

Abstract
The polo-box domain (PBD) of PLK1 determines mitotic substrate recognition and subcellular localization. Compounds that target PLK1 selectively are required due to the tumor suppressor roles of PLK3. A structure-activity analysis of the PBD phosphopeptide binding motif has identified potent peptides that delineate the determinants required for mimicry by non-peptidic inhibitors and provide insights into the structural basis for the selectivity of inhibitors for the PLK1 PBD. Fragment-ligated inhibitory peptides (FLIPs) obtained through REPLACE have been optimized to enhance in vitro binding and a systematic analysis of selectivity for PLK1 vs 3 has been carried out for peptides and peptidomimetics. Furthermore, these more drug-like non-ATP competitive inhibitors had on target engagement in a cellular context as evidenced by stabilization of PLK1 in a thermal shift assay and by inhibition of the phosphorylation of TCTP, a target of PLK1. Investigation in cells expressing a mutant PLK1 showed that these are sensitive to PBD inhibitors but dramatically resistant to clinically investigated ATP competitive compounds. These results provide further validity of targeting the PBD binding site and progress towards PBD-inhibitors active against tumors resistant to ATP-inhibitors.
Keywords: PLK, mitosis, kinase, peptide, protein-protein interaction, inhibitor
Graphical Abstract
The polo-box domain (PBD) of PLK1 determines mitotic substrate recognition and localization. Compounds obtained through REPLACE have been optimized to enhance in binding and undertake systematic analysis of selectivity for PLK1 vs PLK3. These more drug-like inhibitors had on target engagement in a cellular thermal shift assay and by inhibition of the phosphorylation of TCTP, a target of PLK1. Results demonstrate further validity of targeting the PBD binding site and progress inhibitors active against tumors resistant to ATP-inhibitors.
Introduction
The polo-like kinases play key roles in mitosis [1] and inhibition of PLK1 activity leads to anti-proliferative effects [1b, 2]. The four-known human PLKs (with kinase domains, PLK5 does not) have distinct functions despite their sequence similarity especially in the kinase domain. PLK1 is frequently overexpressed in cancer and is a negative prognostic indicator for patient outcomes [3] especially in prostate[4] and colorectal cancers [5]. Furthermore PLK1 has been demonstrated to be a potential therapeutic target for tumors with inactivated p53 [6] with evidence showing that PLK1 is oncogenic when p53 is mutated [7]. The therapeutic rationale for PLK inhibition has been validated in vitro and in vivo [8] and numerous inhibitors of the ATP binding site of PLKs have been identified, with some entering clinical trials after showing significant anti-tumor activity in preclinical models. Results from two compounds suggest acceptable toxicity profiles warranting further investigation in phase II trials [9]. BI-6727 (volasertib) was granted FDA breakthrough therapy designation for Acute Myeloid Leukemia, however apparently did not show good efficacy in subsequent trials. Moreover, there are numerous drawbacks to targeting the ATP cleft, including prominently the inhibition of the three other known members of the mammalian PLKs [10]. Due to its tumor suppressor roles, PLK3 inhibition may lead to diminution of the anti-tumor effect mediated by blocking PLK1 [11] suggesting that inhibiting PLK3 can be deleterious. ATP competitive inhibitors will not necessarily block critical non-catalytic functions of PLK1 necessitating alternative approaches.
The sub-cellular targeting binding site in the polo-box domain (PBD) which interacts with phosphosubstrates such as Cdc25C (a phosphatase activating CDK1 allowing mitotic entry) and PBIP (plays a central role in the assembly of kinetochore proteins and facilitates chromosome segregation), is amenable to small molecule inhibitor development[12] and high-throughput screening approaches have been used to generate small molecule inhibitors of the PBD-peptide interaction. For the most part however, these are either weakly binding or non-drug-like in nature [13] although one compound, Poloxin, has been improved through the addition of a hydrophobic tag (still relatively weak in terms of anti-proliferative activity)[14]. Some inhibitors possess a contrasting phenotype to PLK1 knockdown and catalytic inhibition [10, 15] suggesting that their mechanism is not exclusively through on target activity. Derivatized peptides that occupy a novel site in the PBD binding groove [16] have been reported however are extensive modified, complex molecules and overall are non-drug-like. In addition, the concentrations required for cellular activity indicate inefficient cell uptake.
Peptides however, while non-drug-like can bind selectively to PLK1 and this provide a structural template for the development of compounds that are metabolically stable and cell permeable. REPLACE, a validated strategy for the iterative discovery of non-peptidic protein-protein interaction inhibitors, has been utilized to discover fragment alternatives for the N-terminal hydrophobic motif in a Cdc25C PBD substrate peptide[17]. In this present study structural determinants for peptide binding to the PBDs of PLK1 have been defined, and Fragment Ligated Inhibitory Peptides (FLIPs) with improved binding have been generated. In addition, a detailed evaluation of affinity of ligands for the PBD of PLK3 has been completed and used to generate a selectivity index for PLK1, a novel analysis of selectivity of PBD ligands. Cellular studies with FLIPs demonstrate progress towards obtaining cell permeable compounds that are structurally much less complex than previously described peptidomimetics since they do not require pegylation or masking of the phosphothreonine. These FLIPs engage PLK1 at a cellular level and have antiproliferative activities consistent with PLK1 inhibition while retaining activity against cell lines expressing a mutant PLK1 resistant to ATP directed PLK1 inhibitors. Such compounds make excellent starting points for development as non-ATP competitive PLK1 inhibitors since they preserve selectivity and potency towards the PLK1 PBD while imparting characteristics for drug-likeness.
Results and Discussion
Previous studies have reported initial SAR studies on peptides from the Cdc25C (LLCSpTPNGL) and PBIP (PLHSpTAI) phosphopeptide substrate motifs, two key PBD interacting proteins involved in mitotic regulation[12]. In order to further establish structure-activity relationships for the PBD binding sequence, a peptide library was designed to probe the contributions of the N- and C-terminal residues of the recognition sequences from Cdc25C and PBIP in a systematic fashion and these compounds were tested in a fluorescence polarization (FP) assay to quantify competitive binding of phosphopeptides to the PBD domain of PLK1[17]. Further to this, a similar assay format was developed for the PLK3 PBD to determine the selectivity of PBD inhibitors which has not previously been investigated in depth. Yun et al. looked at the binding of limited number of peptides to PLK2 by ITC but not PLK3[12a]. In the first instance, the binding of the fluorescein labeled tracer peptides used for both assays was evaluated to see if there was a difference in affinity for their respective PBD. A titration curve for each tracer/PLK PBD complex was generated to determine Kd values for each and therefore compare the relative binding affinities. The PLK1 PBD fluorescent tracer was found to bind to the PLK1 PBD with a Kd of 4.6 nM, while the corresponding value for the PLK3 binding tracer to the PLK3 PBD was determined to be 27.2 nM. These results demonstrate that the tracer peptide for the PLK1 PBD bound with a 5-fold higher affinity than the PLK3 tracer to its PBD thereby suggesting that this should be accounted for when comparing the selectivity of peptides and other drug-like PBD inhibitors. A selectivity index was calculated to provide novel insights into the true selectivity of both compounds previously described and those generated in this study (Table 1).
