35 (s, 3H, CH3). HRMS [C15H14F3N4O2+]: calcd, 339.1099; found, 339.1105.
4.1.4.2. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-3-(4-fluoro-1H-pyrazol-1-yl)-2-hydroxy-2-methylpropanamide (10 (UT-34)).
10 was prepared following general procedure A as per Scheme 1. The crude product was purified by a silica gel column using ethyl acetate and hexanes (1:1) as eluents to afford 0.13 g of the titled compound as a white solid. Yield = 32%. 1H NMR (400 MHz, DMSO-d6): δ 10.39 (s, 1H, NH), 8.47 (d, J = 1.6 Hz, 1H, ArH), 8.24 (dd, J = 8.4 Hz, J = 2.0 Hz, 1H, ArH), 8.10 (d, J = 8.4 Hz, 1H, ArH), 7.73 (d, J = 4.4 Hz, 1H, pyrazole-H), 7.41 (d, J = 4.4 Hz, 1H, pyrazole-H), 6.31 (s, 1H, OH), 4.38 (d, J = 14.0 Hz, 1H, CH), 4.21 (d, J = 14.0 Hz, 1H, CH), 1.34 (s, 3H, CH3). HRMS [C15H13F4N4O2+]: calcd, 357.0975; found, 357.0966.
4.1.4.3. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-3-(3-fluoro-1H-pyrazol-1-yl)-2-hydroxy-2-methylpropanamide (16b).
16b was prepared following general procedure A as per Scheme 1. The crude product was purified by a silica gel column using ethyl acetate and hexanes (2:1) as eluents to afford 0.36 g of the titled compound as white needles. Yield = 44%. 1H NMR (400 MHz, DMSO-d6): δ 10.39 (s, 1H, NH), 8.47 (d, J = 2.0 Hz, 1H, ArH), 8.24 (dd, J = 8.8 Hz, J = 2.0 Hz, 1H, ArH), 8.11 (d, J = 8.8 Hz, 1H, ArH), 7.55 (t, J = 3.0 Hz, 1H, yrazole-H), 6.29 (s, 1H, OH), 5.93–5.91 (m, 1H, pyrazole-H), 4.34 (d, J = 13.6 Hz, 1H, CH), 4.15 (d, J = 13.6 Hz, 1H, CH), 1.36 (s, 3H, CH3). HRMS [C15H13F4N4O2+]: calcd, 357.0975; found, 357.0985.
4.1.4.4. (S)-3-(4-Chloro-1H-pyrazol-1-yl)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide (16c).
16c was prepared following general procedure A as per Scheme 1. The crude product was purified by a silica gel column using dichloromethane (DCM) and ethyl acetate (19:1) as eluents to afford 0.30 g of the titled compound as a white solid. Yield = 55%. 1H NMR (400 MHz, DMSO-d6): δ 10.38 (s, 1H, NH), 8.46 (s, 1H, ArH), 8.23 (d, J = 8.6 Hz, J = 1.2 Hz, 1H, ArH), 8.10 (d, J = 8.6 Hz, 1H, ArH), 7.83 (s, 1H, pyrazole-H), 7.47 (s, 1H, pyrazole-H), 6.34 (s, 1H, OH), 4.45 (d, J = 14.0 Hz, 1H, CH), 4.27 (d, J = 14.0 Hz, 1H, CH), 1.36 (s, 3H, CH3). HRMS [C15H13ClF3N4O2+]: calcd, 373.0679; found, 373.0678. Purity: 97.69% (HPLC).
4.1.4.5. (S)-3-(4-Bromo-1H-pyrazol-1-yl)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide (16d).
16d was prepared following general procedure A as per Scheme 1. The crude product was purified by a silica gel column using DCM and ethyl acetate (19:1) as eluents to afford 0.47 g of the titled compound as a white form. Yield = 79.6%. 1H NMR (400 MHz, CDCl3): δ 9.08 (s, 1H, NH), 8.00 (d, J = 2.0 Hz, 1H, ArH), 7.87 (dd, J = 8.4 Hz, J = 2.0 Hz, 1H, ArH), 7.79 (d, J = 8.4 Hz, 1H, ArH), 7.49 (s, 1H, pyrazole-H), 7.47 (s, 1H, pyrazole-H), 5.92 (s, 1H, OH), 4.64 (d, J = 14.0 Hz, 1H, CH), 4.24 (d, J = 14.0 Hz, 1H, CH), 1.47 (s, 3H, CH3). HRMS [C15H13BrF3N4O2+]: calcd, 417.0174; found, 417.0167. Purity: 99.53% (HPLC).
4.1.4.6. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-3-(4-iodo-1H-pyrazol-1-yl)-2-methylpropanamide (16e).
16e was prepared following general procedure A as per Scheme 1. The crude product was purified by a silica gel column using DCM and ethyl acetate (19:1) as eluents to afford 0.25 g of the titled compound as an off-white solid. Yield = 52%. 1H NMR (400 MHz, DMSO-d6): δ 10.36 (s, 1H, NH), 8.45 (s, 1H, ArH), 8.23 (d, J = 8.8 Hz, J = 1.2 Hz, 1H, ArH), 8.10 (d, J = 8.8 Hz, 1H, ArH), 7.78 (s, 1H, pyrazole-H), 7.46 (s, 1H, pyrazole-H), 6.31 (s, 1H, OH), 4.48 (d, J = 14.0 Hz, 1H, CH), 4.31 (d, J = 14.0 Hz, 1H, CH), 1.35 (s, 3H, CH3). HRMS [C15H13F3IN4O2+]: calcd, 465.0035; found, 465.0045.
4.1.4.7. (S)-3-(4-Acetyl-1H-pyrazol-1-yl)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide (16f).
16f was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using DCM and ethyl acetate (19:1) as eluents to afford 70 mg of the titled compound as a yellowish solid. Yield = 20%. 1H NMR (400 MHz, DMSO-d6): δ 10.37 (s, 1H, NH), 8.45 (d, J = 1.2 Hz, 1H, ArH), 8.25 (s, 1H, pyrazole-H), 8.23 (d, J = 8.2 Hz, J = 1.2 Hz, 1H, ArH), 8.10 (d, J = 8.2 Hz, 1H, ArH), 7.86 (s, 1H, pyrazole-H), 6.37 (s, 1H, OH), 4.50 (d, J = 14.0 Hz, 1H, CH), 4.33 (d, J = 14.0 Hz, 1H, CH), 2.34 (s, 3H, CH3), 1.39 (s, 3H, CH3). HRMS [C17H16F3N4O3+]: calcd, 381.1175; found, 381.1178. Purity: 95.66% (HPLC).
4.1.4.8. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methyl-3-(4-(trifluoromethyl)-1H-pyrazol-1-yl)propanamide (16g).
16g was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using DCM and ethyl acetate (19:1) as eluents to afford 0.30 g of the titled compound as a white foam. Yield = 50%. 1H NMR (400 MHz, DMSO-d6): δ 10.38 (s, 1H, NH), 8.45 (d, J = 2.0 Hz, 1H, ArH), 8.25–8.22 (m, 2H, ArH & pyrazole-H), 8.11 (d, J = 8.2 Hz, 1H, ArH), 7.82 (s, 1H, pyrazole-H), 6.39 (s, 1H, OH), 4.55 (d, J = 14.0 Hz, 1H, CH), 4.37 (d, J = 14.0 Hz, 1H, CH), 1.40 (s, 3H, CH3). HRMS [C16H13F6N4O2+]: calcd, 407.0943; found, 407.0945.
4.1.4.9. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methyl-3-(3-(trifluoromethyl)-1H-pyrazol-1-yl)propanamide (16h).
16h was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using ethyl acetate and hexanes (2:1) as eluents to afford 0.31 g of the titled compound as a white solid. Yield = 50%. 1H NMR (400 MHz, DMSO-d6): δ 10.31 (s, 1H, NH), 8.42 (d, J = 2.0 Hz, 1H, ArH), 8.18 (dd, J = 8.8 Hz, J = 2.0 Hz, 1H, ArH), 8.09 (d, J = 8.8 Hz, 1H, ArH), 7.84–7.83 (m, 1H, pyrazole-H), 6.67 (d, J = 2.4 Hz, 1H, pyrazole-H), 6.41 (s, 1H, OH), 4.56 (d, J = 14.0 Hz, 1H, CH), 4.38 (d, J = 14.0 Hz, 1H, CH), 1.40 (s, 3H, CH3). HRMS [C16H13F6N4O2+]: calcd, 407.0943; found, 407.0945.
4.1.4.10. (S)-3-(4-Cyano-1H-pyrazol-1-yl)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide (16i).
16i was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using hexanes and ethyl acetate (1:1–1:2) as eluents to afford 0.18 g of the titled compound as a white solid. Yield = 46%. 1H NMR (400 MHz, DMSO-d6): δ 10.35 (s, 1H, NH), 8.45 (d, J = 1.2 Hz, 1H, ArH), 8.43 (s, 1H, pyrazole-H), 8.22 (d, J = 8.8 Hz, J = 1.2 Hz, 1H, ArH), 8.10 (d, J = 8.8 Hz, 1H, ArH), 7.98 (s, 1H, pyrazole-H), 6.41 (s, 1H, OH), 4.45 (d, J = 14.0 Hz, 1H, CH), 4.36 (d, J = 14.0 Hz, 1H, CH), 1.38 (s, 3H, CH3). HRMS [C16H13F3N5O2+]: calcd, 364.1021; found, 364.1016. Purity: 98.48% (HPLC).
4.1.4.11. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methyl-3-(4-nitro-1H-pyrazol-1-yl)propanamide (16j).
16j was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using hexanes and ethyl acetate (1:1) as eluents to afford 0.15 g of the titled compound as an off-white solid. Yield = 44%. 1H NMR (400 MHz, DMSO-d6): δ 10.36 (s, 1H, NH), 8.69 (s, 1H, pyrazole-H), 8.45 (d, J = 1.2 Hz, 1H, ArH), 8.23 (d, J = 8.8 Hz, J = 1.2 Hz, 1H, ArH), 8.19 (s, 1H, pyrazole-H), 8.11 (d, J = 8.8 Hz, 1H, ArH), 6.47 (s, 1H, OH), 4.56 (d, J = 14.0 Hz, 1H, CH), 4.38 (d, J = 14.0 Hz, 1H, CH), 1.41 (s, 3H, CH3). HRMS [C15H13F3N5O4+]: calcd, 384.0920; found, 384.0932. Purity: 99.58% (HPLC).
4.1.4.12. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-3-(4-methoxy-1H-pyrazol-1-yl)-2-methylpropanamide (16k).
