

Abstract
Background:
Epidemiological studies support the hypothesis that diabetes alters pulmonary responses to air pollutants like ozone (). The mechanism(s) underlying these associations and potential links among diabetes, , and lung inflammation and remodeling are currently unknown.
Objectives:
The goal was to determine whether pulmonary responses to repetitive ozone exposures are exacerbated in murine strains that are hyperglycemic and insulin resistant.
Methods:
Normoglycemic and insulin-sensitive C57BL/6J mice; hyperglycemic, but mildly insulin-resistant, KK mice; and hyperglycemic and markedly insulin-resistant KKAy mice were used for ozone exposure studies. All animals were exposed to filtered air (FA) or repetitive ozone ( , 4 h/d, for 13 consecutive weekdays). Tissue analysis was performed 24 h following the final exposure. This analysis included bronchoalveolar lavage (BAL) for cell and fluid analysis, and tissue for pathology, immunohistology, mRNA, and hydroxyproline.
Results:
Following repetitive exposure, higher bronchoalveolar lavage fluid inflammatory cells were observed in all mice (), with a notable influx of neutrophils and eosinophils in KK and KKAy mice. Although the lungs of -exposed C57BL/6J and KK mice had minimal centriacinar histological changes without fibrosis, the lungs of -exposed KKAy mice contained marked epithelial hyperplasia in proximal alveolar ducts and adjacent alveoli with associated centriacinar fibrosis. Fibrosis in -exposed KKAy lungs was confirmed with immunohistochemistry, tissue hydroxyproline content, and tissue mRNA expression of fibrosis-associated genes (Ccl11, Il13, and Mmp12). Immunofluorescence staining and confocal microscopy revealed alterations in the structure and composition of the airway and alveolar epithelium in regions of fibrosis.
Discussion:
Our results demonstrate that in diabetic animal strains repetitive ambient ozone exposure led to early and exaggerated pulmonary inflammation and remodeling. Changes in distal and interstitial airspaces and the activation of Th2 inflammatory and profibrotic pathways in experimental animals provide a preliminary, mechanistic framework to support the emerging epidemiological associations among air pollution, diabetes, and lung disease. https://doi.org/10.1289/EHP7255
Introduction
Ozone () is a common gaseous air pollutant formed through the photochemical interaction of sunlight with nitrogen oxides and volatile organic compounds commonly produced by combustion sources such as motor vehicles. Exposures to elevated ambient concentrations of have been associated with increases in morbidity and mortality (Cohen et al. 2017; Cromar et al. 2018). In 2015, Cohen et al. (2017) estimated 254,000 deaths and 4.1 million disability-adjusted life years were attributed to global ambient exposures. Additionally, based on chemistry–climate forecast models, -attributable mortality is projected to increase up to 14% by 2030 (Orru et al. 2013; Wilson et al. 2017). This projected increase represents a significant public health concern that must be addressed to protect human health.
Epidemiology suggests that worsens airway-centered diseases such as asthma (Anenberg et al. 2018; Halonen et al. 2010), COPD (Halonen et al. 2010; Medina-Ramón et al. 2006) and cystic fibrosis (Farhat et al. 2013) principally via increasing disease-related symptoms or inciting exacerbations. These epidemiological studies are mechanistically supported by human and rodent exposure studies demonstrating -induced acute airspace inflammation, increased bronchoalveolar lavage fluid (BALF) total protein/albumin, and BALF cytokine release (Birukova et al. 2019; Devlin et al. 1991); delayed increases in airway hyper-responsiveness to methacholine challenge (Birukova et al. 2019; Foster et al. 2000; Que et al. 2011); and changes in lung architecture as evidence by hyperplasia and hypertrophy of the conducting airways and alveolar epithelia (Chang et al. 1992).
In addition to airway-centered disease, recent reports also suggest that affects parenchymal lung diseases, such as idiopathic pulmonary fibrosis (IPF). Separate epidemiological studies by Johannson et al. in a longitudinal South Korean cohort (Johannson et al. 2014) and by Sesé et al. in a French national multicenter longitudinal cohort (Sesé et al. 2018) demonstrated that increases in ambient concentrations were associated with acute exacerbations of IPF. An additional study from a longitudinal cohort at the University of California San Francisco suggests a link between ambient concentrations and baseline forced vital capacity (FVC), a measure of lung restriction/stiffness, in IPF (Johannson et al. 2018). Although these data suggest that may affect the lung parenchyma, there is limited information on mechanistic links that could underlie these epidemiological observations. Studies of chronic exposures [0.5 or (ppm) for 6h/d for 20 months] in F344 rats describe interstitial remodeling with collagen deposition, with alterations in extracellular matrix and evidence of bronchiolar epithelial metaplasia (Last et al. 1993; Stockstill et al. 1995). A more recent study in C57BL/6 mice exposed to for 8h/d for 10 wk found a role for transforming growth factor- () in the accumulations of collagen and smooth muscle actin in conducting airways (Katre et al. 2011). Similarly, paracrine signaling via from -exposed human airway epithelial cells to fibroblasts were associated with proliferation of fibroblasts and collagen synthesis in vitro (Wang et al. 2018). These data support a potential mechanism from environmental exposure to airway remodeling. Less clear is whether and how induces fibrosis and remodeling in distal lung parenchyma.
Data from two cohorts, one a Danish Diet, Cancer and Health cohort (Andersen et al. 2012) and the other a cohort of black women living in Los Angeles, California, USA (Coogan et al. 2012), supported associations between air pollution exposure and increased risk of diabetes. The mechanisms underlying these associations appear to be multifactorial and probably represent a combination of effects on immune system activation, on the metabolic function of adipose tissue and the liver, and on the central nervous system (Rao et al. 2015). Our group has previously demonstrated that subacute exposure altered diabetic responses in mice, where exposure-related reductions in insulin secretion, increases in adipose tissue inflammation, and alterations in insulin resistance were dependent on the strain (Ying et al. 2016; Zhong et al. 2016). Furthermore, strain-dependent responses to particulate matter with aerodynamic diameter () suggested broader effects of air pollution to disrupt metabolic homeostasis in mice with different insulin sensitivities (Sun et al. 2009; Xu et al. 2011). Together these animal studies suggest that air pollution can increase diabetes incidence and alter glucose control but also could augment inflammatory responses to air pollution exposure.
To the best of our knowledge, the mechanistic intersection of diabetes, air pollutant exposure, and pulmonary inflammation and remodeling has not been systematically studied. To address this, we compared metabolic and pulmonary endpoints in three strains of mice (C57BL/6J, KK, and KKAy) to test the hypothesis that exposure would enhance inflammatory and fibrotic lung lesions in spontaneously diabetic mice with preexisting hyperglycemia and insulin-resistance in comparison with normoglycemic, insulin-sensitive mice.
Methods
Animals
Male C57BL/6J, KK, and KKAy (KK.Cg-Ay/J) mice, 4–5 weeks of age, were purchased from The Jackson Laboratory. Mice were housed individually in stainless steel wire cages in whole-body inhalation exposure chambers (H-1000; Lab Products), and allowed to acclimate for 2 wk before initiating inhalation exposures. Mice were monitored daily for signs of stress or discomfort (normal respiration, activity, body weight), throughout the study period. No animals developed adverse health criteria to warrant removal from the study. All experimental protocols were approved by the Institutional Animal Care and Use Committees at Michigan State University, an AAALAC accredited institution.
Exposure Protocol
All strains of mice (8/group) were exposed at the same time to filtered air ( ; air controls) or () for 13 consecutive weekdays [Monday to Friday, 4 h/d, 0900 hours to 1300 hours (9:00 A.M. to 1:00 P.M.)]. Ozone was generated with an OREC Model O3VI-O ozonizer (Ozone Research and Equipment Corp.) using compressed air as a source of oxygen, and chamber ozone concentrations were monitored continuously with Dasibi 1003 AH ozone monitors (Dasibi Environment Corp.). Chamber concentrations of ozone during the 13-d study were (). Food was removed during exposures. Previous studies have reported that it requires 4 to 5 times the concentration of ozone to induce pulmonary inflammatory responses in sedentary laboratory rats that were comparable to those induced in exercising human subjects under controlled acute exposure conditions (Hatch et al. 1994, 2013). As such, the current exposure to in resting rodents is: a) less than ozone concentrations of that caused pulmonary function impairments in exercising adults receiving short-term exposures (Avol et al. 1984; Folinsbee et al. 1984); b) similar to ozone concentrations of that caused pulmonary neutrophilia in resting human adults receiving short-term exposures (Hatch et al. 2013); and c) approximately 4- and 7-fold higher than the 1-h () and 8-h () national ambient air quality standard concentrations for ozone, respectively.
Intraperitoneal Insulin Tolerance Test (IPITT)
Two hours after the last exposure (i.e., when mice have been fasted for 6 h), baseline blood glucose and body weights were measured (0 time). Mice then received an intraperitoneal injection (i.p.) of body weight of insulin. Blood was collected by tail vein puncture at 15, 30, 60, 90, 120, and 130 min post injection. Glucose was measured using a Bayer Contour® glucometer. Food was returned to the animals at completion of the test.
Necropsy
Six hours prior to necropsy, food was removed, and animals were again fasted until necropsy. Approximately 22 h after the last exposure, mice were deeply anesthetized with sodium pentobarbital (, i.p.), blood was collected from the ascending vena cava for plasma isolation, the trachea was cannulated, and the heart and lungs were excised en bloc. A volume of saline was instilled through the tracheal cannula and withdrawn to recover BALF. A second lavage was performed, and pooled BALF was used for cellular and biochemical analyses. Blood was kept on ice until spun at for 15 min at 4°C to collect plasma.