Table 1.
In vitro binding activ ity for PLK PBD Peptides
| Compound | Sequence | PLK1 PBD FP IC 50 [μM] | PLK3 PBD FP IC 50[μM] | Selectivity Index |
|---|---|---|---|---|
| 1 | LLCS[pT]PNGL | 0.17 ± 0.03 | >600 | >17,000 |
| 2 | LLCSTPNGL | >600 | ND | ND |
| 3 | Ac-PLHS[pT]PNGL | 0.06 ± 0.04 | 163.6 | 13,633 |
| 4 | Ac-PLHS[pT]AI | 0.064 ± 0.01 | 270.5 | 21,133 |
| 5 | Ac-LHS[pT]AI | 1.57 ± 0.47 | >600 | >1900 |
| 6 | Ac-PLHS[pT]A | 0.46 ± 0.12 | >600 | >6500 |
| 7 | GPLATS[pT]PKNG | 0.0057 ± 0.002 | 2.2 | 1928 |
| 8 | GPLATS[pT]PNGL | 0.011 ± 0.003 | 6.3 | 2864 |
| 9 | Ac-PLHS[pT]PKNG | 0.032 ± 0.012 | 133.8 | 20,906 |
The affinities of the peptides and FLIPs for PLK1 and PLK3 were subsequently measured by plotting the loss of polarization against increasing concentration of competitor peptide building on previous work through use of an optimized assay and selectivity determination [17]. The native phospho-sequence from Cdc25C was determined to potently bind to the PLK1 PBD (1, IC50 = 0.17 μM) and to be highly selective with no PLK3 PBD binding apparent at the maximum concentration tested (Table 1). As previously shown, removal of the phosphate (2) and introduction of Glu in place of phosphothreonine led to almost complete loss of binding[17]. To probe the contributions of the N and C-terminal peptide regions in both the Cdc25C and PBIP context, a series of chimeric peptides were constructed. These modifications were found to significantly increase binding compared to either individual context (3, IC50 = 0.06 μM). Importantly, this peptide retains strong selectivity for PLK1 over PLK3 (IC50 = 163 μM). The two-residue sequence (Ala- Ile) C-terminal to the phosphothreonine was found to have comparable contribution to binding as the PNGL C-terminal tetrapeptide (4, 0.064 μM), while decreasing binding to PLK3 (270 μM), thus improving selectivity. Removal of the N-terminal Pro from this peptide (5, IC50 = 1.57 μM) resulted in a large potency drop-off for the PLK1 PBD, which also lost all detectable affinity for the PBD of PLK3. Truncation of the C-terminal isoleucine led to a 7-fold reduction in binding (6, IC50 = 0.46 μM). Peptide 7 is a consensus recognition sequence for PLK3 reported in the literature and was initially synthesized as the fluorescein labeled tracer for the PLK3 PBD [13a]. Testing showed that this peptide (without fluorescein) had a measured IC50 of 2.2 μM in the PLK3 PBD competition assay. However, the somewhat surprising observation was made that this peptide is highly potent for the PLK1 PBD (IC50 = 0.0057 μM) while retaining almost 2000-fold preferential binding for PLK1 over PLK3. Two more chimeric molecules were synthesized and tested to examine the consequences of combining the Cdc25C and PLK3 binding motifs. Replacing PKNG of the PLK3 consensus peptide with PNGL from the C-terminus of Cdc25C (8, IC50 = 0.011 μM) was found to reduce affinity for both PLK1 and PLK3 by 2 and 3-fold respectively. Furthermore, combining the N-terminus of the PBIP sequence with that of the PLK3 consensus sequence (9, IC50 = 0.032 μM) modestly reduced affinities for both PBD constructs however, the reduction was more for the PBD of PLK3 thereby significantly improving the selectivity index to 21,000.
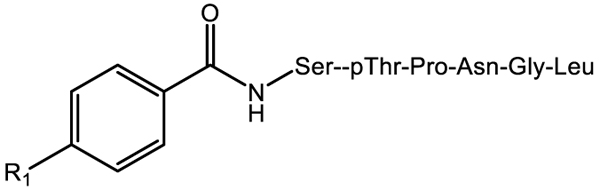
Using REPLACE, low molecular weight fragments are computationally docked into the volume of a binding site known to interact with key peptidic determinants in order to identify more drug-like alternatives and iteratively convert a peptidic compound into a non-peptidic inhibitor [17–18]. Through use of the peptide SAR data, REPLACE was applied to identify fragment alternatives to the N-terminal tripeptide of Cdc25C. This resulted in the identification of PBD inhibitor peptide-small molecule hybrids that, when transfected into cells, recapitulate a PLK1 deficient phenotype[17]. These FLIPs were based on substituted benzamide capped peptides and demonstrated that modification of the 4 n-alkyl substituent contributed to binding with a hydrophobic slot observed in crystal structures of the PBD of PLK1[16a, 16b, 19]. To extend the structure-activity relationship of this series, capping groups containing additional substituents at the 4-position were incorporated into FLIPs by appending onto the C-terminal portion of the Cdc25C sequence, PNGL (Table 2). The PNGL sequence was chosen to maximize affinity of the capped peptide to get the most SAR information. In the first instance, the 4-hydroxybenzamide capped FLIP (10) was completely inactive. The methoxy derivative (11, IC50 = 21.9 μM) had measurable binding to the PBD in the FP assay and therefore subsequent homologs were tested in the series. Extending the alkoxy group by 2 (12) and 3 (13) carbons resulted in subsequent increases in activity to 15.9 and 10.4 μM, respectively, thereby confirming more effective interaction with the hydrophobic groove previously described (Figure 1) and the doubling trend with each successive carbon extension. Two aromatic substituted alkoxy analogs (14, IC50 = 5.0 μM, 15, IC50 = 4.5 μM) had increased binding to the PBD with the 4-phenethoxy (15) being the slightly more effective of the two. In addition to the methoxy derivative, the thiomethyl and the methylamino FLIP derivatives were synthesized and tested (16, IC50 = 3.0 μM, 17, IC50 = 11.2 μM). The propyl homologue of 17 (18, 5.4 μM) increased activity by more than 2-fold, consistent with the improvements observed with alkyl chain length in the alkoxy series. In addition, these were compared with the 4-n-butyl benzamide FLIP previously tested (19, IC50 = 2.5 μM) and revealed that the thiomethyl analog is of comparable potency to the longer alkyl chain. It is therefore apparent in comparison of this isosteric series that the order of S > C > N > O was observed in terms of potency of binding to the PBD. Further validation for extension of the n-alkyl portion of the substituent as a means of improving potency was confirmed by comparison of the propyl amino (18) with 19 and the resulting two-fold potency increase.