16k was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using DCM and ethyl acetate (9:1) as eluents to afford 0.30 g of the titled compound as a white solid. Yield = 60%. 1H NMR (400 MHz, DMSO-d6): δ 10.38 (s, 1H, NH), 8.46 (d, J = 2.0 Hz, 1H, ArH), 8.24 (dd, J = 8.2 Hz, J = 2.0 Hz, 1H, ArH), 8.10 (d, J = 8.2 Hz, 1H, ArH), 7.35 (d, J = 0.8 Hz, 1H, pyrazole-H), 7.15 (d, J = 0.8 Hz, 1H, pyrazole-H), 6.25 (s, 1H, OH), 4.35 (d, J = 14.0 Hz, 1H, CH), 4.18 (d, J = 14.0 Hz, 1H, CH), 3.61 (s, 3H, CH3), 1.36 (s, 3H, CH3). HRMS [C16H16F3N4O3+]: calcd, 369.1175; found, 369.1182. Purity: 99.28% (HPLC).
4.1.4.13. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methyl-3-(4-methyl-1H-pyrazol-1-yl)propanamide (16l).
16l was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using DCM and ethyl acetate (19:1) as eluents to afford 0.28 g of the titled compound as a white solid. Yield = 66%. 1H NMR (400 MHz, DMSO-d6): δ 10.38 (s, 1H, NH), 8.46 (d, J = 2.0 Hz, 1H, ArH), 8.23 (dd, J = 8.8 Hz, J = 2.0 Hz, 1H, ArH), 8.10 (d, J = 8.8 Hz, 1H, ArH), 7.41 (s, 1H, pyrazole-H), 7.17 (s, 1H, pyrazole-H), 6.24 (s, 1H, OH), 4.40 (d, J = 14.0 Hz, 1H, CH), 4.22 (d, J = 14.0 Hz, 1H, CH), 1.97 (s, 3H, CH3), 1.36 (s, 3H, CH3). HRMS [C16H16F3N4O2+]: calcd, 353.1225; found, 353.1232. Purity: 99.75% (HPLC).
4.1.4.14. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methyl-3-(4-phenyl-1H-pyrazol-1-yl)propanamide (16m).
16m was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using ethyl acetate and hexanes (1:2) as eluents to afford 0.90 g of the titled compound as white needles. Yield = 68.5%. 1H NMR (400 MHz, DMSO-d6): δ 10.40 (s, 1H, NH), 8.46 (d, J = 2.0 Hz, 1H, ArH), 8.24 (dd, J = 8.4 Hz, J = 2.0 Hz, 1H, ArH), 8.09 (d, J = 8.4 Hz, 1H, ArH), 8.05 (s, 1H, pyrazole-H), 7.82 (s, 1H, pyrazole-H), 7.52–7.45 (m, 2H, ArH), 7.35–7.31 (m, 2H, ArH), 7.20–7.16 (m, 1H, ArH), 6.33 (s, 1H, OH), 4.50 (d, J = 14.0 Hz, 1H, CH), 4.30 (d, J = 14.0 Hz, 1H, CH), 1.40 (s, 3H, CH3). HRMS [C21H18F3N4O2+], calcd, 415.1382; found, 415.1391.
4.1.4.15. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methyl-3-(3-phenyl-1H-pyrazol-1-yl)propanamide (16n).
16n was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using ethyl acetate and hexanes (1:3 to 1:2) as eluents to afford 0.60 g of the titled compound as white needles. Yield = 41.7%. 1H NMR (400 MHz, DMSO-d6): δ 10.33 (s, 1H, NH), 8.48 (d, J = 2.0 Hz, 1H, ArH), 8.22 (dd, J = 8.2 Hz, J = 2.0 Hz, 1H, ArH), 8.05 (d, J = 8.2 Hz, 1H, ArH), 7.69 (d, J = 2.0 Hz, 1H, ArH), 7.60–7.57 (m, 2H, ArH), 7.28–7.21 (m, 3H, ArH), 6.66 (d, J = 3.0 Hz, 1H, ArH), 6.31 (s, 1H, OH), 4.52 (d, J = 14.6 Hz, 1H, CH), 4.32 (d, J = 14.6 Hz, 1H, CH), 1.43 (s, 3H, CH3). HRMS [C21H18F3N4O2+]: calcd, 415.1382; found, 514.1423.
4.1.4.16. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-3-(4-(4-fluorophenyl)-1H-pyrazol-1-yl)-2-hydroxy-2-methylpropanamide (16o).
16o was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using DCM and ethyl acetate (19:1) as eluents to afford 0.33 g of the titled compound as a white solid. Yield = 62%. 1H NMR (400 MHz, DMSO-d6): δ 10.29 (s, 1H, NH), 8.41 (s, 1H, ArH), 8.21 (d, J = 8.8 Hz, 1H, ArH), 8.05 (d, J = 8.8 Hz, 1H, ArH), 7.68 (s, 1H, pyrazole-H), 7.61 (t, J = 6.4 Hz, 2H, ArH), 7.08 (t, J = 8.4 Hz, 2H, ArH), 6.65 (s, 1H, pyrazole-H), 6.30 (s, 1H, OH), 4.51 (d, J = 14.0 Hz, 1H, CH), 4.31 (d, J = 14.0 Hz, 1H, CH), 1.42 (s, 3H, CH3). HRMS [C21H17F4N4O2+]: calcd, 433.1288; found, 433.1291. Purity: 96.01% (HPLC).
4.1.4.17. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-3-(3-(4-fluorophenyl)-1H-pyrazol-1-yl)-2-hydroxy-2-methylpropanamide (16p).
16p was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using DCM and ethyl acetate (19:1) as eluents to afford 0.27 g of the titled compound as a white solid. Yield = 43%. 1H NMR (400 MHz, DMSO-d6): δ 10.29 (s, 1H, NH), 8.41 (s, 1H, ArH), 8.21 (d, J = 8.8 Hz, 1H, ArH), 8.05 (d, J = 8.8 Hz, 1H, ArH), 7.69 (s, 1H, pyrazole-H), 7.61 (t, J = 6.4 Hz, 2H, ArH), 7.08 (t, J = 8.4 Hz, 2H, ArH), 6.65 (s, 1H, pyrazole-H), 6.30 (s, 1H, OH), 4.51 (d, J = 14.0 Hz, 1H, CH), 4.31 (d, J = 14.0 Hz, 1H, CH), 1.42 (s, 3H, CH3). HRMS [C21H17F4N4O2+]: calcd, 433.1288; found, 433.1290.
4.1.4.18. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-3-(4-ethynyl-1H-pyrazol-1-yl)-2-hydroxy-2-methylpropanamide (16q).
16q was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using DCM and ethyl acetate (95:5) as eluents to afford 0.37 g of the titled compound as a white foam. Yield = 62.7%. 1H NMR (400 MHz, DMSO-d6): δ 10.40 (s, 1H, NH), 8.47 (s, 1H, ArH), 8.24 (d, J = 8.8 Hz, 1H, ArH), 8.11 (d, J = 8.8 Hz, 1H, ArH), 7.91 (s 1H, pyrazole-H), 7.57 (s 1H, pyrazole-H), 6.35 (s, 1H, OH), 4.46 (d, J = 14.4 Hz, 1H, CH), 4.29 (d, J = 14.4 Hz, 1H, CH), 4.00 (s, 1H, CH), 1.35 (s, 3H, CH3). HRMS [C17H14F3N4O2+]: calcd, 363.1069; found, 363.1026. Purity: 99.55% (HPLC).
4.1.4.19. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-3-(4-(4-hydroxybut-1-yn-1-yl)-1H-pyrazol-1-yl)-2-methylpropanamide (16r).
16r was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using DCM and methanol (95:5) as eluents to afford 0.477 g of the titled compound as a yellowish solid. Yield = 20%. 1H NMR (400 MHz, DMSO-d6): δ 12.99 (br s, 1H), 10.47 (s, 1H, NH), 8.55 (s, 1H, ArH), 8.29 (d, J = 8.8 Hz, 1H, ArH), 8.08 (d, J = 8.8 Hz, 1H, ArH), 7.87 (s 1H, pyrazole-H), 7.49 (s 1H, pyrazole-H), 6.00 (s, 1H, OH), 3.64 (d, J = 8.2 Hz, 1H, CH), 4.50 (d, J = 9.6 Hz, 1H, CH), 3.60–3.56 (m, 2H, CH2), 2.59–2.55 (m, 2H, CH2), 1.31 (s, 3H, CH3). HRMS [C19H18F3N4O3+]: calcd, 407.1331; found, 407.1267.
4.1.4.20. (S)-1-(3-((4-Cyano-3-(trifluoromethyl)phenyl)amino)-2-hydroxy-2-methyl-3-oxopropyl)-1H-pyrazole-4-carboxamide (16s).
16s was prepared in two steps. In the first step, (S)-tert-butyl (1-(3-((4-cyano-3-(trifluoromethyl)phenyl)amino)-2-hydroxy-2-methyl-3-oxopropyl)-1H-pyrazol-4-yl)carbamate 16u (an intermediate compound for 16s) was synthesized following general procedure A as per Scheme 1 as described in detail infra. The compound was purified by a silica gel column using hexanes and ethyl acetate (2:1) as eluents to afford the compound as a white solid. Yield = 69%. 1H NMR (400 MHz, CDCl3): δ 9.13 (br s, 1H, NH), 8.18 (d, J = 10.8 Hz, 1H), 8.02 (d, J = 1.2 Hz, 1H), 7.94 (s, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.66 (br s, C(O)NHC(O)), 5.79 (br s, 1H, OH), 4.70 (d, J = 13.8 Hz, 1H), 4.32 (d, J = 13.8 Hz, 1H), 1.52 (s, 3H), 1.50 (s, 9H). 19F NMR (400 MHz, CDCl3): δ −62.20. MS (ESI) m/z: 480.23 [M − H]−.
The second step: To a solution of 16u (0.721 g, 2.05 mmol) in EtOH (10 mL) was added dropwise acetyl chloride (3 mL) at 0 °C and further stirred at rt for 3 h. After removing the solvent under reduced pressure, the mixture was treated with ethyl acetate and hexane (2:1) to afford the desired compound as a yellowish solid. Yield = 95%. UV max 194.45, 270.45. 1H NMR (400 MHz, DMSO-d6): δ 10.39 (br s, 1H, NHC(O)), 8.46 (d, J = 1.6 Hz, 1H), 8.20 (dd, J = 8.6, 1.6 Hz, 1H), 8.08 (d, J = 8.6 Hz, 1H), 8.05 (s, 1H), 7.75 (s, 1H), 7.55 (br s, 2H, C(O)NH2), 6.99 (br s, 1H, OH), 4.45 (d, J = 13.8 Hz, 1H), 4.28 (d, J = 13.8 Hz, 1H), 1.34 (s, 3H). 19F NMR (DMSO-d6, decoupled): δ −61.13. HRMS [C16H15F3N5O3+]: calcd, 382.1127; found, 382.1282. HPLC purity: 98.75%.
4.1.4.21. (S)-3-(4-Amino-1H-pyrazol-1-yl)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide (16t).
