

Abstract
Macroautophagy is a cellular response to various environmental stresses that ensures lysosomal degradation of long-lived and damaged proteins and cellular organelles. It occurs through the formation of an autophagosome, which then fuses with a lysosome to form an autolysosome. Depending on the cellular context, autophagy may promote cancer cell survival or it may serve as a mechanism of tumor suppression. Herein, we show that resveratrol, a natural phytoalexin, induces premature senescence in human A431 SCC cells, and that resveratrol-induced premature senescence is associated with a blockade of autolysosome formation, as assessed by the absence of colocalization of LC3 and Lamp-2, markers for autophagosomes and lysosomes, respectively. Further, we show that resveratrol downregulates the level of Rictor, a component of mTORC2, leading to decreased RhoA-GTPase and altered actin cytoskeleton organization. Exogenous overexpression of Rictor restores RhoA-GTPase activity and actin cytoskeleton network, and decreases resveratrol-induced senescence-associated β-gal activity, indicating a direct role of Rictor in senescence induction. Rictor is overexpressed in UV-induced murine SCCs, whereas its expression is diminished by oral administration of resveratrol. These data indicate that resveratrol attenuates autophagic process via Rictor, and suggest that downregulation of Rictor may be a mechanism of tumor suppression associated with premature senescence.
INTRODUCTION
Nonmelanoma skin cancer (NMSC) including basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) are the most common human cancers in the United States. Solar ultraviolet (UV) B radiation is the major known cause of NMSCs (1). The carcinogenic effects of UVB result from direct structural changes in DNA. The effect of accumulated damage, followed by genetic alterations, ultimately leads to the transformation of normal cells into cancer cells. NMSCs manifest different and distinct mutations: notably, p53 in the case of SCCs and Ptch and p53 in the case of BCCs, which serve as the initiating events in the development of these neoplasms (2). In addition, mutations in the ras oncogene are found in UVB-induced SCCs (3,4). Consequently, aberrations in the PI3K ⁄ Akt ⁄ mTOR pathway are associated with both human and UVB-induced murine SCCs, and the blockade of this pathway using mTOR inhibitors such as everolimus has been shown to substantially reduce the growth of SCCs (5–7). It has also been shown that cyclosporine A (CsA), an immunosuppressive agent used in organ transplant recipients, promotes SCC growth via Akt activation through the downregulation of PTEN expression in immune-deficient mice (8). The incidence of SCCs is 60–100-fold higher in organ transplant recipients receiving cyclosporine CsA, whereas the patients given mTOR inhibitors have a reduced incidence of SCC (9–11). These data collectively demonstrate the importance of mTOR signaling in driving SCC carcinogenesis.
In addition to the diversity of roles played by mTOR in carcinogenesis, which include its regulation of ribosome biogenesis and cell cycle progression, and the activation of prosurvival Akt, mTOR has also been shown to inhibit macroautophagy (henceforth referred to as autophagy), a process of self-degradation (12,13). The autophagic process is characterized by the enclosure within an organelle, termed an autophagosome, of cytoplasmic constituents targeted for degradation, which is followed by the fusion of the autophagosome with a lysosome to form an autolysosome. Lysosomal ⁄ vacuolar hydrolases are available within the autolysosome to degrade the cellular components. In the normal cell, this process serves to maintain homeostasis through the removal of proteins and organelles present in excess or that are aged or damaged (14,15). As a potential cancer therapeutic, excess autophagy can mediate a distinctive form of cancer cell death, known as Type II programed cell death (16). However, although autophagy is a known mechanism of tumor suppression, it has also been shown, at least at certain stages in tumor progression, to promote cancer cell survival (14,17). A prosurvival effect of autophagy has been demonstrated in breast cancer cells following radiation treatment, in which inhibition of autophagy resulted in increased cell death (18). Inhibition of autophagy by cyclophosphamide treatment significantly decreased tumor recurrence and increased cell death (19), consistent with a beneficial role of autophagy in the survival of cancer cells. Nonetheless, significant tumor suppressive activities of autophagy have been described. For example, single allelic mutations in Beclin-1, the mammalian ortholog of the yeast autophagy gene, Apg6, resulted in increased spontaneous tumors in murine models (20). Beclin-1 expression is frequently decreased in human breast cancers and melanomas, and both genetic and epigenetic silencing of the Beclin-1 gene has been shown in human breast tumors. Autophagy has been shown to be required for the efficient killing of tumor cells during cancer chemotherapy (16). Activation of autophagy by inhibiting mTOR with everolimus has also been shown to induce massive reduction of leukemic mass and confer a strong survival advantage in leukemic mice (21). The precise mechanisms of this dual role of autophagy in tumor development have not been elucidated. The cellular context and factors regulating the autophagic process—either activation or inhibition—and mechanisms by which autophagy contributes to carcinogenesis remain to be determined.
We and others have previously demonstrated that resveratrol (RES, 3,5,4-trihydroxystilbene), a natural phytoalexin present in the skin of grapes, nuts and other fruits, effectively suppresses UV-induced SCC development in mice (22–27). We further demonstrated that treatment of human A431 SCC cells with RES resulted in cells that displayed morphological and biochemical characteristics of premature senescence (28). Premature senescence represents an irreversible state of cell cycle arrest; consequently, induction of premature senescence in cancer cells by chemotherapeutic reagents such as adriamycin is considered a mechanism of tumor suppression (29,30). Despite a link between senescence and autophagy, as indicated by accumulation of autophagic vacuoles in senescent cells and the lysosomal origin of senescence-associated β-galactosidase (SA-β-gal; 31), the relevance of autophagic process to RES-induced premature senescence induction and its relation to tumor suppression remains unclear. Herein, we present evidence indicating that RES-mediated premature senescence is associated with attenuated autophagy, and that the mechanism involves downregulation of the mTORC2 component, Rictor, resulting in aberrant actin cytoskeleton organization. Further, we show that Rictor is overexpressed in UVB-induced murine SCCs, but is decreased in SCCs from RES-fed mice.
MATERIALS AND METHODS
Cell lines, reagents and antibodies.
A431 human epidermoid carcinoma cells were obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained in Dulbecco’s modified Eagle medium (DMEM), supplemented with 10% fetal bovine serum and 1% penicillin ⁄ streptomycin, and kept in an atmosphere of 95% air ⁄ 5% CO2 in a 37°C humidified incubator, in accordance with ATCC guidelines. pRK5-myc-Rictor was obtained from Addgene (Cambridge, MA). Transduction of myc-Rictor was performed according to the protocol of the Phoenix retroviral expression system (Orbigen Inc, San Diego, CA). RES and rapamycin were purchased from LKT laboratories Inc. (St. Paul, MN), rhodamine-phalloidin from Molecular Probes (Carlsbad, CA), and bafilomycin A1 and 4-methylumbel-liferyl-β-D-galactopyranoside (MUG) from Sigma (St. Louis, MO). The antibodies against phospho-Akt1 (S473), Rictor, PTEN, p-p70S6K, p70S6K, and myc were purchased from Cell Signaling Technology (Danvers, MA); the antibodies against Lamp-2 and β-actin were from Santa Cruz Biotechnology (Santa Cruz, CA), and antiLC3 antibody was from Novus Biologicals, Inc. (Littleton, CO).
SA-β-gal staining and quantification.