Inflammatory Cells and Cytokines in BALF
Total number of leukocytes in the collected BALF was estimated using a hemocytometer. Cytological slides were prepared from of BALF by centrifugation at for 10 min at room temperature (RT) and stained with Diff-Quick (Siemens, B4132-1A) following manufacturer’s protocol. Differential cell counts of macrophages, neutrophils, eosinophils, and lymphocytes were manually assessed by standard morphology from at least 200 cells in cytospin slides using a light microscope (Olympus BX-40). BALF supernatants were assayed for inflammatory cytokines using BD Biosciences Cytometric Bead Array, which is an ELISA-based detection system. Cytokine-specific Flex Set® reagents and antibodies were used to detect keratinocyte-derived chemokine (KC) (# 558340), (#560232), IL-5 (#558302), IL-6, (#558301), IL-13 (#558349) and IL-17A (#562261) (BD Biosciences), and analysis was performed using a FACSCalibur flow cytometer (BD). Briefly, of BALF was incubated with the antibody-coated bead complexes, and phycoerythrin-conjugated secondary antibodies were then added to form sandwich complexes. After acquisition of sample data using the flow cytometer, cytokine concentrations were calculated based on standard curve data using FCAP Array™ software (BD).
Plasma ELISAs: Insulin, Leptin, Adiponectin
Plasma concentrations of insulin, leptin, and adiponectin were determined using a Mouse Insulin ELISA kit (Crystal Chem Inc.), the Quantikine Mouse Leptin Immunoassay kit, and Quantikine Mouse Adiponectin kit (R&D Systems), per the manufacturers’ instructions. Insulin resistance was determined using the homeostatic model assessment of insulin resistance (HOMA-IR), which assesses the ratio of fasting blood glucose and insulin and is routinely used for human clinic assessment (Brook et al. 2013). HOMA-IR was calculated as follows:
Lung Histopathology
The right caudal lobe was removed and stored in RNAlater for RT-PCR, and the right middle lobe was removed for measurement of tissue hydroxyproline content. The left lung lobe was fixed by instillation with neutral-buffered formalin, and 5-micron-thick paraffin–embedded histological sections were stained with hematoxylin and eosin (H&E) for routine light microscopic examination (VS110, Olympus America), PicroSirius Red (from Sigma-Aldrich, with Fast Green counterstain using the Gladstone Institutes staining protocol) to detect collagen fibers, and rat anti-mouse major basic protein (MBP; #MT3-25.1.1, Mayo Clinic), rat anti-mouse neutrophil marker (#MCA771G, Bio-Rad), rabbit muscle actin (#ab5694, Abcam), polyclonal rabbit anti-club cell secretory protein (CCSP, uteroglobin, #ab40873, Abcam) or rabbit anti-Ym1/2 (#ab192029, Abcam) for immunohistochemical detection of eosinophils, neutrophils, myofibroblasts, club cells, and Ym1/2-postive macrophages, respectively. Either Vectastain ABC (Vector Labs) or Promark (BioCare) secondary detection systems were used for enzyme activation of chromogen as follows: Alkaline phosphatase and Fast Red chromogen (Vector Labs) was used to detect neutrophil- and MBP- positive tissues; peroxidase and Nova Red (Vector Labs) were used for Ym1/2; peroxidase and Romulin (Biocare Medical) were used for muscle actin; and alkaline phosphatase and Warp Red (Biocare) was used to detect CCSP. All immunohistochemically processed slides were counterstained with Gill 2 hematoxylin.
For immunofluorescence staining of epithelial cells and hyaluronan, antigen retrieval was first performed using steamer machine with Na-Citrate buffer (pH 6), and then the slides were allowed to cool for 1 h. Slides were washed in phosphate buffered saline (PBS) three times and incubated with Avidin Block for 15 min, Biotin Block for 15 min, and then MOM block (Vector Labs) for 1 h (all incubations at RT). Following blocking, primary antibodies (rabbit anti-SFPC (ab3786; Abcam) goat anti-CCSP (gift from Barry Stripp, Cedars Sinai, Los Angeles, California, USA), Rh Aggrecan/HA (biotinylated recombinant aggrecan, R&D Systems) were applied to slides and incubated at 4°C overnight. The next day, slides were washed (three times for 5 min) in PBS at RT. Secondary antibodies (Anti-Streptavidin-488 1:500, Anti-rabbit 555 1:500, Anti-Goat 647 1:500, Invitrogen, Thermo Fisher Scientific) were applied for 3 h at RT and then were washed in PBS for 5 min. DAPI staining was performed (dilution 1:1000 for 10 min). The slides were fixed with FluoroSave™ per manufacturer’s recommendations. The slides were imaged on a confocal fluorescent microscope (Olympus Fluoview FV3000).
Measures of Lung Morphometry
Histological slides were scanned and digitized with a slide scanner (VS110, Olympus America), and evaluated via stereological methods with newCAST™ software (VisioPharm). Digitized images of lung sections were selected as regions of interest, and 40% of the lung tissue was captured at 400× magnifications by systematic random sampling. Percentages of neutrophil marker–positive cells, MBP-positive cells, Ym1/2-positive macrophages, total lung macrophages (H&E stained sections), CCSP airway epithelial cells, and parenchymal collagen content in lung tissues were estimated using Stepanizer stereology software with a point grid by dividing number of points hitting areas positive for relevant staining [ positive] by the total number of points falling on all lung tissue [positive and negative staining tissue; reference tissue]. For each region, percent density of the cells of interest were calculated with the equation:
Hydroxyproline Assay
Frozen lung samples (right lung lobes) desiccated, weighed, and hydrolyzed in 6 N hydrochloric acid (HCl) overnight. Samples were diluted in PBS and brought to pH of 7.2. Samples were then incubated with 1.4% chloramine T, 10% n-propanol, and sodium acetate (pH 6.0) for 20 min at RT, followed by incubation with Ehrlich’s solution at 65°C for 15 min. Absorbance was measured at , and the amount of hydroxyproline determined against a standard curve generated using known concentrations of hydroxyproline.
Lung RT-PCR
After necropsy, right caudal lung lobes were stored in RNAlater (Qiagen Inc.) at until further use. Total RNA isolation and cDNA synthesis were performed as described previously (Brandenberger et al. 2013) using RNeasy Mini Kit (Qiagen) and High-Capacity cDNA Reverse Transcription Kit (Qiagen), respectively, per manufacturer’s instructions. Total RNA was quantified using a NanoDrop-1000 Spectrophotometer (Thermo Scientific). Quantitative RT-PCR was carried out using TaqMan Gene Expression Assays (Applied Biosystems). Murine gene targets included Ccl11 (eotaxin), IL13 (interleukin-13), Mcp2 (monocyte chemoattractant protein 2; CCL8), Arg1 (arginase-1), Mmp12 (matrix metallopeptidase 12), Scgb1a1(Club cell secretory protein), Chia (acidic mammalian chitinase), Saa3 (serum amyloid protein A 3), and housekeeping genes (Gusb, Hprt, and Rplp0 mRNA) (Table S1), were analyzed using cDNA in a final reaction volume on a SmartChip Real-Time PCR System (WaferGen) or using cDNA in a final reaction volume on the ABI PRISM 7900HT platform (Applied Biosystems). values of the genes of interest were obtained and normalized by subtracting the geometric mean of Cts from the endogenous controls. Relative gene expression levels were reported as fold change using the method, where .
Statistical Analyses
Data are expressed as the group error. Grubb’s outlier test was used to determine statistical outliers. Data were analyzed using a completely randomized analysis of variance (SigmaStat version 12; Jandel Scientific), with factors of time and exposure for the temporal responses in the insulin tolerance test and with factors of mouse strain and exposure for all other data. Multiple comparisons were made by Student–Newman–Keuls post hoc test. Student’s t-test was used to compare gene expression in air- vs. ozone-exposed groups. Criterion for significance was set at . Each experiment was performed with 8 animals per exposure and per strain, and the results are reported in table form in the online supplement.
Results
Weight Gain, Glycemic Control, and Insulin Resistance in C57BL/6J, KK, and KKAy Mice Exposed to Repetitive FA or Exposure
To explore the role of diabetes on pulmonary responses to multiday exposures, we exposed murine strains with varying diabetic phenotype: normoglycemic and insulin-sensitive C57BL/6J mice; hyperglycemic mice with mild (KK) and marked (KKAy) insulin resistance. Compared with C57BL/6J mice, both KK and KKAy mice exhibited greater body mass and weight gain after the 13-d study period, regardless of exposure to filtered air or (Figure S1A; Table S2). KKAy mice gained more weight than the other strains. Less total weight gain was observed in -exposed KK and KKAy strains but remained higher than was observed in C57BL/6J mice (Figure S1B; Table S3). At necropsy, serum glucose and insulin were measured. Both KK and KKAy mice exposed to filtered air demonstrated hyperglycemia (Figure 1A; Table S4), hyperinsulinemia (Figure 1B; Table S5), and insulin resistance (HOMA-IR; Figure 1C; Table S6) compared with WT mice, with KKAy mice having higher insulin levels and insulin resistance than KK mice. exposure had little effect on blood glucose levels, but it did cause a modest depression in circulating insulin in all three strains of mice (Figure 1B). Insulin resistance was mildly lower in KK mice than KKAy mice but was still 10-fold higher than that observed in C57BL/6 mice (Figure 1C). Differences in HOMA-IR following exposure in C57BL/6J () and KKAy mice () when compared to their air exposure were not significant. We also assessed circulating adipokines. Circulating leptin in air-exposed KK and KKAy mice were 15 and 25 times higher, respectively, than levels found in C57BL/6J mice (Figure S2A; Table S7). Plasma leptin in -exposed KK mice was slightly lower than vehicle-exposed mice, but it was similar between air- and -exposed C57BL/6J or KKAy mice. Alternatively, plasma adiponectin in KK and KKAy mice was about half of the circulating levels in C57BL/6J mice and was largely unaffected by exposure (Figure S2B; Table S8).
Figure 1.