Table 2.
PLK PBD In vitro binding for FLIP compounds (Ncap-S[pT]PNGL)
 | ||
|---|---|---|
| Compound | R1 | PLK1 PBD FP IC50 [μM] |
| 10 | OH | >600 |
| 11 | OCH3 | 21.9 ± 4.3 |
| 12 | OCH2CH3 | 15.9 ± 2.1 |
| 13 | OCH2CH2CH3 | 10.4 ± 2.6 |
| 14 | OCH2C6H4F | 5.0 ± 0.86 |
| 15 | OCH2CH2C6H5 | 4.5 ± 0.72 |
| 16 | SCH3 | 3.0 ± 0.72 |
| 17 | NCH3 | 11.2 ± 2.6 |
| 18 | NCH2CH2CH3 | 5.4 ± 1.8 |
| 19 | CH2CH2CH2CH3 | 2.5 ± 0.96 |
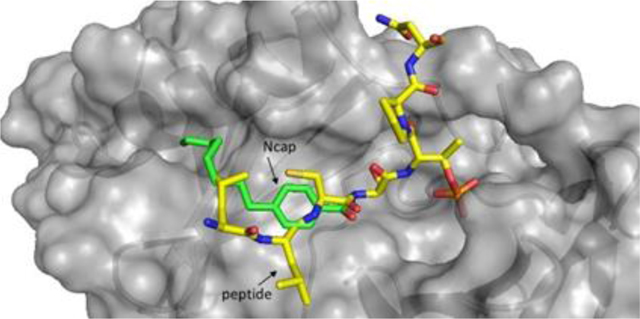
Figure 1.

Interactions of the alkyl group of the benzamide capping group in compound 21 with the PBD of PLK1 (PDB ID: 3RQ7). The hydrophobic groove exploited by the capping group and the interactions of the phosphothreonine are highlighted.
Optimization of 4-n-alkylbenzamido N-cap and truncation studies on residues C-terminal to the phosphothreonine.
As these results (Table 2 and 3) reveal that the 4-position substituent increased potency in this isosteric series[16c], further homologation of the n-alkyl series was the obvious next step in the SAR (Table 3). A FLIP series generated by lengthening the 4-n-alkyl substituent were then tested in the FP assay (19, n-butyl, IC50 = 2.5 μM; 20, n-hexyl, IC50 = 1.0 μM; 21, n-octyl, IC50 = 0.36 μM) and overall resulted in significantly improved affinity for the PLK1 PBD with increasing length of the alkyl substituent. Notably, the n-octyl derivative (21) was found to essentially recapitulate the activity of the native Cdc25C peptide (1). Although 21 measurably bound to the PBD of PLK3 (IC50 = 148 μM), it possesses a 2000-fold selectivity index and thereby retains high specificity for PLK1.
Table 3.
PLK PBD In vitro binding and cellular activity for optimized FLIP compounds.
| Compound | FLIP | Sequence | PLK1 PBD FP IC50 [μM] | PLK3 PBD FP IC50 [μM] | Selectivity Index | MTT IC50 [μM] HeLa | MTT IC50 [μM] PC-3 | MTT IC50 [μM] A549 |
|---|---|---|---|---|---|---|---|---|
| 19 | 4-butyl | S[pT]PNGL | 2.5 ± 0.96 | >600 | >1200 | |||
| 20 | 4-hexyl | S[pT]PNGL | 1.0 ± 0.51 | >600 | >2830 | |||
| 21 | 4-octyl | S[pT]PNGL | 0.36 ± 0.16 | 148.8 ± 38.2 | 2067 | |||
| 22 | 4-octyl | S[pT]A | 15.2 ± 4.74 | 201.9 ± 51.3 | 66 | |||
| 23 | 4-octyl | S[pT]AI | 0.41 ± 0.14 | 152.8 ± 33.5 | 1863 | 128.1 | 55.7 ± 2.7 | 79.45 ± 16.6 |
| 24 | 4-octyl | S[pT]PL | 1.2 ± 0.18 | 620.1 ± 72.5 | 2583 | 142.3 ± 14.4 | 102.7 ± 27.3 | 160.5 ± 61 |
| 25* | 4-octyl | S[pT]AI | 1.49 ± 0.13 | 110.13 ± 5.92 | 369 | 38.9 ± 0.6 | 41.5± 6.5 | |
| 26* | 4-nonyl | S[pT]AI | 1.23 ± 0.81 | 98.45 ± 12.27 | 400 | 38.2 ± 11.7 | 27.0 ± 6.4 | |
C terminal amide
Based on the results for 21 generated as a PNGL capped FLIP, further C-terminal modifications were explored in the context of the octyl-benzamide fragment alternatives (Table 3). Since the peptide SAR results with 4 (0.064 μM) showed that the PNGL sequence could be replaced with two residues without significant potency loss, a 4-octylbenzamide capped S[pT]AI compound was constructed. As expected, this molecule (23, IC50 = 0.41 μM) possessed very similar activity to 21 while retaining its selectivity (SI = 2000). Additionally, the consequences of further truncation were determined by deleting the C-terminal Ile residue. The resulting FLIP (22, IC50 = 15.2 μM) containing a single alanine C-terminal to the phospho-Thr bound to the PLK1 PBD with a ~40-fold weaker affinity revealing a substantial potency loss, which was greater than expected based on the peptide SAR. The binding of 22 to PLK3 was measurable (IC50 = 201 μM) and therefore had dramatically reduced selectivity compared to 21 (SI = 66). When the C-terminal AI of 23 was replaced with proline and leucine, the resulting FLIP (24) bound to both PLK PBDs with 3–4 fold weaker affinity and therefore retained selectivity for PLK1. The C-terminal amide version of 23 was synthesized to determine the impact of removing this charge. Compound 25 was found to have similar competitive binding activity. Furthermore, the n-nonylbenzamide FLIP was synthesized also with a C-terminal amide (26) to further the n-alkyl chain SAR and the resulting assay determination showed equipotency to the octyl derivative (25).