In situ staining of SA-β-gal was performed using a senescence β-galactosidase staining kit (Cell Signaling Technology), according to the manufacturer’s protocol. Cells were viewed by phase contrast on an Axiovert 40 CFL microscope and photographed at 100 and 200× magnification with a Kodak digital color camera. SA-β-gal activities present in cell extracts were measured by the rate of conversion of 4-methylumbelliferyl-β-d-galactopyranoside (MUG) to the fluorescent hydrolysis product 4-methylumbelliferone (4-MU) at pH 6.0 as described previously (32). All experiments were performed in triplicate.
RhoA activity assay.
RhoA activity was determined by Rho Activation Assay Biochem Kit (Cytoskeleton, Denver, CO). Extracts prepared from RES or dimethyl sulfoxide (DMSO) treated cells was incubated with Rhotekin-RBD beads to pull down active RhoA according to the manufacturer’s instruction. Active RhoA was then detected by Western blotting using anti-RhoA antibody.
Western blotting and immunohistochemistry.
Preparation of cell lysates, Western blotting and immunohistochemical analyses were performed as previously described (28,33,34).
Fluorescence microscopy.
A431 cells treated with RES or rapamycin were immediately fixed in 3.7% formaldehyde followed by permeabilization in 0.2% Triton X. Cells were then stained with rhodamine phalloidin followed by visualization of F-actin.
Transmission electron microscopy.
A431 cells transduced with pRK5-myc-Rictor or empty vector were fixed in 4.16% glutaraldehyde ⁄ 2.2% formaldehyde in 0.2 M sodium cacodylate buffer, pH 7.4 overnight. Cells were then postfixed in 2% osmium tetroxide in distilled water for 1 h, dehydrated through a graded acetone series, embedded in EMBed 812 epoxy resin (Electron Microscopy Sciences, Hatfield, PA), cured for 48 h. Sections (100 nm) were cut and collected onto 200 mesh copper TEM grids. Grids were stained with 4% uranyl acetate for 1 h, followed by 0.5% lead citrate for 15 min and operated at 200 kV on a JEOL JEM-2100 transmission electron microscope with digital image collection.
UV-induced SCC carcinogenesis.
Generation of p53+ ⁄ − ⁄ SKH-1 hairless mice and UV light source, and photocarcinogenesis protocol were as described previously (27).
Image analysis.
Images were analyzed using IMAGEJ public domain software (the National Institute of Health, Bethesda, MD; http://rsb.info.nih.gov/ij/) to calculate area and pixel value statistics of Rictor positive areas. Images obtained from 25 SCCs from UV-irradiated, nontreated animals and 25 SCCs from UV-irradiated, RES-treated animals were analyzed.
Statistical analyses.
Statistical analyses were performed using the Student’s t-test (two-tailed): P < 0.05 was considered statistically significant.
RESULTS
LC3II and Lamp-2 accumulate in RES-induced senescence-like A431 cells
Previously, we have shown that RES induces senescence-like G1 cell cycle arrest in A431 cells, which accompanied increased SA-β-gal activity, a biochemical marker for senescence induction (28; Fig. 1A). Because SA-β-gal is located in the lysosome, a critical component of the autophagic process, we determined the effects of RES on autophagy in A431 cells by assessing the levels of two known autophagic proteins, lysosome associated membrane protein (Lamp)-2 and microtubule-associated protein 1 light chain 3 (LC3). The levels of LC3 and Lamp-2 were then correlated with SA-β-gal activity. SA-β-gal activity was measured in A431 cells treated with 50 μM RES for a period of 72 h. Gradual increases in SA-β-gal activity, as measured 8, 16, 24, 48 and 72 h following RES treatment, were observed in an assay using a fluorogenic substrate for β-galactosidase (Fig. 1B). Increased SA-β-gal activity correlated with increases in the levels of Lamp-2, indicative of increases in lysosomal content (Fig. 1B). Similarly, the levels of LC3II gradually increased following RES treatment, peaking at 48–72 h. Because LC3 has been shown to covalently conjugate with phosphatidylethanolamine (PE) to form LC3II during the formation of autophagosomes, the observed increase in LC3II suggested an increase in the amount of autophagosomes in response to RES. The levels of SA-β-gal activity, Lamp-2 and LC3II were maximal at 72 h following RES treatment (Fig. 1B), and the levels found at 72 h were sustained for at least 2 weeks following treatment (data not shown). In contrast, at 72 h, the levels of SA-β-gal, Lamp-2 and LC3II were not significantly altered when A431 cells were treated with rapamycin, a known autophagy inducer, compared with nontreated control cells (Fig. 1C). The RES-mediated autophagic response was compared with that using bafilomycin A1 (BAF), an inhibitor of the vacuolar type H(+)-ATPase (V-ATPase). BAF is known to inhibit the acidification of lysosomes and is thought to prevent autophagosome fusion with lysosomes and ⁄ or LC3II degradation, thereby causing autophagosome accumulation (35). Similar to RES, A431 cells treated with 10 nM BAF for 48 h showed an accumulation of LC3 (Fig. 1D). Cotreatment with both BAF and RES further increased the levels of LC3, whereas rapamycin treatment attenuated RES-induced accumulation of LC3 (Fig. 1D). These results suggest that RES induces aberrations in autophagic processes in A431 cells.
Figure 1.