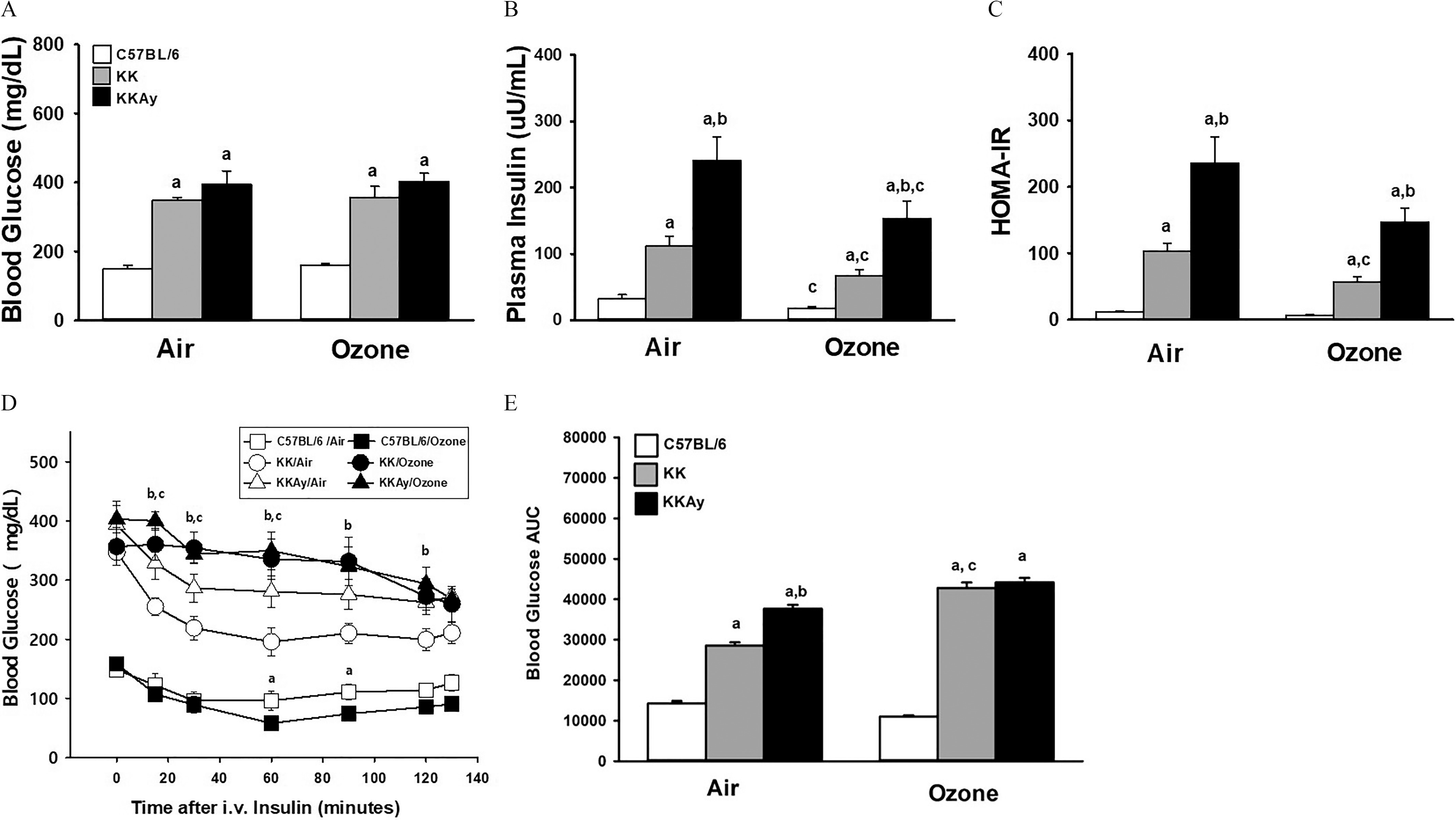
Serum glucose and insulin in fasted mice and following an insulin tolerance test (ITT) in C57BL/6J, KK and KKAy mice after air or exposure for 13 d. Plasma concentrations of glucose (A) and insulin (B) were measured from fasted all mice at the time of necropsy, approximately 22 h after the last exposure. Data of plasma glucose and insulin were used to calculate HOMA-IR (C). Time-dependent changes in blood glucose after a bolus insulin challenge in fasted mice were measured from 15 to 130 min after challenge (D). Effects of exposure within each strain and timepoint were determined. For each experimental group the area under the curve (AUC) was calculated between 0 to 130 min using the trapezoid rule (GraphPad Prism, San Diego, CA) (E). Data are expressed as (). For Figure 1D: (a) shows significant difference between air- and -exposed C57BL/6 mice; (b) shows significant difference between air- and -exposed KK mice. (c) shows significant difference between air- and -exposed KKAy mice. For remaining figures: (a) significantly different from similarly exposed (b) significantly different from similarly exposed KK mice; (c) significantly different from air-exposed mice of the same strain. Note: Significant differences between indicated groups, , were determined using a completely randomized analysis of variance, with factors of time and exposure for the temporal responses in the insulin tolerance test, and with factors of mouse strain and exposure for remaining data. Comparisons of group means were made with the Student–Newman–Keuls post hoc test. Summary data for panels A, B, C, D, and E can be found in Tables S4, S5, S6, S9, and S10, respectively.
To test the effect of exposure on insulin sensitivity, we conducted the ITT in fasted mice to determine changes in circulating glucose in response to an intravenous insulin challenge. Insulin-induced decreases in blood glucose were mildly enhanced in C57BL/6J mice exposed to , with decreases of by 60 min and 90 min after insulin administration, suggesting enhanced sensitivity to insulin in C57BL/6J mice (Figure 1D; Table S9). However, in hyperglycemic KK and KKAy mice, exposure resulted in the opposite effect, with significantly smaller insulin-induced reductions in blood glucose in comparison with that of air-exposed mice. Differences between air- and -exposed KK mice occurred from 15 to 120 min after insulin challenge and by as much as after 60 min. By comparison, in KKAy mice, exposure-related differences in blood glucose occurred from 15 to 60 min after insulin challenge and were as great as by 60 min post challenge (Figure 1D). When the ITT results were expressed as area under the curve (AUC), responses in air-exposed KK and KKAy mice were significantly higher than in C57BL/6J mice, with (Figure 1E; Table S10). exposure increased insulin resistance in KK but not in KKAy mice as the AUC increased by 50% and 19%, respectively.
Bronchoalveolar Lavage Cellular Inflammation and Cytokine Release in C57BL/6J, KK, and KKAy Mice following Repetitive FA or Exposure
To assess alterations in -induced inflammatory responses, we quantified airspace inflammatory cells and cytokine production. In BALF collected 1 d following the last exposure (day 14), total cells and neutrophils were modestly higher in air-exposed KK and KKAy mice in comparison with air-exposed C57BL/6J mice (Figures 2A,D; Tables S11, S14). -exposed mice, on the other hand, had considerably higher BALF total cells, eosinophils, and lymphocytes in comparison with air-exposed mice. This finding was strain-dependent [ (Figures 2A,C,E; Tables S11, S13, S15)]. Numbers of monocytes/macrophages and neutrophils after exposure were similar in KKAy and KK mice (Figures 2B,D; Tables S12, S14), whereas the higher level of cells in C57BL/6J mice after exposure was comparatively modest.
Figure 2.
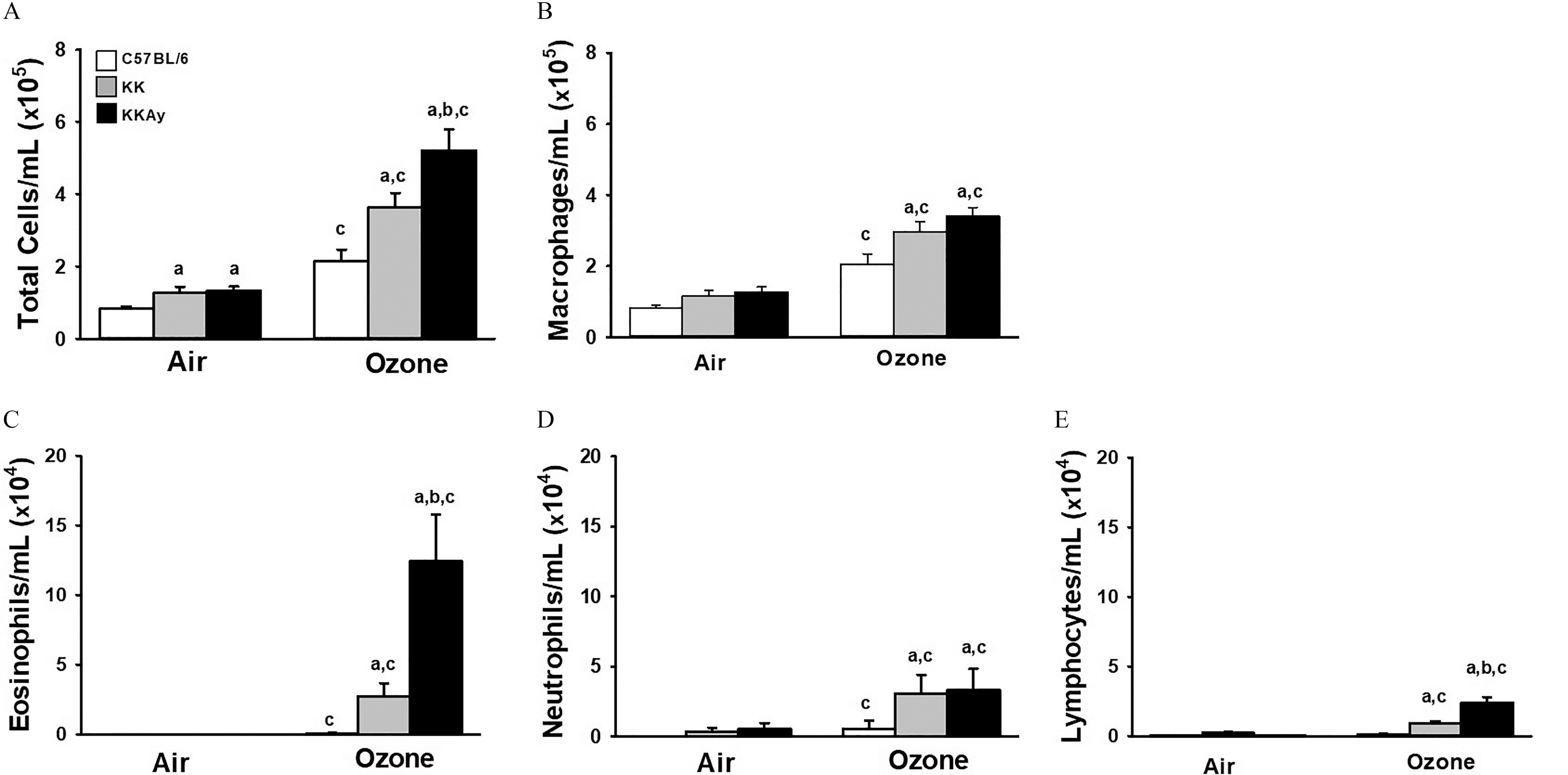
-induced bronchoalveolar lavage cellularity (BALF) cellularity in C57BL/6J, KK, and KKAy mice. Concentration in BALF of total cells (A), macrophages (B), eosinophils (C), neutrophils (D), and lymphocytes (E) were enumerated from cytospin preparations as described in “Methods.” Data are expressed as (). (a) significantly different from similarly exposed ; (b) significantly different from similarly exposed KK mice; (c) significantly different from air-exposed mice of the same strain; . Note: Data were analyzed using a completely randomized analysis of variance with factors of mouse strain and exposure, and comparisons of group means made with the Student–Newman–Keuls post hoc test. Summary data for panels A, B, C, D, and E can be found in Tables S11, S12, S13, S14, and S15, respectively.