To a solution of 16u (see below) (0.815 g, 0.0018 mol) in absolute EtOH (10 mL) was added acetyl chloride (0.4 mL, 5.4 mmol) at 0 °C and further stirred at rt for 3 h. After removing the solvent under vacuum, the resulting mixture was purified by flash column chromatography using hexanes and ethyl acetate (1:1, v/v) to afford the desired compound as a brown solid. Yield = 91%. 1H NMR (400 MHz, DMSO-d6): δ 10.31 (br s, 1H, NH), 10.21 (br s, 2H, NH2), 8.20 (s, 1H), 7.98 (d, J = 7.6 Hz, 1H), 7.77–7.73 (m, 2H), 7.62 (br s, 1H), 7.21 (br s, 1H), 6.28 (br s, 1H, OH), 4.23 (d, J = 14.0 Hz, 1H), 4.04 (d, J = 14.0 Hz, 1H), 1.04 (s, 3H); 19F NMR (acetone-d6, decoupled): δ114.77. MS (ESI) m/z: 354.08 [M + H]+; 351.98 [M − H]−.
4.1.4.22. (S)-tert-Butyl (1-(3-((4-cyano-3-(trifluoromethyl)-phenyl)amino)-2-hydroxy-2-methyl-3-oxopropyl)-1H-pyrazol-4-yl)-carbamate (16u).
16u was prepared following general procedure A as per Scheme 1. The product was purified by a silica gel column using hexanes and ethyl acetate (2:1) as eluents to afford the desired compound as a brown solid. 1H NMR (400 MHz, CDCl3): δ 9.12 (br s, 1H, NH), 8.01 (d, J = 1.6 Hz, 1H), 7.85 (dd, J = 8.4, 1.6 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.63 (br s, 1H), 7.43 (br s, 1H), 6.21 (br s, 1H, HN), 6.17 (br s, 1H, OH), 4.54 (d, J = 14.0 Hz, 1H), 4.17 (d, J = 14.0 Hz, 1H), 1.47 (s, 9H), 1.45 (s, 3H); 19F NMR (CDCl3, decoupled): δ−62.21. MS (ESI) m/z: 452.11 [M − H]−; 454.11 [M + H]+; 476.12 [M + Na]+.
4.1.4.23. (S)-3-(4-Acetamido-1H-pyrazol-1-yl)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide (16v).
Under argon atmosphere, to a solution of 16t (0.17 g, 0.48 mmol) and triethylamine (0.16 mL, 1.15 mmol) in 10 mL of anhydrous DCM was added acetyl chloride (0.04 mL, 0.58 mmol) in an ice–water bath. After stirring for 30 min, the temperature raised to rt and the mixture was stirred for 2 h. The reaction mixture was condensed under reduced pressure and then dispersed into 10 mL of ethyl acetate, washed with water, evaporated, dried over anhydrous MgSO4, and evaporated to dryness. The mixture was purified with flash column chromatography using hexanes and ethyl acetate as eluents (2/1, v/v) to produce the desired compound as a yellow solid. Yield = 92%. 1H NMR (400 MHz, CDCl3): δ 9.08 (br s, 1H, NH), 7.92 (br s, 1H, NH), 7.82–7.80 (m, 2H), 7.69 (d, J = 8.4 Hz, 1H), 7.44 (br s, 1H), 7.15 (br s, 1H), 6.10 (br s, 1H, OH), 4.50 (d, J = 14.0 Hz, 1H), 4.13 (d, J = 14.0 Hz, 1H), 2.04 (s, 3H), 1.39 (s, 3H). 19F NMR (CDCl3, decoupled): δ −62.20. MS (ESI) m/z: 356.11 [M + H]+; 354.06 [M − H]−.
4.1.4.24. (S)-3-(4-(2-Chloroacetamido)-1H-pyrazol-1-yl)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide (16w).
Under argon atmosphere, to a solution of 16t (0.17 g, 0.48 mmol) and triethylamine (0.16 mL, 1.15 mmol) in 10 mL of anhydrous DCM was added 2-chloroacetyl chloride (0.04 mL, 0.58 mmol) in an ice–water bath. After stirring for 30 min, the temperature raised to rt and the mixture was stirred for 2 h. The reaction mixture was condensed under vacuum and then dispersed into 10 mL of ethyl acetate, washed with water, evaporated, dried over anhydrous MgSO4, and evaporated to dryness. The mixture was purified with flash column chromatography using hexanes and ethyl acetate as eluents (2/1, v/v) to produce the titled compound as yellow solids. Yield = 68%. 1H NMR (400 MHz, CDCl3): δ 9.12 (br s, 1H, NH), 8.12 (br s, 1H, NH), 7.99 (d, J = 1.6 Hz, 1H), 7.92 (br s, 1H), 7.88 (dd, J = 8.6, 1.6 Hz, 1H), 7.78 (d, J = 8.6 Hz, 1H), 7.61 (br s, 1H), 6.11 (br s, 1H, OH), 4.60 (d, J = 13.8 Hz, 1H), 4.23 (d, J = 13.8 Hz, 1H), 4.17 (s, 2H), 1.47 (s, 3H); 19F NMR (CDCl3, decoupled): δ −62.19. MS (ESI) m/z: 452.01 [M + Na]+; 428.00 [M − H]−.
4.1.4.25. (S)-Methyl (1-(3-((4-cyano-3-(trifluoromethyl)phenyl)-amino)-2-hydroxy-2-methyl-3-oxopropyl)-1H-pyrazol-4-yl)-carbamate (16x).
Under argon atmosphere, to a solution of 16t (0.17 g, 0.48 mmol) and triethylamine (0.16 mL, 1.15 mmol) in 10 mL of anhydrous DCM was added methyl carbonochloridate (0.04 mL, 0.58 mmol) in an ice–water bath. After stirring for 30 min, the temperature raised to rt and the mixture was stirred for 2 h. The reaction mixture was condensed under vacuum and then dispersed into 10 mL of ethyl acetate, washed with water, evaporated, dried over anhydrous MgSO4, and evaporated to dryness. The mixture was purified with flash column chromatography using hexanes and ethyl acetate as eluents (2/1, v/v) to produce the titled compound as a white solid. Yield = 71%. 1H NMR (400 MHz, CDCl3): δ 9.07 (br s, 1H,C(O)NH), 7.91 (s, 1H, ArH), 7.79 (d, J = 7.2 Hz, 1H, ArH), 7.69 (d, J = 7.2 Hz, 1H, ArH), 7.57 (s, 1H, ArH), 7.40 (s, 1H, ArH), 6.33 (br s, 1H, NH), 6.08 (br s, 1H, OH), 4.50 (d, J = 13.6 Hz, 1H, CH2), 4.12 (d, J = 13.6 Hz, 1H, CH2), 3.67 (s, 3H, NH(CO)OCH3), 1.39 (s, 3H, CH3); 19F NMR (CDCl3, decoupled): δ −62.21. MS (ESI) m/z: 410.30 [M−H]−; 413.21 [M + H]+.
4.1.4.26. (S)-N-(6-Cyano-5-(trifluoromethyl)pyridin-3-yl)-3-(4-fluoro-1H-pyrazol-1-yl)-2-hydroxy-2-methylpropanamide (21a).
21a was prepared following general procedure A as per Scheme 2, where 17 was 5-amino-3-(trifluoromethyl)picolinonitrile. The product was purified by a silica gel column using hexanes and ethyl acetate (1:1) as eluents to afford 0.50 g of the titled compound as a white solid. Yield = 60.2%. 1H NMR (400 MHz, DMSO-d6): δ 10.64 (s, 1H, NH), 9.32 (d, J = 2.0 Hz, 1H, ArH), 8.82 (d, J = 2.0 Hz, 1H, ArH), 7.75 (d, J = 4.8 Hz, 1H, pyrazole-H), 7.40 (d, J = 4.0 Hz, 1H, pyrazole-H), 6.41 (s, 1H, OH), 4.39 (d, J = 14.0 Hz, 1H, CH), 4.22 (d, J = 14.0 Hz, 1H, CH), 1.36 (s, 3H, CH3). HRMS [C14H12F4N5O2+]: calcd, 358.0927; found, 358.0932.
4.1.4.27. (S)-N-(6-Cyano-5-(trifluoromethyl)pyridin-3-yl)-2-hydroxy-2-methyl-3-(4-(trifluoromethyl)-1H-pyrazol-1-yl)-propanamide (21b).
21b was prepared following general procedure A as per Scheme 2, where 17 was 5-amino-3-(trifluoromethyl)-picolinonitrile. The product was purified by a silica gel column using DCM and ethyl acetate (19:1) as eluents to afford 0.18 g of the titled compound as a white solid. Yield = 60%. 1H NMR (400 MHz, DMSO-d6): δ 10.63 (s, 1H, NH), 9.31 (s, 1H, ArH), 8.80 (s, 1H, ArH), 8.32 (s, 1H, pyrazole-H), 7.81 (s, 1H, pyrazole-H), 6.48 (s, 1H, OH), 4.55 (d, J = 14.0 Hz, 1H, CH), 4.37 (d, J = 14.0 Hz, 1H, CH), 1.42 (s, 3H, CH3). HRMS [C15H12F6N5O2+]: calcd, 408.0892; found, 408.0890. Purity: 96.81% (HPLC).
4.1.4.28. (S)-3-(4-Cyano-1H-pyrazol-1-yl)-N-(6-cyano-5-(trifluoromethyl)pyridin-3-yl)-2-hydroxy-2-methylpropanamide (21c).
21c was prepared following general procedure A as per Scheme 2, where 17 was 5-amino-3-(trifluoromethyl)picolinonitrile. The product was purified by a silica gel column using hexanes and ethyl acetate (2:1) as eluents to the titled compound as an off-white solid. Yield = 52%. mp 169.7–169.9 °C; UV max 195.45, 274.45; 1H NMR (400 MHz, CDCl3): δ 9.17 (br s, 1H, NH), 8.83 (s, 1H), 8.67 (d, J = 1.6 Hz, 1H), 7.92 (s, 1H), 7.85 (s, 1H), 5.58 (s, OH), 4.73 (d, J = 14.0 Hz, 1H), 4.34 (d, J = 14.0 Hz, 1H), 1.53 (s, 3H); 19F NMR (CDCl3, decoupled): δ−62.11. HRMS [C15H12F3N6O2+]: calcd, 365.0974; found, 365.0931.
4.1.4.29. (S)-tert-Butyl (1-(3-((6-cyano-5-(trifluoromethyl)pyridin-3-yl)amino)-2-hydroxy-2-methyl-3-oxopropyl)-1H-pyrazol-4-yl)-carbamate (21d).
21d was prepared following general procedure A as per Scheme 2, where 17 was 5-amino-3-(trifluoromethyl)-picolinonitrile. The product was purified by a silica gel column using hexanes and ethyl acetate (2:1) as eluents to afford the desired compound as a brown solid. Yield = 60%. 1H NMR (400 MHz, CDCl3): δ 9.28 (br s, 1H, NH), 8.80 (s, 1H), 7.63 (br s, 1H), 7.43 (br s, 1H), 6.29 (br s, 1H, NH), 6.21 (br s, 1H, OH), 4.55 (d, J = 14.0 Hz, 1H), 4.17 (d, J = 14.0 Hz, 1H), 1.47 (s, 3H); 19F NMR (CDCl3, decoupled): δ −62.11. MS (ESI) m/z: 453.16 [M − H]−; 477.16 [M + Na]+.