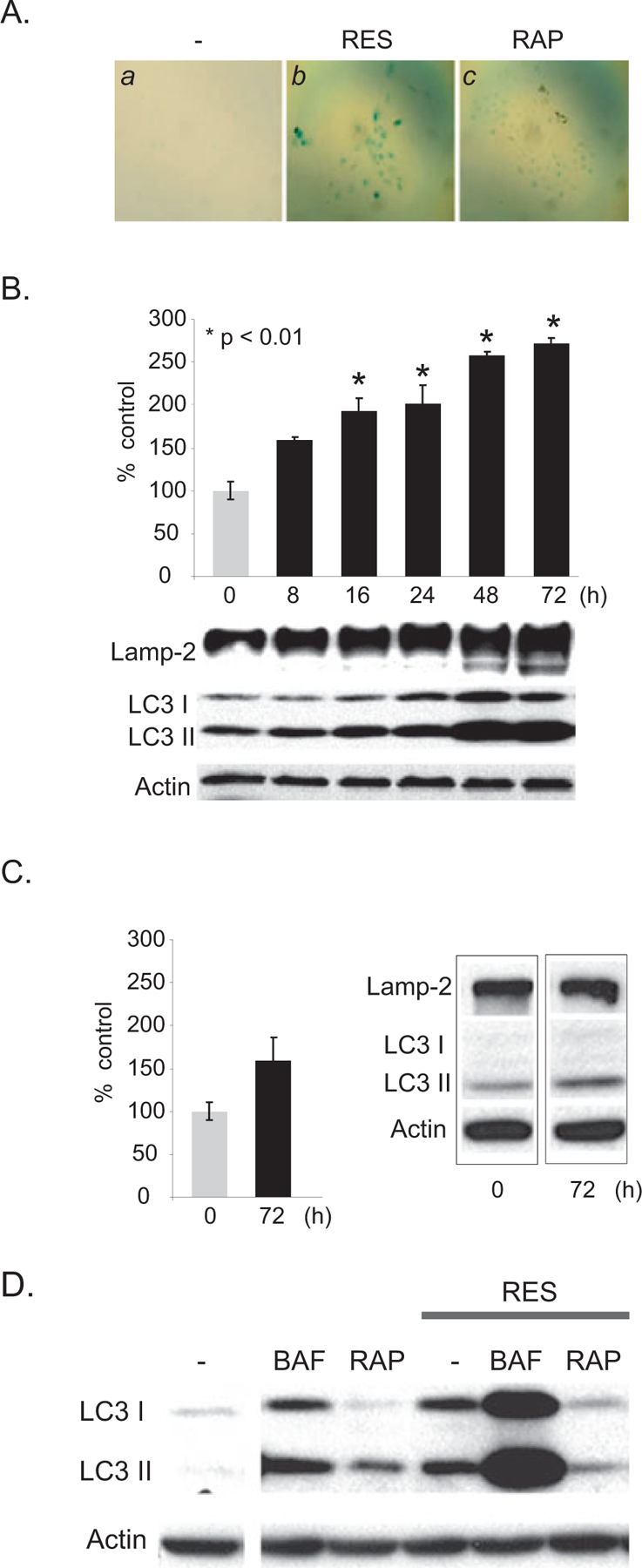
RES-increased SA-β-gal activity results from aberration of autophagic process. (A) SA-β-gal is increased in A431 cells treated with RES, but not in cells treated with rapamycin (RAP). A431 cells were untreated or treated with 50 μM RES or 0.1 μM RAP for 48 h. Senescence induction was detected by staining cells for SA-β-gal activity and was visualized by light microscopy. (B, C) Accumulation of Lamp-2 and LC3 and SA-β-gal activity in RES- (B) and RAP-treated (C) A431 cells. A431 cells were treated with 50 μM RES for a period of 72 h. Cells were harvested at indicated time points and the levels of Lamp-2 and LC3 were assessed by Western blot analysis (80 μg extract was loaded per lane). β-actin was used as an internal loading control. For quantitative SA-β-gal measurement, A431 extracts were incubated with 4-methylumbelliferyl-β-D-galactopyranoside (4-MU) at 37°C. Data were normalized to total protein per assay to correct for differences in extract concentrations and are expressed as percentages of fluorescence levels in untreated control cells. (D) Bafilomycin A1 (BAF) induces LC3II accumulation, whereas RAP suppresses RES-induced LC3II accumulation. A431 cells were untreated or treated with 10 nM BAF, 0.1 μM RAP, 50 μM RES, or combinations as indicated, for 48 h.
RES suppresses autolysosome formation
The colocalization of Lamp-2-positive lysosomes with LC3-positive vesicles implies the formation of autolysosomes, indicating that an active autophagic process is taking place, as was the case when cells were treated with rapamycin, an inducer of autophagy (36). To investigate whether the observed increases in Lamp-2 and LC3II levels in response to RES reflect increased autophagic processes, we assessed the subcellular localization of LC3 and LAMP-2 in RES-induced prematurely senescent A431 cells using immunofluorescence microscopy. We found that RES treatment of A431 cells increased the number of LC3-positive vesicles, which correlated with increased SA-β-gal activity (data not shown). These vesicles, however, did not colocalize with Lamp-2-positive vesicles in RES-treated cells (Fig. 2A, f), whereas colocalization of LC3-positive vesicles and Lamp-2-positive vesicles were found in cells treated with rapamycin (Fig 2A, i). These results indicate a deficiency in autolysosome formation in cells treated with RES. Lysosomes and autophagosomes move along the microtubule network (36,37), and an intact cytoskeleton organization has been shown to be necessary for autophagosome fusion with lysosome (38,39). As shown by fluorescent microscopy of cells stained with rhodamine phalloidin (Fig 2B), unpolarized filopodia (indicated with arrows) were apparent in RES-induced, prematurely senescent A431 cells, suggesting that RES alters cytoskeleton organization.
Figure 2.
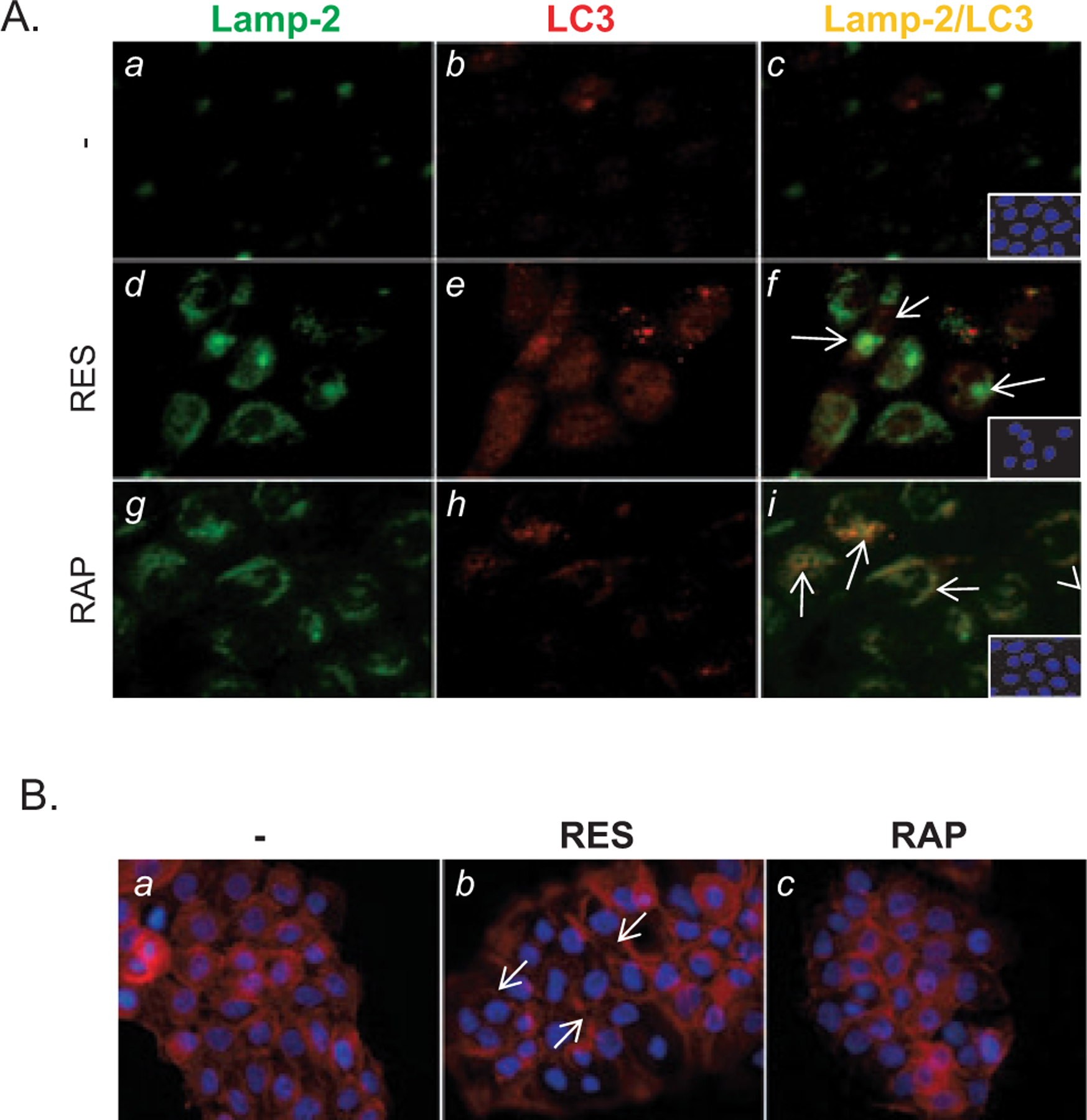
RES blocks the formation of autolysosomes by suppressing actin remodeling. (A) RES inhibits formation of autolysosomes. A431 cells treated as in Fig. 1A followed by subcellular detection of Lamp-2 and LC3 by immunofluorescence microscopy using anti-Lamp-2 and -LC3 antibodies. Green, Lamp-2; red, LC3; yellow, Lamp-2 ⁄ LC3; blue, DAPI. Magnification 200×. (B) Unpolarized filopodia are increased in A431 cells treated with RES as in Fig. 1A (b, white arrows). F-actin was stained using rhodamine-phalloidin. DAPI, blue.