Structural Insights into PLK1 vs. PLK3 selectivity through molecular modeling studies.
In order to shed light into the structural basis for the mimicry of the capping groups and to determine why peptides and FLIPS are selective for PLK1, a homology model for the PBD domain of PLK3 was constructed and compared with crystal structures for the PBD of PLK1 (Figure 2). The PLK3 PBD model was then overlaid with the crystal structure of the Cdc25C peptide used in this study as the starting point for REPLACE (3BZI)[12d]. The striking observation from the sequence and structural alignment is that the majority of residues involved in the phosphopeptide binding interface are strictly conserved between the two homologs and therefore offer little insight into the differential binding of PBD inhibitors. This leads to the conclusion that the dramatically weaker binding of the phosphopeptides largely results from conformational differences between the PBD domains of PLK1 and PLK3. Closer examination of the minor sequence differences however does provide insights into possible reasons for PLK1 selectivity. One of these variations is L491 of PLK1, a residue that directly contacts the phosphothreonine residue in the PBD crystal structure. In the PLK3 PBD, the corresponding residue is M544 and based on the modeling results does not appear to make contacts with the methyl group of the pThr residue that are observed with L491 in PLK1. It is possible that L491 acts as an anchor for the phosphothreonine (critical for high affinity binding) and therefore acts to solidify the many other contacts of this residue. The absence of these contacts would thus result in dramatically lower binding affinity in the PLK3 PBD context. Furthermore, there are conservative differences in the cryptic pocket of the PBD which contacts the hydrophobic tail. These may explain the slightly decreased selectivity of the FLIPs compared to the peptidic sequences.
Figure 2.

Interactions of the Cdc25C phosphopeptide (yellow carbon atoms) with the PBD of both PLK1 (PDB ID: 3BZI) and PLK3 (homology model). PLK1 PBD residues are depicted with green carbon atoms and those of PLK3 are shown with orange carbons.
Cellular Activity of FLIPs
Previously, phosphopeptides and FLIPs from this laboratory were demonstrated to have significant anti-proliferative activity and phenotypes consistent a PLK1 deficient phenotype [17]. In order to transduce these into cells however, a transfection agent was required. In addition, other PBD peptidomimetics have been shown to possess weak cellular activity and PLK1 phenotypes however require structural complexity through pegylation, histidine derivatization and/or masking of the phosphothreonine for this effect[16b–e].
With the increased activity in binding to the PBD and as the next generation FLIPs described here have greater logP values through addition of the 4-octylbenzamide capping group and decreased overall size (after deletion of two C-terminal residues), it was hypothesized that these next generation FLIPs may possess cellular activity without the use of a delivery agent. Accordingly, cell viability was measured using an MTT assay to determine the anti-proliferative activity for 23 and 24. As expected, despite still having peptidic composition and a negatively charged phosphothreonine, these FLIPs demonstrated cellular activity. FLIP 23 had anti-proliferative IC50 values in PTEN deficient prostate cancer (PC3) cells of 55.7 μM, in HeLa cells of 128.1 μM, and Kras mutant (A-549) lung cancers cells of 79.5 μM. The anti-proliferative activity of 24 in the same cells was approximately 2-fold lower yet clearly measurable and consistent with its decreased binding to PLK1 PBD. Interestingly removal of the charge on the C-terminus resulted in significantly increased cellular activity with up to a 3-fold increase being observed with the octyl (25) and nonyl (26) versions of 23 (Table 3). A non-phosphorylated version of 26 (supplementary figure 1) was determined to have no significant anti-proliferative activity in line with known SAR dictating critical charge-charge interactions of the phosphate.
To examine whether the FLIPs interact with and inhibit PLK1 in the cellular context, two different approaches were taken to determine engagement of the target and the anti-target PLK3. First, the stability of PLK1 and PLK3 in cellular extracts was determined by a thermal shift assay (CETSA, supplementary figures 2–7)[20]. Briefly, the thermal shift assay measures the effect of ligands on the thermal stability of target proteins such as PLK1. A positive control dose response with BI-2536 showed an approximately 1.4-fold increase in stabilization of PLK1 against thermal denaturation across a dose range of 0.03 μM to 3.0 μM. Further data (Figure 3) shows that C-terminally amidated FLIP 25 stabilizes PLK1 with an EC50 value of 0.17 μM. PLK3 stabilization was also measured and an effect was seen, albeit to a lower level with the observed EC50 of 0.58 μM. Interestingly FLIP 26 had increased engagement of PLK1 and decreased affinity for PLK3 as shown by the CETSA results (Figure 3). The non-phospho version of 26 as a negative control compound did not engage PLK1 to a significant degree.
Figure 3.

Graph representing densitometry of the western blots of PLK1 and PLK3 across the dose range of the FLIPs listed.
Next, the phosphorylation of a known direct target of PLK1, TCTP (translationally controlled tumor protein), was measured by Western blotting[21]. PC3 cells were synchronized by nocodozole (Noc) block then treated with 25, and p-TCTP was measured with a phosphospecific antibody. The results clearly demonstrate that with increasing dose of 25, decreasing levels of pTCTP are detected, while the total TCTP levels remain unchanged (Figure 4). The catalytic inhibitor BI-2536 served as a positive control for catalytic PLK1 inhibition.
Figure 4.
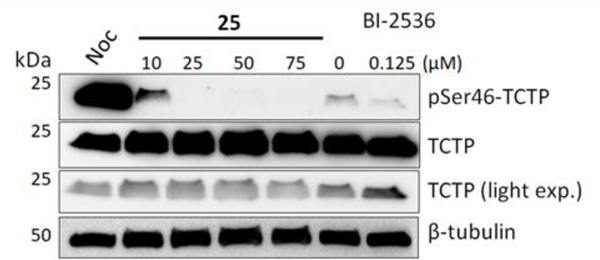
Western blotting analysis of PC-3 cells following 23-hour treatment with 25. Cells were synchronized with nocodazole for 18 h prior to treatment with 25 or BI-2536 for 23 h. Cell lysates were harvested, separated by SDS-PAGE, then immunoblotted for TCTP, p-TCTP, and β-tubulin.