4.1.4.30. (S)-N-(3-Chloro-4-cyanophenyl)-3-(4-fluoro-1H-pyrazol-1-yl)-2-hydroxy-2-methylpropanamide (21e).
21e was prepared following general procedure A as per Scheme 2, where 17 was 4-amino-2-chlorobenzonitrile. The product was purified by a silica gel column using hexanes and ethyl acetate (2:1) as eluents to afford the desired compound as a white solid. Yield = 71%. 1H NMR (400 MHz, CDCl3): δ 8.97 (br s, 1H, NH), 7.87 (d, J = 2.0 Hz, 1H), 7.60 (d, J = 8.4 Hz, 1H), 7.45 (dd, J = 8.4, 2.0 Hz, 1H), 7.36 (dd, J = 8.8, J = 4.0 Hz, 1H), 5.86 (br s, 1H, OH), 4.55 (d, J = 14.0 Hz, 1H), 4.16 (d, J = 14.0 Hz, 1H), 1.46 (s, 3H); 19F NMR (CDCl3, decoupled): δ −176.47. HRMS [C14H13FN4O2+]: calcd, 323.0711; found, 323.0710.
4.1.4.31. (S)-3-(4-Fluoro-1H-pyrazol-1-yl)-2-hydroxy-2-methyl-N-(4-nitro-3-(trifluoromethyl)phenyl)propanamide (21f).
21f was prepared following general procedure A as per Scheme 2, where 17 was 4-nitro-3-(trifluoromethyl)aniline. The product was purified by a silica gel column using hexanes and ethyl acetate (2:1) as eluents to afford the desired compound as a yellowish solid. Yield = 67%. 1H NMR (400 MHz, CDCl3): δ 9.14 (br s, 1H, NH), 8.01 (s, 1H), 7.97–7.91 (m, 2H), 7.38 (d, J = 3.6 Hz, 1H), 7.35 (d, J = 4.4 Hz, 1H), 5.95 (s, 1H, OH), 4.56 (d, J = 14.0 Hz, 1H), 4.17 (d, J = 14.0 Hz, 1H), 1.48 (s, 3H); 19F NMR (CDCl3, decoupled): δ −60.13, −176.47. MS (ESI) m/z: 375.08 [M − H]–; 377.22 [M + H]+; 399.04 [M + Na]+.
4.1.4.32. (S)-5-(3-(4-Fluoro-1H-pyrazol-1-yl)-2-hydroxy-2-methylpropanamido)picolinamide (21g).
21g was prepared following general procedure A as per Scheme 2, where 17 of step a was 5-cyano-6-(trifluoromethyl)picolinamide. In step c, to a solution of 4-fluoro-pyrazole (20; 0.20 g, 0.0023237 mol) in anhydrous THF (5 mL) which was cooled in an ice–water bath under an argon atmosphere was added sodium hydride (60% dispersion in oil, 0.28 g, 0.0069711 mol). After addition, the resulting mixture was stirred for 3 h. (R)-3-Bromo-N-(6-cyanopyridin-3-yl)-2-hydroxy-2-methylpropanamide 18 (0.66 g, 0.0023237 mol) was added to the above solution, and the resulting reaction mixture was allowed to stir overnight at rt under argon atmosphere. The reaction was quenched by water and extracted with ethyl acetate. The organic layer was washed with brine, dried with MgSO4, filtered, and concentrated under vacuum. The product was purified by a silica gel column using DCM and methanol (9:1) as eluents to afford 0.10 g of the titled compound as a white solid. Yield = 14.1%. 1H NMR (400 MHz, DMSO-d6): δ 10.08 (s, 1H, NH), 8.89 (d, J = 2.4 Hz, 1H, ArH), 8.30 (dd, J = 8.2 Hz, J = 2.4 Hz, 1H, ArH), 8.01 (s, 1H, NH), 7.98 (d, J = 8.2 Hz, 1H, ArH), 7.73 (d, J = 4.4 Hz, 1H, pyrazole-H), 7.51 (s, 1H, NH), 7.42 (d, J = 4.0 Hz, 1H, pyrazole-H), 6.24 (s, 1H, OH), 4.38 (d, J = 14.0 Hz, 1H, CH), 4.42 (d, J = 14.0 Hz, 1H, CH), 1.34 (s, 3H, CH3). HRMS [C13H15FN5O3+]: calcd, 308.1159; found, 308.1177.
4.1.4.33. (S)-3-(4-Fluoro-1H-pyrazol-1-yl)-2-hydroxy-2-methyl-N-(quinazolin-6-yl) propanamide (21h).
21h was prepared following general procedure A as per Scheme 2, where 17 of step (a) was quinazolin-6-amine. In step (c), a solution of 4-fluoro-pyrazole (20; 0.20 g, 0.0023237 mol) in anhydrous THF (5 mL), which was cooled in an ice–water bath under an argon atmosphere, was added sodium hydride (60% dispersion in oil, 0.28 g, 0.0069711 mol). After addition, the resulting mixture was stirred for 3 h. (R)-3-Bromo-2-hydroxy-2-methyl-N-(quinazolin-6-yl) propanamide (18; 0.72 g, 0.0023237 mol) was added to the above solution, and the resulting reaction mixture was allowed to stir overnight at rt under argon atmosphere. The reaction was quenched by water and extracted with ethyl acetate. The organic layer was washed with brine, dried with MgSO4, filtered, and concentrated under vacuum. The product was purified by a silica gel column using DCM and methanol (19:1) as eluents to afford 50 mg of the titled compound as a yellow solid. Yield = 13.7%. 1H NMR (400 MHz, DMSO-d6): δ 10.10 (s, 1H, NH), 9.54 (s, 1H, ArH), 9.21 (s, 1H, ArH), 8.64 (d, J = 2.4 Hz, 1H, ArH), 8.22 (dd, J = 8.6 Hz, J = 2.4 Hz, 1H, ArH), 7.97 (d, J = 8.6 Hz, 1H, ArH), 7.75 (d, J = 4.8 Hz, 1H, pyrazole-H), 7.43 (d, J = 4.0 Hz, 1H, pyrazole-H), 6.26 (s, 1H, OH), 4.42 (d, J = 14.0 Hz, 1H, CH), 4.25 (d, J = 14.0 Hz, 1H, CH), 1.36 (s, 3H, CH3). MS (ESI) m/z: 314.05 [M − H]−.
4.1.4.34. (S)-N-(2-Chloropyridin-4-yl)-3-(4-fluoro-1H-pyrazol-1-yl)-2-hydroxy-2-methylpropanamide (21i).
21i was prepared in two steps. In the first step, (R)-3-bromo-N-(2-chloropyridin-4-yl)-2-hydroxy-2-methylpropanamide as an intermediate compound was synthesized following Scheme 2, where 17 was 2-chloropyridin-4-amine. Thionyl chloride (11.2 mL, 0.154 mol) was added dropwise to a cooled solution (less than 4 °C) of 11 (18.3 g, 0.100 mol) in 100 mL of THF under an argon atmosphere. The resulting mixture was stirred for 3 h under the same condition. To this was added Et3N (25.7 mL, 0.185 mol) and stirred for 20 min under the same condition. After 20 min, 2-chloropyridin-4-amine (17; 9.89 g, 0.077 mol) and 100 mL of THF were added and then the mixture was allowed to stir overnight at rt. The solvent was removed under reduced pressure to afford a solid, which was treated with 100 mL of H2O, extracted with ethyl acetate (2 × 50 mL). The combined organic extracts were washed with saturated NaHCO3 solution (2 × 100 mL) and brine (100 mL). The organic layer was dried over MgSO4 and concentrated under reduced pressure to afford a solid, which was purified from column chromatography using ethyl acetate and DCM (80:20) to afford a solid. This solid was recrystallized from DCM and hexane to afford 12.6 g of the intermediate compound as a light-yellow solid. Yield = 43%. 1H NMR (400 MHz, CDCl3): δ 9.06 (br s, 1H, NH), 8.31 (d, J = 5.6 Hz, 1H), 7.77 (d, J = 0.8 Hz, 1H), 7.45 (dd, J = 5.6, 0.8 Hz, 1H), 4.81 (br s, 1H, OH), 3.97 (d, J = 10.6 Hz, 1H), 3.60 (d, J = 10.6 Hz, 1H), 1.64 (s, 3H). MS (ESI) m/z: 295.28 [M + H]+.
In the second step, 21i was prepared following general procedure A as per Scheme 2, where 18 was (R)-3-bromo-N-(2-chloropyridin-4-yl)-2-hydroxy-2-methylpropanamide. The product was purified by a silica gel column using hexanes and ethyl acetate (2:1) as eluents to afford the desired compound as a white solid. Yield = 55%. 1H NMR (400 MHz, CDCl3): δ 8.90 (br s, 1H, NH), 8.26 (d, J = 5.6 Hz, 1H), 7.63 (s, 1H), 7.75 (d, J = 4.2 Hz, 1H), 7.33 (d, J = 4.2 Hz, 1H), 7.31 (dd, J = 5.6, 1.2 Hz, 1H), 5.88 (s, 1H, OH), 4.53 (d, J = 13.6 Hz, 1H), 4.14 (d, J = 13.6 Hz, 1H), 1.45 (s, 3H); 19F NMR (CDCl3, decoupled): δ −176.47. MS (ESI) m/z: 298.98 [M + H]+; 296.96 [M − H]−.
4.1.4.35. (S)-N-(4-Cyano-2-iodo-5-(trifluoromethyl)phenyl)-3-(4-fluoro-1H-pyrazol-1-yl)-2-hydroxy-2-methylpropanamide (21j).
21j was prepared following general procedure A as per Scheme 2, where 17 of step (a) was 4-cyano-2-iodoaniline. In step (c), 20 was 4-fluoro-1H-pyrazole (0.09 g, 0.001048 mol). The product was purified by a silica gel column using hexanes and ethyl acetate (2:1 to 1:1) as eluents to afford 0.32 g of the titled compound as a white solid. Yield = 64%. 1H NMR (400 MHz, CDCl3): δ 9.60 (s, 1H, NH), 8.76 (s, 1H, ArH), 8.69 (s, 1H, ArH), 7.76 (d, J = 4.8 Hz, 1H, pyrazole-H), 7.36 (d, J = 4.4 Hz, 1H, pyrazole-H), 6.85 (s, 1H, OH), 4.39 (d, J = 14.0 Hz, 1H, CH), 4.20 (d, J = 14.0 Hz, 1H, CH), 1.41 (s, 3H, CH3). Mass (ESI) m/z: 481.00 [M − H]−.
4.1.4.36. (S)-3-(4-Bromo-3-fluoro-1H-pyrazol-1-yl)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide (26a).