Rictor expression is attenuated in RES-induced, prematurely senescent A431 cells
Recently, we have shown that RES suppresses epithelial-mesenchymal transition (EMT) in UVB-induced SCC carcinogenesis, and using A431 cells, EMT suppression was mediated via inhibition of phosphorylation at S473 Akt1 (27). The Ser ⁄ Thr protein kinase PKB ⁄ Akt is a key regulator of a wide range of cellular processes including cell growth and survival. Akt1 is phosphorylated by PDK1 at T308, and additionally at S473 by the Pl3K-related kinases, mammalian target of rapamycin complex 2 (mTORC2) or DNA-PK to achieve its full kinase activity, depending on the stimulus and the cellular context (40). mTORC2 is a rapamycin-insensitive complex of mTOR and consists of Rictor, GβL and mTOR (41–43). We found that RES treatment decreased the levels of S473 Akt1 phosphorylation and mTOR signaling components including Rictor in A431 cells (Fig. 3). Because mTORC2 is known to function upstream of Rho-GTPase and regulate the actin cytoskeleton (44), we determined whether RES modulation of Rictor affects Rho activity by measuring Rho-GTPase activity using Rhotekin-RBD bead pulldown assay. A dramatic decrease in GTP-bound Rho was observed in RES-treated A431 cells compared with nontreated and rapamycin-treated cells (Fig. 3). The level of PTEN, a regulator of the Akt pathway, was not altered in response to RES, indicating that suppression of S473 Akt1 phosphorylation in RES-treated cells is PTEN-independent (Fig 3).
Figure 3.
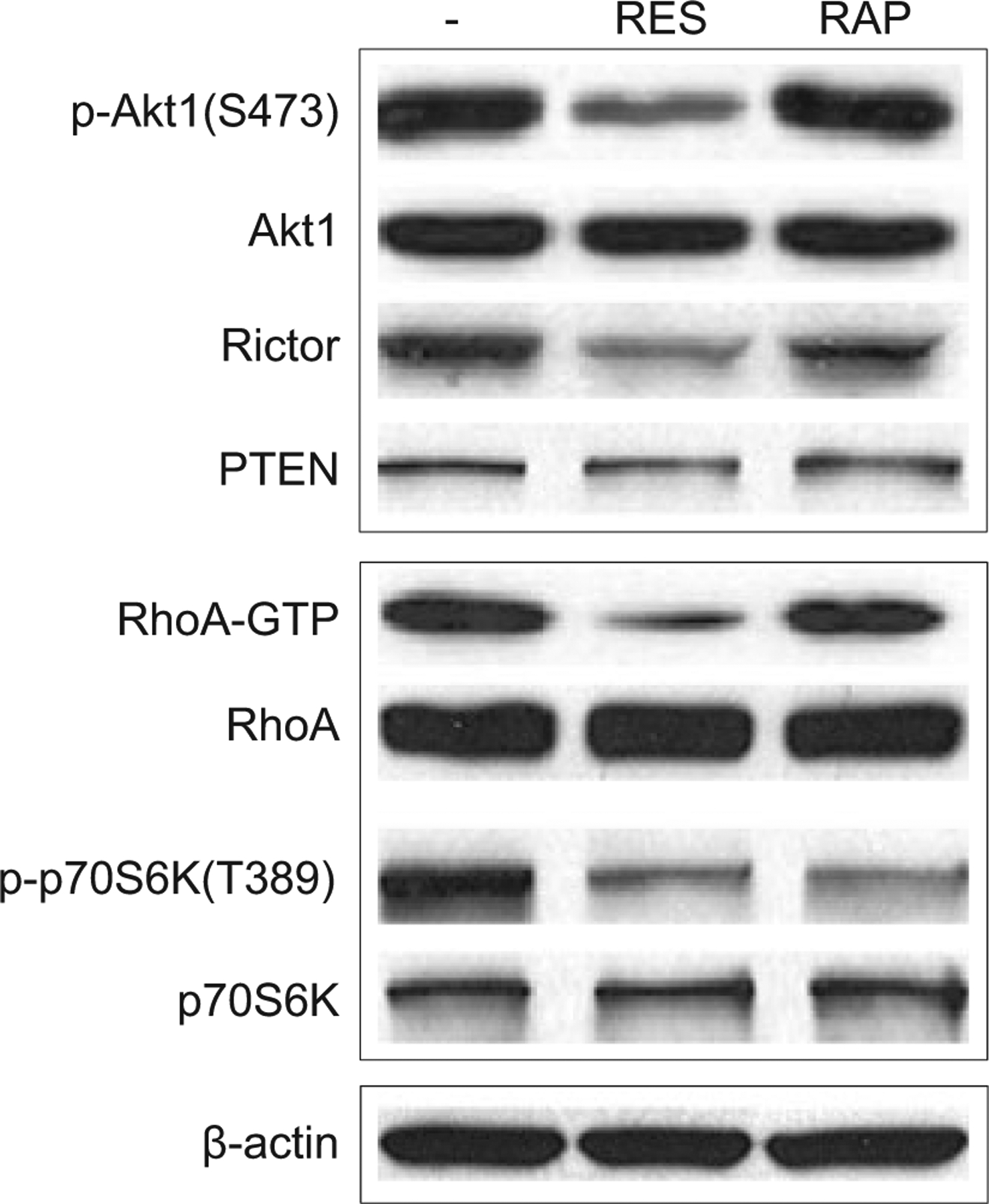
RES inhibits mTORC2 activity by downregulation of Rictor expression. Western blot analysis of extracts prepared from A431 cells that were either untreated or treated with 50 μM RES for 48 h or 0.1 μM RAP for 48 h (80 μg extract was loaded per lane). RhoA-GTP bound to Rhotekin-RBD beads and total cellular RhoA was detected by Western blotting using an anti-RhoA antibody. β-actin was used as an internal loading control. The levels of Akt1, p-Akt1, Rictor, PTEN, p-p70S6K, and p70S6K were assessed by Western blot analysis.