Having confirmed that optimized FLIPs had potent inhibition of the PBD, PLK1 target engagement using two approaches and anti-proliferative activity in tumor cells as a result of improving drug-like properties, their further application in cell lines that have developed resistance to clinically utilized ATP competitive inhibitors of PLK1 was explored. Burkard et al. demonstrated that a single point mutation (C67V) within the ATP-binding domain of PLK1 confers resistance to several structurally unrelated ATP-binding site inhibitors, including BI-2536 [22]. The retinal pigment epithelial cells of this study were obtained and used to test FLIPs in this model of resistance to catalytic inhibitors of PLK1. It was found that, similar to the published results, RPE cells expressing wild-type PLK1 are very sensitive to BI-2536 (IC50 = 21.2 nM) while those expressing the C67V mutant are dramatically resistant to this compound (> 2.5 μM, not shown). The PBD-targeted FLIP 24 inhibited the growth of the PLK1 C67V mutant-expressing cells to an equal or even greater extent as that seen with cells expressing wild-type PLK1 (86.8 μM ± 33.8 versus 158.5 μM ± 8.8, respectively, data not shown). Note the two-fold difference may also result from minor differences in expression between the WT and mutant exogenous PLK1.
The goal of this study was the further development of PBD inhibitors through the REPLACE strategy [17, 23]. Variation of peptide sequences was undertaken to determine the N- and C-terminal contributions within the PBD binding pocket and although peptides are generally not drug-like molecules, it was envisaged that the SAR would facilitate the identification of important residues for substrate binding and selectivity. To this end, a novel detailed investigation of PLK1 vs PLK3 PBD binding was undertaken and revealed key information on the determinants of selectivity. These contributions can then be used to generate FLIP molecules for further development as non-peptidic drug-like small molecules. Therefore, as the precursor to REPLACE, detailed SARs were established for the peptides derived from two major PLK1 substrates and furthermore were evaluated for PLK1 vs PLK3 PBD selectivity. A key objective of this SAR study was to systematically compare chimeric peptides from the different PLK1 substrates to assess contributions of the residues N and C terminal to the S[pT] motif in each context.
Comparison of 1 and 3 revealed that Ac-PLH provides a 3-fold increase relative to LLC and resulted in a highly potent PBD inhibitory peptide with very good PLK1 selectivity. This highlights the contributions of the acetylated N-terminus playing a key role in optimization.
Furthermore, a significant observation in comparing the C-terminus of the Cdc25C and PBIP peptides is that the PBD inhibitory activity of the Cdc25C 9mer could be preserved in a 7-residue peptide as evidenced by the similar potency of 4 and 3. This confirms that the Ala-Ile C-terminal dipeptide can provide sufficient affinity to mimic the interactions of the longer sequence. Importantly the decrease in size also resulted in increased selectivity of 4 for PLK1 (SI of 21,000) thereby providing the basis for optimization of FLIP compounds utilizing this sequence. Further truncation of the sequence of 4 was detrimental to activity in that removal of the isoleucine from the C-terminus led to a 7-fold decrease in PLK1 binding (compare 4 to 6), which suggests an important contribution of this residue in the binding pocket of the PLK1 PBD. Removal of the N-terminal proline from 4 resulted in an even greater loss of binding to PLK1 (~24-fold compared to 5). These studies strongly indicate that the key pharmacophoric elements for potent PLK1 PBD inhibition are contained within the region encompassed by the Ac-PLHSpTAI and that these interactions should be preserved in inhibitor optimization and in the search for fragment alternatives for the N and C-terminal determinants.
After it was determined that 7, the consensus recognition sequence for PLK3, was highly potent for PLK1 with single digit nM IC50, the acetylated N-terminal amino acid sequence of PBIP (6) was combined with the C-terminal PKNG sequence of 7 to probe its relative contribution (9). Comparative binding data demonstrated that PKNG results in a modestly weakened PLK1 affinity (2-fold) relative to PNGL (compare 9 and 8) while imparting significantly improved selectivity for PLK1. The contributions of PKNG observed by this comparison were further corroborated through relative evaluations with the PNGL C-terminal motif in the PLK3 N-terminal sequence (GPLAT, 7 vs. 8) and a similar 2-fold relative increase was observed.
Initial studies with FLIPs incorporating the 4-alkyl substituted N-terminal benzamide capping group identified these as promising starting structures for the development of PBD-inhibitors as chemical biology probes and anti-tumor therapeutics[17]. In this present study, the results clearly demonstrate the optimization of this capping group through extension of the alkyl chain to 6 carbons (20) and that the potency of the native Cdc25C peptide can be recapitulated in the FLIP molecule with sub-micromolar activity by incorporating a 4-octyl substituent (21). Molecular modeling showed that the linear alkyl chain exploits a hydrophobic groove (also known as the cryptic pocket) and that extension of this chain interacts to a greater extent (Figure 1). While this groove has been exploited in previously described compounds, the 4-alkylbenzamide structure represents a low MW and simple strategy to obtain highly potent but more drug-like PBD inhibitors. Furthermore, the peptide SAR knowledge was applied in the FLIP context to generate capped peptides with only 4 residues (23) and which preserve the activity of the 6mers ligated to the benzamide groups (21 vs. 23) while maintaining high levels of selectivity for PLK1 vs PLK3 (SI = >1800) and confirmed that the C-terminal Ile is critical for potent FLIP activity.
Peptides and particularly phosphopeptides are generally not active in cells due to their instability and lack of permeability. Previous PBD peptidomimetics have required extensive modification including pegylation, histidine alkylation and/or phosphothreonine masking to generate compounds with cellular activity[16d, 16e]. The two FLIPS that were optimized in terms of potency and decreased MW (23 and 24) displayed a measurable anti-proliferative effect without such modification or the use of or a drug delivery agent. The cells utilized were chosen due to reported synthetic lethal interactions with PLK1 inhibition (PC-3 PTEN deficient, A549 K-RAS mutant) [24].[24]. The hypothesis that neutralizing the negative charge on the C-terminus might improve cell permeability was proven by the increased potency of 25 and 26 on cells with a respectable level of activity for a phosphate containing molecule being observed. Confirmation of on target anti-proliferative activity was obtained through the CETSA assay and revealed potent engagement of PLK1 at the cellular level. Interestingly the EC50 for engagement was greater than the competitive binding observed in the FP binding assay suggesting that interaction with the isolated PBD was not reflective of the intact full length PLK1 protein. It also indicates that the FLIPs do not get into cells efficiently since the difference between the cellular IC50 and the PLK1 engagement is 2 orders of magnitude. Furthermore, in the intact PLK context, the FLIPs appear to be somewhat less selective. Compound 25 had only 3-fold selectivity for PLK1 vs 3 in the CETSA assay although 26 had considerably enhanced specificity. It is possible that its extra carbon in the alkyl chain prevents potent interaction with the hydrophobic groove of the cryptic pocket in the PLK3 context. As a negative control to further determine the validity of these results, a non-phosphorylated version of 26 was found to have no appreciable anti-proliferative activity and also did not lead to significant stabilization of PLK1 in the CETSA assay.
