

Abstract
Motivation
Kalign is an efficient multiple sequence alignment (MSA) program capable of aligning thousands of protein or nucleotide sequences. However, current alignment problems involving large numbers of sequences are exceeding Kalign’s original design specifications. Here we present a completely re-written and updated version to meet current and future alignment challenges.
Results
Kalign now uses a SIMD (single instruction, multiple data) accelerated version of the bit-parallel Gene Myers algorithm to estimate pairwise distances, adopts a sequence embedding strategy and the bi-secting K-means algorithm to rapidly construct guide trees for thousands of sequences. The new version maintains high alignment accuracy on both protein and nucleotide alignments and scales better than other MSA tools.
Availability and implementation
The source code of Kalign and code to reproduce the results are found here: https://github.com/timolassmann/kalign.
1 Introduction
Multiple sequence alignment (MSA) remains an important task in biological sequence analysis. MSA programs can be divided into consistency and progressive methods. The latter estimate pairwise sequence distances, construct a guide tree and align sequences following the order of the guide tree. Consistency-based methods tend to be more accurate than compared with progressive methods but are orders of magnitude slower and therefore not practical when aligning thousands of sequences. Kalign (Lassmann et al., 2008) is a progressive alignment method striking a good balance between accuracy and speed compared with other alignment programs on a range of popular benchmark datasets (see e.g. Sievers et al., 2011). Despite having aged well Kalign was not designed to handle the tens of thousands of sequences frequently encountered today. In particular, the original Kalign program uses the unweighted pair group method with arithmetic mean (UPGMA) algorithm to construct a guide tree resulting in quadratic time complexity. More recent alignment programs have overcome this hurdle by implementing heuristics to construct guide trees (Blackshields et al., 2010; Katoh and Toh, 2006).
Here we present a new version of Kalign, introducing a SIMD (single instruction, multiple data) accelerated version of Gene Myers’ bit-parallel algorithm (Myers, 1999) to estimate pairwise sequence distances and adopting the sequence embedding strategy introduced by Blackshields et al. (2010) to speed up the construction of guide trees.
2 Materials and methods
We replaced the fast string matching algorithm used in Kalign2 (Muth and Manber, 1996) with a new implementation of Gene Myers’ approximate string matching algorithm. The algorithm calculates the exact edit distance between two strings using bit-parallel instructions. In the standard implementation the maximum length of a query is equivalent to the size of a computer word (64 characters on 64 bit architectures). However the algorithm lends itself to further parallelization using SIMD instructions including the AVX and AVX2 instructions available on all modern computers. Using these instructions it becomes possible to compare sequences of length 256. Although the implementation of the Gene Myers algorithm is fairly straight forward using AVX instructions some operations are absent from the AVX instruction set and had to be implemented separately. A stand-alone implementation of the algorithm is distributed together with Kalign to facilitate downstream adoption and development.
To estimate pairwise sequence distances Kalign scans the first 256 characters of the shorter sequence across the longer sequence. The distance is defined as the number of edits required to turn one sequence into an exact match in the longer sequence. For distantly related protein sequences the sequence similarity is too low for the algorithm to detect meaningful distances. Therefore, following the method by Steinegger and Söding (2018), Kalign converts all protein sequences into a reduced alphabet by merging (L, M), (I, V), (K, R), (E, Q), (A, S, T), (N, D) and (F, Y) for the purpose of the distance calculation.
Kalign adopts the guide tree construction methods used in clustal omega (Sievers et al., 2011). A number of seed sequences are selected and all sequences are compared against those forming for each sequence a vector of distances to all seeds. The bi-secting k-means algorithm is used to cluster sequences based on the Euclidean distance between these vectors until clusters containing fewer than 100 sequences are found. Here Kalign again uses AVX instructions to accelerate the distance calculation. Finally, the UPGMA method is used to cluster the remaining sequences.
Since the bi-secting k-means algorithm is not guaranteed to discover the optimal split of sequences into two clusters Kalign runs the algorithm 50 times using randomly selected sequences to seed the calculation.
3 Results
We compared the performance of Kalign against two other popular progressive alignment methods muscle (Edgar, 2004) and clustal omega (Sievers et al., 2011). We used the Balibase (Thompson et al., 1999), Quantest2 (Sievers and Higgins, 2019), Bralibase (Gardner et al., 2005) and HomFam benchmark datasets (Fig. 1). Clustal omega and Muscle were run with parameters recommended for large alignments on the BaliFam dataset (Clustal:-threads = 8 -MAC-RAM = 48 000 -iterations = 2; Muscle: -maxiters 2), but otherwise default parameters were used.
Fig. 1.