Rictor overexpression attenuates SA-β-gal activity in RES-treated A431 cells
A marked downregulation of Rictor expression observed in RES-induced, prematurely senescent A431 cells suggested that Rictor may have a role in regulating senescence. To assess the role of Rictor in senescence, A431 cells were transduced with either myc-tagged Rictor cDNA or empty vector. The resulting myc-Rictor expressing or vector-transduced A431 cells were treated with RES. Rictor overexpression restored the levels of S473 Akt1 phosphorylation and RhoA-GTP compared with cells transduced with empty vector alone in RES-treated cells (Fig. 4A, lanes 2 and 4). Furthermore, Rictor overexpression substantially reduced the formation of unpolarized filopodia following RES treatment (Fig 4B, d), whereas they were readily visible in RES-treated A431 cells transduced with empty vector (Fig. 4B, b, white arrows). Colocalization of Lamp-2 and LC3 was evident in RES-treated A431 cells overexpressing Rictor, suggesting that Rictor modulates autophagosome-lysosome fusion (Fig. 4C). To further assess the effect of overexpression of Rictor on autophagic activity, we analyzed autophagic structures by transmission electron microscopy (TEM) in RES-treated A431 cells and RES-treated A431 cells overexpressing Rictor. The presence of large mitochondria has been shown to be associated with senescent cells (45), and blocking autophagy using 3-ethyladenine (3-MA) increases the number of large mitochondria (46). In RES-treated A431 cells, the presence of autophagosomes (Fig. 4D, left panel, indicated as AG, yellow) and large mitochondria (data not shown) was evident. In contrast, increases in autolysosomes were apparent in Rictor-overex-pressing A431 cells (Fig. 4D, right panel, AL, blue). Further, we demonstrated that Rictor overexpression suppressed RES-induced SA-β-gal activity (Fig. 4E). Taken together, these data indicate that Rictor has a role in the formation of autolysosomes, and that RES-mediated decreases in Rictor expression and subsequent RhoA-GTP activity may lead to aberrant cytoskeleton organization, thereby hindering the autophagic process in RES-induced, prematurely senescent A431 cells.
Figure 4.
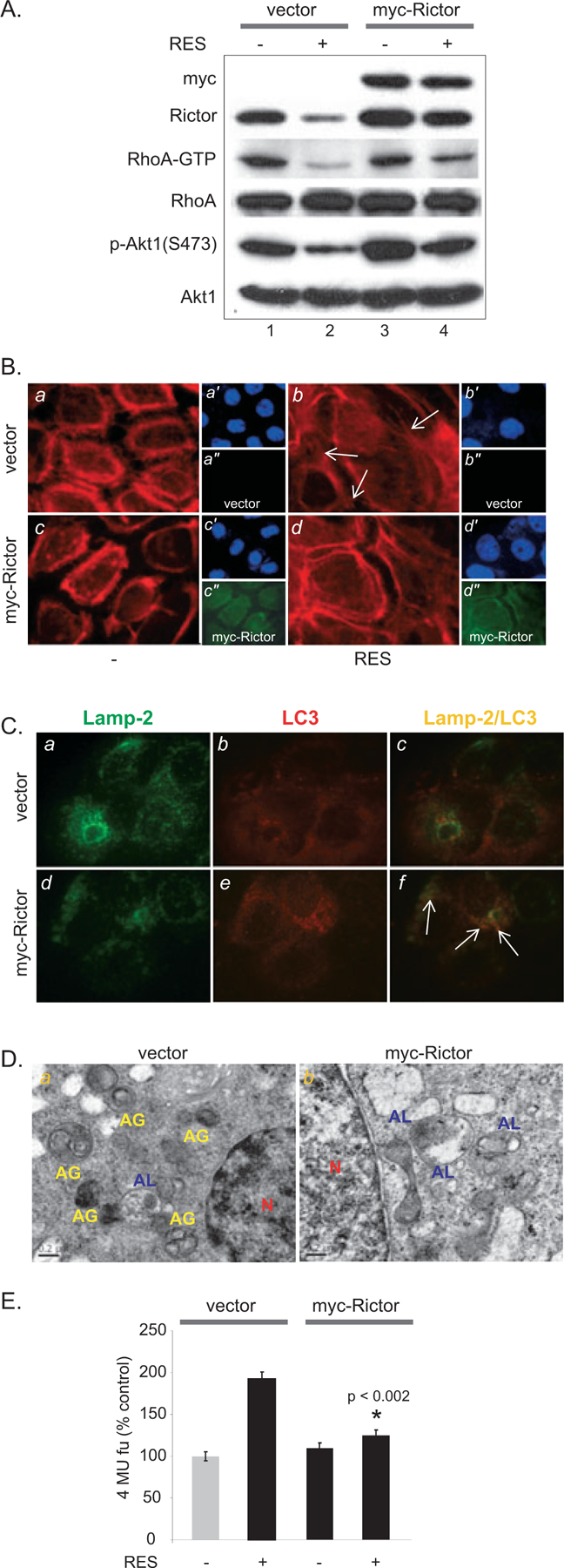
Exogenous Rictor expression restores RhoA-GTP activity and attenuates RES-induced SA-β-gal activity. (A) Exogenous Rictor restores RhoA-GTP activity. Cells were incubated with 50 μM RES for 48 h following viral transduction of myc-Rictor. (B) Actin remodeling is restored by exogenous Rictor expression in RES-treated A431 cells. F-actin was detected by rhodamine-phalloidin (red). Myc-Rictor expression is detected using anti-myc antibody (green, a″, b″, c″, d″). DAPI (blue, a′, b′, c′, d′). (C) Colocalization of Lamp-2 and LC3 is increased by exogenous Rictor expression. Green, Lamp-2; red, LC3, yellow, Lamp-2 ⁄ LC3. (D) EM images of A431 cells transduced with empty vector (a) or myc-Rictor (b) followed by RES treatment. AG, yellow, autophagosomes; AL, blue, autolysosomes; N, red, nucleus. (E) Rictor overexpression reduces RES-induced SA-β-gal activity.
Rictor is overexpressed in UV-induced SCCs and its expression is decreased following RES treatment
To determine whether RES regulates Rictor expression in vivo, the tissue distribution of Rictor was analyzed in SCCs-induced in UV-irradiated p53+ ⁄ − ⁄ SKH-1 mice (27). Rictor was substantially expressed in tumors, but not in nontumor bearing tumor adjacent skin, assessed by immunohistochemistry (Fig. 5A, upper panel). Oral administration of RES significantly decreased the levels of Rictor in SCCs (Fig. 5A, bottom panel; Fig. 5B, n = 25, P < 0.01). Furthermore, increased SA-β-gal staining (Fig. 5C) and activity (Fig. 5D) were detected in SCCs harvested from mice treated with RES, compared with nontreated SCCs. These data indicate that Rictor is overexpressed in UV-induced murine SCCs, and that RES-mediated suppression of UV-induced SCC carcinogenesis may involve premature senescence induction in tumors in p53+ ⁄ − ⁄ SKH-1 mice, which is likely mediated by downregulating Rictor and Rictor-mediated regulatory processes.
Figure 5.