To determine strain-dependent effects on lung tissue inflammation, we performed staining and morphometric assessments of pulmonary neutrophils, eosinophils, and macrophages (Figure S3; Tables S16–S19). Air-exposed KK and KKAy mice had higher lung neutrophil density vs. C57BL/6 mice (4.5- and 8.5-fold, respectively), with KKAy having higher density than KK mice (Figure S3A; Table S16). exposure caused higher accumulation of neutrophils in lungs of KKAy mice (1.6-fold), but not in KK or C57BL/6J mice. There were few eosinophils found in the lungs from any strain of air-exposed-mice (Figure S3B; Table S17). -exposed C57BL/6J mice, when compared to their FA controls, had similar accumulation of eosinophils (as a percent of total lung tissue). Alternatively, -exposed KK and KKAy mice exhibited higher accumulation of pulmonary eosinophils, accounting for 0.6% and 3.2% of total lung tissue, respectively (Figure S3E). In comparison, neutrophil density in -exposed mice was 0.5% and 1.2% in KK and KKAy mice, respectively (Figure S3A; Table S16). Macrophage density in lungs of air-exposed KK mice was significantly lower than in those of C57BL/6J mice (Figure S3C; Table S18). There were no strain-dependent differences among -exposed mice, because macrophage density was lower in C57BL/6J mice and higher in KK mice compared with air-exposed strain controls. The percentage of pulmonary macrophages staining for Ym1/2 protein were significantly higher in air-exposed KKAy mice in comparison with C57BL/6J mice (56% vs. 36%; Figure S3D; Table S19). exposure led to higher proportions of Ym1/2-positive macrophages in C57BL/6J and KK mice, but the ratio was similar to FA controls in KKAy mice.
From the BALF, we assessed cytokine release into the airspace. exposure led to a higher strain-dependent BALF IL-5 concentration, with a relatively higher amount in (Figure 3A; Table S20). IL-13 was undetectable in C57BL/6J mice but was present in both KK and KKAy mice exposed to filtered air (Figure 3B; Table S21). Only KKAy mice responded to , with an 80% higher IL-13. BALF IL-6 was higher only in the KK and KKAy strains, though it was below the level of detection in the BALF from C57BL/6J mice (Figure 3C, Table S22). Air-exposed KK and KKAy mice had higher KC but lower IL-17A in BALF in comparison with C57BL/6J mice (Figures 3D,E; Tables S23–S24). exposure caused higher BALF IL-17A in KKAy mice, but KC levels were decreased by 50% in comparison with filtered air–exposed mice. Concentrations of in BALF were similar among filtered air-exposed strains. No significant differences were observed in comparison with exposure in any strain, although levels in exposed KK mice were significantly less than levels found in C57BL/6J or KKAy mice (Figure 3F; Table S25).
Figure 3.
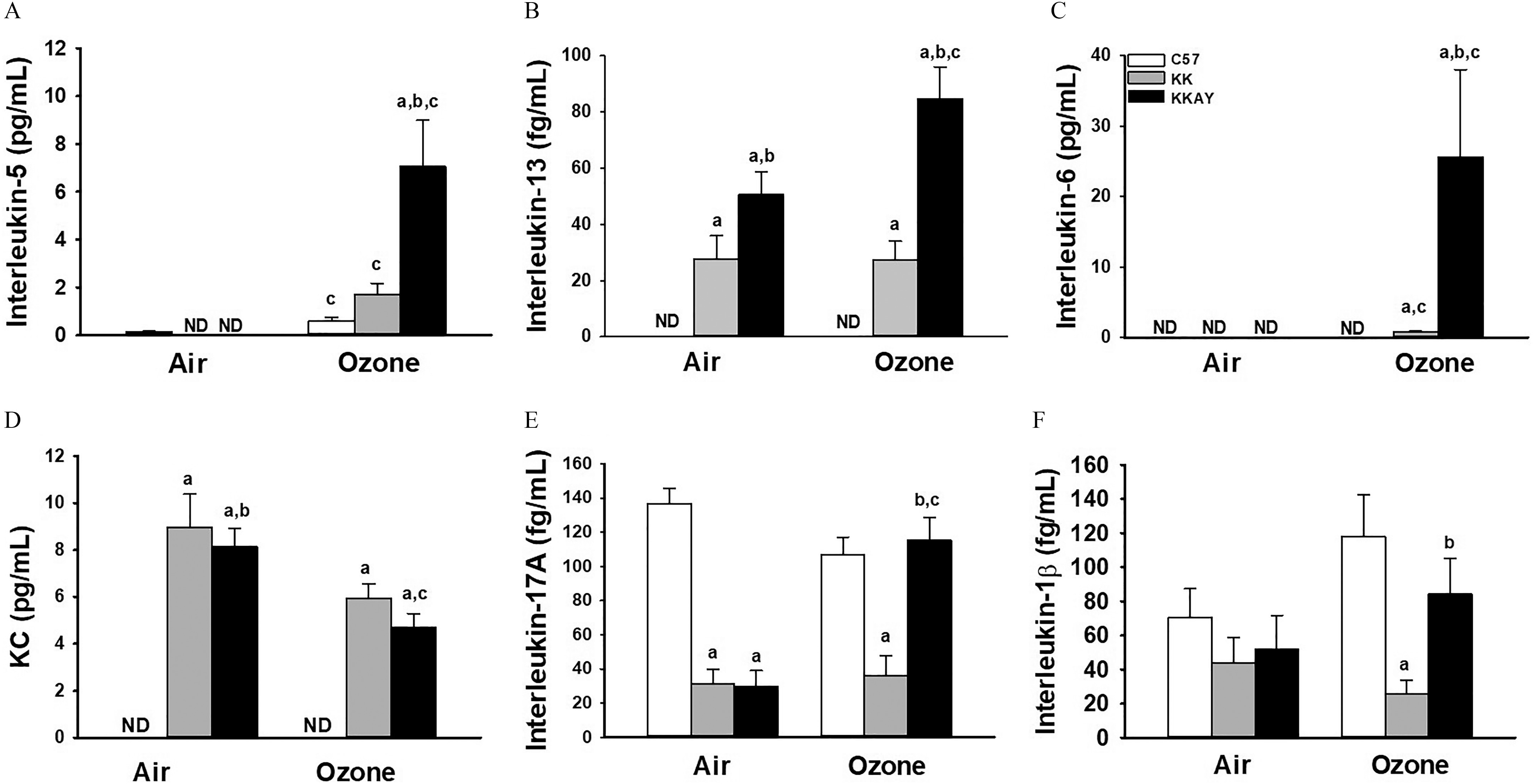
BALF cytokines in C57BL/6J, KK and KKAy mice after exposure to air or . Concentration in BALF of interleukin-5 (A), , (B), interleukin-17A (C), and KC (D) were determined by ELISA as described in “Methods.” Data are expressed as (). (a) significantly different from similarly exposed ; (b) significantly different from similarly exposed KK mice; (c) significantly different from air-exposed mice of the same strain; . Note: Data were analyzed using a completely randomized analysis of variance with factors of mouse strain and exposure, and comparisons of group means made with the Student–Newman–Keuls post hoc test. Summary data for panels A, B, C, D, E, and F can be found in Tables S20, S21, S22, S23, S24, and S25, respectively. ND, not detected.
Pulmonary Histology, Collagen Content, and Pro-Fibrotic Gene Expression in C57BL/6J, KK and KKAy Mice following Repetitive FA or Exposure
Given the evidence of altered inflammation and cytokine release in a strain-dependent fashion, we were interested whether evidence of altered pulmonary pathology might be observed. In the air-exposed groups, there was no overt pulmonary pathology (Figure 4 and data not shown). Alternatively, clear differences were observed in the -exposed strains. C57BL/6J mice demonstrated some minimal thickening of the alveolar duct interstitium consistent with prior observations (Michaudel et al. 2018). Alveolar duct thickening with the addition of cellular inflammation was observed in the KK mice. An interesting finding was that the -exposed KKAy mice developed focal regions of alveolar cell hypertrophy, interstitial thickening, and cellular inflammation in the regions adjacent to the alveolar ducts. These centriacinar lesions were not appreciated in the C57BL/6J or the KK strains.
Figure 4.

Histology of terminal airways in C57BL/6J, KK and KKAy mice. Light photomicrographs of centriacinar lesions (arrows, asterisk) in the lungs of KKAy, KK, and C57BL/6 mice exposed to . Tissues stained with hematoxylin and eosin. Note: Representative sections from the representative groups. a, alveolus; ad, alveolar duct; tb, terminal bronchiole.