The increased cellular potency also allowed further investigation of the FLIPs to examine PLK1 phosphorylation markers that are reflective of inhibition of the PBD. TCTP (translationally controlled tumor protein) is phosphorylated on serine 46 by PLK1 in vitro and this site has been validated as a direct readout of PLK1[21a].[21a]. PLK1 phosphorylation of TCTP results in its nuclear localization and mitotic regulation through inducing an increase in microtubule dynamics. Western blotting analysis showed that treatment of PC-3 cells with FLIP 25 resulted in a dose-dependent decrease in the amount of pTCTP thus confirming cellular inhibition of PLK1 activity. This observation is novel since it has not previously been shown that blocking the PBD results in inhibition of PLK1 mediated phosphorylation of TCTP.
Cell cycle distribution experiments demonstrate that the optimized FLIPs induce a presumptive mitotic arrest that phenocopies the mode of action of BI2536 and other methods of down regulating PLK1 activity (supplementary figure 3).
In addition to these results, further development potential was shown by results obtained with a cell line expressing the mutant PLK1 (C67V) and which are resistant to catalytic inhibition [25]. These cells are sensitive to FLIPs, thereby demonstrating that blocking the PBD could be a synergistic approach with ATP blockers currently in clinical development. Future studies will address the effectiveness of combination therapy using ATP-based and PBD-targeted inhibitors.
Conclusion
In summary, this study further validates the use of the REPLACE method to develop more drug like small molecule PBD-inhibitors that have a high binding affinity for PLK1 while retaining selectivity against other PLKs including PLK3, a known tumor suppressor. It should be noted that this study is the first to experimentally measure and report PLK3 binding for peptides and peptidomimetics and explicitly compare PLK1 versus PLK3 selectivity both in vitro and at the cellular level. Peptide SAR confirmed the critical determinants of the PBD motif that require successful mimicry and a promising drug-like molecular fragment that replaces the N-terminal residues has been identified and further optimized. SAR and molecular modeling studies in both the peptide and FLIP contexts demonstrate that both the N and C-termini contribute to the high selectivity of the PBIP peptide and that truncated compounds lose both potency and selectivity for PLK1. Cellular studies with the optimized FLIPs show significant anti-proliferative activity, potent on target engagement and promise against drug resistant tumors thus providing impetus for medicinal chemistry efforts aimed at incorporation of further drug-like properties through REPLACE mediated optimization. Future studies will focus on further application of the REPLACE strategy to the C-terminus and to generating phosphate prodrugs of the FLIPs to improve potency and anti-tumor efficacy of the more drug-like PBD inhibitors.
Experimental Section
Peptide & FLIP Synthesis
Phospho-peptides were synthesized and purified using standard Fmoc chemistry by GenScript (Piscataway, NJ) and unless stated otherwise, all peptides were synthesized with an N-terminal amino group and a C-terminal carboxyl group. HPLC and MS were used to confirm the purity and structure of each peptide (see Supplementary Information Table 1). Peptides and FLIPs generated are based on endogenously binding ligands for the PBD and therefore there is not issue with them being false positives from aggregation or spurious florescence signals. This is further unlikely due to the sequence degeneracy of the peptides studied and also the availability of crystal structures for the parent motifs studied.
Fluorescent Polarization Binding Assay
FLIPs and peptides to be tested were dissolved in DMSO (10 mM) and diluted to working concentrations in assay buffer (a maximum of 600 μM, which equates to a maximum 6% DMSO tolerance determined for the assay). Assays were optimized following standard guidelines (http://www.ncbi.nlm.nih.gov/books/NBK92000/). The PLK1 PBD (367–603) and PLK3 PBD (335–646) proteins were obtained from BPS Bioscience Inc. (San Diego, CA); 41.4 nM PLK1 and 245 nM PLK3 were used per reaction. The fluorescein-tracer phospho-peptides (MAGPMQS[pT]PLNGAKK for PLK1, and GPLATS[pT]PKNG for PLK3) were used at a final concentration of 10 nM. Incubation was carried out at room temperature for 45 min on a shaker. Fluorescence was measured using either a DTX 880 plate reader and Multimode Analysis software (Beckman Coulter, now Molecular Devices, Brea, CA) or a SpectraMax i3 (Molecular Devices, Brea, CA). The polarization values in millipolarization (mP) units were measured at an excitation wavelength of 488 nm and an emission wavelength of 535 nm. Each data point was performed in triplicate for every experiment, and experiments were performed at least three times. An IC50 value for each compound was calculated from non-linear regression analysis of the plots of mP values relative to PBD-tracer mP values alone versus FLIP/peptide concentrations (Supplementary Figures 8, 9).
Cell Culture
HeLa cervical cancer cells and PC-3 prostate cancer cells were obtained from ATCC (Manassas, VA) and were not authenticated by the authors for this study. HeLa and PC-3 cells were maintained in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% Nu-serum (BD Bioscience, Franklin Lakes, NJ) and 1% penicillin/streptomycin (Invitrogen) in a humidified incubator and 5% CO2 at 37 °C.
Cell Viability Assay
Cells were maintained in 10% FBS and cell proliferation assays are conducted in 10% Nu Serum. Exponentially growing cells were plated in 96- well plates for 24 hours prior to treatment with FLIPs for 72 hours. Following treatment, cell viability was measured using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) colorimetric assay. Cells were incubated for 4 hours with MTT and absorbance at 595 nm was measured on a DTX 880 plate reader. IC50 values were generated from normalized absorbance data using GraphPad Prism.