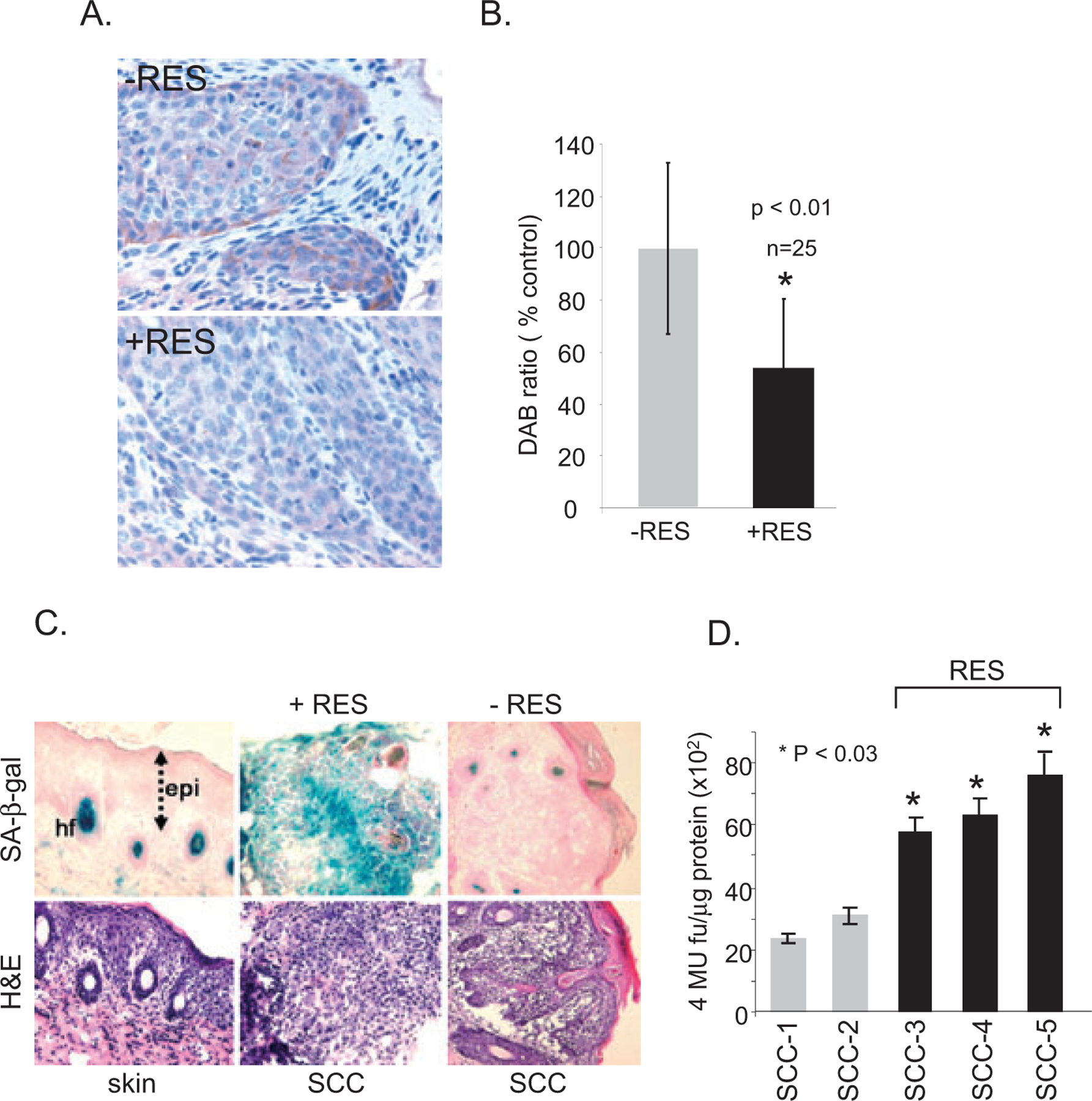
Rictor expression in UV-induced SCCs. (A) Immunohistochemical distribution of Rictor in UV-induced SCCs harvested from nontreated (upper panel) and RES-treated (bottom panel) p53+ ⁄ − ⁄ SKH-1 mice. Magnification 200×. (B) Immunohistochemical image analysis of Rictor expression determined using the IMAGEJ program. n = 25 per group, *P < 0.01. (C, D) RES-induced senescence in UV-induced SCCs from p53+ ⁄ − ⁄ SKH-1 mouse skin. (C) Detection of SA-β-gal in UV-irradiated SCCs harvested from nontreated and RES-treated mice. n = 25 per group. Representative pictures are shown. Magnification 100×. epi, epidermis; hf, hair follicle; H&E, hematoxylin and eosin. (D) SA-β-gal activity in randomly selected UV-induced SCCs from p53+ ⁄ − ⁄ SKH-1 mice, treated (n = 3) or nontreated (n = 2) with RES. Quantification of SA-β-gal activity was performed in triplicate per sample.
DISCUSSION
Resveratrol has been implicated as an autophagy inducer, as evidenced by increases in the amount of autophagosomes in various cell lines (47–49). This notion is further strengthened by the observation that NAD-dependent deacetylase SIRT1, a known target of RES, induces autophagy by deacetylating autophagy genes Atg5, Atg7 and Atg8 in mouse embryonic fibroblasts (50). However, the facts that persistent, excess autophagic activity will inevitably cause cell death due to excessive degradation of cellular materials and that prematurely senescent cells are viable albeit with enlarged cell morphology, suggested that RES-induced premature senescence induction is associated with aberrant autophagy signaling. We observed increased LC3II in RES-treated A431 cells, although its level was unaltered in cells treated with rapamycin. Rapamycin is a known inducer of autophagy and is typically used in concentrations between 20 and 200 nM in the related studies (39,51); however, higher concentrations of rapamycin have also been used as in the study by Groth-Pedersen et al. (2.5 μM as compared with 0.1 μM in our study; 36). As they used HeLa as well as MCF-7 cells in their study, the effective doses may vary according to the cell types. In A431 cells, the dose of 0.1 μM seems effective as it blocked RES-induced accumulation of LC3 (Fig. 1D). We also observed that cotreatment with BAF, which prevents the fusion of autophagosomes with lysosomes, thereby causing LC3II accumulation, further enhanced LC3II accumulation compared with accumulation in cells treated with BAF or RES alone. Together with the data showing a lack of colocalization of LC3II and Lamp-2 in RES-treated cells, these results suggested that, although RES enhances autophagosome formation, the autophagosomes so formed are not likely to proceed to autolysosome formation. Previously, it has been shown that treatment of MCF-7 breast carcinoma cells with vincristine, a microtubule-destabilizing antimitotic drug, induces a senescent-like morphology via inhibition of autolysosome formation (36). Despite the increases in LC3-positive vesicles and Lamp-2 levels, we found that LC3-positive vesicles and Lamp-2-positive vesicles did not colocalize. We further showed that RES-induced senescence involved downregulation of Rictor, which in turn decreased RhoA-GTPase activity. Overexpression of Rictor suppressed RES-induced increases in SA-β-gal activity and actin stress fibers. Furthermore, overexpression of Rictor was able to increase colocalization of Lamp-2 and LC3II in RES-treated A431 cells. The involvement of F-actin assembly in stimulating autophagosomes-lysosome fusion has been shown previously (52). Lovastatin, a 3-hydroxy 3-methylglutary coenzyme A reductase inhibitor, was recently shown to induce senescence. Lovastatin-induced senescence was shown to involve the inhibition of RhoA activity, whereas constitutively active RhoA reversed lovastatin-induced senescence in the PC3 prostate cancer cell line (53). These data are in accordance with our results, which suggest the importance of actin cytoskeleton organization in guiding autophagic processes. Our data further suggest the significance of Rictor-mediated RhoA-GTPase activity in the autophagic process, as overexpression of Rictor attenuated SA-β-gal activity, suggesting a link between Rictor-mediated signaling and the senescence phenotype. Rictor has been shown to be required for cancer progression in a prostate cancer model driven by PTEN loss. The level of Rictor was elevated in glioma cell lines and primary tumor cells as compared with normal brain tissue (54,55). Elevated levels of Rictor were found in UV-induced murine SCCs, but levels were reduced in SCCs harvested from mice receiving oral administration of RES. It remains to be elucidated whether Rictor downregulation constitutes an in vivo mechanism of RES-induced tumor suppression. For our in vivo study, we have used 200 mg kg−1 RES, 3× a week (27), and found in vivo senescence induction in tumors (Fig. 5). Because physiological doses of RES depend on several factors such as bioavailability, absorption, metabolism and tissue distribution, the measurement of RES concentration in the SCCs would be useful to determine the actual physiological concentration inducing senescence in the target tissue.