Because these centriacinar lesions appeared to demonstrate alveolar interstitial thickening, we stained the lung tissue sections with PicroSirius Red to identify collagen deposition (Figure 5A–F). Quantification of PicroSirius Red staining in parenchymal collagen demonstrated that -exposed KK and KKAy mice were 1.7- and 3.2-fold higher, respectively (Figure 5G; Table S26). Alternatively, in -exposed C57BL/6J mice, lung collagen staining was not considerably different from that of filtered air. staining was performed in air and -exposed KKAy mice, revealing positive staining within the centriacinar lesions of -exposed KKAy mice consistent with presence of myofibroblasts (Figure S4A–SB). To confirm these histological observations, collagen content was determined biochemically using the hydroxyproline assay. The hydroxyproline content in lungs from C57BL/6J mice (Figure 5I; Table S27) or KK mice (Figure 5H; Table S28) was similar to strain matched filtered-air mice, but in the KKAy mice it was higher by 39% when compared with filtered-air KKAy mice.
Figure 5.
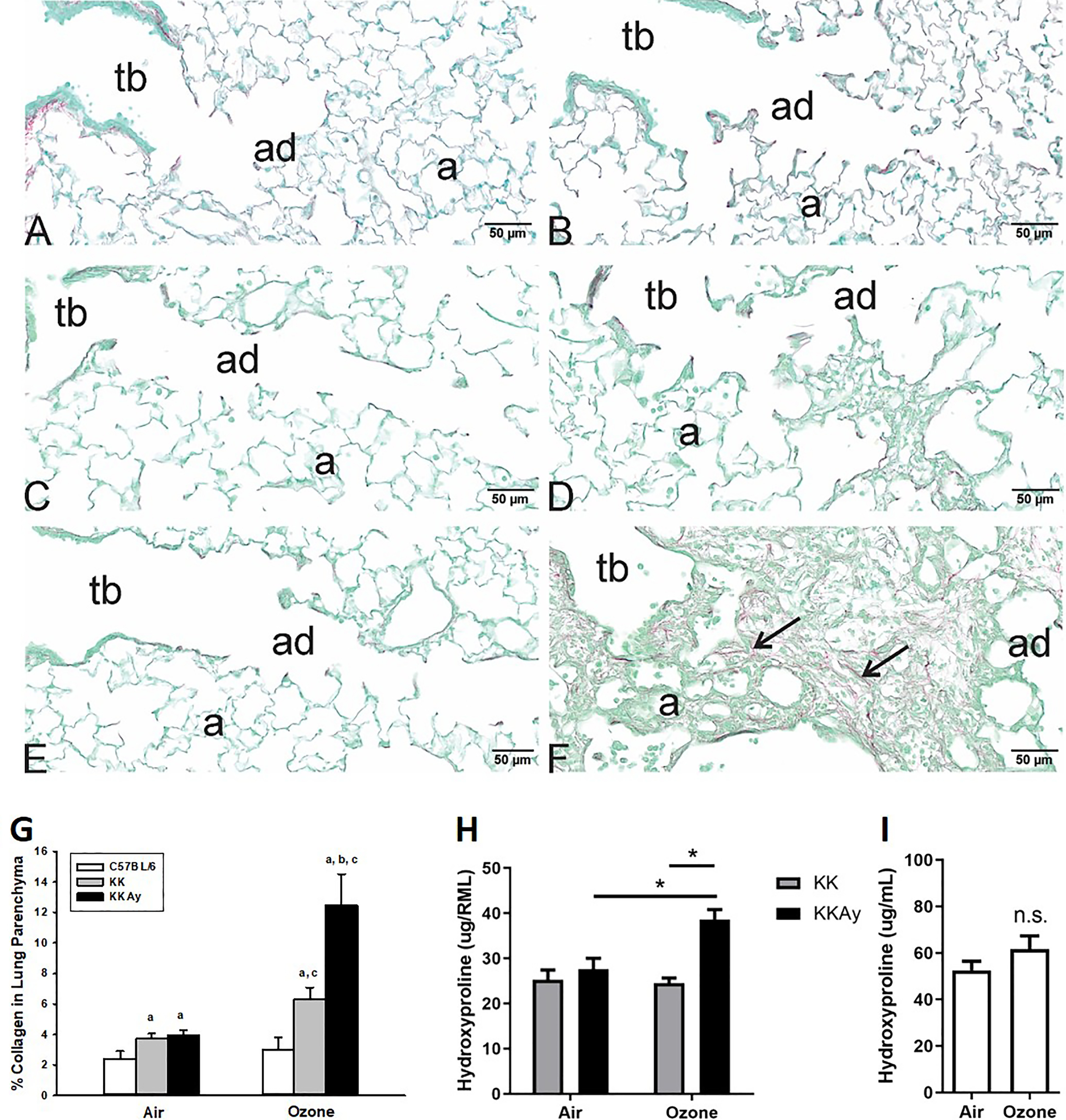
Assessment of lung fibrosis following air or exposure in C57BL/6J, KK, and KKAy mice. PicroSirius Red staining was performed in lung tissue sections from air-exposed (A) C57BL/6, (C) KK, and (E) KKAy mice. Staining was also performed in tissue sections from repetitive -exposed (B) C57BL/6, (D) KK, and (F) KKAy mice. In the -exposed KKAy mice, centriacinar areas of epithelial cell hyperplasia and interstitial thickening demonstrate enhanced staining for collagen by PicroSirius Red staining (F, large arrows). Collagen density in PicroSirius Red–stained lungs was quantified using morphometric approaches as described in “Methods,” and data were analyzed using a completely randomized analysis of variance with factors of mouse strain and exposure and the Student–Newman–Keuls post hoc test. (G). Lung content of hydroxyproline was assessed in right middle lung of (I) C57BL/6 and (H) KK and KKAy mice; by one-way analysis of variance with Tukey’s Multiple Comparison Testing. Data are expressed as (). (a) significantly different from similarly exposed ; (b) significantly different from similarly exposed KK mice; (c) significantly different from air-exposed mice of the same strain; . Note: Summary data for panels G, H, and I can be found in Tables S26, S28, and S27, respectively. a, alveoli; ad, alveolar duct; ND, not detected; n.s., not significant; tb, terminal bronchial.
To characterize the fibrotic response in KKAy mice, we performed a gene expression analysis on the strains. KKAy mice responded to repetitive exposures with the expression of Th2 and fibrotic-associated genes encoding for eotaxin, IL-13, monocyte chemotactic protein 2 (Mcp2), and arginase-1 (Arg1) (Figures 6A–D; Tables S29–S32). also elicited higher Mmp12 and lower Scgb1a1 [club cell secretory protein (CCSP)] in KKAy mice (Figures 6E,F; Tables S33–S34). KK mice also responded to with higher expression of Arg1 and Mmp12. In comparison, -exposed C57BL/6J had lower expression of Arg1 but higher expression of Chia (acidic mammalian chitinase) and Saa3 (serum amyloid protein) (Figure 6G,H; Tables S35–S36). Higher Saa3 in KKAy mice was significantly less than in lungs from C57BL/6J mice.
Figure 6.
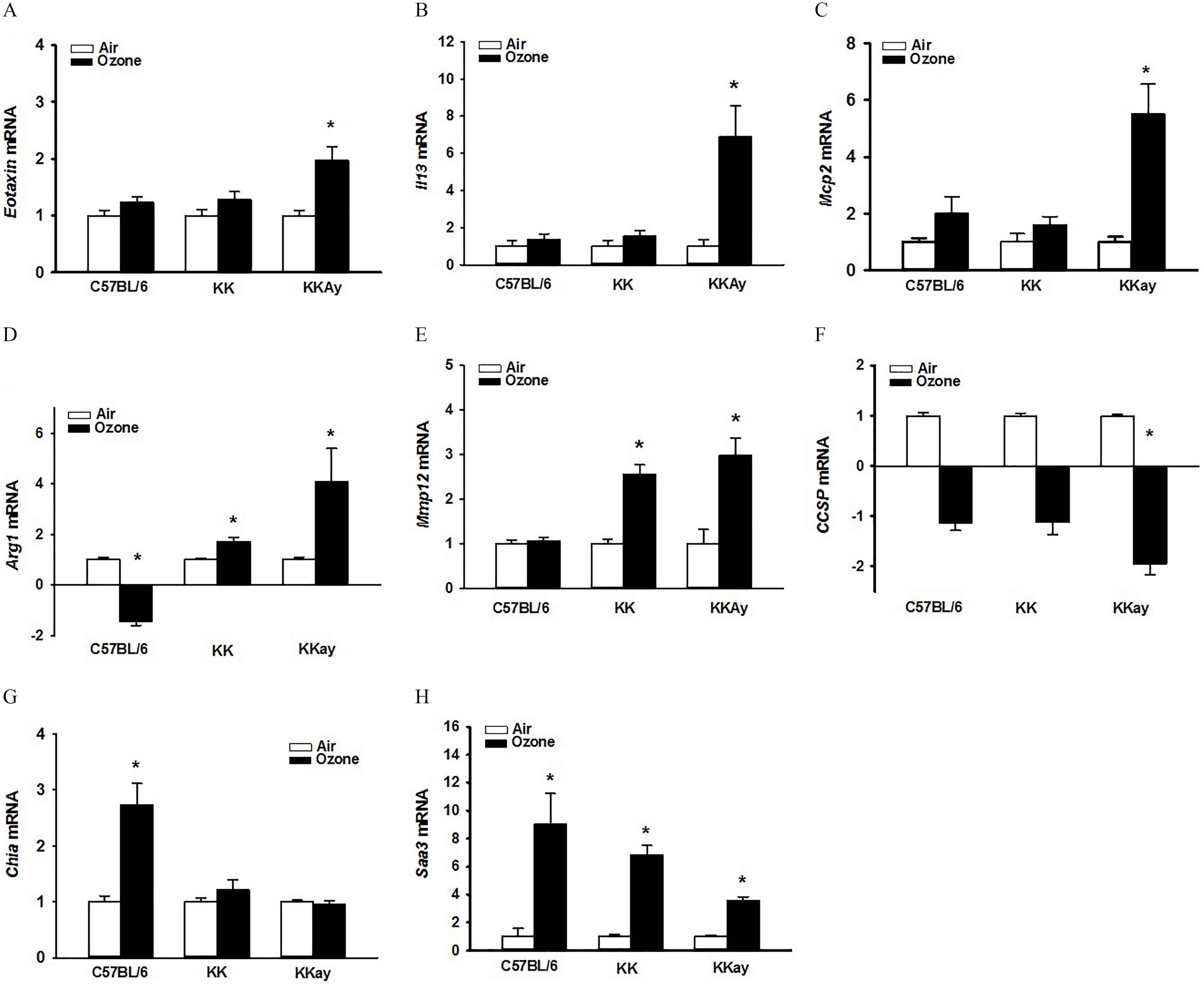
Strain-dependent whole-lung gene expression after exposure to air or . Relative fold increases in mRNA transcripts were analyzed in lung tissues for (A) Ccl11 (eotaxin); (B) IL13; (C) Mcp2 (monocyte chemoattractant protein 2; Ccl8); (D) Arg1 (arginase-1), (E) Mmp12 (matrix metallopeptidase 12), (F) Scgb1a1 (Club cell secretory protein), (G) Chia (acidic mammalian chitinase), (H) Saa3 (serum amyloid protein A 3). Data are expressed as fold changes relative to their respective air-exposed (). Student’s t-test was used to compare gene expression in air- vs. ozone-exposed groups. Summary data for panels A, B, C, D, E, F, G, and H can be found in Tables S29, S30, S31, S32, S33, S34, S35, and S36, respectively. *Genes showing statistical significances (air vs. /same mouse strain, ).