Western Blotting
Cells were synchronized in prometaphase by treatment with 100 ng/ml nocodazole for 18 hours. Detached cells were collected, centrifuged and rinsed with PBS. Attached cells were also rinsed with PBS. Media containing test compounds was added to the cells and incubated for 23 hours. Cells were harvested, centrifuged, rinsed with PBS supplemented with protease and phosphatase inhibitors (# A32959 and A32961, Pierce, Rockford, IL, USA) and centrifuged again. Cells were suspended in RIPA buffer supplemented with protease and phosphatase inhibitors and then sonicated on ice. The lysate was shaken for 20 min at 4 °C then centrifuged at 12,000 rpm for 20 min. Supernatant was aspirated and the cell pellet transferred into fresh chilled tubes. Protein quantity was determined using the BCA protein assay (Pierce, Rockford, IL, USA). Proteins (20 μg) were separated by 4–15% SDS-PAGE. Immunoblotting analysis was done using following antibodies: anti-PLK1 (#05–844, EMD Millipore, Temecula, CA, USA), anti-phospho-TCTP-Ser46 (#5251, Cell Signaling Technology, Danvers, MA, USA), anti-TCTP (#8441, Cell Signaling Technology), anti-β-tubulin (#NB600–936, Novus Biologicals, Centennial, CO, USA), mouse HRP-conjugated secondary antibody (#NA931, GE Healthcare, Little Chalfont, Buckinghamshire, UK) and mouse HRP-conjugated secondary antibody (#NA934, GE Healthcare). Immunoblotting was performed following the company instructions. ECL Western Blotting Substrate and SuperSignal (Pierce) were used for detection.
Cellular Thermal Shift Assay (CETSA)
Cellular thermal shift assay (CETSA) was performed with PC3 cells cultured in RPMI medium supplemented with 10% FBS. For an initial determination of the melting profile of PLK1 AND PLK3, cells dispensed into 96-well PCR plates in the above medium (6000 cells/well in 50 μl) were subjected to temperature gradient (38–60 °C) for 10 min. Cold non-denaturing lysis buffer (PBS supplemented with 0.1% TritonX-100 and 1X protease inhibitors) was added to wells, and the plate was rocked, then incubated for 15 min on ice. Centrifugation was performed at 14,000 rpm to sediment the denatured protein content. Supernatant was collected and subjected to SDS-PAGE, and immuno-detection was performed using anti-PLK1 and anti-PLK3 antibodies. PLK1 and PLK3 bands were quantified on a LI-COR C-Digit Blot Scanner, and Tagg(50) and Tagg(75) values were calculated for both proteins. In subsequent experiments, cells were treated at increasing doses (3.0, 1.0, 0.3, 0.1, 0.03 μM) of inhibitors together with DMSO control, for 2 hours. Cells were then subjected to heat shock at Tagg(50) for 10 min, and unstable protein was removed by centrifugation step. Following immuno-blotting, bands of stable PLK1 and PLK3 were quantified, normalized to loading control and plotted using GraphPad Prism software. EC50 values for inhibitors were calculated.
In an initial heat gradient, PLK1 displayed a temperature-dependent decay with a Tagg(50) of 56.2 °C, whereas PLK3 displayed a temperature-dependent decay with a Tagg(50) at 48.0 °C. Subsequently, dose-dependent potency of each inhibitor was tested at Tagg(50), the temperature point at which 50% of the target protein is denatured. The EC50 of the peptides were determined from the curve fit of the dose response.
Supplementary Material
Acknowledgements
Retinal Pigment Epithelial (RPE) cells expressing wild-type or C67V PLK1 were kindly provided by Dr. Prasad V. Jallepalli (Memorial Sloan-Kettering Cancer Center, New York, NY). We wish to thank the National Institutes of Health for Funding (Grant numbers NIH/NCI Grants R41 CA213711-01 and NIH/NCRR Grants UL1RR029882 and UL1 TR000062).
References
- [1].a) Xie S, Xie B, Lee MY, Dai W, Oncogene 2005, 24(2), 277–286; [DOI] [PubMed] [Google Scholar]; b) Craig SN, Wyatt MD, McInnes C, Expert Opin Drug Discov 2014, 9(7), 773–789. [DOI] [PubMed] [Google Scholar]
- [2].McInnes C, Wyatt MD, Drug Discov Today 2011, 16(13–14), 619–625. [DOI] [PubMed] [Google Scholar]
- [3].a) Yuan J, Hoerlin A, Hock B, Stutte HJ, Ruebsamen-Waigmann H, Strebhardt K, American Journal of Pathology 1997, 150(4), 1165–1172; [PMC free article] [PubMed] [Google Scholar]; b) Wolf G, Elez R, Doermer A, Holtrich U, Ackermann H, Stutte HJ, Altmannsberger H-M, Rübsamen-Waigmann H, Strebhardt K, Oncogene 1997, 14, 543–549; [DOI] [PubMed] [Google Scholar]; c) Tokumitsu Y, Mori M, Tanaka S, Akazawa K, Nakano S, Niho Y, International Journal of Oncology 1999, 15(4), 687–692. [DOI] [PubMed] [Google Scholar]
- [4].Weichert W, Schmidt M, Gekeler V, Denkert C, Stephan C, Jung K, Loening S, Dietel M, Kristiansen G, The Prostate 2004, 60(3), 240–245. [DOI] [PubMed] [Google Scholar]
- [5].Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K, Kimura M, Okano Y, Saji S, Cancer Science 2003, 94(2), 148–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA Jr., Kinzler KW, Vogelstein B, Papadopoulos N, Proc Natl Acad Sci U S A 2009, 106(10), 3964–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].McKenzie L, King S, Marcar L, Nicol S, Dias SS, Schumm K, Robertson P, Bourdon JC, Perkins N, Fuller-Pace F, Meek DW, Cell cycle (Georgetown, Tex 2010, 9(20), 4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Elez R, Piiper A, Kronenberger B, Kock M, Brendel M, Hermann E, Pliquett U, Neumann E, Zeuzem S, Oncogene 2003, 22(1), 69–80; [DOI] [PubMed] [Google Scholar]; b) Spankuch-Schmitt B, Wolf G, Solbach C, Loibl S, Knecht R, Stegmuller M, von Minckwitz G, Kaufmann M, Strebhardt K, Oncogene 2002, 21(20), 3162–3171. [DOI] [PubMed] [Google Scholar]