DNA damage also has been shown to induce autophagy. DNA damage induced by doxorubicin has recently been shown to accompany autophagy, and inhibition of autophagy potentiated doxorubicin-induced cell death, suggesting that autophagy acts as a protective mechanism against DNA damage-induced apoptosis (51,56). RES has been shown to induce DNA damage via ROS generation (28). RES-generated ROS is likely to cause damage to proteins and organelles, which may have to be removed to prevent further cellular damage (57). In this respect, RES may initially induce autophagy; the levels of LC3 and Lamp-2 remained unchanged up to 16 h following RES treatment, which may act as a cell survival mechanism. On the other hand, the lack of colocalization of autophagic components in prematurely senescent A431 cells might also indicate a mechanism of cell survival by preventing autophagocytosis of prematurely senescent A431 cells. In this scenario, RES-induced, prematurely senescent cells may escape from autophagic cell death via suppression of autolysosome formation. Along with macro-autophagy, chaperone-mediated autophagy (CMA), a stress-induced pathway, selectively degrades proteins in lysosomes (58). Because CMA-deficient cells have been shown to be dramatically sensitive to oxidative stress (59), treatment with pharmacological inhibitors of lysosome along with DNA damage-inducing agents may increase cancer cell death (36,59). Given the possibility of cancer cells escaping from premature senescence, targeting the CMA pathway to eliminate prematurely senescent (but viable) cancer cells may be a beneficial therapeutic option.
Acknowledgements—
This work was partially supported by Irving Scholar Award and 5P30 ES009089.
Abbreviations:
- Akt
prtotein kinase B
- LC3
wild-type human microtubule-associated protein 1 light chain 3
- LC3–1
soluble nonlipidated form of LC3
- LC3II
LC3-phospholipid conjugate
- mTOR
mammalian target of rapamycin
- PI3K
Phosphatidylinositol 3-kinases
- PTEN
Phosphatase and tensin homolog
- Ras
RAt sarcoma
- RES
resveratrol
- SA-β-gal
senescence-associated β-galactosidase
Footnotes
This paper is part of the Special Issue in Commemoration of the 70th birthday of Dr. David R. Bickers.
REFERENCES
- 1.Woodhead AD, Setlow RB and Tanaka M (1999) Environmental factors in nonmelanoma and melanoma skin cancer. J. Epidemiol 9, S102–S114. [DOI] [PubMed] [Google Scholar]
- 2.Saldanha G (2001) The Hedgehog signalling pathway and cancer. J. Pathol 193, 427–432. [DOI] [PubMed] [Google Scholar]
- 3.Amstad PA and Cerutti PA (1995) Ultraviolet-B-light-induced mutagenesis of C-H-ras codons 11 and 12 in human skin fibroblasts. Int. J. Cancer 63, 136–139. [DOI] [PubMed] [Google Scholar]
- 4.Nishigori C (2006) Cellular aspects of photocarcinogenesis. Photochem. Photobiol. Sci 5, 208–214. [DOI] [PubMed] [Google Scholar]
- 5.Jimeno A, Kulesza P, Wheelhouse J, Chan A, Zhang X, Kincaid E, Chen R, Clark DP, Forastiere A and Hidalgo M (2007) Dual EGFR and mTOR targeting in squamous cell carcinoma models, and development of early markers of efficacy. Br. J. Cancer 96, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khariwala SS, Kjaergaard J, Lorenz R, Van Lente F, Shu S and Strome M (2006) Everolimus (RAD) inhibits in vivo growth of murine squamous cell carcinoma (SCC VII). Laryngoscope 116, 814–820. [DOI] [PubMed] [Google Scholar]
- 7.Feldmeyer L, Hofbauer GF, Boni T, French LE and Hafner J (2012) Mammalian target of rapamycin (mTOR) inhibitors slow skin carcinogenesis, but impair wound healing. Br. J. Dermatol 166, 422–424. [DOI] [PubMed] [Google Scholar]
- 8.Han W, Ming M, He TC and He YY (2010) Immunosuppressive cyclosporin A activates AKT in keratinocytes through PTEN suppression: implications in skin carcinogenesis. J. Biol. Chem 285, 11369–11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monaco AP (2009) The role of mTOR inhibitors in the management of posttransplant malignancy. Transplantation 87, 157–163. [DOI] [PubMed] [Google Scholar]
- 10.Ulrich C, Kanitakis J, Stockfleth E and Euvrard S (2008) Skin cancer in organ transplant recipients—where do we stand today? Am. J. Transplant 8, 2192–2198. [DOI] [PubMed] [Google Scholar]
- 11.Jensen P, Hansen S, Moller B, Leivestad T, Pfeffer P, Geiran O, Fauchald P and Simonsen S (1999) Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J. Am. Acad. Dermatol 40, 177–186. [DOI] [PubMed] [Google Scholar]
- 12.Schmelzle T and Hall MN (2000) TOR, a central controller of cell growth. Cell 103, 253–262. [DOI] [PubMed] [Google Scholar]
- 13.Rubinsztein DC, Gestwicki JE, Murphy LO and Klionsky DJ (2007) Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov 6, 304–312. [DOI] [PubMed] [Google Scholar]
- 14.Kondo Y and Kondo S (2006) Autophagy and cancer therapy. Autophagy 2, 85–90. [DOI] [PubMed] [Google Scholar]
- 15.Shintani T and Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306, 990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Notte A and Leclere LC (2011) Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem. Pharmacol 82, 427–434. [DOI] [PubMed] [Google Scholar]
- 17.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH and Lenardo MJ (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304, 1500–1502. [DOI] [PubMed] [Google Scholar]
- 18.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D and Yahalom J (2001) A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 61, 439–444. [PubMed] [Google Scholar]
- 19.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A and Thompson CB (2007) Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Invest 117, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yue Z, Jin S, Yang C, Levine AJ and Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl Acad. Sci. USA 100, 15077–15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crazzolara R, Bradstock KF and Bendall LJ (2009) RAD001 (Everolimus) induces autophagy in acute lymphoblastic leukemia. Autophagy 5, 727–728. [DOI] [PubMed] [Google Scholar]
- 22.Athar M, Back JH, Kopelovich L, Bickers DR and Kim AL (2009) Multiple molecular targets of resveratrol: anti-carcinogenic mechanisms. Arch. Biochem. Biophys 486, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goswami SK and Das DK (2009) Resveratrol and chemoprevention. Cancer Lett 284, 1–6. [DOI] [PubMed] [Google Scholar]
- 24.Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S and Takada Y (2004) Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res 24, 2783–2840. [PubMed] [Google Scholar]
- 25.Aziz MH, Afaq F and Ahmad N (2005) Prevention of ultraviolet-B radiation damage by resveratrol in mouse skin is mediated via modulation in survivin. Photochem. Photobiol 81, 25–31. [DOI] [PubMed] [Google Scholar]
- 26.Reagan-Shaw S, Afaq F, Aziz MH and Ahmad N (2004) Modulations of critical cell cycle regulatory events during chemoprevention of ultraviolet B-mediated responses by resveratrol in SKH-1 hairless mouse skin. Oncogene 23, 5151–5160. [DOI] [PubMed] [Google Scholar]