26a was prepared following general procedure A as per Scheme 3. To a solution of 4-bromo-3-fluoro-pyrazole (25; 0.30 g, 0.001819 mol) in anhydrous THF (10 mL), which was cooled in an ice–water bath under an argon atmosphere, was added sodium hydride (60% dispersion in oil, 0.26 g, 0.006365 mol). After addition, the resulting mixture was stirred for 3 h. 23 (where X is CH; 0.64 g, 0.001819 mol) was added to the above solution, and the resulting reaction mixture was allowed to stir overnight at rt under argon atmosphere. The reaction was quenched with water and extracted with ethyl acetate. The organic layer was washed with brine, dried with MgSO4, filtered, and concentrated under vacuum. The product was purified by a silica gel column using ethyl acetate and hexanes (2:1) as eluents to afford 0.34 g of the titled compound as a pinkish solid. Yield = 34%. mp 110–112 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.38 (s, 1H, NH), 8.45 (d, J = 2.0–1.6 Hz, 1H, ArH), 8.23 (dd, J = 8.2 Hz, J = 2.0 Hz, 1H, ArH), 8.11 (d, J = 8.2 Hz, 1H, ArH), 7.82 (d, J = 2.0 Hz, 1H, pyrazole-H), 6.35 (s, 1H, OH), 4.35 (d, J = 14.0 Hz, 1H, CH), 4.04 (d, J = 14.0 Hz, 1H, CH), 1.37 (s, 3H, CH3). HRMS [C15H12BrF4N4O2+]: calcd, 435.0080; found, 435.0080. Purity: 96.98% (HPLC).
4.1.4.37. (S)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-3-(3-fluoro-4-(4-fluorophenyl)-1H-pyrazol-1-yl)-2-hydroxy-2-methylpropanamide (26b).
26b was prepared by Suzuki reaction mixing 26a (0.20 g, 0.4596 mmol), 4-fluoro boronic acid (77 mg, 0.5515 mmol), Pd(II)(OAc)2 (2–3 mg, 0.009192 mmol), PPh3 (7–8 mg, 0.02758 mmol), and K2CO3 (0.13 g, 0.965 mmol) into ACN (4–5 mL) and H2O (2–3 mL). The mixture was degassed and refilled with argon three times. The resulting reacting mixture was heated at reflux for 3 h under argon atmosphere. The product was purified by a silica gel column using hexanes and ethyl acetate (2:1–1:1) as eluents to afford 51 mg of the titled compound as a yellowish solid. Yield = 25%. 1H NMR (400 MHz, CDCl6): δ 9.12 (s, 1H, NH), 8.06 (d, J = 1.6 Hz, 1H, ArH), 7.85 (dd, J = 8.2 Hz, J = 1.6 Hz, 1H, ArH), 7.77 (d, J = 8.2 Hz, 1H, ArH), 7.51 (d, J = 3.0 Hz, 1H, pyrazole-H), 7.43–7.40 (m, 2H, ArH), 7.08–7.04 (m, 2H, ArH), 4.57 (d, J = 10.5 Hz, 1H, CH), 4.17 (d, J = 10.5 Hz, 1H, CH), 1.26 (s, 3H, CH3). HRMS [C21H16F5N4O2+]: calcd, 451.1193; found, 451.1196.
4.1.4.38. (S)-3-(3-Bromo-4-cyano-1H-pyrazol-1-yl)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide (26c).
26c was prepared following general procedure A as per Scheme 3, where X of 22 (step a) is CH and 25 of step c is 3-bromo-4-cyano-pyrazole. The product was purified by a silica gel column using ethyl acetate and hexanes (2:1) as eluents to afford 0.10 g of the titled compound as an off-white solid. Yield = 20%. 1H NMR (400 MHz, DMSO-d6): δ 10.32 (s, 1H, NH), 8.50 (s 1H, pyrazole-H), 8.41 (s, 1H, ArH), 8.20 (d, J = 8.4 Hz, 1H, ArH), 8.11 (d, J = 8.4 Hz, 1H, ArH), 6.47 (s, 1H, OH), 4.52 (d, J = 13.6 Hz, 1H, CH), 4.33 (d, J = 13.6 Hz, 1H, CH), 1.41 (s, 3H, CH3). HRMS [C16H12BrF3N5O2+]: calcd, 442.0126; found, 442.0109. Purity: 98.84% (HPLC).
4.1.4.39. (S)-3-(3-Chloro-4-methyl-1H-pyrazol-1-yl)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide (26d).
26d was prepared following general procedure A as per Scheme 3, where X of 22 (step a) is CH and 25 of step c is 3-chloro-4-methylpyrazole. The product was purified by a silica gel column using DCM and ethyl acetate (98:2 to 95:5) as eluents to afford 0.27 g of the titled compound as a white solid. Yield = 54%. 1H NMR (400 MHz, DMSO-d6): δ 10.33 (s, 1H, NH), 8.42 (d, J = 0.8 Hz, 1H, ArH), 8.21 (dd, J = 8.4 Hz, J = 0.8 Hz, 1H, ArH), 8.10 (d, J = 8.2 Hz, 1H, ArH), 7.50 (s 1H, pyrazole-H), 6.29 (s, 1H, OH), 4.36 (d, J = 14.4 Hz, 1H, CH), 4.18 (d, J = 14.4 Hz, 1H, CH), 1.91 (s, 3H, CH3), 1.35 (s, 3H, CH3). HRMS [C16H15ClF3N4O2+]: calcd, 387.0836; found, 387.0839. Purity: 97.07% (HPLC).
4.1.4.40. (S)-3-(3-Bromo-4-chloro-1H-pyrazol-1-yl)-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-2-methylpropanamide (26e).
26e was prepared following general procedure A as per Scheme 3, where X of 22 (step a) is CH and 25 of step c is 3-bromo-4-chloro-pyrazole. The product was purified by a silica gel column using DCM and ethyl acetate (95:5) as eluents to afford 0.25 g of the titled compound as a white solid. Yield = 50%. 1H NMR (400 MHz, DMSO-d6): δ 10.34 (s, 1H, NH), 8.41 (s, 1H, ArH), 8.20 (d, J = 8.8 Hz, 1H, ArH), 8.11 (d, J = 8.8 Hz, 1H, ArH), 7.93 (s 1H, pyrazole-H), 6.39 (s, 1H, OH), 4.43 (d, J = 14.0 Hz, 1H, CH), 4.25 (d, J = 14.0 Hz, 1H, CH), 1.38 (s, 3H, CH3). HRMS [C15H12BrClF3N4O2+]: calcd, 450.9784; found, 450.9807. Purity: 96.55% (HPLC).
4.1.4.41. (S)-3-(4-Bromo-3-fluoro-1H-pyrazol-1-yl)-N-(6-cyano-5-(trifluoromethyl)pyridin-3-yl)-2-hydroxy-2-methylpropanamide (26f).
26f was prepared following general procedure A as per Scheme 3, where X of 22 (step a) is N and 25 of step c is 4-bromo-3-fluoro-pyrazole. The product was purified by a silica gel column using hexanes and ethyl acetate (2:1 to 1:1) as eluents to afford 0.28 g of the titled compound as a white solid. Yield = 54%. 1H NMR (400 MHz, DMSO-d6): δ 10.67 (s, 1H, NH), 9.32 (d, J = 2.0 Hz, 1H, ArH), 8.82 (d, J = 2.0 Hz, 1H, ArH), 7.85 (d, J = 2.0 Hz 1H, pyrazole-H), 6.47 (s, 1H, OH), 4.35 (d, J = 14.0 Hz, 1H, CH), 4.17 (d, J = 14.0 Hz, 1H, CH), 1.39 (s, 3H, CH3). HRMS [C15H12BrClF3N4O2+]: calcd, 434.9954; found, 435.9997. Purity: 93.41% (HPLC).
4.1.4.42. (S)-3-(3-Bromo-4-cyano-1H-pyrazol-1-yl)-N-(6-cyano-5-(trifluoromethyl)pyridin-3-yl)-2-hydroxy-2-methylpropanamide (26g).
26g was prepared following general procedure A as per Scheme 3, where X of 22 (step a) is N and 25 of step c is 3-bromo-4-cyano-pyrazole. The product was purified by a silica gel column using hexanes and ethyl acetate (2:1) as eluents to afford the titled compound as a white solid. Yield = 81%. mp 172.5–173.6 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.60 (br s, 1H, NH), 9.29 (s, 1H), 8.79 (s, 1H), 8.53 (s, 1H), 6.59 (s, OH), 4.50 (d, J = 14.0 Hz, 1H), 4.32 (d, J = 14.0 Hz, 1H), 1.43 (s, 3H); 19F NMR (CDCl3, decoupled): δ −61.25. HRMS [C15H11BrF3N6O2+]: calcd, 443.0079; found, 443.0083.
4.1.4.43. (S)-3-(4-Cyano-3-phenyl-1H-pyrazol-1-yl)-N-(6-cyano-5-(trifluoromethyl)pyridin-3-yl)-2-hydroxy-2-methylpropanamide (26h).
A flask equipped with a reflux condenser, a septum inlet, and a magnetic stirring bar charged with 26g (0.053 g, 0.23 mmol), tetrakis(triphenylphosphine) palladium (0) (9 mg, 0.07 mmol), and phenyl boronic acid (35 mg, 0.28 mmol) in THF/MeOH (5 mL/1 mL) with sodium carbonate (50 mg, 0.48 mmol) in deoxygenated water (1 mL) was stirred and heated to reflux for 2 h until the starting material was not detectable on TLC. The mixture was cooled to rt and the solvent was removed in vacuo, then poured into ethyl acetate (10 mL), and extracted with ethyl acetate. The combined organic layers were washed with sat. NH4Cl and water and dried over MgSO4. The solvent was removed in vacuo and then purified by flash column chromatography on silica gel using hexanes and ethyl acetate (1:1) as eluents to afford 36 mg of the targeted compound as a yellowish solid. Yield = 69%. mp 112.3–124.4 °C; 1H NMR (400 MHz, CDCl3): δ 9.17 (br s, 1H, NH), 8.76 (s, 1H), 8.60 (s, 1H), 7.77 (s, 1H), 7.57–7.52 (m, 3H), 7.18 (d, J = 8.8 Hz, 2H), 5.32 (s, OH), 4.60 (d, J = 14.0 Hz, 1H), 4.23 (d, J = 14.0 Hz, 1H), 1.47 (s, 3H). 19F NMR (CDCl3, decoupled): δ −62.09. HRMS [C21H16F3N6O2+]: calcd, 441.1287; found, 441.1291.
4.1.4.44. (R)-N-(4-Cyano-3-(trifluoromethyl)phenyl)-3-(4-fluoro-1H-pyrazol-1-yl)-2-hydroxy-2-methylpropanamide (29a).
29a was prepared following general procedure A as per Scheme 1 as exemplified above for 10, except that instead of 11, the opposite isomer was used [(S)-11 or (S)-3-bromo-2-hydroxy-2-methylpropanoic acid]. The product was purified by a silica gel column using hexanes and ethyl acetate (1:1) as eluents to afford the titled compound as a yellowish solid. Yield = 64%. +126.7° (c 1.0, MeOH); 1H NMR (400 MHz, CDCl3): δ 9.07 (br s, 1H, NH), 8.01 (d, J = 2.0 Hz, 1H), 7.95 (dd, J = 8.4, J = 2.0 Hz, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.38 (d, J = 4.0 Hz, 1H), 7.34 (d, J = 4.4 Hz, 1H), 5.92 (s, OH), 4.54 (d, J = 14.0 Hz, 1H), 4.16 (d, J = 14.4 Hz, 1H), 1.47 (s, 3H); 19F NMR (CDCl3, decoupled): δ−62.23, −176.47. HRMS [C15H13F4N4O2+]: calcd, 357.0975; found, 357.0984.