- [9].a) Jimeno A, Li J, Messersmith WA, Laheru D, Rudek MA, Maniar M, Hidalgo M, Baker SD, Donehower RC, J Clin Oncol 2008, 26(34), 5504–5510; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mross K, Frost A, Steinbild S, Hedbom S, Rentschler J, Kaiser R, Rouyrre N, Trommeshauser D, Hoesl CE, Munzert G, J Clin Oncol 2008, 26(34), 5511–5517. [DOI] [PubMed] [Google Scholar]
- [10].Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM, Curr Biol 2007, 17(4), 304–315. [DOI] [PubMed] [Google Scholar]
- [11].a) Yang Z, Waldman AS, Wyatt MD, Biochem Pharmacol 2008, 76, 987–996; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang Q, Xie S, Chen J, Fukasawa K, Naik U, Traganos F, Darzynkiewicz Z, Jhanwar-Uniyal M, Dai W, Molecular and cellular biology 2002, 22(10), 3450–3459; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yang Y, Bai J, Shen R, Brown SA, Komissarova E, Huang Y, Jiang N, Alberts GF, Costa M, Lu L, Winkles JA, Dai W, Cancer Res 2008, 68(11), 4077–4085; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Xu D, Yao Y, Jiang X, Lu L, Dai W, J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Yun SM, Moulaei T, Lim D, Bang JK, Park JE, Shenoy SR, Liu F, Kang YH, Liao C, Soung NK, Lee S, Yoon DY, Lim Y, Lee DH, Otaka A, Appella E, McMahon JB, Nicklaus MC, Burke TR Jr., Yaffe MB, Wlodawer A, Lee KS, Nature structural & molecular biology 2009, 16(8), 876–882; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Elia AE, Cantley LC, Yaffe MB, Science 2003, 299(5610), 1228–1231; [DOI] [PubMed] [Google Scholar]; Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB, Cell 2003, 115(1), 83–95; [DOI] [PubMed] [Google Scholar]; c) Garcia-Alvarez B, de Carcer G, Ibanez S, Bragado-Nilsson E, Montoya G, Proc Natl Acad Sci U S A 2007, 104(9), 3107–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Reindl W, Yuan J, Kramer A, Strebhardt K, Berg T, Chemistry & biology 2008, 15(5), 459–466; [DOI] [PubMed] [Google Scholar]; b) Watanabe N, Sekine T, Takagi M, Iwasaki J, Imamoto N, Kawasaki H, Osada H, J Biol Chem 2009, 284(4), 2344–2353; [DOI] [PubMed] [Google Scholar]; c) Chen Y, Li Z, Liu Y, Lin T, Sun H, Yang D, Jiang C, Bioorg Chem 2018, 81, 278–288. [DOI] [PubMed] [Google Scholar]
- [14].Rubner S, Scharow A, Schubert S, Berg T, Angewandte Chemie (International ed 2018, 57(52), 17043–17047. [DOI] [PubMed] [Google Scholar]
- [15].a) van Vugt MATM, van de Weerdt BCM, Vader G, Janssen H, Calafat J, Klompmaker R, Wolthuis RMF, Medema RH, J. Biol. Chem 2004, 279(35), 36841–36854; [DOI] [PubMed] [Google Scholar]; b) Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, Grauert M, Adolf GR, Kraut N, Peters JM, Rettig WJ, Curr Biol 2007, 17(4), 316–322. [DOI] [PubMed] [Google Scholar]
- [16].a) Liu F, Park JE, Qian WJ, Lim D, Scharow A, Berg T, Yaffe MB, Lee KS, Burke TR, ACS Chem Biol 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu F, Park JE, Qian WJ, Lim D, Graber M, Berg T, Yaffe MB, Lee KS, Burke TR Jr., Nature chemical biology 2011, 7(9), 595–601; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Qian WJ, Park JE, Lim D, Lai CC, Kelley JA, Park SY, Lee KW, Yaffe MB, Lee KS, Burke TR Jr., Biopolymers 2014, 102(6), 444–455; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Srinivasrao G, Park JE, Kim S, Ahn M, Cheong C, Nam KY, Gunasekaran P, Hwang E, Kim NH, Shin SY, Lee KS, Ryu E, Bang JK, PloS one 2014, 9(9), e107432; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Ahn M, Han YH, Park JE, Kim S, Lee WC, Lee SJ, Gunasekaran P, Cheong C, Shin SY Sr., Kim HY, Ryu EK, Murugan RN, Kim NH, Bang JK, J Med Chem 2015, 58(1), 294–304. [DOI] [PubMed] [Google Scholar]
- [17].McInnes C, Estes K, Baxter M, Yang Z, Farag DB, Johnston P, Lazo JS, Wang J, Wyatt MD, Molecular Cancer Therapeutics 2012, 11(8), 1683–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Andrews MJ, Kontopidis G, McInnes C, Plater A, Innes L, Cowan A, Jewsbury P, Fischer PM, Chembiochem 2006, 7(12), 1909–1915; [DOI] [PubMed] [Google Scholar]; b) Premnath PN, Craig SN, Liu S, Anderson EL, Grigoroudis AI, Kontopidis G, Perkins TL, Wyatt MD, Pittman DL, McInnes C, J Med Chem 2015, 58(1), 433–442; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu S, Premnath PN, Bolger JK, Perkins TL, Kirkland LO, Kontopidis G, McInnes C, J Med Chem 2013, 56(4), 1573–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sharma P, Mahen R, Rossmann M, Stokes JE, Hardwick B, Huggins DJ, Emery A, Kunciw DL, Hyvonen M, Spring DR, McKenzie GJ, Venkitaraman AR, Sci Rep 2019, 9(1), 15930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Babic I, Kesari S, Nurmemmedov E, Future medicinal chemistry 2018, 10(14), 1641–1644. [DOI] [PubMed] [Google Scholar]
- [21].a) Cucchi U, Gianellini LM, De Ponti A, Sola F, Alzani R, Patton V, Pezzoni A, Troiani S, Saccardo MB, Rizzi S, Giorgini ML, Cappella P, Beria I, Valsasina B, Anticancer Res 2010, 30(12), 4973–4985; [PubMed] [Google Scholar]; b) Johnson TM, Antrobus R, Johnson LN, Biochemistry 2008, 47(12), 3688–3696. [DOI] [PubMed] [Google Scholar]
- [22].Burkard ME, Santamaria A, Jallepalli PV, ACS Chem Biol 2012, 7(6), 978–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].McInnes C, in Annual Reports in Medicinal Chemistry, Vol. 47 (Ed.: Bernstein M Desai P), Elsevier, 2012, pp. 459–474 [Google Scholar]
- [24].a) Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KW, Elledge SJ, Cell 2009, 137, 835–848; [DOI] [PMC free article] [PubMed] [Google Scholar]; a) Jiang BH, Liu LZ, Advances in cancer research 2009, 102, 19–65; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu XS, Song B, Elzey BD, Ratliff TL, Konieczny SF, Cheng L, Ahmad N, Liu X, The Journal of Biological Chemistry 2011, 286(41), 35795–35800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Burkard ME, Santamaria A, Jallepalli PV, ACS Chemical Biology 2012, 7, 978–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.