Imaging of Respiratory Epithelium at the Alveolar Duct Junction in C57BL/6J, KK, and KKAy Mice in Response to Repetitive FA vs. Exposure
To characterize the response in the alveolar tissue at the alveolar duct, we performed immunofluorescence staining to characterize the airway and alveolar epithelium and underlying matrix. Tissue sections from the C57BL/6J, KK and KKAy mice were obtained in the filtered air and -exposed groups. These were stained for club cell secretory protein (CCSP; a marker of club cells in the airway epithelium), surfactant protein C (SFTPC; a marker of type 2 alveolar epithelial cells), and hyaluronan (HA; a component of the extracellular matrix associated with fibrosis). Consistent with normal terminal murine airways, C57BL/6J mice demonstrated CCSP-expressing secretory cells in the distal airways, extending all of the way to the bronchoalveolar duct junction (BADJ) (Figure 7A; asterisks in images). Following repetitive exposure, there was more subepithelial HA deposition extending to the alveolar space just distal to the BADJ (Figure 7A) but no clear alteration in the airway composition of CCSP-expression. Distal airways of air-exposed KK mice had evidence of CCSP-expressing secretory cells, but many of these cells had decreased height and several expressed very low levels of CCSP (Figure 7B; see arrowheads). Following exposure in KK mice, there remained similar low numbers of CCSP-expressing cells proximal to the BADJ (Figure 7B). Some of these cells appeared squamous in morphology and expressed SFTPC (arrowheads), an alveolar marker not typically observed in murine airways. Furthermore, HA deposition was increased in the alveolar space of KK mice after exposure. In air-exposed KKAy mice, airway regions proximal to the BADJ were characterized by pronounced stretches of epithelium that lacked normal CCSP expression (Figure 7C,D; arrowheads). Many of the distal airways of KKAy mice were devoid of normal CCSP-expressing secretory cells; there were large stretches of distal airway covered by irregular and flattened epithelium that expressed low levels of CCSP and some SFTPC. Following exposure, regions of fibrosis adjacent to the airway were enriched with type 2 pneumocytes (Figure 7C; arrows), and the subepithelial space had become thickened with increased HA deposition (Figure 7D). To better show these regions in the KK and KKAy strains, enlarged images are provided for KK and KKAy filter air and tissue sections (Figure S5).
Figure 7.

Immunohistological assessment of distal airways and proximal alveoli in C57BL/6J, KK, and KKAy mice. Lung tissue sections were obtained from air- and -exposed C57BL/6, KK, and KKAy mice and underwent immunofluorescence staining for surfactant protein C (SFTPC; red), club cell secretory protein (CCSP; green), hyaluronan [Hyaluronic Acid Binding Protein (HABP); white], and 4′,6-diamidino-2-phenylindole (DAPI; blue). Representative images were obtained of the terminal airways and alveolar regions adjacent to the terminal airways. These included filtered air images from (A) C57BL/6, (B) KK, and (C, D) KKAy mice and ozone images from (A′) C57Bl/6, (B′) KK, and (C′,D′) KKAy mice. Within images, asterisks define regions of normal airway and alveolar regions, arrowheads defined regions of abnormal respiratory epithelium, and arrows define regions of fibrotic alveolar regions enriched with HA and type 2 pneumocytes. Images are representative of six mice per group with two slides analyzed per mouse.
To confirm the observations of reduced CCSP in the air-exposed strains, RT-PCR and histochemical staining for CCSP was performed. Compared with air-exposed C57BL/6J mice, whole lung tissue CCSP expression (fold change compared to C57BL/6 by RT-PCR) was lower in KK (, mice) and in the KKAy (, mice) strains. Immunohistochemical CCSP staining of the alveolar duct regions and morphometry assessment of the whole airway (Figure S6A–S6F), demonstrating lower CCSP in the KK and KKAy strains when compared to the C57BL/6J strain both in the air- and -exposed groups (Figure S6G; Table S37).
Discussion
The goal of our study was to test the hypothesis that abnormal glucose metabolism can exacerbate -induced pulmonary inflammation and remodeling. To address this hypothesis, our study was designed to characterize pulmonary responses to short-term repetitive ozone exposures and define how they were altered in diabetes-prone animal strains. First, we assessed changes in inflammation and in structural remodeling of airways and interstitium. Following repetitive exposure, we demonstrated that diabetes-prone KK and KKAy mice exhibited enhanced inflammatory responses in the airspace and lung parenchyma that associated with the degree of insulin resistance and hyperglycemia. Furthermore, we observed that -exposed insulin-resistant KKAy mice develop centriacinar fibrotic lesions composed of hyperplastic epithelial cells, myofibroblasts, and infiltrating immune cells. We confirmed enhanced fibrosis by histochemical and biochemical measures of collagen deposition. To the best of our knowledge, this study is the first description of exacerbated pulmonary inflammation and remodeling to short-term repetitive exposures in insulin-resistant rodent strain and supports prior epidemiology and animal studies suggesting enhanced susceptibility to ambient air pollution in diabetes.
A central observation of our study was the early and exacerbated pulmonary inflammation and remodeling in the diabetes-prone strains. The inflammatory response was characterized by increased airspace and parenchymal inflammation, including both neutrophil and eosinophils as well as macrophages. Remodeling was associated with an alteration in epithelial cell morphology, infiltration of myofibroblasts, and evidence of parenchymal fibrosis in centriacinar regions. Other animal studies have suggested a link between diabetes and air pollution responses focused on air pollution exposure driving to hyperglycemia and insulin resistance (Bass et al. 2013; Miller et al. 2016). Beyond induction of hyperglycemia and insulin resistance, how diabetes-prone animal strains exhibit augmented pulmonary inflammation and remodeling is less well understood. Our prior work in diabetes-prone KK mice revealed that repetitive exposures altered baseline metabolic states and enhance pulmonary sensitivity to air pollution (Ying et al. 2016; Zhong et al. 2016). Concentrated ambient exposures (5–8 wk) in KKAy mice have also been associated with worsened insulin resistance and systemic inflammation (Liu et al. 2014), but as far as we are aware, no studies have looked at pulmonary inflammation in KKAy mice. Our data suggest consideration of hyperglycemia and insulin resistance as a factor that may increase pulmonary sensitivity to ambient air pollution.
In addition to the evidence of exacerbated inflammation, we observed pronounced parenchymal centriacinar fibrosis in the KKAy mice at an early time point in response to short-term repetitive exposures. Our observation was distinct from other prior rodent studies assessing fibrosis induced by subchronic and/or chronic exposures because those prior studies used high cumulative doses (higher than ambient conditions) (Last et al. 1993; Stockstill et al. 1995) and have focused on peribronchiolar, as opposed to parenchymal collagen deposition (Katre et al. 2011; Michaudel et al. 2018). For the present studies, we used an exposure model (0.5 ppm for 4h/d for 13 consecutive workdays), reflecting frequently encountered ambient conditions with a focus on lung parenchymal changes. The lack of fibrotic changes we observed in the C57BL/6J strain with our exposure regimen is consistent with Katre et al., where a longer exposure duration (5 cycles of , where a cycle was for 8h/d during weekdays) did not result in measurable differences in fibrosis and rather required a longer regimen (Katre et al. 2011). We suggest that the lack of fibrosis in the C57BL/6J strain in our exposure model is due to their normoglycemic state. Alternatively, it is possible that fibrosis could be observed in other normoglycemic -exposed strains, because strain variation clearly can alter phenotypic results (Kleeberger et al. 1997; Savov et al. 2004). Additional studies using multiple stains and exposure conditions will be required to confirm that hyperglycemic, insulin-resistant conditions enhance centriacinar fibrosis to air pollution.
The mechanisms driving the enhanced sensitivity to -induced remodeling in diabetes-prone strains were not clearly elucidated in this work. However, our data suggest that changes in immune polarization and baseline airway epithelial composition might contribute to the observed pathology. We found that the insulin-resistant KKAy strain had an augmented immune response defined by increased macrophages and eosinophils in the airspaces and parenchyma. This influx was associated with increased Th2 cytokines and profibrotic factors such as MMP12. Recent advances in single cell and transcriptomic analytical approaches confirm a central role of airspace and interstitial macrophages in the pathogenesis of human fibrotic lung disease and rodent experimental pulmonary fibrosis models (Aran et al. 2019; Misharin et al. 2017b; Reyfman et al. 2019; Yu et al. 2016). In particular, macrophages recruited to the lung during experimental fibrosis appeared to drive these profibrotic responses (Aran et al. 2019; Misharin et al. 2017a). Our prior work in C57BL/6J strains suggests that macrophages are not recruited to the lung following acute challenge (Birukova et al. 2019; Tighe et al. 2011). However, it is not clear whether there is macrophage recruitment with repetitive exposures and how this is modified with insulin resistance and hyperglycemia. Future studies will explore this relationship as a potential mechanism where the lung microenvironment, based on recruitment of macrophages and other immune cells, favors remodeling.