- 27.Kim KH, Back JH, Zhu Y, Arbesman J, Athar M, Kopelovich L, Kim AL and Bickers DR (2011) Resveratrol targets transforming growth factor-beta2 signaling to block UV-induced tumor progression. J. Invest. Dermatol 131, 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Back JH, Rezvani HR, Zhu Y, Guyonnet-Duperat V, Athar M, Ratner D and Kim AL (2011) Cancer cell survival following DNA damage-mediated premature senescence is regulated by mammalian target of rapamycin (mTOR)-dependent inhibition of sirtuin 1. J. Biol. Chem 286, 19100–19108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mooi WJ and Peeper DS (2006) Oncogene-induced cell senescence—halting on the road to cancer. N. Engl. J. Med 355, 1037–1046. [DOI] [PubMed] [Google Scholar]
- 30.Campisi J (2005) Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120, 513–522. [DOI] [PubMed] [Google Scholar]
- 31.Gerland LM, Peyrol S, Lallemand C, Branche R, Magaud JP and French M (2003) Association of increased autophagic inclusions labeled for beta-galactosidase with fibroblastic aging. Exp. Gerontol 38, 887–895. [DOI] [PubMed] [Google Scholar]
- 32.Gary RK and Kindell SM (2005) Quantitative assay of senescence-associated beta-galactosidase activity in mammalian cell extracts. Anal. Biochem 343, 329–334. [DOI] [PubMed] [Google Scholar]
- 33.Kim AL, Athar M, Bickers DR and Gautier J (2002) Ultraviolet-B-induced G1 arrest is mediated by downregulation of cyclin-dependent kinase 4 in transformed keratinocytes lacking functional p53. J. Invest. Dermatol 118, 818–824. [DOI] [PubMed] [Google Scholar]
- 34.Kim AL, Athar M, Bickers DR and Gautier J (2002) Stage-specific alterations of cyclin expression during UVB-induced murine skin tumor development. Photochem. Photobiol 75, 58–67. [DOI] [PubMed] [Google Scholar]
- 35.Wu YC, Wu WK, Li Y, Yu L, Li ZJ, Wong CC, Li HT, Sung JJ and Cho CH (2009) Inhibition of macroautophagy by bafilomycin A1 lowers proliferation and induces apoptosis in colon cancer cells. Biochem. Biophys. Res. Commun 382, 451–456. [DOI] [PubMed] [Google Scholar]
- 36.Groth-Pedersen L, Ostenfeld MS, Hoyer-Hansen M, Nylandsted J and Jaattela M (2007) Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome-destabilizing siramesine. Cancer Res 67, 2217–2225. [DOI] [PubMed] [Google Scholar]
- 37.Kochl R, Hu XW, Chan EY and Tooze SA (2006) Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 7, 129–145. [DOI] [PubMed] [Google Scholar]
- 38.Bursch W, Hochegger K, Torok L, Marian B, Ellinger A and Hermann RS (2000) Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J. Cell Sci 113(Pt 7), 1189–1198. [DOI] [PubMed] [Google Scholar]
- 39.Reggiori F, Monastyrska I, Shintani T and Klionsky DJ (2005) The actin cytoskeleton is required for selective types of autophagy, but not nonspecific autophagy, in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 16, 5843–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bozulic L and Hemmings BA (2009) PIKKing on PKB: regulation of PKB activity by phosphorylation. Curr. Opin. Cell Biol 21, 256–261. [DOI] [PubMed] [Google Scholar]
- 41.Martin DE and Hall MN (2005) The expanding TOR signaling network. Curr. Opin. Cell Biol 17, 158–166. [DOI] [PubMed] [Google Scholar]
- 42.Sarbassov DD, Ali SM and Sabatini DM (2005) Growing roles for the mTOR pathway. Curr. Opin. Cell Biol 17, 596–603. [DOI] [PubMed] [Google Scholar]
- 43.Sarbassov DD, Guertin DA, Ali SM and Sabatini DM (2005) Phosphorylation and regulation of Akt ⁄ PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- 44.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A and Hall MN (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol 6, 1122–1128. [DOI] [PubMed] [Google Scholar]
- 45.Brunk UT and Terman A (2002) The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem 269, 1996–2002. [DOI] [PubMed] [Google Scholar]
- 46.Terman A, Dalen H, Eaton JW, Neuzil J and Brunk UT (2003) Mitochondrial recycling and aging of cardiac myocytes: the role of autophagocytosis. Exp. Gerontol 38, 863–876. [DOI] [PubMed] [Google Scholar]
- 47.Opipari AW, Tan L, Boitano AE, Sorenson DR, Aurora A and Liu JR (2004) Resveratrol-induced autophagocytosis in ovarian cancer cells. Cancer Res 64, 696–703. [DOI] [PubMed] [Google Scholar]
- 48.Trincheri NF, Follo C, Nicotra G, Peracchio C, Castino R and Isidoro C (2008) Resveratrol-induced apoptosis depends on the lipid kinase activity of Vps34 and on the formation of auto-phagolysosomes. Carcinogenesis 29, 381–389. [DOI] [PubMed] [Google Scholar]
- 49.Armour SM, Baur JA, Hsieh SN, Land-Bracha A, Thomas SM and Sinclair DA (2009) Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging (Albany NY) 1, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW and Finkel T (2008) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl Acad. Sci. USA 105, 3374–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Munoz-Gamez JA, Rodriguez-Vargas JM, Quiles-Perez R, Aguilar-Quesada R, Martin-Oliva D, de Murcia G, de Murcia J, Almendros A, de Almodovar M and Oliver FJ (2009) PARP-1 is involved in autophagy induced by DNA damage. Autophagy 5, 61–74. [DOI] [PubMed] [Google Scholar]
- 52.Lee JY, Koga Y, Kawaguchi W, Tang E, Wong YS, Gao UB, Pandey S, Kaushik E, Tresse E, Lu J, Taylor JP, Cuervo AM and Yao TP (2010) HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 29, 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee J, Lee I, Park C and Kang WK (2006) Lovastatin-induced RhoA modulation and its effect on senescence in prostate cancer cells. Biochem. Biophys. Res. Commun 339, 748–754. [DOI] [PubMed] [Google Scholar]
- 54.Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, Sheen JH, Mullholland DJ, Magnuson MA, Wu H and Sabatini DM (2009) mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 15, 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Masri J, Bernath A, Martin J, Jo OD, Vartanian R, Funk A and Gera J (2007) mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res 67, 11712–11720. [DOI] [PubMed] [Google Scholar]
- 56.Huang Q and Shen HM (2009) To die or to live: the dual role of poly(ADP-ribose) polymerase-1 in autophagy and necrosis under oxidative stress and DNA damage. Autophagy 5, 273–276. [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez-Rocha H, Garcia-Garcia A, Panayiotidis MI and Franco R (2011) DNA damage and autophagy. Mutat. Res 711, 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaushik S and Cuervo AM (2006) Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol. Aspects Med 27, 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Massey AC, Kaushik S, Sovak G, Kiffin R and Cuervo AM (2006) Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl Acad. Sci. USA 103, 5805–5810. [DOI] [PMC free article] [PubMed] [Google Scholar]