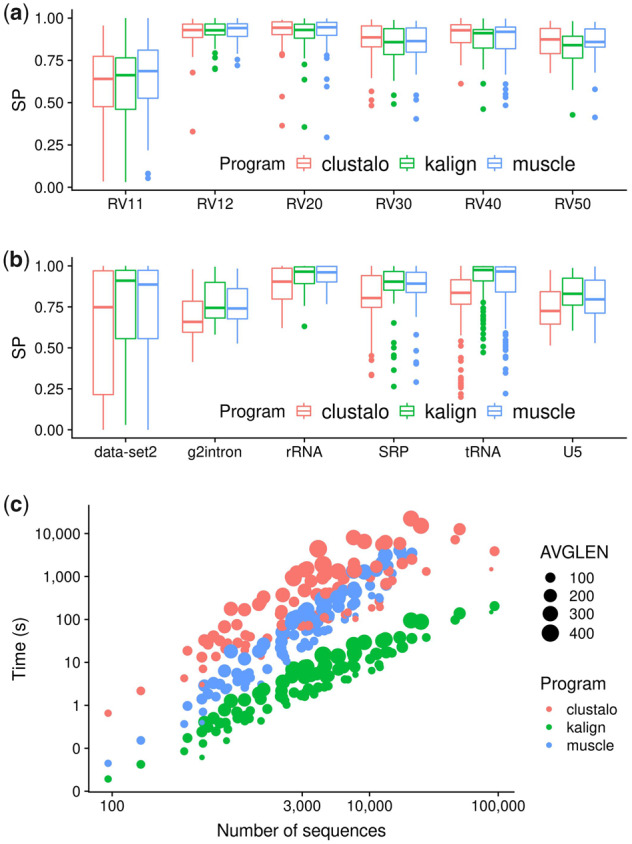
Benchmark results. (a) Sum of pairs scores (SP) of all tested alignment programs on Balibase protein alignment datasets. (b) SP scores of RNA bralibase alignments. (c) Computational performance assessed on the HomFam dataset
Kalign’s performance on all six Balibase categories is statistically indistinguishable from the other two programs (two sample t-test, corrected P < 0.05). Likewise there is no statistical difference in alignment accuracy on the Quantest2 benchmark dataset (results not shown). Kalign’s mean performance is significantly better compared with the other two programs in two out of the six Bralibase alignment categories. However, we note that the performance of all algorithms can vary dramatically depending on the specific alignment case (see Fig. 1, box plot error bars and outliers). Therefore, we do not assume that good performance on an MSA benchmark sets generalizes and recommend users to manually inspect their alignments and compare the results of different alignment programs.
Kalign compares favorably to the other two programs in terms of running times and scalability on the Balifam dataset (Fig. 1c). In all alignment cases Kalign is one to two orders of magnitude quicker and compared with clustal omega only uses a single CPU core.
4 Conclusion
We present a new version of Kalign that outperforms other programs in terms of running times while sacrificing little in terms of accuracy. This combination makes Kalign especially attractive in large alignment problems.
Acknowledgements
I would like to thank Max Burroughs for providing feedback on Kalign.
Funding
T.L. is supported by a fellowship from the Feilman Foundation.
Conflict of Interest: none declared.
References
- Blackshields G. et al. (2010) Sequence embedding for fast construction of guide trees for multiple sequence alignment. Algorithms Mol. Biol., 5, 21.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. (2004) Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res., 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner P. et al. (2005) A benchmark of multiple sequence alignment programs upon structural RNAs. Nucleic Acids Res., 33, 2433–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Toh H. (2007) Parttree: an algorithm to build an approximate tree from a large number of unaligned sequences. Bioinformatics, 23, 372–374. [DOI] [PubMed] [Google Scholar]
- Lassmann T. et al. (2009) Kalign2: high-performance multiple alignment of protein and nucleotide sequences allowing external features. Nucleic Acids Res., 37, 858–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth R., Manber U. (1996) Approximate multiple string search. In: Annual Symposium on Combinatorial Pattern Matching, pp. 75–86.
- Myers G. (1999) A fast bit-vector algorithm for approximate string matching based on dynamic programming. J. ACM, 46, 395–415. [Google Scholar]
- Sievers F., Higgins D.G. (2019) Quantest2: benchmarking multiple sequence alignments using secondary structure prediction. Bioinformatics, doi: 10.1093/bioinformatics/btz552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F. et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol., 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M., Söding J. (2018) Clustering huge protein sequence sets in linear time. Nat. Commun., 9, 2542.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D. et al. (1999) Balibase: a benchmark alignment database for the evaluation of multiple alignment programs. Bioinformatics, 15, 87–88. [DOI] [PubMed] [Google Scholar]