In addition to the immune cell infiltration, we also observed that diabetes-prone rodents exhibited baseline abnormalities in the cellular composition of their terminal airways. In the air-exposed strains, KK and KKAy mice had reduced numbers of CCSP-expressing airway cells when compared with C57BL/6J mice. This finding was confirmed by immunofluorescent/immunohistochemical staining, RT-PCR for CCSP and morphometry quantification of airway CCSP. In the air-exposed C57BL/6J mice, CCSP was present in the airway epithelial cells all the way to the alveolar duct junction. In -exposed C57BL/6J mice, CCSP staining persisted in the airway epithelium and appeared largely unchanged in composition and morphology. Alternatively, in the air-exposed KK and KKAy mice, the airway epithelium had lower numbers of CCSP expressing cells. Instead we observed cells lacking CCSP or SFTPC expression exhibiting a squamoid appearance. We also observed cells in the airway that appear to stain positive for SFTPC. These observations were largely unchanged in the -exposed KK and KKAy groupings, where large sections of the terminal airways remained devoid of CCSP expressing cells. The -exposed KK and KKAy mice had evidence of increased matrix deposition in the alveolar spaces adjacent to these terminal airways (as shown by hyaluronan staining) associated with areas of SFTPC-positive alveolar epithelial cells (i.e., type 2 pneumocytes). Together, these observations raise the question regarding whether diabetes caused modifications to the terminal airway structures, enhancing vulnerability of the distal airway and adjacent alveolar spaces, thereby resulting in centriacinar epithelial cell hyperplasia, inflammation, and fibrosis.
Prior research suggests potential mechanisms for how baseline alterations in terminal airway structures could result in enhanced sensitivity to . In particular, the reduction of CCSP expressing airway epithelial cells in the terminal airways. Plopper et al., using CCSP-deficient mice () exposed to (1 ppm for 8 h), observed significant injury to the airway epithelium as defined by electron microscopy (Plopper et al. 2006). In the mice, the intact cells following were noted to be very low in height, with some exhibiting a squamoid appearance. This finding was consistent with the observed phenotype in the KK and KKAy strains. Distinct from this prior research, we observed airway epithelial cell morphological alterations in the air-exposed KK and KKAy strains, whereas these were not observed in air-exposed mice. In separate studies, CCSP-null mice exposed to (various doses from for 2–8 h) exhibit increased cytokine and oxidant stress responses when compared with CCSP-sufficient mice (Johnston et al. 1999; Mango et al. 1998). This finding suggests that the loss of CCSP in the KK and KKAy strains could drive the early and exaggerated response. To the best of our knowledge, no clear links have been explored between diabetes and the CCSP. However, it is intriguing to speculate that reductions in CCSP in diabetics may be a mechanism underlying human epidemiology studies suggesting associations between diabetes (Klein et al. 2010), insulin resistance (Sagun et al. 2015), and the metabolic syndrome (Chen et al. 2014) and reduced lung function (FEV1 and FVC) and increased incidence of restrictive lung disease (Sonoda et al. 2018).
In summary, the present study demonstrated that an insulin-resistant hyperglycemic mouse strain (KKAy) developed early and exacerbated inflammation and fibrosis in response to repetitive exposure not observed in ozone-sensitive, normoglycemic C57BL/6J mice. Lesions were characterized by increased immune cell influx into areas of epithelial hyperplasia and fibrosis adjacent to terminal airspaces. Limitations include strain comparisons of genetically unrelated KK and C57 strains and the pilot characterization of parenchymal remodeling. Nevertheless, these novel findings in an animal model describe a unique interaction of air pollution, metabolic status, and pulmonary inflammation and fibrosis, and the findings propose a mechanistic framework to support the emerging epidemiological associations among air pollution, diabetes, and lung disease.
Supplementary Material
Acknowledgments
The authors appreciate funding support provided by the National Institutes of Health R01 ES013611, K08 HL105537, and R01 ES027574 (to R.M.T.); Burroughs Wellcome Fund Career Award for Medical Scientists (to C.E.B.); U.S. EPA RD834797 (J.G.W. and J.R.H.), USDA-NIFA MICL02649 (J.G.W.); and the Albert C. and Lois E. Dehn Endowment (J.R.H.). The authors thank A. Porter, A. Allen, and J. L. Neumann of the Michigan State University Laboratory for Investigative Histopathology for their assistance with the histotechnology. Author contributions: A.V. and C.E.B. performed with immunohistology of the terminal airway, interpreted the results, and assisted with drafting the manuscript. R.P.L. performed experiments and assisted with experimental design and interpretation. R.M.T., J.W., and J.R.H. conceived of the area of investigation, led studies, performed experiments, collected and analyzed data, and drafted/assisted with drafting the manuscript.
References
- Andersen ZJ, Raaschou-Nielsen O, Ketzel M, Jensen SS, Hvidberg M, Loft S, et al. 2012. Diabetes incidence and long-term exposure to air pollution: a cohort study. Diabetes Care 35(1):92–98, PMID: 22074722, 10.2337/dc11-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anenberg SC, Henze DK, Tinney V, Kinney PL, Raich W, Fann N, et al. 2018. Estimates of the global burden of ambient , ozone, and on asthma incidence and emergency room visits. Environ Health Perspect 126(10):107004, PMID: 30392403, 10.1289/EHP3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. 2019. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20(2):163–172, PMID: 30643263, 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avol EL, Linn WS, Venet TG, Shamoo DA, Hackney JD. 1984. Comparative respiratory effects of ozone and ambient oxidant pollution exposure during heavy exercise. J Air Pollut Control Assoc 34(8):804–809, PMID: 6481003, 10.1080/00022470.1984.10465814. [DOI] [PubMed] [Google Scholar]
- Bass V, Gordon CJ, Jarema KA, MacPhail RC, Cascio WE, Phillips PM, et al. 2013. Ozone induces glucose intolerance and systemic metabolic effects in young and aged brown Norway rats. Toxicol Appl Pharmacol 273(3):551–560, PMID: 24103449, 10.1016/j.taap.2013.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova A, Cyphert-Daly J, Cumming RI, Yu YR, Gowdy KM, Que LG, et al. 2019. Sex modifies acute ozone-mediated airway physiologic responses. Toxicol Sci 169(2):499–510, PMID: 30825310, 10.1093/toxsci/kfz056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandenberger C, Rowley NL, Jackson-Humbles DN, Zhang Q, Bramble LA, Lewandowski RP, et al. 2013. Engineered silica nanoparticles act as adjuvants to enhance allergic airway disease in mice. Part Fibre Toxicol 10:26, PMID: 23815813, 10.1186/1743-8977-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook RD, Xu X, Bard RL, Dvonch JT, Morishita M, Kaciroti N, et al. 2013. Reduced metabolic insulin sensitivity following sub-acute exposures to low levels of ambient fine particulate matter air pollution. Sci Total Environ 448:66–71, PMID: 22901427, 10.1016/j.scitotenv.2012.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LY, Huang Y, Stockstill BL, Graham JA, Grose EC, Menache MG, et al. 1992. Epithelial injury and interstitial fibrosis in the proximal alveolar regions of rats chronically exposed to a simulated pattern of urban ambient ozone. Toxicol Appl Pharmacol 115(2):241–252, PMID: 1641858, 10.1016/0041-008X(92)90329-Q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WL, Wang CC, Wu LW, Kao TW, Chan JY, Chen YJ, et al. 2014. Relationship between lung function and metabolic syndrome. PLoS One 9(10):e108989, PMID: 25299452, 10.1371/journal.pone.0108989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AJ, Brauer M, Burnett R, Anderson HR, Frostad J, Estep K, et al. 2017. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet 389(10082):1907–1918, PMID: 28408086, 10.1016/S0140-6736(17)30505-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coogan PF, White LF, Jerrett M, Brook RD, Su JG, Seto E, et al. 2012. Air pollution and incidence of hypertension and diabetes mellitus in black women living in Los Angeles. Circulation 125(6):767–772, PMID: 22219348, 10.1161/CIRCULATIONAHA.111.052753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromar KR, Gladson LA, Ghazipura M, Ewart G. 2018. Estimated excess morbidity and mortality associated with air pollution above American Thoracic Society-recommended Standards, 2013-2015. American Thoracic Society and Marron Institute Report. Ann Am Thorac Soc 15(5):542–551, PMID: 29425050, 10.1513/AnnalsATS.201710-785EH. [DOI] [PubMed] [Google Scholar]
- Devlin RB, McDonnell WF, Mann R, Becker S, House DE, Schreinemachers D, et al. 1991. Exposure of humans to ambient levels of ozone for 6.6 hours causes cellular and biochemical changes in the lung. Am J Respir Cell Mol Biol 4(1):72–81, PMID: 1846079, 10.1165/ajrcmb/4.1.72. [DOI] [PubMed] [Google Scholar]
- Farhat SCL, Almeida MB, Silva-Filho L, Farhat J, Rodrigues JC, Braga ALF. 2013. Ozone is associated with an increased risk of respiratory exacerbations in patients with cystic fibrosis. Chest 144(4):1186–1192, PMID: 23493973, 10.1378/chest.12-2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folinsbee LJ, Bedi JF, Horvath SM. 1984. Pulmonary function changes after 1 h continuous heavy exercise in 0.21 ppm ozone. J Appl Physiol Respir Environ Exerc Physiol 57(4):984–988, PMID: 6501039, 10.1152/jappl.1984.57.4.984. [DOI] [PubMed] [Google Scholar]