4.1.4.45. N-(4-Cyano-3-(trifluoromethyl)phenyl)-3-(4-fluoro-1H-pyrazol-1-yl)-2-methylpropanamide (29b).
29b was prepared following general procedure A as per Scheme 4, where Y in 27 is a tertiary carbon [CH(CH3)] instead of quaternary [C(OH)(CH3)] like all previous compounds. To a solution of 4-fluoro-pyrazole (20; 0.20 g, 0.0023237 mol) in anhydrous THF (5 mL), which was cooled in an ice–water bath under an argon atmosphere, was added sodium hydride (60% dispersion in oil, 0.28 g, 0.0069711 mol). After addition, the resulting mixture was stirred for 3 h. 3-Bromo-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-methylpropanamide (28; 0.78 g, 0.0023237 mol) was added to the above solution, and the resulting reaction mixture was allowed to stir overnight at rt under argon. The reaction was quenched with water and extracted with ethyl acetate. The organic layer was washed with brine, dried with MgSO4, filtered, and concentrated under vacuum. The product was purified by a silica gel column using hexanes and ethyl acetate (1:1) as eluents to afford 0.050 g of the titled compound as a yellowish solid. Yield = 6.3%. 1H NMR (400 MHz, DMSO-d6): δ 10.77 (s, 1H, NH), 8.25 (s, 1H, ArH), 8.10 (d, J = 8.2 Hz, 1H, ArH), 7.96 (d, J = 8.2 Hz, 1H, ArH), 7.85 (d, J = 4.4 Hz, 1H, pyrazole-H), 7.47 (d, J = 4.4 Hz, 1H, pyrazole-H), 4.35–4.30 (m, 1H, CH), 4.12–4.07 (m, 1H, CH), 3.12–3.10 (m, 1H, CH), 1.22 (d, J = 6.8 Hz, 3H, CH3). MS (ESI) m/z: 341.14 [M + H]+.
4.1.4.46. N-(4-Cyano-3-(trifluoromethyl)phenyl)-3-(4-fluoro-1H-pyrazol-1-yl)propanamide (29c).
29c was prepared following general procedure A as per Scheme 4, where Y in 27 is secondary carbon [CH2]] instead of quarternary [C(OH)(CH3)]. To a solution of 4-fluoro-pyrazole (20; 0.20 g, 0.0023237 mol) in anhydrous THF (5 mL), which was cooled in an ice–water bath under an argon atmosphere, was added sodium hydride (60% dispersion in oil, 0.28 g, 0.0069711 mol). After addition, the resulting mixture was stirred for 3 h. 3-Bromo-N-(4-cyano-3-(trifluoromethyl)phenyl)propanamide (28; 0.75 g, 0.0023237 mol) was added to the above solution, and the resulting reaction mixture was allowed to stir overnight at rt under argon atmosphere. The reaction was quenched with water and extracted with ethyl acetate. The organic layer was washed with brine, dried with MgSO4, filtered, and concentrated under vacuum. The product was purified by a silica gel column using DCM and methanol (19:1) as eluents to afford 0.75 mg of the titled compound as a white solid. Yield = 10%. 1H NMR (400 MHz, DMSO-d6): δ 10.81 (s, 1H, NH), 8.25 (d, J = 2.4 Hz, 1H, ArH), 8.10 (dd, J = 8.8 Hz, J = 2.4 Hz, 1H, ArH), 7.95 (d, J = 8.8 Hz, 1H, ArH), 7.88 (s, 1H, pyrazole-H), 7.46 (s, 1H, pyrazole-H), 4.35 (t, J = 6.0 Hz, 2H, CH2), 2.79 (t, J = 6.0 Hz, 2H, CH2). MS (ESI) m/z: 325.03 [M − H]−.
4.1.4.47. (S)-4-(5-((4-Fluoro-1H-pyrazol-1-yl)methyl)-5-methyl-2,4-dioxooxazolidin-3-yl)-2-(trifluoromethyl)benzonitrile (29d).
Preparation of 29d proceeded by cyclization of the 2-methyl-2-hydroxy-propanamide linker of 10 form an oxazolidinedione ring system. To a solution of 10 (0.234 g, 0.0006568 mol) in anhydrous pyridine (8 mL) was added 1,1′-carbonyldiimidazole (CDI) (0.16 g, 0.0009825 mol). After addition, the resulting mixture was allowed to stir overnight at rt under argon atmosphere. The reaction was quenched with water and extracted with ethyl acetate. The organic layer was washed with brine, dried with MgSO4, filtered, and concentrated under vacuum. The product was purified by a silica gel column using hexanes and ethyl acetate (2:1) as eluents to afford 0.134 g of the titled compound as a white foam. Yield = 42%. 1H NMR (400 MHz, DMSO-d6): δ 8.41 (d, J = 8.0 Hz, 1H, ArH), 7.98 (s, 1H, ArH), 7.94 (d, J = 4.0 Hz, 1H, pyrazole-H), 7.85 (d, J = 8.2 Hz, 1H, ArH), 7.58 (d, J = 4.4 Hz, 1H, pyrazole-H), 4.78 (d, J = 14.8 Hz, 1H, CH), 4.69 (d, J = 14.8 Hz, 1H, CH), 1.71 (s, 3H, CH3). HRMS [C16H11F4N4O3+]: calcd, 383.0767; found, 383.0726. Purity: 97.64% (HPLC).
4.1.4.48. (S)-3-(4-Amino-1H-pyrazol-1-yl)-1-((4-cyano-3-(trifluoromethyl)phenyl)amino)-2-methyl-1-oxopropan-2-yl 2-chloroacetate (29e).
Under argon atmosphere, to a solution of 16t (0.17 g, 0.48 mmol) and triethylamine (0.16 mL, 1.15 mmol) in 10 mL of anhydrous DCM was added 2-chloroacetyl chloride (0.04 mL, 0.58 mmol) in an ice–water bath. After stirring for 30 min, the temperature was raised to rt and the mixture was stirred for 2 h. The reaction mixture was condensed under vacuum and then dispersed into 10 mL of EtOAc, washed with water, evaporated, dried over anhydrous MgSO4, and evaporated to dryness. The mixture was purified with flash column chromatography using hexanes and ethyl acetate as eluents (2/1, v/v) to produce the titled compound as yellow solids. Yield = 19%. 1H NMR (400 MHz, CDCl3): δ 9.22 (br s, 1H, NH), 8.10 (br s, 2H, NH2), 7.93 (d, J = 1.6 Hz, 1H), 7.89–7.86 (m, 2H), 7.78 (d, J = 8.4 Hz, 1H), 7.53 (br s, 1H), 5.16 (d, J = 14.8 Hz, 1H), 4.61 (d, J = 14.8 Hz, 1H), 4.15 (s, 2H), 1.78 (s, 3H); 19F NMR (CDCl3, decoupled): δ −62.19. MS (ESI) m/z: 452.01 [M + Na]+; 428.03 [M − H]−.
4.1.4.49. (S)-N-(3-((4-Cyano-3-(trifluoromethyl)phenyl)amino)-2-hydroxy-2-methyl-3-oxopropyl)-1H-pyrazole-4-carboxamide (29f).
29f was prepared following general procedure A as per Scheme 4. The product was purified by a silica gel column using DCM and methanol (19:1) as an eluent to afford the titled compound as a brown solid. Yield = 43%. 1H NMR (400 MHz, acetone-d6): δ 9.92 (br s, 1H, NHCO), 8.44 (d, J = 1.8 Hz, 1H), 8.24 (dd, J = 8.8, J = 1.8 Hz, 1H), 8.12 (s, 1H), 8.03 (d, J = 1.8 Hz, 1H), 7.84 (s, 1H), 7.11 (br s, 1H, NHCO), 6.38 (br s, 1H, NH), 5.74 (s, OH), 4.67 (d, J = 14.0 Hz, 1H), 4.39 (d, J = 14.0 Hz, 1H), 1.50 (s, 3H). 19F NMR (acetone-d6, decoupled): δ 114.69. HRMS [C16H15F3N5O3+]: calcd, 382.1127; found, 382.1051.
4.2. Biological Methods.
4.2.1. Competitive Ligand-Binding Assay.
AR ligand-binding assay was performed as described previously using purified AR-LBD cloned from rat prostate.1,51
4.2.2. AR Transactivation Assay.
HEK-293 cells plated in 24-well plates at 70,000 cells/well were transfected using the lipofectamine transfection reagent (Life Technologies, Carlsbad, CA). Cells were transfected with 0.25 μg of GRE-LUC, 25 ng of CMV-hAR, and 10 ng of CMV-LUC. Cells were treated 24 h after transfection and the luciferase assay performed 48 h after transfection. Firefly luciferase assay values were normalized to Renilla luciferase assay numbers.
4.2.3. Mutant AR (F876L) and wt PR Transactivation Assay.
COS cells were plated at 70,000 cells/well of a 24-well plate in DME plus 5% csFBS without phenol red. Cells were transfected with 0.25 μg of GRE-LUC, 10 ng of CMV-renilla LUC, and 50 ng of pCR3.1-hPR(wt) or F876L AR using the lipofectamine transfection reagent in an optiMEM medium. Medium was changed 24 h after transfection to DME +5% csFBS without phenol red and treated with a dose response of various drugs (1 pM to 10 μM) in the presence or absence of 0.1 nM progesterone (PR) or R1881 (F876L AR). The luciferase assay was performed 24 h after treatment on a Biotek synergy 4 plate reader. Firefly luciferase values were normalized to Renilla luciferase values.
4.2.4. AR-Dependent Gene Expression in LNCaP Cells (Figure 4).
LNCaP cells were plated in 96-well plates in RPMI plus 1% csFBS without phenol red. Cells were maintained in this medium for 2 days and treated in the presence of 0.1 nM R1881. Twenty-four hours after treatment, the cells were harvested, RNA was isolated, and cDNA was prepared using Cells-to-CT kit (Life Technologies). Expression of genes was measured using real-time PCR using TaqMan primers and probe (Life Technologies).
4.2.5. Cellular Proliferation Assays in MR49F LNCaP Cells (Figure 5).
MR49F cells were plated in 96-well plates in RPMI plus 1% csFBS without phenol red. Cells were treated in this medium in the presence of 0.1 nM R1881 for 6 days with a medium change and retreatment after 3 days. The number of viable cells was measured using CellTiter-Glo (Promega).
4.2.6. Western Blot.
Indicated cell lines were treated for 24 h. Cells were harvested and protein-extracted, and Western blot for AR, AR-SV, and GAPDH was performed using an AR PG-21 rabbit polyclonal antibody that binds to the N-terminus of the AR.1,51
4.2.7. In Vitro Metabolism Assays.
In vitro metabolism assays were performed as described before.1,51 Metabolism assays were performed in MLM, RLM, and HLM as described before.