- Foster WM, Brown RH, Macri K, Mitchell CS. 2000. Bronchial reactivity of healthy subjects: 18-20 h postexposure to ozone. J Appl Physiol (1985) 89(5):1804–1810, PMID: 11053329, 10.1152/jappl.2000.89.5.1804. [DOI] [PubMed] [Google Scholar]
- Halonen JI, Lanki T, Tiittanen P, Niemi JV, Loh M, Pekkanen J. 2010. Ozone and cause-specific cardiorespiratory morbidity and mortality. J Epidemiol Community Health 64(9):814–820, PMID: 19854743, 10.1136/jech.2009.087106. [DOI] [PubMed] [Google Scholar]
- Hatch GE, McKee J, Brown J, McDonnell W, Seal E, Soukup J, et al. 2013. Biomarkers of dose and effect of inhaled ozone in resting versus exercising human subjects: comparison with resting rats. Biomark Insights 8:53–67, PMID: 23761957, 10.4137/BMI.S11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch GE, Slade R, Harris LP, McDonnell WF, Devlin RB, Koren HS, et al. 1994. Ozone dose and effect in humans and rats. A comparison using oxygen-18 labeling and bronchoalveolar lavage. Am J Respir Crit Care Med 150(3):676–683, PMID: 8087337, 10.1164/ajrccm.150.3.8087337. [DOI] [PubMed] [Google Scholar]
- Johannson KA, Vittinghoff E, Lee K, Balmes JR, Ji W, Kaplan GG, et al. 2014. Acute exacerbation of idiopathic pulmonary fibrosis associated with air pollution exposure. Eur Respir J 43(4):1124–1131, PMID: 24176998, 10.1183/09031936.00122213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannson KA, Vittinghoff E, Morisset J, Wolters PJ, Noth EM, Balmes JR, et al. 2018. Air pollution exposure is associated with lower lung function, but not changes in lung function, in patients with idiopathic pulmonary fibrosis. Chest 154(1):119–125, PMID: 29355549, 10.1016/j.chest.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston CJ, Finkelstein JN, Oberdörster G, Reynolds SD, Stripp BR. 1999. Clara cell secretory protein-deficient mice differ from wild-type mice in inflammatory chemokine expression to oxygen and ozone, but not to endotoxin. Exp Lung Res 25(1):7–21, PMID: 10027076, 10.1080/019021499270394. [DOI] [PubMed] [Google Scholar]
- Katre A, Ballinger C, Akhter H, Fanucchi M, Kim DK, Postlethwait E, et al. 2011. Increased transforming growth factor beta 1 expression mediates ozone-induced airway fibrosis in mice. Inhal Toxicol 23(8):486–494, PMID: 21689010, 10.3109/08958378.2011.584919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeberger SR, Levitt RC, Zhang LY, Longphre M, Harkema J, Jedlicka A, et al. 1997. Linkage analysis of susceptibility to ozone-induced lung inflammation in inbred mice. Nat Genet 17(4):475–478, PMID: 9398854, 10.1038/ng1297-475. [DOI] [PubMed] [Google Scholar]
- Klein OL, Krishnan JA, Glick S, Smith LJ. 2010. Systematic review of the association between lung function and Type 2 diabetes mellitus. Diabet Med 27(9):977–987, PMID: 20722670, 10.1111/j.1464-5491.2010.03073.x. [DOI] [PubMed] [Google Scholar]
- Last JA, Gelzleichter T, Harkema J, Parks WC, Mellick P. 1993. Effects of 20 months of ozone exposure on lung collagen in Fischer 344 rats. Toxicology 84(1–3):83–102, PMID: 8266340, 10.1016/0300-483x(93)90110-e. [DOI] [PubMed] [Google Scholar]
- Liu C, Bai Y, Xu X, Sun L, Wang A, Wang TY, et al. 2014. Exaggerated effects of particulate matter air pollution in genetic type II diabetes mellitus. Part Fibre Toxicol 11:27, PMID: 24886175, 10.1186/1743-8977-11-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mango GW, Johnston CJ, Reynolds SD, Finkelstein JN, Plopper CG, Stripp BR. 1998. Clara cell secretory protein deficiency increases oxidant stress response in conducting airways. Am J Physiol 275(2):L348–356, PMID: 9700096, 10.1152/ajplung.1998.275.2.L348. [DOI] [PubMed] [Google Scholar]
- Medina-Ramón M, Zanobetti A, Schwartz J. 2006. The effect of ozone and PM10 on hospital admissions for pneumonia and chronic obstructive pulmonary disease: a national multicity study. Am J Epidemiol 163(6):579–588, PMID: 16443803, 10.1093/aje/kwj078. [DOI] [PubMed] [Google Scholar]
- Michaudel C, Fauconnier L, Julé Y, Ryffel B. 2018. Functional and morphological differences of the lung upon acute and chronic ozone exposure in mice. Sci Rep 8(1):10611, PMID: 30006538, 10.1038/s41598-018-28261-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DB, Snow SJ, Henriquez A, Schladweiler MC, Ledbetter AD, Richards JE, et al. 2016. Systemic metabolic derangement, pulmonary effects, and insulin insufficiency following subchronic ozone exposure in rats. Toxicol Appl Pharmacol 306:47–57, PMID: 27368153, 10.1016/j.taap.2016.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. 2017a. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 214(8):2387–2404, PMID: 28694385, 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. 2017b, PMID: 28694385, 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orru H, Andersson C, Ebi KL, Langner J, Aström C, Forsberg B. 2013. Impact of climate change on ozone-related mortality and morbidity in Europe. Eur Respir J 41(2):285–294, PMID: 22743679, 10.1183/09031936.00210411. [DOI] [PubMed] [Google Scholar]
- Plopper CG, Mango GW, Hatch GE, Wong VJ, Toskala E, Reynolds SD, et al. 2006. Elevation of susceptibility to ozone-induced acute tracheobronchial injury in transgenic mice deficient in Clara cell secretory protein. Toxicol Appl Pharmacol 213(1):74–85, PMID: 16226776, 10.1016/j.taap.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Que LG, Stiles JV, Sundy JS, Foster WM. 2011. Pulmonary function, bronchial reactivity, and epithelial permeability are response phenotypes to ozone and develop differentially in healthy humans. J Appl Physiol (1985) 111(3):679–687, PMID: 21700892, 10.1152/japplphysiol.00337.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao X, Patel P, Puett R, Rajagopalan S. 2015. Air pollution as a risk factor for type 2 diabetes. Toxicol Sci 143(2):231–241, PMID: 25628401, 10.1093/toxsci/kfu250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. 2019. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med 199(12):1517–1536, PMID: 30554520, 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagun G, Gedik C, Ekiz E, Karagoz E, Takir M, Oguz A. 2015. The relation between insulin resistance and lung function: a cross sectional study. BMC Pulm Med 15:139, PMID: 26542243, 10.1186/s12890-015-0125-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savov JD, Whitehead GS, Wang J, Liao G, Usuka J, Peltz G, et al. 2004. Ozone-induced acute pulmonary injury in inbred mouse strains. Am J Respir Cell Mol Biol 31(1):69–77, PMID: 14975936, 10.1165/rcmb.2003-0001OC. [DOI] [PubMed] [Google Scholar]
- Sesé L, Nunes H, Cottin V, Sanyal S, Didier M, Carton Z, et al. 2018. Role of atmospheric pollution on the natural history of idiopathic pulmonary fibrosis. Thorax 73(2):145–150, PMID: 28798214, 10.1136/thoraxjnl-2017-209967. [DOI] [PubMed] [Google Scholar]
- Sonoda N, Morimoto A, Tatsumi Y, Asayama K, Ohkubo T, Izawa S, et al. 2018. A prospective study of the impact of diabetes mellitus on restrictive and obstructive lung function impairment: the Saku study. Metabolism 82:58–64, PMID: 29288691, 10.1016/j.metabol.2017.12.006. [DOI] [PubMed] [Google Scholar]
- Stockstill BL, Chang LY, Ménache MG, Mellick PW, Mercer RR, Crapo JD. 1995. Bronchiolarized metaplasia and interstitial fibrosis in rat lungs chronically exposed to high ambient levels of ozone. Toxicol Appl Pharmacol 134(2):251–263, PMID: 7570602, 10.1006/taap.1995.1191. [DOI] [PubMed] [Google Scholar]
- Sun Q, Yue P, Deiuliis JA, Lumeng CN, Kampfrath T, Mikolaj MB, et al. 2009. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation 119(4):538–546, PMID: 19153269, 10.1161/CIRCULATIONAHA.108.799015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tighe RM, Li Z, Potts EN, Frush S, Liu N, Gunn MD, et al. 2011. Ozone inhalation promotes CX3CR1-dependent maturation of resident lung macrophages that limit oxidative stress and inflammation. J Immunol 187(9):4800–4808, PMID: 21930959, 10.4049/jimmunol.1101312. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Wang Y, Tan M, Ouyang H, Deng L. 2018. Effects of ozone stimulation of bronchial epithelial cells on proliferation and collagen synthesis of co-cultured lung fibroblasts. Exp Ther Med 15(6):5314–5322, PMID: 29896220, 10.3892/etm.2018.6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Reich BJ, Nolte CG, Spero TL, Hubbell B, Rappold AG. 2017. Climate change impacts on projections of excess mortality at 2030 using spatially varying ozone-temperature risk surfaces. J Expo Sci Environ Epidemiol 27(1):118–124, PMID: 27005744, 10.1038/jes.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Liu C, Xu Z, Tzan K, Zhong M, Wang A, et al. 2011. Long-term exposure to ambient fine particulate pollution induces insulin resistance and mitochondrial alteration in adipose tissue. Toxicol Sci 124(1):88–98, PMID: 21873646, 10.1093/toxsci/kfr211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Allen K, Zhong J, Chen M, Williams KM, Wagner JG, et al. 2016. Subacute inhalation exposure to ozone induces systemic inflammation but not insulin resistance in a diabetic mouse model. Inhal Toxicol 28(4):155–163, PMID: 26986950, 10.3109/08958378.2016.1146808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YR, Hotten DF, Malakhau Y, Volker E, Ghio AJ, Noble PW, et al. 2016. Flow cytometric analysis of myeloid cells in human blood, bronchoalveolar lavage, and lung tissues. Am J Respir Cell Mol Biol 54(1):13–24, PMID: 26267148, 10.1165/rcmb.2015-0146OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Allen K, Rao X, Ying Z, Braunstein Z, Kankanala SR, et al. 2016. Repeated ozone exposure exacerbates insulin resistance and activates innate immune response in genetically susceptible mice. Inhal Toxicol 28(9):383–392, PMID: 27240593, 10.1080/08958378.2016.1179373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.