4.2.8. In Vivo PK in Rats.
PK studies were conducted at Covance using standard methods as briefly discussed below.
4.2.9. Animal Husbandry and Experimental Design.
Male Sprague Dawley rats from Envigo RMS, Inc. were acclimated to study conditions for 5 days prior to initial dose administration. At initial dosing, the animals were 12 weeks of age. The animals were group-housed (up to three animals/cage/group) in polycarbonate cages with hardwood chip bedding. Certified Rodent Diet #2016C (Envigo RMS, Inc.) was provided ad libitum. Water was provided fresh daily, ad libitum. Environmental controls for the animal room were set to maintain a temperature of 20–26 °C, a relative humidity of 50 ± 20%, and a 12 h light/12 h dark cycle. As necessary, the 12-h dark cycle was interrupted to accommodate study procedures. The test article was prepared in 15% dimethyl sulfoxide (DMSO)/85% polyethylene glycol (PEG) 300 by Covance. Individual doses were calculated based on body weights recorded on day 1 and day 7 of dose administration. A single oral daily dose was administered via a gavage needle on seven consecutive days, and blood was sampled as described below. A single iv dose was administered via a tail vein and blood sample on day 1.
Additional detailed information for the 26a experiment, including the groups, number of animals per group, dose (oral 5, 10, 20, and 30 mg/kg per day for 7 days; iv 10 mg/kg on day 1), and route, is given in the Experimental Design section. Animals were observed for mortality and signs of pain and distress twice daily (a.m. and p.m.), and cage side observations for general health and appearance were done once daily. Animals were weighed at the time of animal selection and on day 1 and day 7 of dose administration.
4.2.10. Sample Collection.
Blood (approximately 0.5 mL) was collected via the jugular vein via a syringe and a needle and transferred into tubes containing K3EDTA on days 1 and 7 from three animals/group predose (day 7 only) and at approximately 0.083, 0.25, 0.5, 1, 3, 6, 12, and 24 h postdose. For iv group, blood (approximately 0.5 mL) was collected via the jugular vein at approximately 0.083, 0.25, 0.5, 1, 3, 6, 12, and 24 h postdose. Blood was maintained in chilled cryoracks prior to centrifugation to obtain plasma. Centrifugation began within 1 h of collection. Plasma was placed into 96-well tubes with barcode labels. Plasma was maintained on dry ice prior to storage at approximately −70 °C. Drug concentrations were measured by established chromatography/mass spectrometry (LC–MS/MS) methods.
4.2.11. Hershberger Assay.
Male rats (6–8 weeks old) were randomized into groups based on body weight. Animals were treated with drugs by oral administration as indicated in the figures for 14 days. Animals were sacrificed, prostate and SVs were weighed, and organ weights were normalized to body weight. Male rats (n = 5/group) were left intact for 13 days. Intact rats were treated with the indicated compounds at the indicated dose by mouth daily for 13 days. Rats were sacrificed on day 14 of treatment and prostate and SVs organs were removed and weighed. Organ weights were normalized to body weight. This 20 mg/kg fixed dose screening Hershberger, which was performed for 10, 21a, 16i (toxic so no data), and 26a. The goal of the experiments was to find compounds with in vivo antiandrogen efficacies greater than 10.
4.2.12. Xenograft Studies.
Xenograft studies were performed at Hera Biolabs (Lexington, KY). Enz-R VCaP (MDVR VCaP; licensed from Dr. Donald McDonnell, Duke University, Durham, NC) cells were implanted subcutaneously in SRG rats (n = 5–7/group) (Hera Biolabs). Once the tumors grow to 1000–2000 mm3, the animals were randomized and treated with the indicated drugs. Tumor volume was measured thrice weekly. Thirty days after treatment, the animals were sacrificed, tumors were weighed, and stored for further analysis.
4.2.13. Animal Studies.
All animal studies were conducted under UTHSC Animal Care and Use Committee approved protocols and in accordance to the UTHSC guidelines. Xenograft studies were conducted at HERA biolabs under ACUC protocols approved by the University of Kentucky ACUC committee.
Supplementary Material
ACKNOWLEDGMENTS
We thank GTx, Inc., and Oncternal Therapeutics, Inc., for supporting this project and Dr. Dejian Ma of UTHSC, College of Pharmacy, for assistance with HPLC purity and HRMS experiments.
Funding
This work was funded in part by a research grant from GTx, Inc., and Oncternal Therapeutics, Inc. (RN and DDM), and by a research grant from National Cancer Institute (NCI) to RN (1R01CA229164).
ABBREVIATIONS
- AR
androgen receptor
- PC
prostate cancer
- CRPC
castration-resistant prostate cancer
- SARDs
selective androgen receptor degraders
- DMPK
distribution, metabolism, and pharmacokinetic
- Enz-R
enzalutamide-resistant
- SV
splice variant
- NTD
N-terminal domain
- DBD
DNA binding domain
- LBD
ligand binding domain
- AF-1
an activation function
- AF-2
an external (solvent exposed) binding site termed activation function-2
- DHT
5α-dihydrotestosterone
- SAR
structure–activity relationship
- PD
pharmacodynamics
- PK
pharmacokinetic
- ADT
androgen deprivation therapy
- CSPC
castration sensitive PC
- wt
wild-type
- UPS
ubiquitin proteasome system
- SV
seminal vesicles
- VP
ventral prostate
- FL
full length
- EWGs
withdrawing groups
- SARM
selective androgen receptor modulator
- GR
glucocorticoid receptor
- ER
estrogen receptor
- PR
progesterone receptor
- MLM
mouse liver microsomes
- HLM
human liver microsomes
- RLM
rat liver microsomes
- RLU
relative light units
- MIB
mibolerone
- PROTACs
proteolysis-targeting chimaeras
- csFBS
charcoal-stripped fetal bovine serum
- GRE
glucocorticoid response element
- NSG
NOD SCID gamma
- TGI
tumor growth inhibitio
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00943.
Additional information on compound characterization, additional biological experiments, and figures (PDF)
Molecular formula strings (CSV)
PK method in rats for 21a (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c00943
Contributor Information
Yali He, Department of Pharmaceutical Sciences, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States.
Dong-Jin Hwang, Department of Pharmaceutical Sciences, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States.
Suriyan Ponnusamy, Department of Medicine, College of Medicine, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States.
Thirumagal Thiyagarajan, Department of Medicine, College of Medicine, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States.
Michael L. Mohler, Department of Pharmaceutical Sciences, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States
Ramesh Narayanan, Department of Medicine, College of Medicine, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States.
Duane D. Miller, Department of Pharmaceutical Sciences, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States.
REFERENCES
- 1.Ponnusamy S; Coss CC; Thiyagarajan T; Watts K; Hwang D-J; He Y; Selth LA; McEwan IJ; Duke CB; Pagadala J; Singh G; Wake RW; Ledbetter C; Tilley WD; Moldoveanu T; Dalton JT; Miller DD; Narayanan R Novel selective agents for the degradation of androgen receptor variants to treat castration-resistant prostate cancer. Cancer Res 2017, 77, 6282–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ponnusamy S; He Y; Hwang D-J; Thiyagarajan T; Houtman R; Bocharova V; Sumpter BG; Fernandez E; Johnson D; Du Z; Pfeffer LM; Getzenberg RH; McEwan IJ; Miller DD; Narayanan R Orally bioavailable androgen receptor degrader, potential next-generation therapeutic for enzalutamide-resistant prostate cancer. Clin. Cancer Res 2019, 25, 6764–6780. [DOI] [PubMed] [Google Scholar]
- 3.Aragon-Ching JB The evolution of prostate cancer therapy: targeting the androgen receptor. Front. Oncol 2014, 4, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel RL; Miller KD; Jemal A Cancer statistics, 2015. Ca-Cancer J. Clin 2015, 65, 5–29. [DOI] [PubMed] [Google Scholar]
- 5.Chen CD; Welsbie DS; Tran C; Baek SH; Chen R; Vessella R; Rosenfeld MG; Sawyers CL Molecular determinants of resistance to antiandrogen therapy. Nat. Med 2003, 10, 33. [DOI] [PubMed] [Google Scholar]
- 6.Torre LA; Bray F; Siegel RL; Ferlay J; Lortet-tieulent J; Jemal A Global cancer statistics, 2012. Ca-Cancer J. Clin 2015, 65, 87–108. [DOI] [PubMed] [Google Scholar]
- 7.Tian X; He Y; Zhou J Progress in antiandrogen design targeting hormone binding pocket to circumvent mutation based resistance. Front. Pharmacol 2015, 6, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delaere KP; Van Thillo EL Flutamide monotherapy as primary treatment in advanced prostatic carcinoma. Semin. Oncol 1991, 18, 13–18. [PubMed] [Google Scholar]
- 9.Kolvenbag GJCM; Blackledge GR; Gotting-Smith K Bicalutamide (casodex) in the treatment of prostate cancer: history of clinical development. Prostate 1998, 34, 61–72. [DOI] [PubMed] [Google Scholar]
- 10.Scher HI; Beer TM; Higano CS; Anand A; Taplin M-E; Efstathiou E; Rathkopf D; Shelkey J; Yu EY; Alumkal J; Hung D; Hirmand M; Seely L; Morris MJ; Danila DC; Humm J; Larson S; Fleisher M; Sawyers CL Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet 2010, 375, 1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chong J; Oh W; Liaw B Profile of apalutamide in the treatment of metastatic castration-resistant prostate cancer: evidence to date. OncoTargets Ther 2018, 11, 2141–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dellis AE; Papatsoris AG Apalutamide: The established and emerging roles in the treatment of advanced prostate cancer. Expet Opin. Invest. Drugs 2018, 27, 553–559. [DOI] [PubMed] [Google Scholar]
- 13.Sugawara T; Baumgart SJ; Nevedomskaya E; Reichert K; Steuber H; Lejeune P; Mumberg D; Haendler B Darolutamide is a potent androgen receptor antagonist with strong efficacy in prostate cancer models. Int. J. Cancer 2019, 145, 1382–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bastos DA; Antonarakis ES Darolutamide for castration-resistant prostate cancer. OncoTargets Ther 2019, 12, 8769–8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Attard G; Reid AHM; Auchus RJ; Hughes BA; Cassidy AM; Thompson E; Oommen NB; Folkerd E; Dowsett M; Arlt W; de Bono JS Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J. Clin. Endocrinol. Metab 2012, 97, 507–516. [DOI] [PubMed] [Google Scholar]
- 16.Chi KN; Agarwal N; Bjartell A; Chung BH; Pereira de Santana Gomes AJ; Given R; Juárez Soto Á; Merseburger AS; Özgüroğlu M; Uemura H; Ye D; Deprince K; Naini V; Li J; Cheng S; Yu MK; Zhang K; Larsen JS; McCarthy S; Chowdhury S; TITAN Investigators. Apalutamide for metastatic, castration-sensitive prostate cancer. N. Engl. J. Med 2019, 381, 13–24. [DOI] [PubMed] [Google Scholar]
- 17.Heidegger I; Brandt MP; Heck MM Treatment of non-metastatic castration resistant prostate cancer in 2020: What is the best? Urol. Oncol.: Semin. Orig. Invest 2020, 38, 129–136. [DOI] [PubMed] [Google Scholar]
- 18.Nuhn P; De Bono JS; Fizazi K; Freedland SJ; Grilli M; Kantoff PW; Sonpavde G; Sternberg CN; Yegnasubramanian S; Antonarakis ES Update on systemic prostate cancer therapies: management of metastatic castration-resistant prostate cancer in the era of precision oncology. Eur. Urol 2019, 75, 88–99. [DOI] [PubMed] [Google Scholar]
- 19.Kwan EM; Thangasamy IA; Teh J; Alghazo O; Sathianathen NJ; Lawrentschuk N; Azad AA Navigating systemic therapy for metastatic castration-naïve prostate cancer. World J. Urol 2020, 38 DOI: 10.1007/s00345-019-03060-7. [DOI] [PubMed] [Google Scholar]
- 20.Scher HI; Sawyers CL Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol 2005, 23, 8253–8261. [DOI] [PubMed] [Google Scholar]
- 21.Ito Y; Sadar MD Enzalutamide and blocking androgen receptor in advanced prostate cancer: lessons learnt from the history of drug development of antiandrogens. Res. Rep. Urol 2018, 10, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harris WP; Mostaghel EA; Nelson PS; Montgomery B Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol 2009, 6, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan ME; Li J; Xu HE; Melcher K; Yong E.-l. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin 2014, 36, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Visakorpi T; Hyytinen E; Koivisto P; Tanner M; Keinänen R; Palmberg C; Palotie A; Tammela T; Isola J; Kallioniemi O-P In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet 1995, 9, 401. [DOI] [PubMed] [Google Scholar]
- 25.Linja MJ; Savinainen KJ; Sarämaki OR; Tammela TLJ; Vessella RL; Visakorpi T Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res 2001, 61, 3550–3555. [PubMed] [Google Scholar]
- 26.Yamaoka M; Hara T; Kusaka M Overcoming persistent dependency on androgen signaling after progression to castration-resistant prostate cancer. Clin. Cancer Res 2010, 16, 4319–4324. [DOI] [PubMed] [Google Scholar]
- 27.Veldscholte J; Ris-Stalpers C; Kuiper GGJM; Jenster G; Berrevoets C; Claassen E; van Rooij HCJ; Trapman J; Brinkmann AO; Mulder E A mutation in the ligand binding domain of the androgen receptor of human INCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem. Biophys. Res. Commun 1990, 173, 534–540. [DOI] [PubMed] [Google Scholar]
- 28.Grasso CS; Wu Y-M; Robinson DR; Cao X; Dhanasekaran SM; Khan AP; Quist MJ; Jing X; Lonigro RJ; Brenner JC; Asangani IA; Ateeq B; Chun SY; Siddiqui J; Sam L; Anstett M; Mehra R; Prensner JR; Palanisamy N; Ryslik GA; Vandin F; Raphael BJ; Kunju LP; Rhodes DR; Pienta KJ; Chinnaiyan AM; Tomlins SA The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scher HI; Sayers CL Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol 2005, 23, 8253. [DOI] [PubMed] [Google Scholar]
- 30.Gregory CW; He B; Johnson RT; Ford OH; Mohler JL; French FS; Wilson EM A Mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res 2001, 61, 4315–4319. [PubMed] [Google Scholar]
- 31.Wen Y; Hu MC; Makino K; Spohn B; Bartholomeusz G; Yan DH; Hung MC HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res 2000, 60, 6841. [PubMed] [Google Scholar]
- 32.Majumder PK; Sellers WR Akt-regulated pathways in prostate cancer. Oncogene 2005, 24, 7465–7474. [DOI] [PubMed] [Google Scholar]
- 33.Ueda T; Sadar MD; Suzuki H; Akakura K; Sakamoto S; Shimbo M; Suyama T; Imamoto T; Komiya A; Yukio N; Ichikawa T Interleukin-4 in patients with prostate cancer. Anticancer Res 2005, 25, 4595–4598. [PubMed] [Google Scholar]
- 34.Ueda T; Mawji NR; Bruchovsky N; Sadar MD Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J. Biol. Chem 2002, 277, 38087–38094. [DOI] [PubMed] [Google Scholar]
- 35.Culig Z; Hobisch A; Cronauer MV; Radmayr C; Trapman J; Hittmair A; Bartsch G; Klocke RH Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res 1994, 54, 5447–5448. [PubMed] [Google Scholar]
- 36.Antonarakis ES; Chandhasin C; Osbourne E; Luo J; Sadar MD; Perabo F Targeting the N-Terminal Domain of the Androgen Receptor: A New Approach for the Treatment of Advanced Prostate Cancer. Oncologist 2016, 21, 1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dutt SS; Gao AC Molecular mechanisms of castration-resistant prostate cancer progression. Future Oncol 2009, 5, 1403–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Maeseneer DJ; Van Praet C; Lumen N; Rottey S Battling resistance mechanisms in antihormonal prostate cancer treatment: novel agents and combinations. Urol. Oncol.: Semin. Orig. Invest 2015, 33, 310–321. [DOI] [PubMed] [Google Scholar]
- 39.Taplin ME; Bubley GJ; Ko Y-J; Small EJ; Upton M; Rajeshkumar B; Balk SP Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res 1999, 59, 2511–2515. [PubMed] [Google Scholar]
- 40.Borgmann H; Lallous N; Ozistanbullu D; Beraldi E; Paul N; Dalal K; Fazli L; Haferkamp A; Lejeune P; Cherkasov A; Gleave ME Moving towards precision urologic oncology: targeting enzalutamide-resistant prostate cancer and mutated forms of the androgen receptor using the novel inhibitor darolutamide (ODM-201). Eur. Urol 2018, 73, 4–8. [DOI] [PubMed] [Google Scholar]
- 41.Li Y; Yang R; Henzler CM; Ho Y; Passow C; Auch B; Carreira S; Nava Rodrigues D; Bertan C; Hwang TH; Quigley DA; Dang HX; Morrissey C; Fraser M; Plymate SR; Maher CA; Feng FY; de Bono JS; Dehm SM Diverse AR gene rearrangements mediate resistance to androgen receptor inhibitors in metastatic prostate cancer. Clin. Cancer Res 2020, 26, 1965–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sadar MD Small molecule inhibitors targeting the ‘Achilles’ heel of androgen receptor activity. Cancer Res 2011, 71, 1208–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crews CM Inducing Protein Degradation as a Therapeutic Strategy. J. Med. Chem 2018, 61, 403–404. [DOI] [PubMed] [Google Scholar]
- 44.Han X; Wang C; Qin C; Xiang W; Fernandez-Salas E; Yang C-Y; Wang M; Zhao L; Xu T; Chinnaswamy K; Delproposto J; Stuckey J; Wang S Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem 2019, 62, 941–964. [DOI] [PubMed] [Google Scholar]
- 45.Churcher I Protac-induced protein degradation in drug discovery: breaking the rules or just making new ones? J. Med. Chem 2018, 61, 444–452. [DOI] [PubMed] [Google Scholar]
- 46.Shibata N; Nagai K; Morita Y; Ujikawa O; Ohoka N; Hattori T; Koyama R; Sano O; Imaeda Y; Nara H; Cho N; Naito M Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands. J. Med. Chem 2018, 61, 543–575. [DOI] [PubMed] [Google Scholar]
- 47.Kim DH; Rossi JJ Strategies for silencing human disease using RNA interference. Nat. Rev. Genet 2007, 8, 173. [DOI] [PubMed] [Google Scholar]
- 48.Davidson BL; McCray PB Jr. Current prospects for RNA interference-based therapies. Nat. Rev. Genet 2011, 12, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salami J; Alabi S; Willard RR; Vitale NJ; Wang J; Dong H; Jin M; McDonnell DP; Crew AP; Neklesa TK; Crews CM Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol 2018, 1, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stone L UT-34: a promising new AR degrader. Nat. Rev. Urol 2019, 16, 640. [DOI] [PubMed] [Google Scholar]
- 51.Hwang D-J; He Y; Ponnusamy S; Mohler ML; Thiyagarajan T; McEwan IJ; Narayanan R; Miller DD New generation of selective androgen receptor degraders: our initial design, synthesis, and biological evaluation of new compounds with enzalutamide-resistant prostate cancer activity. J. Med. Chem 2019, 62, 491–511. [DOI] [PubMed] [Google Scholar]
- 52.de Jesus Cortez F; Nguyen P; Truillet C; Tian B; Kuchenbecker KM; Evans MJ; Webb P; Jacobson MP; Fletterick RJ; England PM Development of 5N-bicalutamide, a high-affinity reversible covalent antiandrogen. ACS Chem. Biol 2017, 12, 2934–2939. [DOI] [PubMed] [Google Scholar]
- 53.Okegawa T; Ninomiya N; Masuda K; Nakamura Y; Tambo M; Nutahara K AR-V7 in circulating tumor cells cluster as a predictive biomarker of abiraterone acetate and enzalutamide treatment in castration-resistant prostate cancer patients. Prostate 2018, 78, 576–582. [DOI] [PubMed] [Google Scholar]
- 54.Hershberger LG; Shipley EG; Meyer RK Myotrophic activity of 19-nortestosterone and other steroids determined by modified levator ani muscle method. Proc. Soc. Exp. Biol. Med 1953, 83, 175–180. [DOI] [PubMed] [Google Scholar]
- 55.Youson JH Absorption and transport of ferritin and exogenous horseradish peroxidase in the opisthonephric kidney of the sea lamprey. I. The renal corpuscle. Can. J. Zool 1975, 53, 571–581. [DOI] [PubMed] [Google Scholar]
- 56.Clegg NJ; Wongvipat J; Joseph JD; Tran C; Ouk S; Dilhas A; Chen Y; Grillot K; Bischoff ED; Cai L; Aparicio A; Dorow S; Arora V; Shao G; Qian J; Zhao H; Yang G; Cao C; Sensintaffar J; Wasielewska T; Herbert MR; Bonnefous C; Darimont B; Scher HI; Smith-Jones P; Klang M; Smith ND; De Stanchina E; Wu N; Ouerfelli O; Rix PJ; Heyman RA; Jung ME; Sawyers CL; Hager JH ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res 2012, 72, 1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moilanen A-M; Riikonen R; Oksala R; Ravanti L; Aho E; Wohlfahrt G; Nykanen PS; Tormakangas OP; Palvimo JJ; Kallio PJ Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci. Rep 2015, 5, 12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clegg NJ; Wongvipat J; Joseph JD; Tran C; Ouk S; Dilhas A; Chen Y; Grillot K; Bischoff ED; Cai L; Aparicio A; Dorow S; Arora V; Shao G; Qian J; Zhao H; Yang G; Cao C; Sensintaffar J; Wasielewska T; Herbert MR; Bonnefous C; Darimont B; Scher HI; Smith-Jones P; Klang M; Smith ND; De Stanchina E; Wu N; Ouerfelli O; Rix PJ; Heyman RA; Jung ME; Sawyers CL; Hager JH ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res 2012, 72, 1494–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.